2.1. Horseradish Peroxidase As a Reporter
The first reporter for endo/lysosomal escape that was investigated in this study was horseradish peroxidase (HRP). For this purpose, saporin (recombinantly expressed and purified by Ni- nitrilotriacetic acid (NTA) chromatography; see Figure S1 in the Supplementary) was chemically conjugated to HRP via a covalent linkage, and the reaction mixture was analysed by SDS-PAGE (
Figure 1A). A diffused band with high molecular mass was identified by the reaction mixture in the gel. This band in all probability contained conjugates of saporin-HRP with different molecular ratios of saporin to HRP. Furthermore, a band with the same molecular mass as HRP was observed, while no unconjugated saporin was detected in the reaction mixture.
To remove unconjugated HRP, the reaction mixture was purified by Ni-NTA chromatography. The flow-through, wash and elution fractions were analysed by SDS-PAGE (
Figure 1B). Unconjugated HRP that did not contain the 6× his-tag was directly eluted in the flow-through and during the washing steps. A certain amount of saporin-HRP was eluted in the flow-through and subsequent washing steps, apparently because the cross-linking reaction of saporin to HRP resulted in the partial masking of the 6× his-tag and prevented the affinity binding of a subset of saporin-HRP to the column. However, saporin-HRP with accessible 6× his-tag bound strongly to the column and was eluted in Fractions 1–3 at 62 mM imidazole (see arrow in
Figure 1B). Fractions containing purified saporin-HRP were separately dialyzed and concentrated. The total yield for saporin-HRP was around 1.4 mg of conjugate, and the relative outcome was 14% of the protein input (saporin plus HRP).
Figure 1.
Chemical conjugation, purification and characterization of saporin-HRP. (A) Cross-linkage of saporin and HRP. The reaction mixture was directly analysed by SDS-PAGE. Saporin and HRP served as unconjugated controls; (B) Purification of saporin-HRP by Ni-NTA chromatography with increasing concentrations of imidazole in the elution buffer. All fractions were assessed by SDS-PAGE. Saporin-HRP was eluted in Fractions 1–3 at 62 mM imidazole (see arrow); (C) Validation of the chemical conjugation of saporin and HRP. Saporin-HRP was analysed by SDS-PAGE, and the conjugate was visualized in the gel (see arrow). Saporin-HRP was analysed by Western blot with a primary polyclonal antibody against saporin. Saporin and the conjugate (see arrow) were specifically detected in the membrane; (D) Peroxidase activity of saporin-HRP. A directly proportional correlation between absorbance and concentration was observed from 0.1 to 10 nM saporin-HRP. Each data point is the mean ± SD, n = 3; Comparison of the cytotoxicity of (E) saporin and (F) saporin-HRP in the presence of SA1641. ECV-304 cells (4000 cells/well) were treated with saporin or saporin-HRP in a concentration range from 0.01 to 100 nM alone or in combination with SA1641 (final concentration of 5 µg/mL) for 48 h. Cell viability was determined by the MTT assay. Data represents the mean ± SD, n = 4.
Figure 1.
Chemical conjugation, purification and characterization of saporin-HRP. (A) Cross-linkage of saporin and HRP. The reaction mixture was directly analysed by SDS-PAGE. Saporin and HRP served as unconjugated controls; (B) Purification of saporin-HRP by Ni-NTA chromatography with increasing concentrations of imidazole in the elution buffer. All fractions were assessed by SDS-PAGE. Saporin-HRP was eluted in Fractions 1–3 at 62 mM imidazole (see arrow); (C) Validation of the chemical conjugation of saporin and HRP. Saporin-HRP was analysed by SDS-PAGE, and the conjugate was visualized in the gel (see arrow). Saporin-HRP was analysed by Western blot with a primary polyclonal antibody against saporin. Saporin and the conjugate (see arrow) were specifically detected in the membrane; (D) Peroxidase activity of saporin-HRP. A directly proportional correlation between absorbance and concentration was observed from 0.1 to 10 nM saporin-HRP. Each data point is the mean ± SD, n = 3; Comparison of the cytotoxicity of (E) saporin and (F) saporin-HRP in the presence of SA1641. ECV-304 cells (4000 cells/well) were treated with saporin or saporin-HRP in a concentration range from 0.01 to 100 nM alone or in combination with SA1641 (final concentration of 5 µg/mL) for 48 h. Cell viability was determined by the MTT assay. Data represents the mean ± SD, n = 4.
![Toxins 06 01644 g001]()
Although chemical conjugation was already validated by the specific binding of the conjugate (containing the 6× his-tag in the saporin moiety) to the Ni-NTA agarose and detection of the high molecular mass conjugate after SDS-PAGE (see
Figure 1B), validation of the cross-linking reaction was confirmed again by western blot with a primary polyclonal antibody against saporin (
Figure 1C). Both saporin (intense band in the unconjugated control) and saporin-HRP (slight band) were specifically detected. Unconjugated saporin was not detected in the conjugate sample.
To determine if the peroxidase activity of HRP had been affected by the chemical conjugation process and to define the sensitivity of the conjugate, peroxidase activity of saporin-HRP was measured at a series of concentrations ranging from 0.00001 to 100 nM (
Figure 1D). In the lower concentrations (0.00001 to 0.01 nM), the enzymatic activity of saporin-HRP was not detectible. The first detectable signals in the presence of saporin-HRP appeared at concentrations between 0.01 and 0.1 nM of the conjugate. Then, a linear correlation between the absorbance and concentration of the conjugate was observed in the range from 0.1 to 10 nM saporin-HRP. Finally, for concentrations higher than 10 nM, a saturation of the absorbance signal was recorded.
The N-glycosidase activity of saporin-HRP was compared to that of saporin to investigate if the enzymatic activity of the toxic moiety had been influenced by the chemical conjugation process. Saporin-HRP released 46.3 pmol adenine/pmol toxin/h after incubation with herring sperm DNA. Since the adenine release of saporin was calculated to 117.6 pmol adenine/pmol toxin/h, the N-glycosidase activity of saporin-HRP was reduced to 39.3%. However, saporin-HRP still presented enzymatic activity and was therefore suitable for further studies.
The effect of triterpenoidal saponins (SA1641) on the enhancement of the endo/lysosomal escape of saporin-HRP was compared to that on saporin on ECV-304 cells. In the case of saporin (
Figure 1E), the toxin alone showed cytotoxic effects and caused a reduction of the viability to 61% at a concentration of 100 nM. In combination with SA1641 (5 µg/mL), saporin cytotoxicity was drastically enhanced, causing a reduction of cell viability to 54% at a concentration of 0.1 nM.
In the case of saporin-HRP (
Figure 1F), no cytotoxicity was shown up to a concentration of 100 nM. However, in the presence of SA1641, saporin-HRP exerted cytotoxicity at the two highest concentrations tested: cell viability was reduced to 51% at 10 nM and to 16% at 100 nM. It is important to note that the cytotoxicity enhancement also took place when saporin was chemically coupled to HRP, therefore indicating that the endo/lysosomal escape of saporin in combination with triterpenoidal saponins was coherent, even after coupling to the reporter, and that saporin has a general potential as a molecular drag.
In order to measure the specific augmentation of the endo/lysosomal release of saporin-HRP mediated by triterpenoidal saponins, cells were first treated either with saporin-HRP (100 nM) or with the combination of saporin-HRP and SA1641 (5 µg/mL). After an incubation time of 6 h and cell fractionation, the endo/lysosomal release of the toxin-reporter conjugate was evaluated by measuring peroxidase activity in the cytosolic fraction and in the lysosomal fraction. The ratio of the peroxidase activity in the cytosolic fraction and the peroxidase activity in the lysosomal fraction (CF/LF) is proportional to the relative amount of saporin-HRP released to the cytosol.
Unfortunately, the CF/LF value mirrored contradictory results in repeated experiments. Peroxidase activity detected in the cytosolic fractions was very close to the detection limit (A450–490 nm = 0.018–0.030), and therefore, small variations in the absorbance values greatly influenced the CF/LF ratio. In one example, the CF/LF value of the cells treated with SA1641 (0.082) was higher than that of the cells treated without the triterpenoidal saponin (0.072). However, in another example, the CF/LF value of the cells treated with SA1641 (0.107) was lower than that of the cells treated without the triterpenoidal saponin (0.151). In short, the measurements close to the detection limit resulted in a bad reproducibility and prevented the precise calculation of ratios representing the endo/lysosomal escape of saporin-HRP in the presence or absence of triterpenoidal saponins.
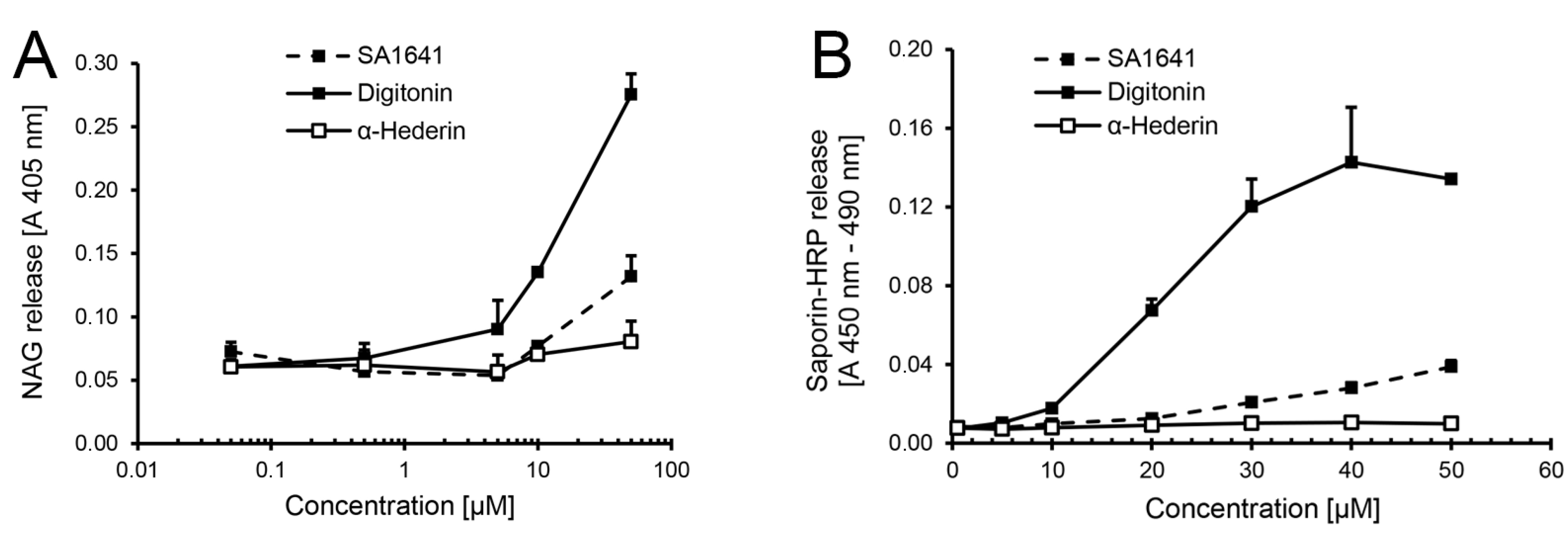
In an alternative approach, the endo/lysosomal escape of saporin-HRP was evaluated in isolated organelles preloaded with the conjugate. To determine the highest non-permeabilizing concentration of triterpenoidal saponins, short-term permeabilizing effects of SA1641 on lysosomal membranes from isolated organelles were measured using the lysosomal enzyme β
-N-acetylglucosaminidase (NAG) release assay (
Figure 2A). In the case of digitonin (positive control, highly lytic saponin isolated from
Digitalis purpurea L.), slight membrane permeabilizing effects on the lysosomal membrane were first observed at a concentration of 5 µM (6.15 µg/mL). The permeabilizing effects of digitonin progressively increased as the concentration was augmented to 50 µM (61.47 µg/mL). In the case of α-hederin (negative control, non-lytic saponin isolated from
Hedera helix L.), no membrane permeabilizing effects were observed on the lysosomal membrane. Even at the highest concentration tested, only a very slight release of NAG was measured. SA1641 had substantial membrane permeabilizing effects at a concentration of 50 µM (82.05 µg/mL). However, at lower concentrations, SA1641 did not present any permeabilizing effects on lysosomal membranes, and the absorbance values were comparable to those of α-hederin.
In the case of a specific augmentation of the endo/lysosomal release of saporin-HRP in the presence of SA1641, higher amounts of the conjugate are expected to be released at low non-membrane permeabilizing concentrations of SA1641 (lower than 50 µM or 82.05 µg/mL). In such a case, saporin-HRP will be released from the lysosomes, due to a specific interaction with SA1641.
Nevertheless, no such specific release of saporin-HRP was observed in the presence of SA1641 (
Figure 2B). Although cytotoxicity enhancement is already observed at 5 µg/mL of SA1641 (
Figure 1F), no specific endo/lysosomal release of saporin-HRP was observed at this concentration. The endo/lysosomal release of saporin-HRP increased slightly as the concentration was augmented to 30, 40 and 50 µM (49.23, 64.64 and 82.05 µg/mL, respectively), most probably attributed to unspecific membrane permeabilizing effects. In the case of digitonin, the high endo/lysosomal release of saporin-HRP was shown at 30, 40 and 50 µM (36.88, 49.17 and 61.47 µg/mL, respectively), due to disruption of lysosomal membranes. In the case of α-hederin, no endo/lysosomal release of saporin-HRP was observed at all the concentrations tested. Regrettably, the reporter assay for endo/lysosomal escape using HRP is not suitable to quantify the endo/lysosomal release of protein-based therapeutics.
Figure 2.
HRP as a reporter for endo/lysosomal escape in isolated organelles. Lysosomes were isolated by cell fractionation form ECV-304 cells (3 × 10
7 cells). (
A) First, the permeabilizing effects of SA1641 on the lysosomal membranes were evaluated by the β
-N-acetylglucosaminidase (NAG) release assay.The permeabilizing effects of digitonin (highly lytic saponin) and α-hederin (non-lytic saponin) were simultaneously determined as controls; (
B) Thereafter, the endo/lysosomal escape enhancement of saporin-HRP observed in the cytotoxicity assay in
Figure 1F was tried, to monitor in isolated lysosomes loaded with saporin-HRP (from cells previously treated with the conjugate at 100 nM and 37 °C for 6 h) by determination of the peroxidase activity. Saponin concentrations are shown in µM to allow a better comparison. Each data point represents the mean ± SD,
n = 3.
Figure 2.
HRP as a reporter for endo/lysosomal escape in isolated organelles. Lysosomes were isolated by cell fractionation form ECV-304 cells (3 × 10
7 cells). (
A) First, the permeabilizing effects of SA1641 on the lysosomal membranes were evaluated by the β
-N-acetylglucosaminidase (NAG) release assay.The permeabilizing effects of digitonin (highly lytic saponin) and α-hederin (non-lytic saponin) were simultaneously determined as controls; (
B) Thereafter, the endo/lysosomal escape enhancement of saporin-HRP observed in the cytotoxicity assay in
Figure 1F was tried, to monitor in isolated lysosomes loaded with saporin-HRP (from cells previously treated with the conjugate at 100 nM and 37 °C for 6 h) by determination of the peroxidase activity. Saponin concentrations are shown in µM to allow a better comparison. Each data point represents the mean ± SD,
n = 3.
It is known from previous studies that the release of diphtheria toxin’s catalytic (C) domain from endosomes into the cytosol requires a host cell cytosolic translocation factor complex, which consists of the chaperone heat shock protein 90 (Hsp 90) and thioredoxin reductase [
18]. Furthermore, host cell Hsp 90 is also essential for the translocation of
Clostridium botulinum C2 toxin from endosomes into the cytosol [
19] and for the transfer of the cholera toxin A1 subunit from the endoplasmic reticulum to the cytosol [
20]. Similarly, since a certain amount of saporin-HRP must definitively escape from the endo/lysosomes in the presence of triterpenoidal saponins, because cytotoxicity was observed (see the cytotoxicity enhancement at 3.05 µM or 5 µg/mL of SA1641 in
Figure 1F), a possible explanation for the lack of saporin-HRP release from the isolated organelles (see saporin-HRP release at 5 µM or 8.21 µg/mL SA1641 in
Figure 2B) is the necessity of a certain cytosolic machinery to efficiently mediate the specific endo/lysosomal release of saporin (or saporin conjugates, such as saporin-HRP) in the presence of triterpenoidal saponins. An alternative hypothesis is that the amount of saporin-HRP that escapes from the isolated endo/lysosomes is so low that its measurement remains under the limit of detection, but it is high enough to cause cell death.
2.2. Alexa Fluor 488 As a Reporter
The second reporter that was investigated was Alexa Fluor 488. Recombinantly expressed saporin was labelled with Alexa Fluor 488, and the conjugate (
Alexasaporin) was administered to cells in the presence of SA1641 to visualize the endo/lysosomal escape of saporin by live cell imaging (
Figure 3).
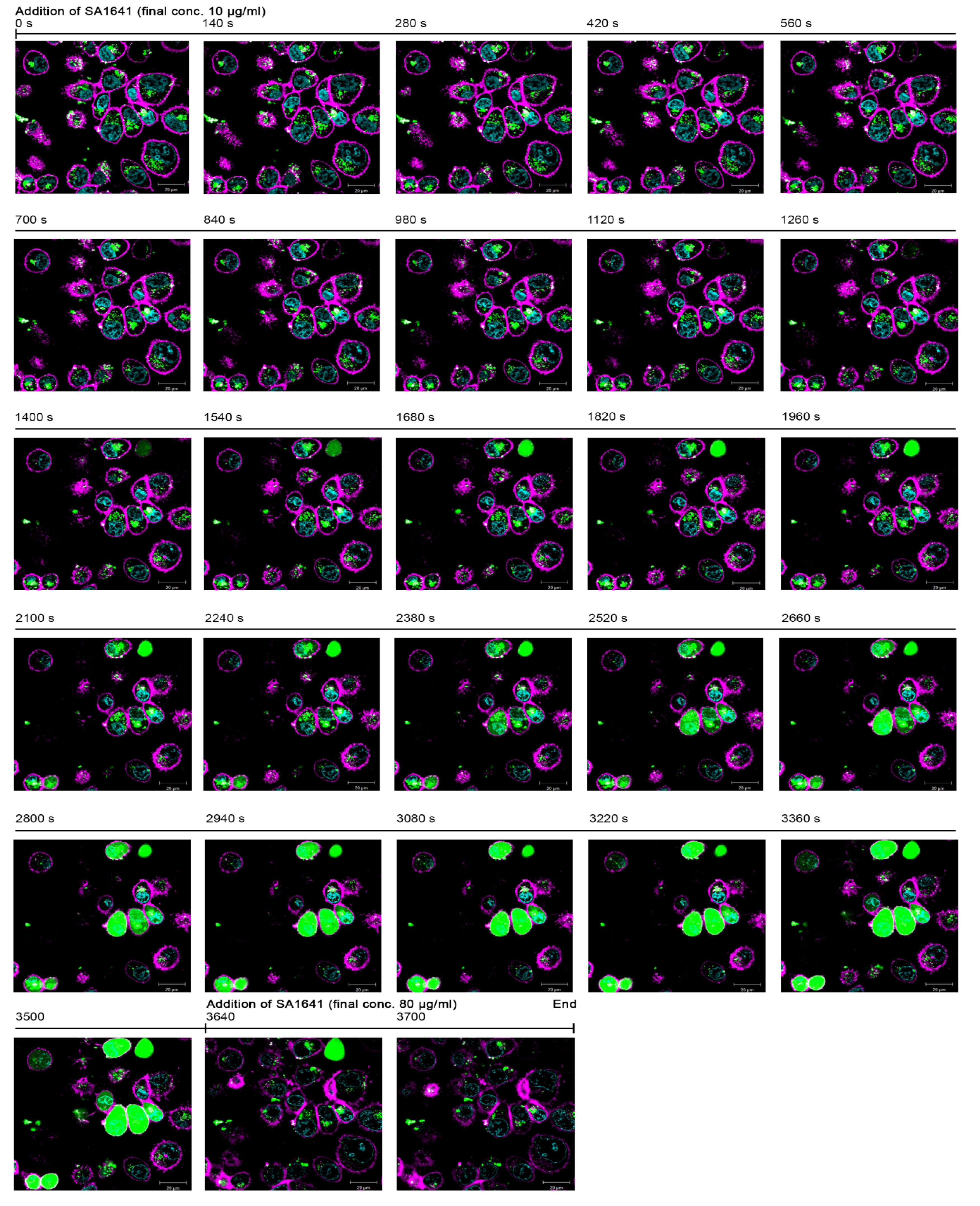
After the incubation of cells with
Alexasaporin for 3 h, only the accumulation of the labelled protein (emitting green fluorescence) was observed inside the cell, but no endo/lysosomal escape was detected. Cells treated with
Alexasaporin under the same conditions for 6 h indicated that the toxin accumulates in acidic organelles, namely late endosomes and lysosomes, but is not released to the cytosol [
13]. Furthermore, in the present experiment,
Alexasaporin did presumably not have any cytotoxic effects in the short incubation time of 3 h, because the morphology of the cells was intact. At that moment (t = 0 s), triterpenoidal SA1641 was added to the cells to a final concentration of 10 µg/mL, and initially, no changes were observed. However, after 1400 s of addition of SA1641, a diffusion of green fluorescence started to become visible in a cell at the top right of the fluorescence microscopy picture. This diffusion was limited to the intracellular space and was associated with the endo/lysosomal escape of
Alexasaporin.
Within the next 300 s (from t = 1400 s to t = 1680 s), the intensity of fluorescence in the cell increased rapidly and reached a saturation point, indicating a complete release of
Alexasaporin into the cytosol. Furthermore, after 2100 s, fluorescence diffusion appeared in two more cells: one at the top centre and another one at the bottom left of the fluorescence image. After 2800 s, the fluorescence diffusion was observed in seven cells, and after 3500 s, a total of nine cells exhibited diffused fluorescence in the cytosol. It is remarkable that the diffusion of the fluorescence occurred only in the intracellular space, but did not cross the cell membrane, indicating that SA1641 did not cause unspecific cell membrane permeabilization, but specifically mediated the endo/lysosomal escape of
Alexasaporin. Only after the addition of SA1641 at a high concentration (80 µg/mL), cell membranes were unspecifically permeabilized, and fluorescence diffusion disappeared quickly, due to the leakage of
Alexasaporin from the cytosol to the extracellular space. A video comprising the whole sequence of images is available in the Supplementary (Video S1). The single cell analysis derived from the fluorescence images in the Video S1, but not the fluorescence images themselves, was previously published [
13].
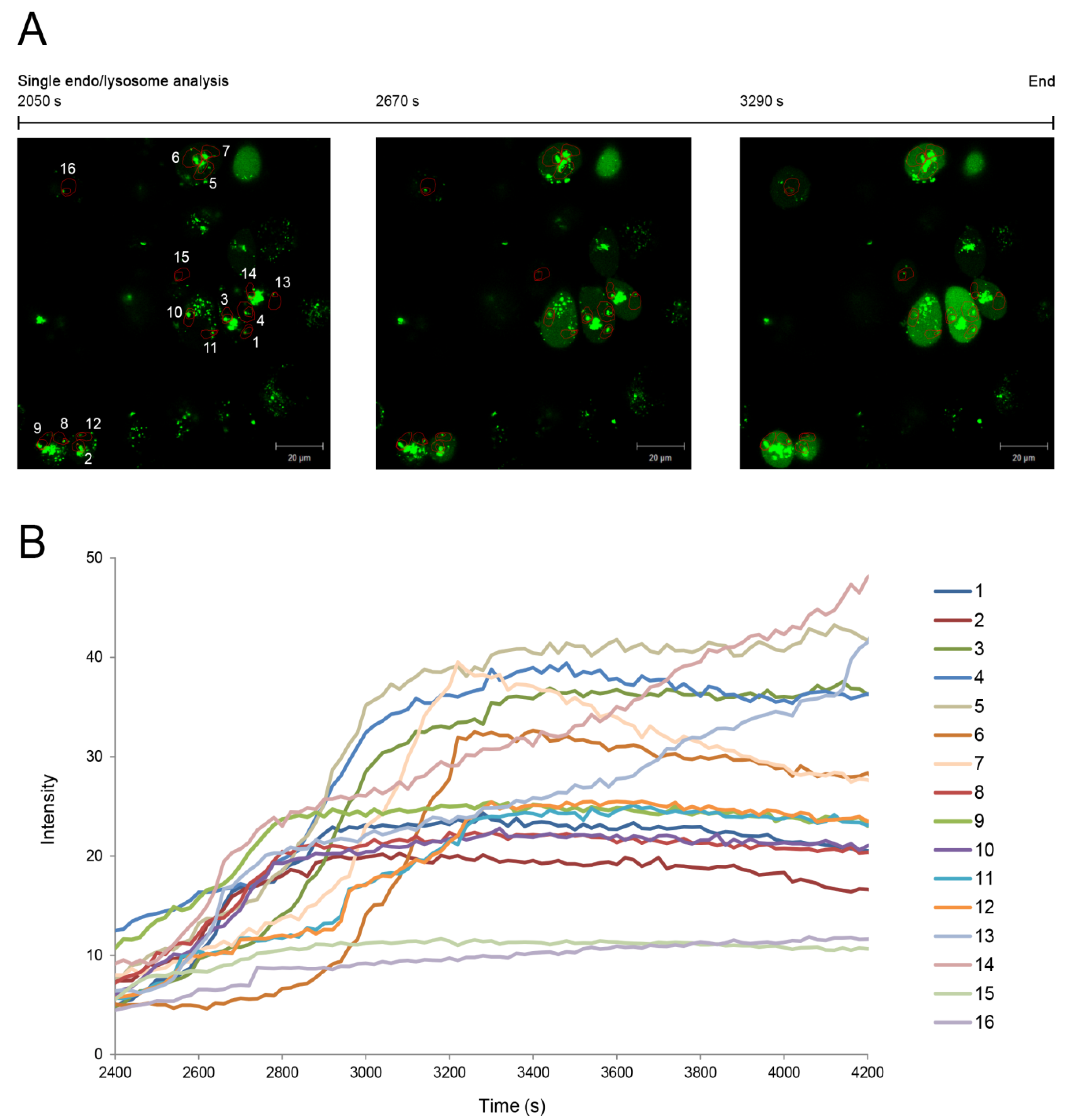
In addition, the endo/lysosomal escape was analysed in this study for 16 single endo/lysosomes identified in the fluorescence images from 2050 s to 3290 s at a final concentration of 10 µg/mL (
Figure 4A). Regions of interest were defined for each endo/lysosome, and the amount of green fluorescence diffusion that corresponds to the released amount of
Alexasaporin was individually evaluated in the case of each endo/lysosome. Furthermore, in a second experiment, where cells were monitored for 7200 s after the addition of SA1641, the fluorescence intensity of
Alexasaporin released from individual endo/lysosomes was plotted against the time (from t = 2400 s to t = 4200 s) (
Figure 4B). A curve for each of the 16 endo/lysosomes was obtained and every curve presented a particular behaviour, showing the differences in the release rate of
Alexasaporin from one endo/lysosome to another.
Figure 3.
Alexa Fluor 488 as a reporter for endo/lysosomal escape in live cell imaging. The report of the naturally occurring phenomenon of the endo/lysosomal escape enhancement of saporin (in this case Alexasaporin) was assayed by constant measurement of fluorescence. ECV-304 cells were incubated with 1000 nM Alexasaporin at 37 °C for 3 h. Afterwards, most of the internalized toxin accumulated in intracellular vesicles. Alexasaporin is visualized in green fluorescence, cell membranes in magenta and cell nuclei in cyan. The addition of 10 µg/mL SA1641 (non-cytotoxic concentration) was defined as t = 0 s. Green colouring of the cells reflects the release of Alexasaporin (from t = 1400 s to t = 3500 s). To finish the experiment, SA1641 was added (t = 3640 s) to a final concentration of 80 µg/mL (cytotoxic concentration), which results in total cell membrane disruption (from t = 3640 s to t = 3700 s).
Figure 3.
Alexa Fluor 488 as a reporter for endo/lysosomal escape in live cell imaging. The report of the naturally occurring phenomenon of the endo/lysosomal escape enhancement of saporin (in this case Alexasaporin) was assayed by constant measurement of fluorescence. ECV-304 cells were incubated with 1000 nM Alexasaporin at 37 °C for 3 h. Afterwards, most of the internalized toxin accumulated in intracellular vesicles. Alexasaporin is visualized in green fluorescence, cell membranes in magenta and cell nuclei in cyan. The addition of 10 µg/mL SA1641 (non-cytotoxic concentration) was defined as t = 0 s. Green colouring of the cells reflects the release of Alexasaporin (from t = 1400 s to t = 3500 s). To finish the experiment, SA1641 was added (t = 3640 s) to a final concentration of 80 µg/mL (cytotoxic concentration), which results in total cell membrane disruption (from t = 3640 s to t = 3700 s).
Figure 4.
Single endo/lysosome analysis. The endo/lysosomal escape of
Alexasaporin in the presence of SA1641 was monitored in the case of single endo/lysosomes. (
A) ECV-304 cells were incubated with
Alexasaporin at 1000 nM and 37 °C for 3 h, washed, and then, 10 µg/mL SA1641 were added to cells as indicated in
Figure 3.
AlexaSaporin is visualized in green fluorescence. A total of 16 single endo/lysosomes was selected and constantly monitored from t = 2050 s to t = 3290 s. For each endo/lysosome, a region of interest (numbered 1–16 in the fluorescence picture) comprised of the surroundings of the endo/lysosomes, but excluding the vesicle itself, was defined; (
B) ECV-304 cells were continuously monitored for 7200 s after the addition of 10 µg/mL of SA1641. Single endo/lysosome analysis is presented as a relation of time (from t = 2400 s to t = 4200 s)
vs. the fluorescence intensity of the released
Alexasaporin. Each curve represents the fluorescence intensity increase in each region of interest, corresponding to the amount of
Alexasaporin that escapes from each single endo/lysosome.
Figure 4.
Single endo/lysosome analysis. The endo/lysosomal escape of
Alexasaporin in the presence of SA1641 was monitored in the case of single endo/lysosomes. (
A) ECV-304 cells were incubated with
Alexasaporin at 1000 nM and 37 °C for 3 h, washed, and then, 10 µg/mL SA1641 were added to cells as indicated in
Figure 3.
AlexaSaporin is visualized in green fluorescence. A total of 16 single endo/lysosomes was selected and constantly monitored from t = 2050 s to t = 3290 s. For each endo/lysosome, a region of interest (numbered 1–16 in the fluorescence picture) comprised of the surroundings of the endo/lysosomes, but excluding the vesicle itself, was defined; (
B) ECV-304 cells were continuously monitored for 7200 s after the addition of 10 µg/mL of SA1641. Single endo/lysosome analysis is presented as a relation of time (from t = 2400 s to t = 4200 s)
vs. the fluorescence intensity of the released
Alexasaporin. Each curve represents the fluorescence intensity increase in each region of interest, corresponding to the amount of
Alexasaporin that escapes from each single endo/lysosome.
![Toxins 06 01644 g004]()
While
Figure 1E demonstrates that saporin at 1000 nM in combination with SA1641 produces close to 100% total cell death, in
Figure 3, only a portion of cells treated by saporin at the same concentration in the presence of SA1641 displays the cytosolic fluorescence indicative of the toxin release to the cytosol. This discrepancy may be related to the timing of the experiments and also to the detection limits of the techniques.
Firstly, regarding the timing of the experiments, results presented in
Figure 1E refer to cells treated for 48 h, whereas results in
Figure 3 represent cells treated first with saporin alone for 3 h and thereafter with the combination of saporin and SA1641 for 2 h more. It is therefore expected that a major number of cells will be affected in the first case, since cells have been in contact with both compounds for a longer period of time.
Secondly, concerning the detection limits, the sensitivity of the cytotoxicity assay reaches much lower concentrations than that of confocal microscopy. In
Figure 1E, cytotoxicity is observed down to a concentration of 0.1 nM. However, the fluorescence diffusion of toxins after endo/lysosomal escape is clearly observed only at 1000 nM (see
Figure 3). When an immunotoxin consisting of saporin chemically coupled to the therapeutic antibody, Trastuzumab (Herceptin
®), was labelled with Alexa Fluor 488, its endo/lysosomal escape was also observed at a toxin concentration of 100 nM, but the diffusion of fluorescence to the cytosol was not as clear as in the case of
Alexasaporin at 1000 nM [
16]. To sum up, the effects of saporin are detected in a major number of cells in the cytotoxicity assay, also probably due to the higher sensitivity of the method.
Alexa Fluor 488 was previously used as a reporter for endo/lysosomal escape of saporin in the presence of triterpenoidal saponins. First, cells were treated with
Alexasaporin in combination with SA1641 [
13]. Later,
Alexasaporin was administered to cells in the presence of SO1861 [
21]. In both cases, the endo/lysosomal escape of the toxin was observed at a toxin concentration of 1000 nM. Based on the previous experiments, the limit of detection with the reporter system for endo/lysosomal escape based on Alexa Fluor 488 is approximated to 100–1000 nM. A novelty of the present study is the single endo/lysosome analysis of cells incubated with
Alexasaporin (also at 1000 nM). To summarize the results of this section, a concentration of 1000 nM was a suitable concentration for single-endo/lysosome analysis and for the usage of Alexa Fluor 488 as a reporter for the endo/lysosomal escape of protein-based therapeutics.
2.3. Ricin A-Chain As a Reporter
The third reporter for the endo/lysosomal escape considered in this investigation was the ricin A-chain (RTA). In order to study the usage of RTA as a reporter, it was chemically conjugated to an enzymatically inactive variant of saporin (saporin-KQ), previously reported in the literature [
17]. Saporin-KQ was cloned into an expression vector, and the presence of the insert was confirmed by DNA sequencing. Then, saporin-KQ was heterologously expressed and purified by Ni-NTA chromatography (Figure S2A in the Supplementary). The identity of saporin-KQ after recombinant expression and purification was substantiated by Western blot with a primary polyclonal antibody against saporin (Figure S2B, Supplementary).
After chemical conjugation of saporin-KQ and RTA, a first diffused band with an approximate molecular mass of 60 kDa and a second diffused band with high molecular mass were observed in the reaction mixture by SDS-PAGE (
Figure 5A). The first band contained most likely one single saporin-KQ coupled to a single molecule of RTA. In contrast, the second band may probably contain conjugates of saporin-KQ-RTA with different molecular ratios of saporin-KQ conjugated to RTA. Furthermore, a band of approximately 30 kDa was detected, as well, in the reaction mixture, but since saporin-KQ and RTA have a very similar molecular mass, the presence of either one or the other protein cannot be distinguished.
To purify the saporin-KQ-RTA conjugates from the unconjugated saporin-KQ and unconjugated RTA, Ni-NTA chromatography was carried out, and all the fractions were analysed by SDS-PAGE (
Figure 5B). On the one hand, unconjugated RTA was directly eluted in the flow-through, as it lacks the 6× his-tag. On the other hand, unconjugated saporin-KQ (presenting the 6× his-tag) was eluted in Fractions 1–5 at 62 mM and 1–2 at 125 mM imidazole. In the case of saporin-KQ-RTA (also presenting the 6× his-tag), the conjugate was eluted in Fractions 5 at 125 mM and 1–5 at 250 mM imidazole. All fractions that contained the purified conjugate were pooled together, desalted and concentrated. The final amount of saporin-KQ-RTA was 0.26 mg of conjugate, and the relative yield of the chemical conjugation and subsequent purification was 18% of protein input (saporin-KQ plus RTA).
In order to check that saporin-KQ was enzymatically inactive, the
N-glycosidase activity of saporin was compared to that of saporin-KQ. In the case of saporin, the release of adenine after incubation with herring sperm DNA was 117.6 pmol adenine/pmol toxin/h (see
Section 2.1). However, in the case of saporin-KQ, no adenine release was observed after the incubation of the inactive mutant with the substrate. In brief, the enzymatic inactivity of saporin-KQ was confirmed.
The effect of triterpenoidal saponins (SO1861 at 1–2 µg/mL) on the enhancement of the endo/lysosomal escape of saporin-KQ was compared to that on saporin and RTA. Saporin alone presented cytotoxicity at a concentration of 100 nM and reduced the viability to 76% (
Figure 5C). In combination with SO1861, the cytotoxicity of saporin was tremendously augmented, and cell viability was reduced to 53% at a concentration of 0.1 nM. These results are comparable to those obtained for the combination of saporin and SA1641 in
Figure 1E. Both triterpenoidal saponins (SA1641 and SO1861) have been reported for the specific enhancement of the endo/lysosomal escape of saporin (SA1641 in [
13] and SO1861 in [
21]), and in both cases, this phenomenon has been elucidated by cytotoxicity assays, as well as by confocal microscopy. The endo/lysosomal escape of saporin mediated by both triterpenoidal saponins is consequently based on the same molecular mechanism, and this allows the comparison between the different reporters presented in this study.
In the case of RTA (
Figure 5D), the toxin alone was already very cytotoxic at 1 and 10 nM (cell viability of 31% and 14%, respectively). RTA at a final concentration of 0.1 nM was also cytotoxic, and cell viability was reduced to 73%. When RTA was applied in combination with SO1861, the toxin presented very similar cytotoxic effects as before with no significant differences. The drastic cytotoxicity augmentation of saporin in the presence of triterpenoidal saponins was not observed for RTA. These results are in accordance with the literature that indicates that SO1861 specifically modulates the intracellular trafficking of saporin [
13] and that RTA and saporin follow different intracellular routes to enter the cytosol [
22].
Figure 5.
Ricin A-chain (RTA) as a reporter for endo/lysosomal escape. The report of the endo/lysosomal escape enhancement of saporin-KQ (an enzymatically inactive saporin in order to avoid interferences in the readout of the experiment) was attempted by measuring the cytotoxic activity of RTA. (A) Saporin-KQ and RTA were chemically cross-linked. The reaction mixture was analysed by SDS-PAGE, and both unconjugated saporin-KQ and RTA were included as controls; (B) Saporin-KQ-RTA was purified by Ni-NTA chromatography. Proteins were eluted with increasing concentrations of imidazole (31, 62, 125 and 250 mM), and an SDS-PAGE analysis was effectuated for the fractions.Saporin-KQ-RTA was eluted in Fractions 5 at 125 mM and 1–5 at 250 mM imidazole (see arrows); Cytotoxicity of (C) saporin; (D) RTA; (E) saporin-KQ; and (F) saporin-KQ-RTA in combination with SO1861. ECV-304 (4000 cells/well) were treated with saporin, RTA, saporin-KQ or saporin-KQ-RTA in a concentration range from 0.01 to 1000 nM in the presence or absence of SO1861 (final concentration of 1–2 µg/mL) for 48 h. The viability of cells was evaluated by the MTT assay. Data is the mean ± SD, n = 4.
Figure 5.
Ricin A-chain (RTA) as a reporter for endo/lysosomal escape. The report of the endo/lysosomal escape enhancement of saporin-KQ (an enzymatically inactive saporin in order to avoid interferences in the readout of the experiment) was attempted by measuring the cytotoxic activity of RTA. (A) Saporin-KQ and RTA were chemically cross-linked. The reaction mixture was analysed by SDS-PAGE, and both unconjugated saporin-KQ and RTA were included as controls; (B) Saporin-KQ-RTA was purified by Ni-NTA chromatography. Proteins were eluted with increasing concentrations of imidazole (31, 62, 125 and 250 mM), and an SDS-PAGE analysis was effectuated for the fractions.Saporin-KQ-RTA was eluted in Fractions 5 at 125 mM and 1–5 at 250 mM imidazole (see arrows); Cytotoxicity of (C) saporin; (D) RTA; (E) saporin-KQ; and (F) saporin-KQ-RTA in combination with SO1861. ECV-304 (4000 cells/well) were treated with saporin, RTA, saporin-KQ or saporin-KQ-RTA in a concentration range from 0.01 to 1000 nM in the presence or absence of SO1861 (final concentration of 1–2 µg/mL) for 48 h. The viability of cells was evaluated by the MTT assay. Data is the mean ± SD, n = 4.
![Toxins 06 01644 g005]()
The cytotoxicity of saporin-KQ was also evaluated alone or in combination with SO1861 to assure that the enzymatically inactive saporin was non-cytotoxic (
Figure 5E). According to its mutation, which turns saporin-KQ into an enzymatically inactive protein, saporin-KQ did not present any cytotoxicity up to 1000 nM. Nevertheless, in the presence of SO1861, saporin-KQ caused the inhibition of cell growth at the two highest concentrations tested (presumably due to known mechanisms unrelated to enzymatic activity; see below), and cell viability was reduced to 39% (100 nM) and 23% (1000 nM), indicating an endo/lysosomal escape enhancement of saporin-KQ in the presence of triterpenoidal saponins. As cells cannot be killed by the enzymatic activity of saporin-KQ (inactive mutant), it is hypothesized that saporin-KQ induces caspase-dependent apoptosis via the mitochondrial or intrinsic pathway, independently of translation inhibition [
23].
Finally, the cytotoxicity of saporin-KQ-RTA was evaluated in the presence of SO1861 to find out whether the endo/lysosomal escape of saporin-KQ was efficiently reported by RTA (
Figure 5F). When saporin-KQ-RTA was tested alone, it was cytotoxic only at the highest concentration tested (100 nM), reducing cell viability to 59%. In contrast, in the presence of SO1861, saporin-KQ-RTA was cytotoxic already at a concentration of 10 nM lowering cell viability to 65% and also at 100 nM, decreasing cell viability to 34%. Remarkably, the cytotoxicity of saporin-KQ-RTA was enhanced in the presence of triterpenoidal saponins. Considering that the cytotoxicity of the reporter RTA is not enhanced by SO1861 (see
Figure 5D) and that the cytotoxic effects of saporin-KQ in the presence of SO1861 only appear with concentrations higher than 100 nM (see also
Figure 5E), the cytotoxicity enhancement of saporin-KQ-RTA at 10 nM mediated by SO1861 can only be explained by the endo/lysosomal escape enhancement of the saporin-KQ moiety and the cytotoxicity of the RTA reporter once the conjugate has reached the cytosol. Thus, the reporter assay based on RTA was able to detect the endo/lysosomal escape with a sensitivity down to 10 nM.
Theoretically, since it has been reported that the presence of a single molecule of ricin in the cytosol is enough to trigger apoptosis and thus kill a cell [
24], the strategy of using RTA as a reporter would allow the achievement of sensitivities even lower than 10 nM. From the present investigations, it can be certainly concluded that RTA is only an appropriate reporter for protein therapeutics affected by structurally-specific triterpenoidal saponins. However, we hypothesize that RTA could be further used as an endo/lysosomal escape reporter for protein therapeutics that are affected by compounds (endo/lysosomal escape enhancers) that do not cause any effects on RTA and for proteins therapeutics that achieve the endo/lysosomal escape in concentrations lower than the cytotoxicity of RTA alone (<0.1 nM, see
Figure 5D). This methodology confers the possibility of detecting small quantitative differences, which only take place at very low concentrations, in the endo/lysosomal escape of a protein-based therapeutic under different conditions.


 and
and 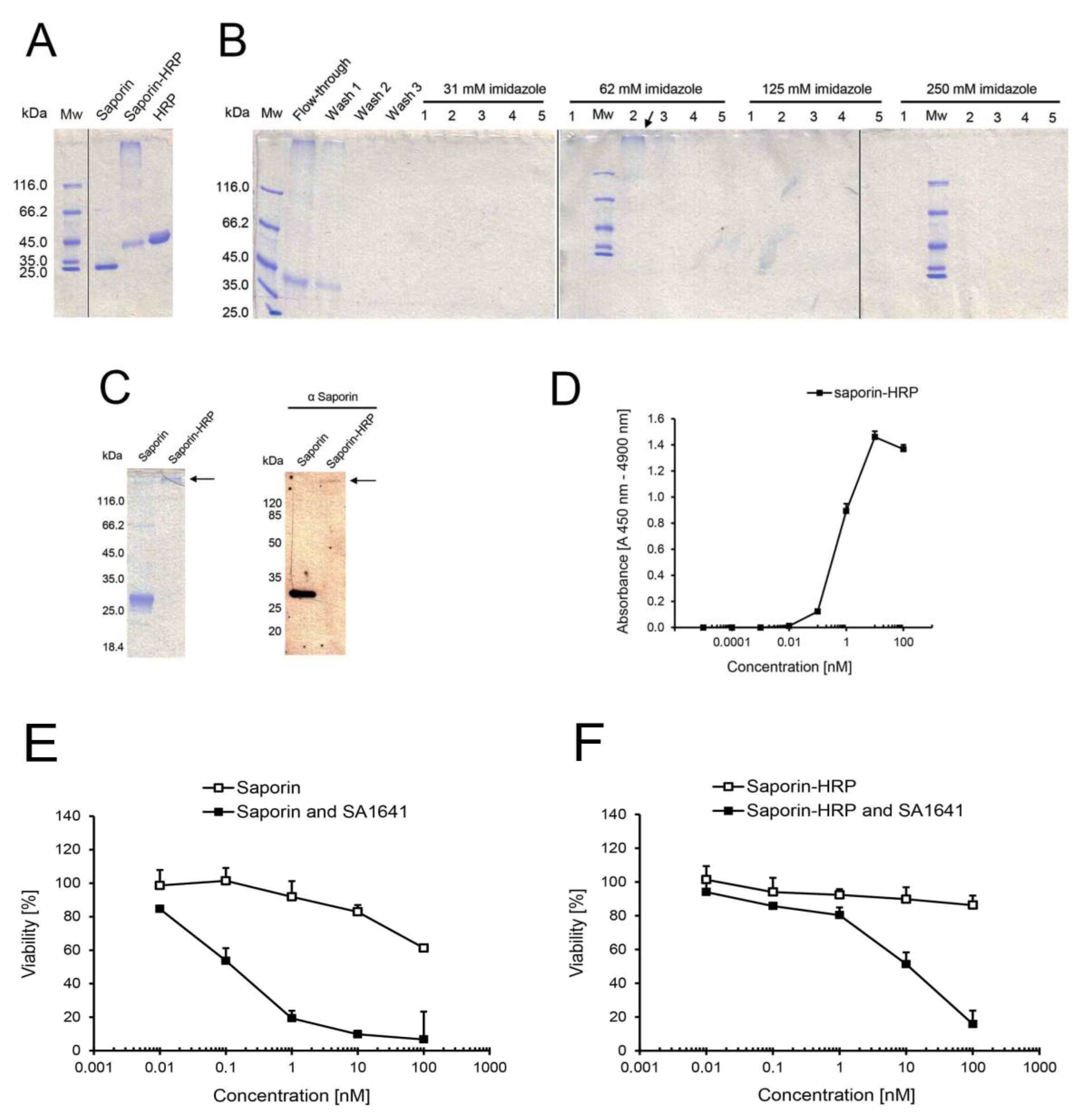{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




