
Modified Lipids and Lipoproteins in Chronic Kidney Disease: A New Class of Uremic Toxins

Abstract
:1. Introduction
2. Uremic Lipoproteins, Evidences of Toxicity
2.1. Dyslipidemia in CKD, A Unique Phenotype
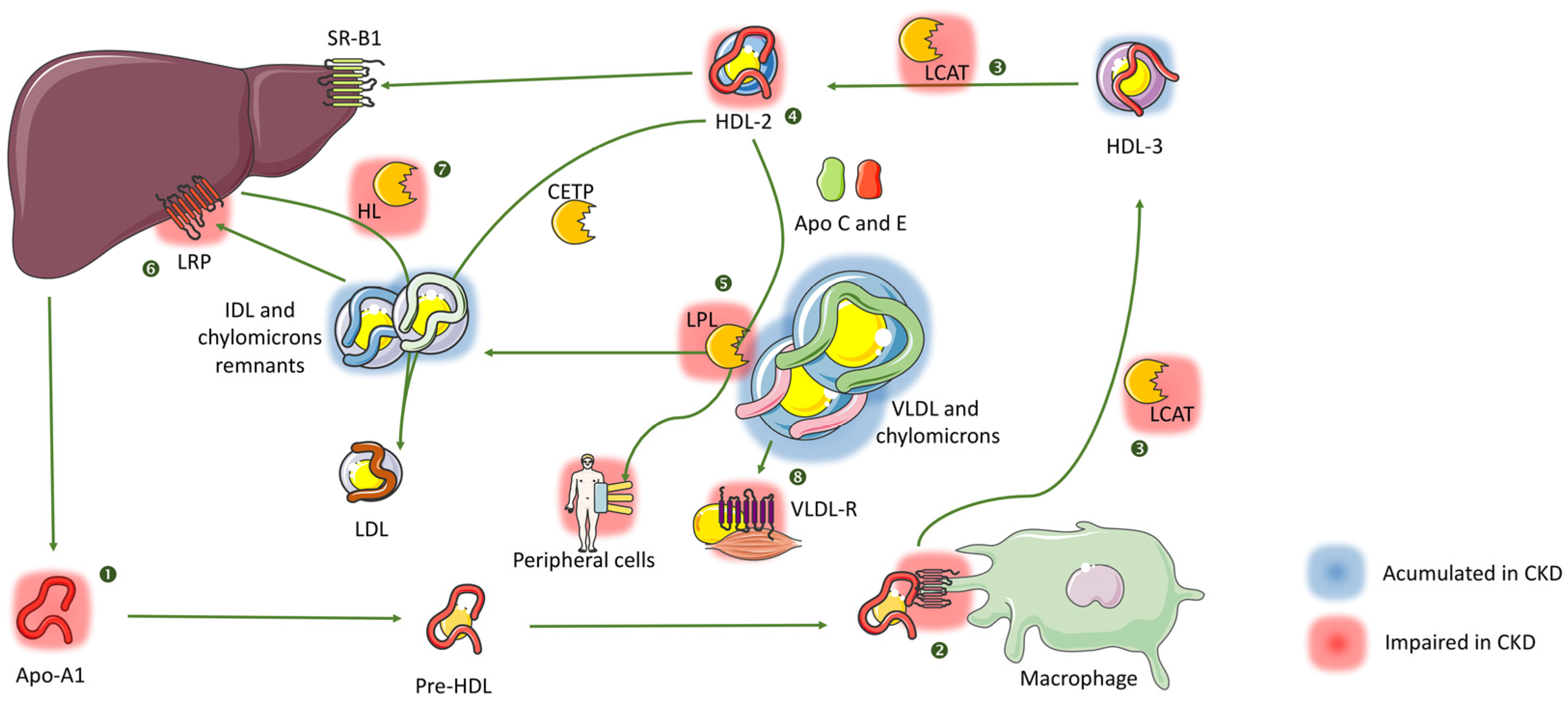
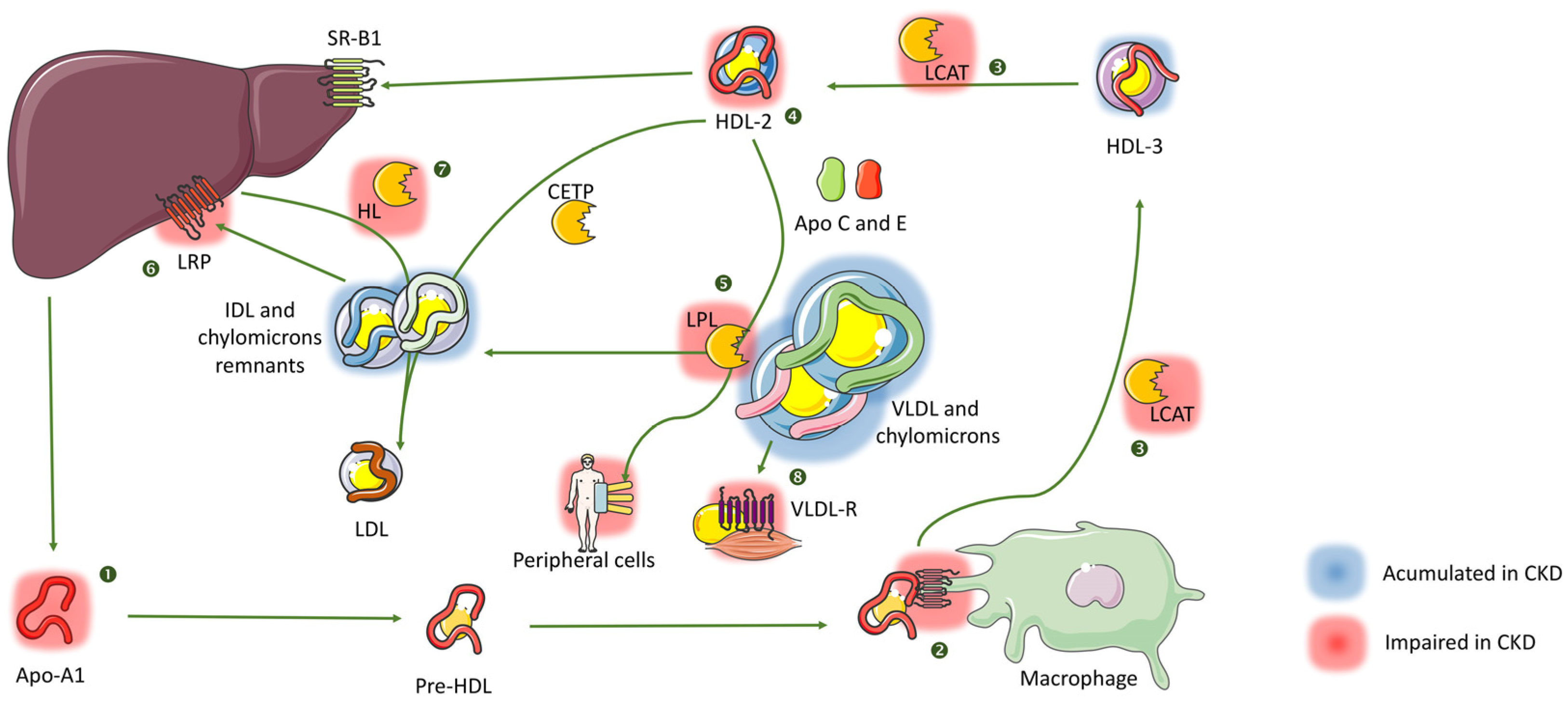
2.2. Very Low Density, Intermediate Density Lipoproteins (VLDL, IDL) and Chylomicrons
2.3. Low Density Lipoproteins (LDL)
2.4. High Density Lipoproteins (HDL)
2.5. Lipoprotein A (Lp(a))
3. Oxidative Stress/Non-Oxidative Modifications of Lipids and Lipoproteins in CKD
3.1. Oxidative Stress, Lipid Peroxidation and Antioxidant Defenses
3.2. Oxidative Stress in CKD
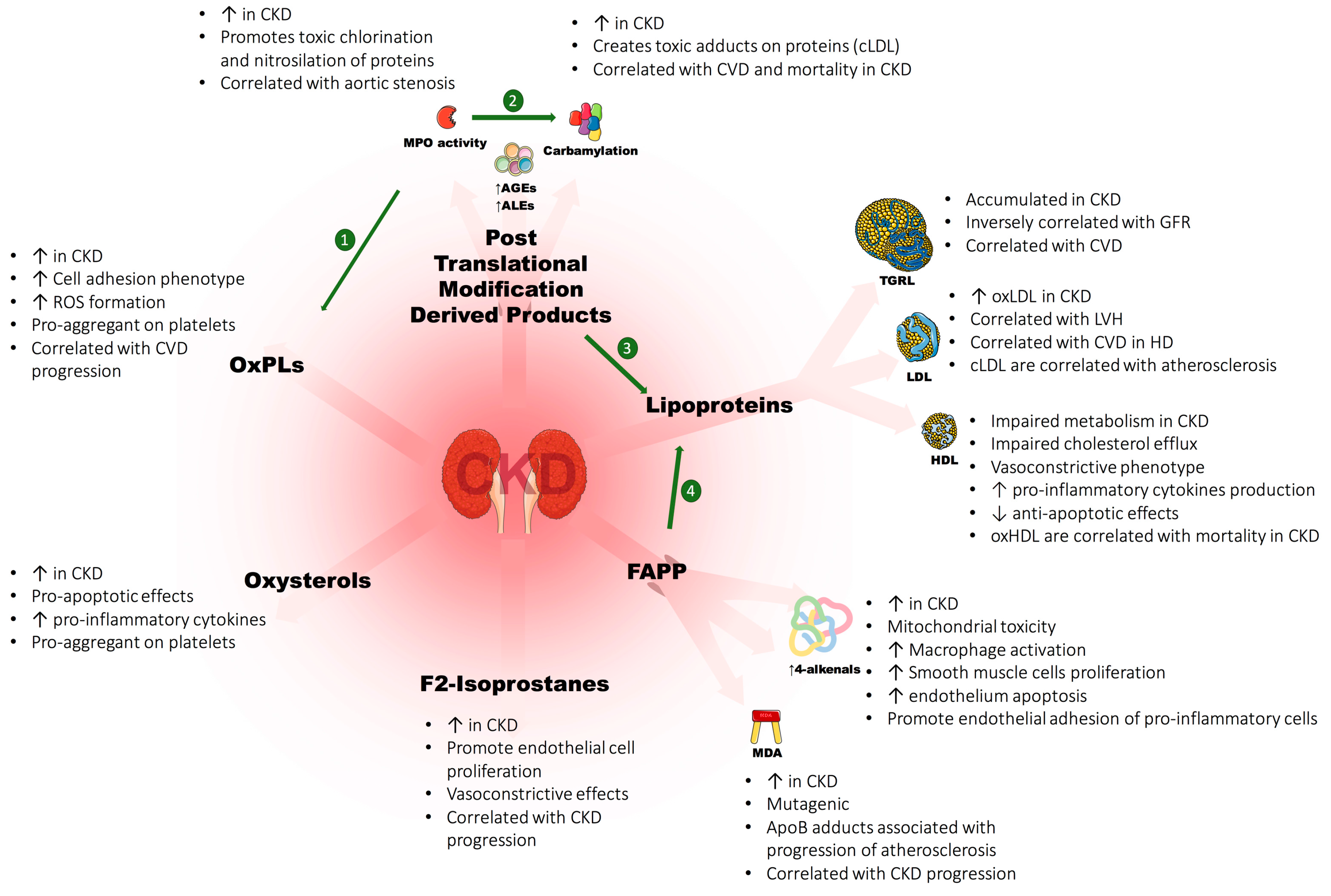
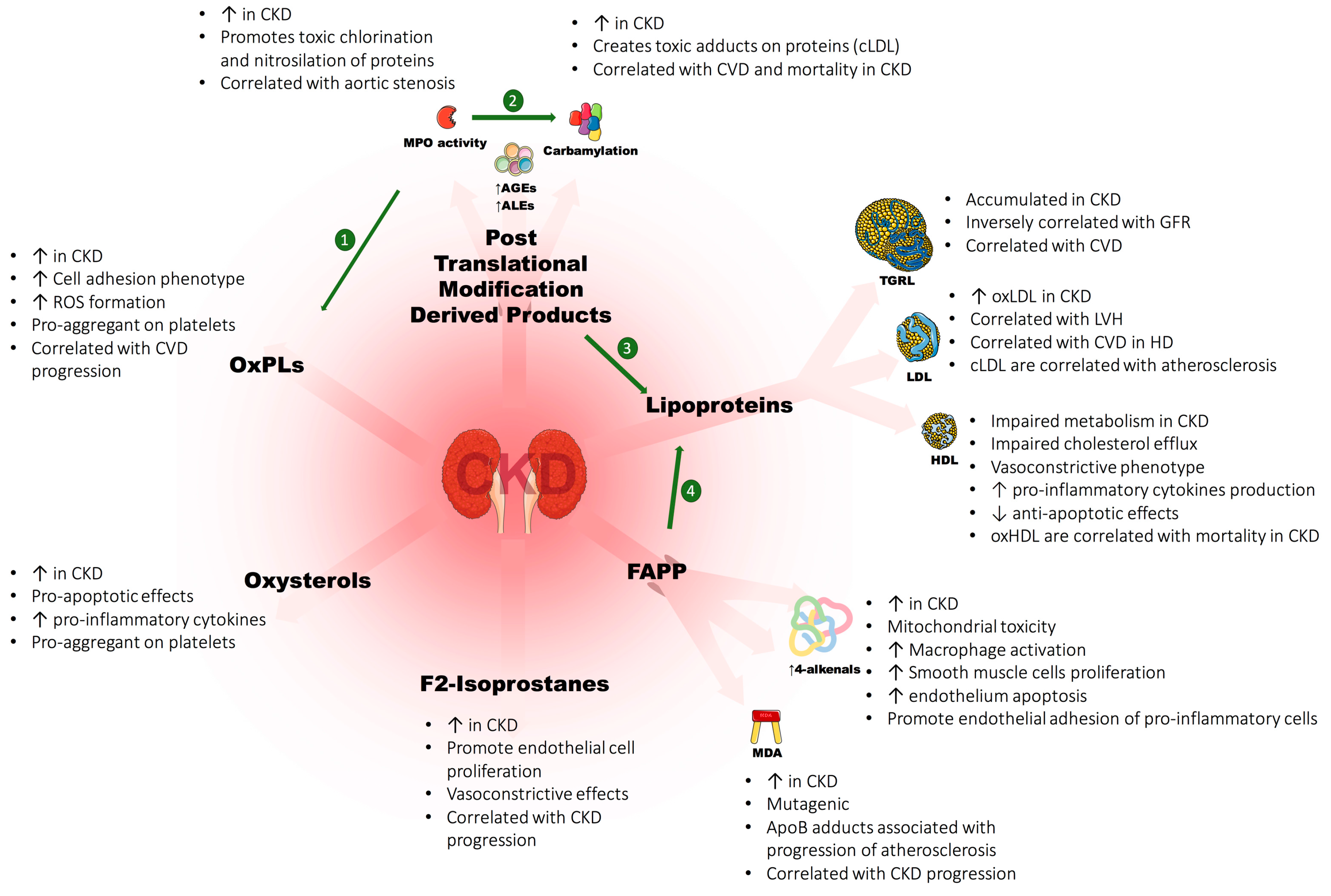
3.3. Post-Translational Modification Derived Products (PTMDPs) of Lipoprotein and Covalent Modifications of Lipids
4. Oxidized Lipids in CKD: Evidences of Toxicity
4.1. Cholesterol and Oxysterols
4.2. Oxidized Phospholipids
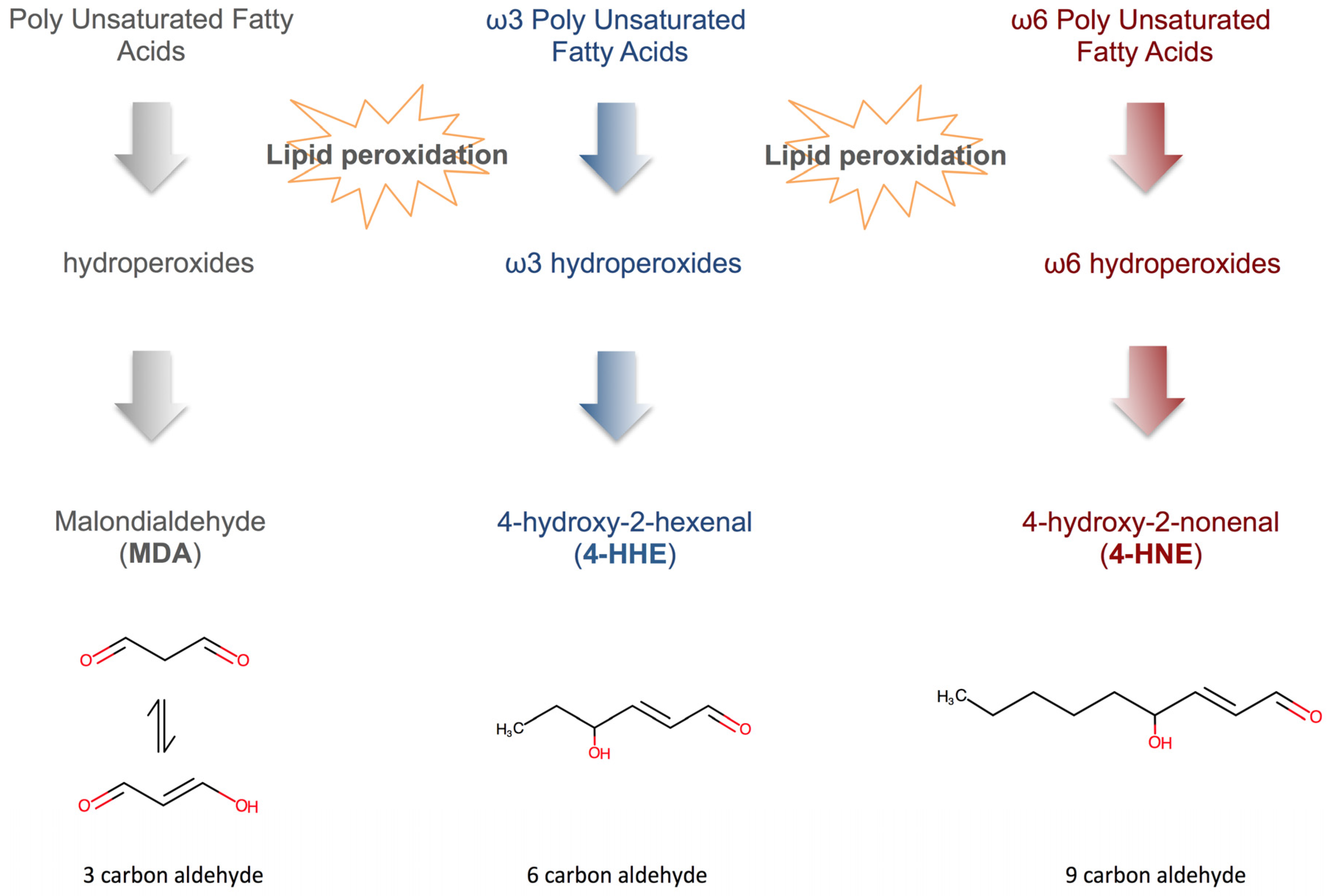
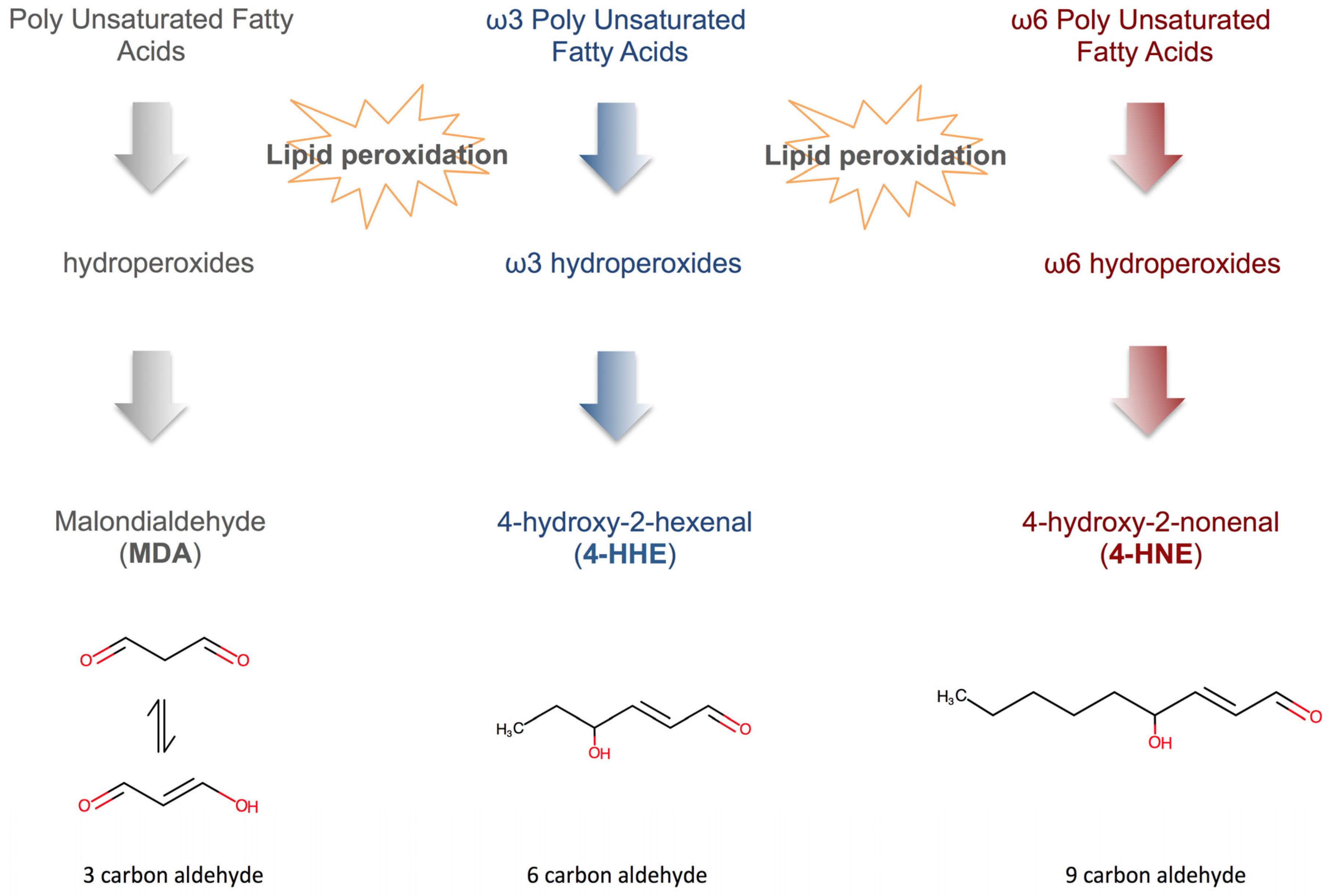
4.3. Fatty Acid Peroxidation Products (FAPP)
4.4. F2-Isoprostanes
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ABCA-1 | ATP-biding cassette 1 |
| ACAT | acetyl-CoA acetyltransferase-1 |
| AGEs | advanced glycation end-products |
| ALEs | advanced lipoxidation end-products |
| ApoA/B/C/E | apolipoprotein A/B/C/E |
| CETP | cholesterol-ester transfer protein |
| CKD | chronic kidney disease |
| cLDL | carbamylated low-density lipoprotein |
| CV | cardiovascular |
| CVD | cardiovascular disease |
| ESRD | end-stage renal disease |
| EuTox | European uremic toxin work group |
| FAPPs | fatty acid peroxidation products |
| GFR | glomerular filtration rate |
| GPX | gluthatione peroxidase |
| HD | hemodialysis |
| HDL | high density lipoproteins |
| 4-HNE | 4-hydroxy-2-nonenal |
| 4-HHE | 4-hydroxy-2-hexenal |
| HOCL | hypochlorous acid |
| ICAM-1 | intercellular adhesion molecule 1 |
| IL-1ß | interleukine 1ß |
| LCAT | lecithin-cholesterol acyltransferase |
| LCFA | long chain fatty acids |
| LDL | low-density lipoproteins |
| LOX-1 | lectin-like oxidized lox density receptor 1 |
| Lp(a) | lipoprotein a |
| LRP | LDL receptor protein |
| LVH | left ventricle hypertrophy |
| MCP-1 | monocyte chemoattractant protein 1 |
| MDA | malondialdehyde |
| MPO | myeloperoxidase |
| NO | nitric oxide |
| oxPLs | oxidized phospholipids |
| oxLDL | oxidized low-density lipoproteins |
| PLs | phospholipids |
| PON1 | paraoxonase 1 |
| PTMDPs | post translational modification derived products |
| PUFAs | polyunsaturated fatty acids |
| RAS | renin-angiotensin system |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| SOD | superoxide dismutase |
| SR-B1 | scavenger receptor class B member 1 |
| TBARS | thiobarbituric acid reactive species |
| TGRL | triglyceride-rich lipoproteins |
| TNF-α | tumor necrosis factor α |
| TxA2-R | thromboxane A2 receptor |
| VCAM-1 | vascular cell adhesion molecule 1 |
References
- Go, A.S.; Chertow, G.M.; Fan, D.; McCulloch, C.E.; Hsu, C.-Y. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N. Engl. J. Med. 2004, 351, 1296–1305. [Google Scholar] [CrossRef] [PubMed]
- McCullough, P.A.; Agrawal, V.; Danielewicz, E.; Abela, G.S. Accelerated atherosclerotic calcification and Monckeberg’s sclerosis: A continuum of advanced vascular pathology in chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2008, 3, 1585–1598. [Google Scholar] [CrossRef] [PubMed]
- Schiffrin, E.L.; Lipman, M.L.; Mann, J.F.E. Chronic kidney disease: Effects on the cardiovascular system. Circulation 2007, 116, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Cozzolino, M.; Brancaccio, D.; Gallieni, M.; Slatopolsky, E. Pathogenesis of vascular calcification in chronic kidney disease. Kidney Int. 2005, 68, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Moe, S.M.; Chen, N.X. Pathophysiology of vascular calcification in chronic kidney disease. Circ. Res. 2004, 95, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Kovesdy, C.P.; Trivedi, B.K.; Anderson, J.E. Association of kidney function with mortality in patients with chronic kidney disease not yet on dialysis: A historical prospective cohort study. Adv. Chronic Kidney Dis. 2006, 13, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Muntner, P.; He, J.; Astor, B.C.; Folsom, A.R.; Coresh, J. Traditional and nontraditional risk factors predict coronary heart disease in chronic kidney disease: Results from the atherosclerosis risk in communities study. J. Am. Soc. Nephrol. 2005, 16, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Dounousi, E.; Papavasiliou, E.; Makedou, A.; Ioannou, K.; Katopodis, K.P.; Tselepis, A.; Siamopoulos, K.C.; Tsakiris, D. Oxidative stress is progressively enhanced with advancing stages of CKD. Am. J. Kidney Dis. 2006, 48, 752–760. [Google Scholar] [CrossRef] [PubMed]
- Duranton, F.; Cohen, G.; De Smet, R.; Rodriguez, M.; Jankowski, J.; Vanholder, R.; Argiles, A.; on behalf of the European Uremic Toxin Work Group. Normal and pathologic concentrations of uremic toxins. J. Am. Soc. Nephrol. 2012, 23, 1258–1270. [Google Scholar] [CrossRef] [PubMed]
- Cottone, S.; Mulè, G.; Guarneri, M.; Palermo, A.; Lorito, M.C.; Riccobene, R.; Arsena, R.; Vaccaro, F.; Vadalà, A.; Nardi, E.; et al. Endothelin-1 and F2-isoprostane relate to and predict renal dysfunction in hypertensive patients. Nephrol. Dial. Transplant. 2009, 24, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D. Role of dyslipidemia in impairment of energy metabolism, oxidative stress, inflammation and cardiovascular disease in chronic kidney disease. Clin. Exp. Nephrol. 2014, 18, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Wanner, C.; Krane, V.; März, W.; Olschewski, M.; Mann, J.F.E.; Ruf, G.; Ritz, E.; German Diabetes and Dialysis Study Investigators. Atorvastatin in patients with type 2 diabetes mellitus undergoing hemodialysis. N. Engl. J. Med. 2005, 353, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Fellström, B.C.; Jardine, A.G.; Schmieder, R.E.; Holdaas, H.; Bannister, K.; Beutler, J.; Chae, D.-W.; Chevaile, A.; Cobbe, S.M.; Grönhagen-Riska, C.; et al. Rosuvastatin and cardiovascular events in patients undergoing hemodialysis. N. Engl. J. Med. 2009, 360, 1395–1407. [Google Scholar] [CrossRef] [PubMed]
- Barter, P. Lipoprotein metabolism and CKD: Overview. Clin. Exp. Nephrol. 2014, 18, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Norris, K. Lipid disorders and their relevance to outcomes in chronic kidney disease. Blood Purif. 2011, 31, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Massy, Z.A. The role of lipids and uremic toxins in cardiovascular disease in CKD. Clin. Exp. Nephrol. 2014, 18, 255–256. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D. HDL abnormalities in nephrotic syndrome and chronic kidney disease. Nat. Rev. Nephrol. 2015, 12, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Keane, W.F.; Tomassini, J.E.; Neff, D.R. Lipid Abnormalities in Patients with Chronic Kidney Disease: Implications for the Pathophysiology of Atherosclerosis. J. Atheroscler. Thromb. 2013, 20, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Maduell, F.; Moreso, F.; Pons, M.; Ramos, R.; Mora-Macia, J.; Carreras, J.; Soler, J.; Torres, F.; Campistol, J.M.; Martinez-Castelao, A.; et al. High-efficiency postdilution online hemodiafiltration reduces all-cause mortality in hemodialysis patients. J. Am. Soc. Nephrol. 2013, 24, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Sever, P.S.; Dahlöf, B.; Poulter, N.R.; Wedel, H.; Beevers, G. Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial—Lipid Lowering Arm (ASCOT-LLA): A multicentre randomised controlled trial. Lancet 2003, 361, 1149–1158. [Google Scholar] [PubMed]
- Tonelli, M.; Moye, L.; Sacks, F.M.; Kiberd, B. Pravastatin for secondary prevention of cardiovascular events in persons with mild chronic renal insufficiency. Ann. Intern. Med. 2003, 138, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, M.; Moyé, L.; Sacks, F.M.; Cole, T.; Curhan, G.C.; Cholesterol and recurrent events trial investigators. Effect of pravastatin on loss of renal function in people with moderate chronic renal insufficiency and cardiovascular disease. J. Am. Soc. Nephrol. 2003, 14, 1605–1613. [Google Scholar] [CrossRef] [PubMed]
- Fathi, R.; Isbel, N.; Short, L.; Haluska, B.; Johnson, D.; Marwick, T.H. The effect of long-term aggressive lipid lowering on ischemic and atherosclerotic burden in patients with chronic kidney disease. Am. J. Kidney Dis. 2004, 43, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Palmer, S.C.; Navaneethan, S.D.; Craig, J.C. HMG CoA reductase inhibitors (statins) for dialysis patients. Cochrane Database Syst. Rev. 2013. [Google Scholar] [CrossRef]
- Navaneethan, S.D.; Pansini, F.; Perkovic, V. HMG CoA reductase inhibitors (statins) for people with chronic kidney disease not requiring dialysis. Cochrane Database Syst. Rev. 2009. [Google Scholar] [CrossRef]
- Palmer, S.C.; Navaneethan, S.D.; Craig, J.C. HMG CoA reductase inhibitors (statins) for kidney transplant recipients. Cochrane Database Syst. Rev. 2014. [Google Scholar] [CrossRef]
- Oda, H.; Keane, W.F. Recent advances in statins and the kidney. Kidney Int. 1999, 56, 2–5. [Google Scholar] [CrossRef]
- Zoja, C.; Corna, D.; Rottoli, D.; Cattaneo, D.; Zanchi, C. Effect of combining ACE inhibitor and statin in severe experimental nephropathy. Kidney Int. 2002, 61, 1635–1645. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, M.P.; Kasiske, B.L.; Kim, Y.; Atluru, D.; Keane, W.F. Lovastatin inhibits proliferation of rat mesangial cells. J. Clin. Investig. 1993, 91, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.C.; Han, J.M.; Kwon, S.; Jee, J.H.; Yu, T.Y.; Lee, M.K.; Kim, J.H. LDL-C/apoB and HDL-C/apoA-1 ratios predict incident chronic kidney disease in a large apparently healthy cohort. Atherosclerosis 2016, 251, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Nestel, P.J.; Fidge, N.H.; Tan, M.H. Increased lipoprotein-remnant formation in chronic renal failure. N. Engl. J. Med. 1982, 307, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Okubo, M.; Hanada, H.; Matsui, M.; Hidaka, Y.; Masuda, D.; Sakata, Y.; Yamashita, S. Serum apolipoprotein B-48 concentration is associated with a reduced estimated glomerular filtration rate and increased proteinuria. J. Atheroscler. Thromb. 2014, 21, 974–982. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Hirano, T.; Taira, T.; Tokuno, A.; Mori, Y.; Koba, S.; Adachi, M. Remarkable increase of apolipoprotein B48 level in diabetic patients with end-stage renal disease. Atherosclerosis 2008, 197, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Cofan, M.; Cofan, F.; Campos, B.; Guerra, R.; Campistol, J.M.; Oppenheimer, F. Effect of apolipoprotein B polymorphism in kidney transplantation. Transplant. Proc. 2005, 37, 3794–3795. [Google Scholar] [CrossRef] [PubMed]
- Moradi, H.; Pahl, M.V.; Elahimehr, R.; Vaziri, N.D. Impaired antioxidant activity of high-density lipoprotein in chronic kidney disease. Transl. Res. 2009, 153, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Shoji, T.; Nishizawa, Y.; Nishitani, H.; Yamakawa, M. Impaired metabolism of high density lipoprotein in uremic patients. Kidney Int. 1992, 41, 1653–1661. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Liang, K.; Parks, J.S. Down-regulation of hepatic lecithin: Cholesterol acyltransferase gene expression in chronic renal failure. Kidney Int. 2001, 59, 2192–2196. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Liang, K. Down-regulation of tissue lipoprotein lipase expression in experimental chronic renal failure. Kidney Int. 1996, 50, 1928–1935. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Wang, X.Q.; Liang, K. Secondary hyperparathyroidism downregulates lipoprotein lipase expression in chronic renal failure. Am. J. Physiol. 1997, 273, 925–930. [Google Scholar]
- Vaziri, N.D.; Yuan, J.; Ni, Z.; Nicholas, S.B.; Norris, K.C. Lipoprotein lipase deficiency in chronic kidney disease is accompanied by down-regulation of endothelial GPIHBP1 expression. Clin. Exp. Nephrol. 2012, 16, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Vaziri, N.D. Down-regulation of hepatic LDL receptor-related protein (LRP) in chronic renal failure. Kidney Int. 2005, 67, 1028–1032. [Google Scholar] [CrossRef] [PubMed]
- Hirowatari, Y.; Homma, Y.; Yoshizawa, J.; Homma, K. Increase of electronegative-LDL-fraction ratio and IDL-cholesterol in chronic kidney disease patients with hemodialysis treatment. Lipids Health Dis. 2012, 11, 1. [Google Scholar] [CrossRef] [PubMed]
- Klin, M.; Smogorzewski, M.; Ni, Z.; Zhang, G. Abnormalities in hepatic lipase in chronic renal failure: Role of excess parathyroid hormone. J. Clin. Investig. 1996, 97, 2167–2173. [Google Scholar] [CrossRef] [PubMed]
- Liang, K.; Vaziri, N.D. Down-regulation of hepatic lipase expression in experimental nephrotic syndrome. Kidney Int. 1997, 51, 1933–1937. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Liang, K.; Vaziri, N.D. Protein restriction and AST-120 improve lipoprotein lipase and VLDL receptor in focal glomerulosclerosis. Kidney Int. 2003, 64, 1780–1786. [Google Scholar] [CrossRef] [PubMed]
- Liang, K.; Vaziri, N.D. Acquired VLDL receptor deficiency in experimental nephrosis. Kidney Int. 1997, 51, 1933–1937. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Liang, K.; Vaziri, N.D. Down-regulation of lipoprotein lipase and VLDL receptor in rats with focal glomerulosclerosis. Kidney Int. 2002, 64, 1780–1786. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D. Dyslipidemia of chronic renal failure: The nature, mechanisms, and potential consequences. Am. J. Physiol. Ren. Physiol. 2006, 290, 262–272. [Google Scholar] [CrossRef] [PubMed]
- DeGoulet, P.; LeGrain, M.; Reach, I.; Aime, F.; Devries, C.; Rojas, P.; Jacobs, C. Mortality risk factors in patients treated by chronic hemodialysis. Nephron 1982, 31, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Lowrie, E.G.; Lew, N.L. Death risk in hemodialysis patients: The predictive value of commonly measured variables and an evaluation of death rate differences between facilities. Am. J. Kidney Dis. 1990, 15, 458–482. [Google Scholar] [CrossRef]
- Liu, Y.; Coresh, J.; Eustace, J.A.; Longenecker, J.C.; Jaar, B.; Fink, N.E.; Tracy, R.P.; Powe, N.R.; Klag, M.J. Association between cholesterol level and mortality in dialysis patients: Role of inflammation and malnutrition. JAMA 2004, 291, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Levitan, I.; Volkov, S. Oxidized LDL: Diversity, patterns of recognition, and pathophysiology. Antioxid. Redox 2010, 13, 39–75. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, S.; Faria, M.D.S.; Silva, G.; Nascimento, H.; Rocha-Pereira, P.; Miranda, V.; Vieira, E.; Santos, R.; Mendonça, D.; Quintanilha, A.; et al. Oxidized low-density lipoprotein and lipoprotein(a) levels in chronic kidney disease patients under hemodialysis: Influence of adiponectin and of a polymorphism in the apolipoprotein(a) gene. Hemodial. Int. 2012, 16, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Samouilidou, E.C.; Karpouza, A.P.; Kostopoulos, V.; Bakirtzi, T.; Pantelias, K.; Petras, D.; Tzanatou-Exarchou, H.; Grapsa, E. Lipid abnormalities and oxidized LDL in chronic kidney disease patients on hemodialysis and peritoneal dialysis. Ren. Fail. 2012, 34, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Lobo, J.; Santos, F.; Grosso, D.; Lima, R.; Barreira, A.L.; Leite, M., Jr.; Mafra, D.; Abdalla, D.S.P. Electronegative LDL and lipid abnormalities in patients undergoing hemodialysis and peritoneal dialysis. Nephron Clin. Pract. 2008, 108, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Bossola, M.; Tazza, L.; Merki, E.; Giungi, S.; Luciani, G.; Miller, E.R.; Lin, E.B.; Tortorelli, A.; Tsimikas, S. Oxidized low-density lipoprotein biomarkers in patients with end-stage renal failure: Acute effects of hemodialysis. Blood Purif. 2007, 25, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Drożdż, D.; Kwinta, P.; Sztefko, K.; Kordon, Z.; Drożdż, T.; Łątka, M.; Miklaszewska, M.; Zachwieja, K.; Rudziński, A.; Pietrzyk, J.A. Oxidative stress biomarkers and left ventricular hypertrophy in children with chronic kidney disease. Oxid. Med. Cell. Longev. 2016. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, T.; Takahashi, K.; Kobayashi, T.; Oshima, E.; Iwasaki, S.; Suzuki, H. Oxidized low density lipoprotein (Ox-LDL) as a marker of atherosclerosis in hemodialysis (HD) patients. Clin. Nephrol. 2002, 58, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, K.; Myśliwiec, M.; Pawlak, D. Oxidized LDL to autoantibodies against oxLDL ratio—The new biomarker associated with carotid atherosclerosis and cardiovascular complications in dialyzed patients. Atherosclerosis 2012, 224, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Kalantar-Zadeh, K.; Brennan, M.-L.; Hazen, S.L. Serum myeloperoxidase and mortality in maintenance hemodialysis patients. Am. J. Kidney Dis. 2006, 48, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Kitabayashi, C.; Naruko, T.; Sugioka, K.; Yunoki, K.; Nakagawa, M.; Inaba, M.; Ohsawa, M.; Konishi, Y.; Imanishi, M.; Inoue, T.; et al. Positive association between plasma levels of oxidized low-density lipoprotein and myeloperoxidase after hemodialysis in patients with diabetic end-stage renal disease. Hemodial. Int. 2013, 17, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Hazen, S.L.; Heinecke, J.W. 3-Chlorotyrosine, a specific marker of myeloperoxidase-catalyzed oxidation, is markedly elevated in low density lipoprotein isolated from human atherosclerotic intima. J. Clin. Investig. 1997, 99, 2075–2081. [Google Scholar] [CrossRef] [PubMed]
- Pirillo, A.; Norata, G.D.; Catapano, A.L. LOX-1, OxLDL, and Atherosclerosis. Mediat. Inflamm. 2013, 2013, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Himmelfarb, J.; McMenamin, M.E.; Loseto, G.; Heinecke, J.W. Myeloperoxidase-catalyzed 3-chlorotyrosine formation in dialysis patients. Free Radic. Biol. Med. 2001, 31, 1163–1169. [Google Scholar] [CrossRef]
- Podrez, E.A.; Schmitt, D.; Hoff, H.F.; Hazen, S.L. Myeloperoxidase-generated reactive nitrogen species convert LDL into an atherogenic form in vitro. J. Clin. Investig. 1999, 103, 1547–1560. [Google Scholar] [CrossRef] [PubMed]
- Shiu, S.W.M.; Xiao, S.M.; Wong, Y.; Chow, W.S.; Lam, K.S.L.; Tan, K.C.B. Carbamylation of LDL and its relationship with myeloperoxidase in Type 2 diabetes mellitus. Clin. Sci. 2013, 126, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Exner, M.; Hermann, M.; Hofbauer, R.; Hartmann, B.; Kapiotis, S.; Gmeiner, B. Thiocyanate catalyzes myeloperoxidase-initiated lipid oxidation in LDL. Free Radic. Biol. Med. 2004, 37, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Apostolov, E.O.; Ok, E.; Burns, S.; Nawaz, S.; Savenka, A.; Shah, S.V.; Basnakian, A.G. Carbamylated-oxidized LDL: Proatherosclerotic effects on endothelial cells and macrophages. J. Atheroscler. Thromb. 2013, 20, 878–892. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.M. CD36, a scavenger receptor implicated in atherosclerosis. Exp. Mol. Med. 2014, 46, e99. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, M.; Bryl, E.; Marzec, L.; Aleksandrowicz, E.; Witkowski, J.M.; Rutkowski, B. Expression of scavenger receptor CD36 in chronic renal failure patients. Artif. Organs 2005, 29, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, J.; Merino, A.; Briceno, C.; Soriano, S.; Buendia, P.; Calleros, L.; Rodriguez, M.; Martin-Malo, A.; Aljama, P.; Ramirez, R. Carbamylated low-density lipoprotein induces oxidative stress and accelerated senescence in human endothelial progenitor cells. FASEB J. 2011, 25, 1314–1322. [Google Scholar] [CrossRef] [PubMed]
- Speer, T.; Owala, F.O.; Holy, E.W.; Zewinger, S.; Frenzel, F.L.; Stähli, B.E.; Razavi, M.; Triem, S.; Cvija, H.; Rohrer, L.; et al. Carbamylated low-density lipoprotein induces endothelial dysfunction. Eur. Heart J. 2014, 35, 3021–3032. [Google Scholar] [CrossRef] [PubMed]
- Holy, E.W.; Akhmedov, A.; Speer, T.; Camici, G.G.; Zewinger, S.; Bonetti, N.; Beer, J.H.; Lüscher, T.F.; Tanner, F.C. Carbamylated low-density lipoproteins induce a prothrombotic state via lox-1: Impact on arterial thrombus formation in vivo. J. Am. Coll. Cardiol. 2016, 68, 1664–1676. [Google Scholar] [CrossRef] [PubMed]
- Son, J.N.; Lho, Y.; Shin, S.; Kwon, S.-H.; Moon, K.C.; Ha, E. Carbamylated low-density lipoprotein increases reactive oxygen species (ROS) and apoptosis via lectin-like oxidized LDL receptor (LOX-1) mediated pathway in human umbilical vein endothelial cells. Int. J. Cardiol. 2011, 146, 428–430. [Google Scholar] [CrossRef] [PubMed]
- Apostolov, E.O.; Ray, D.; Savenka, A.V.; Shah, S.V.; Basnakian, A.G. Chronic Uremia Stimulates LDL Carbamylation and Atherosclerosis. J. Am. Soc. Nephrol. 2010, 21, 1852–1857. [Google Scholar] [CrossRef] [PubMed]
- Apostolov, E.O.; Basnakian, A.G.; Ok, E.; Shah, S.V. Carbamylated low-density lipoprotein: Nontraditional risk factor for cardiovascular events in patients with chronic kidney disease. J. Ren. Nutr. 2012, 22, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Kamanna, V.S.; Kashyap, M.L.; Pai, R.; Bui, D.T. Uremic serum subfraction inhibits apolipoprotein AI production by a human hepatoma cell line. J. Am. Soc. Nephrol. 1994, 5, 193–200. [Google Scholar] [PubMed]
- Shah, G.M.; Lin, Z.L.; Kamanna, V.S.; Pai, R.; Bassa, B. Effect of serum subfractions from peritoneal dialysis patients on Hep-G2 cell apolipoprotein AI and B metabolism. Kidney Int. 1996, 50, 2079–2087. [Google Scholar] [CrossRef] [PubMed]
- Shao, B.; Oda, M.N.; Oram, J.F.; Heinecke, J.W. Myeloperoxidase: An inflammatory enzyme for generating dysfunctional high density lipoprotein. Curr. Opin. Cardiol. 2006, 21, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Liang, K.; Vaziri, N.D. Upregulation of acyl-CoA: Cholesterol acyltransferase in chronic renal failure. Am. J. Physiol. Endocrinol. Metab. 2002, 283, 676–681. [Google Scholar] [CrossRef] [PubMed]
- Holzer, M.; Schilcher, G.; Curcic, S.; Trieb, M.; Ljubojevic, S.; Stojakovic, T.; Scharnagl, H.; Kopecky, C.M.; Rosenkranz, A.R.; Heinemann, A.; et al. Dialysis modalities and HDL composition and function. J. Am. Soc. Nephrol. 2015, 26, 2267–2276. [Google Scholar] [CrossRef] [PubMed]
- Hewing, B.; Parathath, S.; Barrett, T.; Chung, W.K.K.; Astudillo, Y.M.; Hamada, T.; Ramkhelawon, B.; Tallant, T.C.; Yusufishaq, M.S.S.; DiDonato, J.A.; et al. Effects of native and myeloperoxidase-modified apolipoprotein A-I on reverse cholesterol transport and atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Shao, B.; Tang, C.; Sinha, A.; Mayer, P.S.; Davenport, G.D.; Brot, N.; Oda, M.N.; Zhao, X.-Q.; Heinecke, J.W. Humans with atherosclerosis have impaired ABCA1 cholesterol efflux and enhanced high-density lipoprotein oxidation by myeloperoxidase. Circ. Res. 2014, 114, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Holzer, M.; Zangger, K.; El-Gamal, D.; Binder, V.; Curcic, S.; Konya, V.; Schuligoi, R.; Heinemann, A.; Marsche, G. Myeloperoxidase-derived chlorinating species induce protein carbamylation through decomposition of thiocyanate and urea: Novel pathways generating dysfunctional high-density lipoprotein. Antioxid. Redox Signal. 2012, 17, 1043–1052. [Google Scholar] [CrossRef] [PubMed]
- Holzer, M.; Gauster, M.; Pfeifer, T.; Wadsack, C.; Fauler, G.; Stiegler, P.; Koefeler, H.; Beubler, E.; Schuligoi, R.; Heinemann, A.; et al. Protein carbamylation renders high-density lipoprotein dysfunctional. Antioxid. Redox Signal. 2011, 14, 2337–2346. [Google Scholar] [CrossRef] [PubMed]
- Undurti, A.; Huang, Y.; Lupica, J.A.; Smith, J.D.; DiDonato, J.A.; Hazen, S.L. Modification of high density lipoprotein by myeloperoxidase generates a pro-inflammatory particle. J. Biol. Chem. 2009, 284, 30825–30835. [Google Scholar] [CrossRef] [PubMed]
- Honda, H.; Ueda, M.; Kojima, S.; Mashiba, S.; Michihata, T.; Takahashi, K.; Shishido, K.; Akizawa, T. Oxidized high-density lipoprotein as a risk factor for cardiovascular events in prevalent hemodialysis patients. Atherosclerosis 2012, 220, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Kalantar-Zadeh, K.; Kopple, J.D.; Kamranpour, N.; Fogelman, A.M.; Navab, M. HDL-inflammatory index correlates with poor outcome in hemodialysis patients. Kidney Int. 2007, 72, 1149–1156. [Google Scholar] [CrossRef] [PubMed]
- Holzer, M.; Birner-Gruenberger, R.; Stojakovic, T.; El-Gamal, D.; Binder, V.; Wadsack, C.; Heinemann, A.; Marsche, G. Uremia Alters HDL Composition and Function. J. Am. Soc. Nephrol. 2011, 22, 1631–1641. [Google Scholar] [CrossRef] [PubMed]
- Mangé, A.; Goux, A.; Badiou, S.; Patrier, L.; Canaud, B.; Maudelonde, T.; Cristol, J.-P.; Solassol, J. HDL proteome in hemodialysis patients: A quantitative nanoflow liquid chromatography-tandem mass spectrometry approach. PLoS ONE 2012. [Google Scholar] [CrossRef] [PubMed]
- Khera, A.V.; Cuchel, M.; la Llera-Moya, M.; Rodrigues, A.; Burke, M.F.; Jafri, K.; French, B.C.; Phillips, J.A.; Mucksavage, M.L.; Wilensky, R.L.; et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N. Engl. J. Med. 2011, 364, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Kopecky, C.; Haidinger, M.; Birner-Grunberger, R.; Darnhofer, B.; Kaltenecker, C.C.; Marsche, G.; Holzer, M.; Weichhart, T.; Antlanger, M.; Kovarik, J.J.; et al. Restoration of Renal Function Does Not Correct Impairment of Uremic HDL Properties. J. Am. Soc. Nephrol. 2015, 26, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Kaseda, R.; Jabs, K.; Hunley, T.E.; Jones, D.; Bian, A.; Allen, R.M.; Vickers, K.C.; Yancey, P.G.; Linton, M.F.; Fazio, S.; et al. Dysfunctional high-density lipoproteins in children with chronic kidney disease. Metabolism 2015, 64, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Yancey, P.G.; Ikizler, T.A.; Jerome, W.G.; Kaseda, R.; Cox, B.; Bian, A.; Shintani, A.; Fogo, A.B.; Linton, M.R.F.; et al. Dysfunctional high-density lipoprotein in patients on chronic hemodialysis. J. Am. Coll. Cardiol. 2012, 60, 2372–2379. [Google Scholar] [CrossRef] [PubMed]
- Shroff, R.; Speer, T.; Colin, S.; Charakida, M.; Zewinger, S.; Staels, B.; Chinetti-Gbaguidi, G.; Hettrich, I.; Rohrer, L.; O’Neill, F.; et al. HDL in children with ckd promotes endothelial dysfunction and an abnormal vascular phenotype. J. Am. Soc. Nephrol. 2014, 25, 2658–2668. [Google Scholar] [CrossRef] [PubMed]
- Kopecky, C.; Ebtehaj, S.; Genser, B.; Drechsler, C.; Krane, V.; Antlanger, M.; Kovarik, J.J.; Kaltenecker, C.C.; Parvizi, M.; Wanner, C.; et al. HDL cholesterol efflux does not predict cardiovascular risk in hemodialysis patients. J. Am. Soc. Nephrol. 2016, in press. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Sato, K.; Malchinkhuu, E.; Tomura, H.; Tamama, K.; Kuwabara, A.; Murakami, M.; Okajima, F. High-density lipoprotein stimulates endothelial cell migration and survival through sphingosine 1-phosphate and its receptors. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1283–1288. [Google Scholar] [CrossRef] [PubMed]
- Litvinov, D.; Mahini, H.; Garelnabi, M. Antioxidant and anti-inflammatory role of paraoxonase 1: Implication in arteriosclerosis diseases. N. Am. J. Med. Sci. 2012, 4, 523–532. [Google Scholar] [PubMed]
- Dantoine, T.F.; Debord, J.; Charmes, J.P.; Merle, L.; Marquet, P.; Lachatre, G.; Leroux-Robert, C. Decrease of serum paraoxonase activity in chronic renal failure. J. Am. Soc. Nephrol. 1998, 9, 2082–2088. [Google Scholar] [PubMed]
- Kennedy, D.J.; Tang, W.H.W.; Fan, Y.; Wu, Y.; Mann, S.; Pepoy, M.; Hazen, S.L. Diminished antioxidant activity of high-density lipoprotein-associated proteins in chronic kidney disease. J. Am. Heart Assoc. 2013. [Google Scholar] [CrossRef] [PubMed]
- Kotani, K.; Kimura, S.; Gugliucci, A. Paraoxonase-1 and ischemia-modified albumin in patients with end-stage renal disease. J. Physiol. Biochem. 2011, 67, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Tsimikas, S.; Brilakis, E.S.; Miller, E.R.; McConnell, J.P.; Lennon, R.J.; Kornman, K.S.; Witztum, J.L.; Berger, P.B. Oxidized phospholipids, Lp(a) lipoprotein, and coronary artery disease. N. Engl. J. Med. 2005, 353, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Tsimikas, S.; Kiechl, S.; Willeit, J.; Mayr, M.; Miller, E.R.; Kronenberg, F.; Xu, Q.; Bergmark, C.; Weger, S.; Oberhollenzer, F.; et al. Oxidized phospholipids predict the presence and progression of carotid and femoral atherosclerosis and symptomatic cardiovascular disease: Five-year prospective results from the Bruneck study. J. Am. Coll. Cardiol. 2006, 47, 2219–2228. [Google Scholar] [CrossRef] [PubMed]
- Tsimikas, S.; Hall, J.L. Lipoprotein(a) as a potential causal genetic risk factor of cardiovascular disease: A rationale for increased efforts to understand its pathophysiology and develop targeted therapies. J. Am. Coll. Cardiol. 2012, 60, 716–721. [Google Scholar] [CrossRef] [PubMed]
- Tsimikas, S.; Viney, N.J.; Hughes, S.G.; Singleton, W.; Graham, M.J.; Baker, B.F.; Burkey, J.L.; Yang, Q.; Marcovina, S.M.; Geary, R.S.; et al. Antisense therapy targeting apolipoprotein(a): A randomised, double-blind, placebo-controlled phase 1 study. Lancet 2015, 386, 1472–1483. [Google Scholar] [CrossRef]
- Kollerits, B.; Drechsler, C.; Krane, V.; Lamina, C.; März, W.; Dieplinger, H.; Ritz, E.; Wanner, C.; Kronenberg, F.; German Diabetes and Dialysis Study Investigators. Lipoprotein(a) concentrations, apolipoprotein(a) isoforms and clinical endpoints in haemodialysis patients with type 2 diabetes mellitus: Results from the 4D Study. Nephrol. Dial. Transplant. 2016, 31, 1901–1908. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, T.X.; McCormick, S.P.; Tsimikas, S.; Bro, S.; Nielsen, L.B. Lipoprotein(a) accelerates atherosclerosis in uremic mice. J. Lipid Res. 2010, 51, 2967–2975. [Google Scholar] [CrossRef] [PubMed]
- Frischmann, M.E.; Kronenberg, F.; Trenkwalder, E.; Schaefer, J.R.; Schweer, H.; Dieplinger, B.; Koenig, P.; Ikewaki, K.; Dieplinger, H. In vivo turnover study demonstrates diminished clearance of lipoprotein(a) in hemodialysis patients. Kidney Int. 2007, 71, 1036–1043. [Google Scholar] [CrossRef] [PubMed]
- Tzanatos, H.A.; Agroyannis, B.; Chondros, C.; Kapetanaki, A.; Fourtounas, C.; Soubassi, L.; Kopelias, I. Cytokine release and serum lipoprotein (a) alterations during hemodialysis. Artif. Organs 2000, 24, 329–333. [Google Scholar] [CrossRef] [PubMed]
- De Sain-Van Der Velden, M.G.; Reijngoud, D.J.; Kaysen, G.A.; Gadellaa, M.M.; Voorbij, H.; Stellaard, F.; Koomans, H.A.; Rabelink, T.J. Evidence for increased synthesis of lipoprotein(a) in the nephrotic syndrome. J. Am. Soc. Nephrol. 1998, 9, 1474–1481. [Google Scholar] [PubMed]
- Kronenberg, F.; König, P.; Neyer, U.; Auinger, M.; Pribasnig, A.; Lang, U.; Reitinger, J.; Pinter, G.; Utermann, G.; Dieplinger, H. Multicenter study of lipoprotein(a) and apolipoprotein(a) phenotypes in patients with end-stage renal disease treated by hemodialysis or continuous ambulatory peritoneal dialysis. J. Am. Soc. Nephrol. 1995, 6, 110–120. [Google Scholar] [PubMed]
- Kronenberg, F. Causes and consequences of lipoprotein(a) abnormalities in kidney disease. Clin. Exp. Nephrol. 2014, 18, 234–237. [Google Scholar] [CrossRef] [PubMed]
- Boaz, M.; Iuliano, L.; Himmelfarb, J.; Matas, Z.; Micheletta, F.; McMonagle, E.; Friedman, V.; Natoli, S.; Gvirtz, G.; Biro, A.; et al. Baseline oxysterols and other markers of oxidative stress, inflammation and malnutrition in the Vitamin E and Intima media thickness Progression in End-stage Renal disease (VIPER) cohort. Nephron Clin. Pract. 2005, 100, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Siems, W.; Quast, S.; Peter, D.; Augustin, W.; Carluccio, F.; Grune, T.; Sevanian, A.; Hampl, H.; Wiswedel, I. Oxysterols are increased in plasma of end-stage renal disease patients. Kidney Blood Press. Res. 2005, 28, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Bossola, M.; Tazza, L.; Luciani, G.; Tortorelli, A.; Tsimikas, S. OxPL/apoB, lipoprotein(a) and OxLDL biomarkers and cardiovascular disease in chronic hemodialysis patients. J. Nephrol. 2011, 24, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Samouilidou, E.; Grapsa, E. Effect of dialysis on plasma total antioxidant capacity and lipid peroxidation products in patients with end-stage renal failure. Blood Purif. 2003, 21, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Núñez, J.M.; Ghais, Z.; Bustamante, J. Evaluation of oxidant-antioxidant balance in patients on maintenance haemodialysis: A comparative study of dialyzers membranes. Nephron Clin. Pract. 2010, 114, 67–73. [Google Scholar]
- Ajala, M.O.; Ogunro, P.S.; Odun, A. Effect of hemodialysis on total antioxidant status of chronic renal failure patients in government hospitals in Lagos Nigeria. Niger J. Clin. Pract. 2011, 14, 154–158. [Google Scholar] [PubMed]
- Wiswedel, I.; Peter, D.; Gardemann, A.; Carluccio, F.; Hampl, H.; Siems, W. Serum concentrations of F2-isoprostanes and 4-Hydroxynonenal in hemodialysis patients in relation to inflammation and renal anemia. Biomark. Insights 2008, 3, 419–428. [Google Scholar] [PubMed]
- Odetti, P.; Traverso, N.; Monacelli, F.; Menini, S.; Vazzana, J.; Tasso, B.; Pronzato, M.A.; Robaudo, C.; Deferrari, G. Vitamin E-coated filter decreases levels of free 4-hydroxyl-2-nonenal during haemodialysis sessions. Free Radic. Res. 2006, 40, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Guichardant, M.; Bacot, S.; Moliere, P.; Lagarde, M. Hydroxy-alkenals from the peroxidation of n-3 and n-6 fatty acids and urinary metabolites. Prostaglandins Leukot. Essent. Fat. Acids 2006, 75, 179–182. [Google Scholar] [CrossRef] [PubMed]
- Handelman, G.J.; Walter, M.F.; Adhikarla, R.; Gross, J.; Dallal, G.E.; Levin, N.W.; Blumberg, J.B. Elevated plasma F2-isoprostanes in patients on long-term hemodialysis. Kidney Int. 2001, 59, 1960–1966. [Google Scholar] [CrossRef] [PubMed]
- Ikizler, T.A.; Morrow, J.D.; Roberts, L.J.; Evanson, J.A.; Becker, B.; Hakim, R.M.; Shyr, Y.; Himmelfarb, J. Plasma F2-isoprostane levels are elevated in chronic hemodialysis patients. Clin. Nephrol. 2002, 58, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Himmelfarb, J.; Phinney, S.; Ikizler, T.A.; Kane, J.; McMonagle, E.; Miller, G. Gamma-tocopherol and docosahexaenoic acid decrease inflammation in dialysis patients. J. Ren. Nutr. 2007, 17, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Kamgar, M.; Zaldivar, F.; Vaziri, N.D.; Pahl, M.V. Antioxidant therapy does not ameliorate oxidative stress and inflammation in patients with end-stage renal disease. J. Natl. Med. Assoc. 2009, 101, 336–344. [Google Scholar] [CrossRef]
- Harman, S.M.; Liang, L.; Tsitouras, P.D.; Gucciardo, F.; Heward, C.B.; Reaven, P.D.; Ping, W.; Ahmed, A.; Cutler, R.G. Urinary excretion of three nucleic acid oxidation adducts and isoprostane F(2)alpha measured by liquid chromatography-mass spectrometry in smokers, ex-smokers, and nonsmokers. Free Radic. Biol. Med. 2003, 35, 1301–1309. [Google Scholar] [CrossRef] [PubMed]
- Morrow, J.D.; Zackert, W.E.; Yang, J.P.; Kurhts, E.H.; Callewaert, D.; Dworski, R.; Kanai, K.; Taber, D.; Moore, K.; Oates, J.A.; et al. Quantification of the major urinary metabolite of 15-F2t-isoprostane (8-iso-PGF2α) by a stable isotope dilution mass spectrometric assay. Anal. Biochem. 1999, 269, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Morena, M.; Jaussent, I.; Chalabi, L.; Bargnoux, A.-S.; Dupuy, A.-M.; Badiou, S.; Rakic, C.; Thomas, M.; Canaud, B.; Cristol, J.-P. Biocompatibility of heparin-grafted hemodialysis membranes: Impact on monocyte chemoattractant protein-1 circulating level and oxidative status. Hemodial. Int. 2010, 14, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Bonomini, F.; Tengattini, S.; Fabiano, A.; Bianchi, R. Atherosclerosis and oxidative stress. Histol. Histopathol. 2008, 23, 381–390. [Google Scholar] [PubMed]
- Cadenas, E.; Davies, K.J.A. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Vignais, P.V. The superoxide-generating NADPH oxidase: Structural aspects and activation mechanism. Cell. Mol. Life Sci. 2002, 59, 1428–1459. [Google Scholar] [CrossRef] [PubMed]
- De Minicis, S.; Bataller, R.; Brenner, D.A. NADPH oxidase in the liver: Defensive, offensive, or fibrogenic? Gastroenterology 2006, 131, 272–275. [Google Scholar] [CrossRef] [PubMed]
- Novo, E.; Parola, M. Redox mechanisms in hepatic chronic wound healing and fibrogenesis. Fibrogenesis Tissue Repair 2008, 1, 1. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D. Nox enzymes, ROS, and chronic disease: An example of antagonistic pleiotropy. Free Radic. Biol. Med. 2007, 43, 332–347. [Google Scholar] [CrossRef] [PubMed]
- Grotto, D.; Santa Maria, L.D.; Boeira, S.; Valentini, J.; Charão, M.F.; Moro, A.M.; Nascimento, P.C.; Pomblum, V.J.; Garcia, S.C. Rapid quantification of malondialdehyde in plasma by high performance liquid chromatography-visible detection. J. Pharm. Biomed. Anal. 2007, 43, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Guichardant, M.; Chantegrel, B.; Deshayes, C.; Doutheau, A.; Moliere, P.; Lagarde, M. Specific markers of lipid peroxidation issued from n-3 and n-6 fatty acids. Biochem. Soc. Trans. 2004, 32, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.L.; Johnson, B.H. Biological activities of oxysterols. Free Radic. Biol. Med. 1989, 7, 285–332. [Google Scholar] [CrossRef]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 48, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Peters, T.J. All about Albumin; Academic Press, Inc.: San Diego, CA, USA, 1996. [Google Scholar]
- Jacobsen, C. Lysine residue 240 of human serum albumin is involved in high-affinity binding of bilirubin. Biochem. J. 1978, 171, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Neužil, J.; Stocker, R. Bilirubin attenuates radical-mediated damage to serum albumin. FEBS Lett. 1993, 331, 281–284. [Google Scholar] [CrossRef]
- Neužil, J.; Stocker, R. Free and albumin-bound bilirubin are efficient co-antioxidants for alpha-tocopherol, inhibiting plasma and low density lipoprotein lipid peroxidation. J. Biol. Chem. 1994, 269, 16712–16719. [Google Scholar] [PubMed]
- Gutteridge, J.M.C. Antioxidant properties of the proteins caeruloplasmin, albumin and transferrin. A study of their activity in serum and synovial fluid from patients with rheumatoid arthritis. Biochim. Biophys. Acta 1986, 869, 119–127. [Google Scholar] [CrossRef]
- Oettl, K.; Stauber, R.E. Physiological and pathological changes in the redox state of human serum albumin critically influence its binding properties. Br. J. Pharmacol. 2007, 151, 580–590. [Google Scholar] [CrossRef] [PubMed]
- Del Vecchio, L.; Locatelli, F.; Carini, M. What we know about oxidative stress in patients with chronic kidney disease on dialysis—Clinical effects, potential treatment, and prevention. Semin. Dial. 2011, 24, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Tucker, P.S.; Dalbo, V.J.; Han, T.; Kingsley, M.I. Clinical and research markers of oxidative stress in chronic kidney disease. Biomarkers 2013, 18, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Lahera, V.; Goicoechea, M.; de Vinuesa, S.G.; Oubiña, P.; Cachofeiro, V.; Gómez-Campderá, F.; Amann, R.; Luño, J. Oxidative stress in uremia: The role of anemia correction. J. Am. Soc. Nephrol. 2006, 17, S174–S177. [Google Scholar] [CrossRef] [PubMed]
- Ratliff, B.B.; Abdulmahdi, W.; Pawar, R.; Wolin, M.S. Oxidant mechanisms in renal injury and disease. Antioxid. Redox Signal. 2016, 25, 119–146. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.-T.; Chiang, C.-K. Uremic toxins, oxidative stress, and renal fibrosis: An interwined complex. J. Ren. Nutr. 2015, 25, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Modaresi, A.; Nafar, M.; Sahraei, Z. Oxidative stress in chronic kidney disease. Iran. J. Kidney Dis. 2015, 9, 165–179. [Google Scholar] [PubMed]
- Popolo, A.; Autore, G.; Pinto, A.; Marzocco, S. Oxidative stress in patients with cardiovascular disease and chronic renal failure. Free Radic. Res. 2013, 47, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Libetta, C.; Sepe, V.; Esposito, P.; Galli, F.; Dal Canton, A. Oxidative stress and inflammation: Implications in uremia and hemodialysis. Clin. Biochem. 2011, 44, 1189–1198. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D. Oxidative stress in uremia: Nature, mechanisms, and potential consequences. Semin. Nephrol. 2004, 24, 469–473. [Google Scholar] [CrossRef] [PubMed]
- Himmelfarb, J.; Stenvinkel, P.; Ikizler, T.A.; Hakim, R.M. The elephant in uremia: Oxidant stress as a unifying concept of cardiovascular disease in uremia. Kidney Int. 2002, 62, 1524–1538. [Google Scholar] [CrossRef] [PubMed]
- Ramos, L.F.; Shintani, A.; Ikizler, T.A.; Himmelfarb, J. Oxidative stress and inflammation are associated with adiposity in moderate to severe CKD. J. Am. Soc. Nephrol. 2008, 19, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y.; Sasaki, S.; Nakagawa, K.; Matsuura, H.; Oshima, T.; Chayama, K. Endothelial function and oxidative stress in renovascular hypertension. N. Engl. J. Med. 2002, 346, 1954–1962. [Google Scholar] [CrossRef] [PubMed]
- Touyz, R.M. Reactive oxygen species, vascular oxidative stress, and redox signaling in hypertension: What is the clinical significance? Hypertension 2004, 44, 248–252. [Google Scholar] [CrossRef] [PubMed]
- Zalba, G.; San José, G.; Moreno, M.U.; Fortuño, M.A.; Fortuño, A.; Beaumont, F.J.; Díez, J. Oxidative stress in arterial hypertension: Role of NAD(P)H oxidase. Hypertension 2001, 38, 1395–1399. [Google Scholar] [CrossRef] [PubMed]
- Moh, A.; Sakata, N.; Takebayashi, S.; Tateishi, K.; Nagai, R.; Horiuchi, S.; Chihara, J. Increased production of urea hydrogen peroxide from Maillard reaction and a UHP-Fenton pathway related to glycoxidation damage in chronic renal failure. J. Am. Soc. Nephrol. 2004, 15, 1077–1085. [Google Scholar] [CrossRef] [PubMed]
- Fatouros, I.G.; Pasadakis, P.; Sovatzidis, A. Acute exercise may exacerbate oxidative stress response in hemodialysis patients. Nephron Clin. Pract. 2008, 109, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Coaccioli, S.; Standoli, M.L.; Biondi, R.; Panaccione, A.; Landucci, P.; Del Giorno, R.; Paladini, A.; Standoli, M.; Puxeddu, A. Open comparison study of oxidative stress markers between patients with chronic renal failure in conservative therapy and patients in haemodialysis. Clin. Ter. 2010, 161, 435–439. [Google Scholar] [PubMed]
- Kuchta, A.; Pacanis, A.; Kortas-Stempak, B.; Çwiklińska, A.; Ziętkiewicz, M.; Renke, M.; Rutkowski, B. Estimation of oxidative stress markers in chronic kidney disease. Kidney Blood Press. Res. 2011, 34, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Biasioli, S.; Schiavon, R.; Petrosino, L.; Cavallini, L.; De Fanti, E.; Zambello, A.; Borin, D.; Targa, L. Do different dialytic techniques have different atherosclerotic and antioxidant activities? ASAIO J. 2001, 47, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Shurtz-Swirski, R.; Mashiach, E.; Kristal, B.; Shkolnik, T.; Shasha, S.M. Antioxidant enzymes activity in polymorphonuclear leukocytes in chronic renal failure. Nephron 1995, 71, 176–179. [Google Scholar] [CrossRef] [PubMed]
- Atamer, A.; Kocyigit, Y.; Ecder, S.A.; Selek, S.; Iihan, N. Effect of oxidative stress on antioxidant enzyme activities, homocysteine and lipoproteins in chronic kidney disease. J. Nephrol. 2008, 21, 924–930. [Google Scholar] [PubMed]
- Kuo, H.-T.; Kuo, M.-C.; Chiu, Y.-W.; Chang, J.-M.; Guh, J.-Y.; Chen, H.-C. Increased glomerular and extracellular malondialdehyde levels in patients and rats with focal segmental glomerulosclerosis. Eur. J. Clin. Investig. 2005, 35, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Inal, M.; Kanbak, G.; Sen, S.; Akyüz, F.; Sunal, E. Antioxidant status and lipid peroxidation in hemodialysis patients undergoing erythropoietin and erythropoietin-vitamin E combined therapy. Free Radic. Res. 1999, 31, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Vural, A.; Yilmaz, M.I.; Caglar, K.; Aydin, A.; Sonmez, A.; Eyileten, T.; Acikel, C.; Gulec, B.; Kozak, O.; Oner, K. Assessment of oxidative stress in the early posttransplant period: Comparison of cyclosporine A and tacrolimus-based regimens. Am. J. Nephrol. 2005, 25, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Ongajooth, L.; Ongajyooth, S.; Likidlilid, A.; Chantachum, Y.; Shayakul, C.; Nilwarangkur, S. Role of lipid peroxidation, trace elements and anti-oxidant enzymes in chronic renal disease patients. J. Med. Assoc. Thail. 1996, 79, 791–800. [Google Scholar] [PubMed]
- Romeu, M.; Nogues, R.; Marcas, L.; Sánchez-Martos, V.; Mulero, M.; Martinez-Vea, A.; Mallol, J.; Giralt, M. Evaluation of oxidative stress biomarkers in patients with chronic renal failure: A case control study. BMC Res. Notes 2010, 3, 20. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, P.; Malgorzewicz, S.; Slominska, E. Interrelationship between uremic toxicity and oxidative stress. J. Ren. Nutr. 2006, 16, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, P.; Słominska, E.M.; Szołkiewicz, M.; Aleksandrowicz, E.; Smolenski, R.T.; Wołyniec, W.; Renke, M.; Wisterowicz, K.; Swierczynski, J.; Rutkowski, B. Relationship between uremic toxins and oxidative stress in patients with chronic renal failure. Scand. J. Urol. Nephrol. 2007, 41, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Suehiro, T.; Itahara, T.; Inui, Y.; Chikazawa, H.; Inoue, M.; Arii, K.; Hashimoto, K. Human serum paraoxonase concentration predicts cardiovascular mortality in hemodialysis patients. Clin. Nephrol. 2007, 67, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Weiss, M.F.; Erhard, P.; Kader-Attia, F.A.; Wu, Y.C. Mechanisms for the formation of glycoxidation products in end-stage renal disease. Kidney Int. 2000, 57, 2571–2585. [Google Scholar] [CrossRef] [PubMed]
- Miyata, T.; Kurokawa, K. Advanced glycation and lipoxidation end products role of reactive carbonyl compounds generated during carbohydrate and lipid metabolism. J. Am. Soc. Nephrol. 2000, 11, 1744–1752. [Google Scholar] [PubMed]
- Stadtman, E.R.; Levine, R.L. Free radical-mediated oxidation of free amino acids and amino acid residues in proteins. Amino Acids 2003, 25, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Basta, G.; Schmidt, A.M.; De Caterina, R. Advanced glycation end products and vascular inflammation: Implications for accelerated atherosclerosis in diabetes. Cardiovasc. Res. 2004, 63, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Bucala, R.; Mitchell, R.; Arnold, K.; Innerarity, T.; Vlassara, H.; Cerami, A. Identification of the major site of Apolipoprotein B modification by advanced glycosylation end products blocking uptake by the low density lipoprotein receptor. J. Biol. Chem. 1995, 270, 10828–10832. [Google Scholar] [PubMed]
- Klein, R.L.; Laimins, M.; Lopes-Virella, M.F. Isolation, characterization, and metabolism of the glycated and nonglycated subfractions of low-density lipoproteins isolated from Type I diabetic patients and nondiabetic subjects. Diabetes 1995, 44, 1093–1098. [Google Scholar] [CrossRef] [PubMed]
- Negre Salvayre, A.; Coatrieux, C.; Ingueneau, C.; Salvayre, R. Advanced lipid peroxidation end products in oxidative damage to proteins. Potential role in diseases and therapeutic prospects for the inhibitors. Br. J. Pharmacol. 2008, 153, 6–20. [Google Scholar] [CrossRef] [PubMed]
- Kalousova, M.; Zima, T.; Tesař, V.; Štípek, S.; Sulková, S. Advanced glycation end products in clinical nephrology. Kidney Blood Press. Res. 2004, 27, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Kalousova, M.; Zima, T.; Tesař, V.; Lachmanová, J. Advanced glycation end products and advanced oxidation protein products in hemodialyzed patients. Blood Purif. 2003, 20, 531–536. [Google Scholar] [CrossRef]
- Schinzel, R.; Münch, G.; Heidland, A.; Sebekova, K. Advanced glycation end products in end-stage renal disease and their removal. Nephron 2001, 87, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Canaud, B.; Cristol, J.P.; Morena, M.; Leray-Moragues, H.; Bosc, J.Y.; Vaussenat, F. Imbalance of oxidants and antioxidants in haemodialysis patients. Blood Purif. 1999, 17, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, F.; Canaud, B.; Eckardt, K.U.; Stenvinkel, P.; Wanner, C.; Zoccali, C. Oxidative stress in end-stage renal disease: An emerging threat to patient outcome. Nephrol. Dial. Transplant. 2003, 18, 1272–1280. [Google Scholar] [CrossRef] [PubMed]
- Verbrugge, F.H.; Tang, W.; Hazen, S.L. Protein carbamylation and cardiovascular disease. Kidney Int. 2015, 88, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Koeth, R.A.; Kalantar-Zadeh, K.; Wang, Z.; Fu, X.; Tang, W.H.W.; Hazen, S.L. Protein carbamylation predicts mortality in ESRD. J. Am. Soc. Nephrol. 2013, 24, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Berg, A.H.; Drechsler, C.; Wenger, J.; Buccafusca, R.; Hod, T.; Kalim, S.; Ramma, W.; Parikh, S.M.; Steen, H.; Friedman, D.J.; et al. Carbamylation of serum albumin as a risk factor for mortality in patients with kidney failure. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Mehta, J.L.; Basnakian, A.G. Interaction of carbamylated LDL with LOX-1 in the induction of endothelial dysfunction and atherosclerosis. Eur. Heart J. 2014, 35, 2996–2997. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, S.J. Myeloperoxidase and cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1102–1111. [Google Scholar] [CrossRef] [PubMed]
- Rutgers, A.; Heeringa, P.; Kooman, J.P.; van der Sande, F.M.; Cohen Travaert, J.W. Peripheral blood myeloperoxidase activity increases during hemodialysis. Kidney Int. 2003, 64, 760–762. [Google Scholar] [CrossRef] [PubMed]
- Stenvinkel, P. Statin treatment and diabetes affect myeloperoxidase activity in maintenance hemodialysis patients. Clin. J. Am. Soc. Nephrol. 2006, 1, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Wada, S.; Sugioka, K.; Naruko, T.; Kato, Y.; Shibata, T.; Inoue, T.; Inaba, M.; Ohsawa, M.; Yoshiyama, M.; Ueda, M. Myeloperoxidase and progression of aortic valve stenosis in patients undergoing hemodialysis. J. Heart Valve Dis. 2013, 22, 640–647. [Google Scholar] [PubMed]
- Murphy, R.C.; Johnson, K.M. Cholesterol, reactive oxygen species, and the formation of biologically active mediators. J. Biol. Chem. 2008, 283, 15521–15525. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.J.; Jessup, W. Oxysterols and atherosclerosis. Atherosclerosis 1999, 142, 1–28. [Google Scholar] [CrossRef]
- Hodis, H.N.; Crawford, D.W.; Sevanian, A. Cholesterol feeding increases plasma and aortic tissue cholesterol oxide levels in parallel: Further evidence for the role of cholesterol oxidation in atherosclerosis. Atherosclerosis 1991, 89, 117–126. [Google Scholar] [CrossRef]
- Garcia-Cruset, S.; Carpenter, K.L.; Guardiola, F.; Stein, B.K.; Mitchinson, M.J. Oxysterol profiles of normal human arteries, fatty streaks and advanced lesions. Free Radic. Res. 2001, 35, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Lizard, G.; Monier, S.; Cordelet, C.; Gesquière, L.; Deckert, V.; Gueldry, S.; Lagrost, L.; Gambert, P. Characterization and comparison of the mode of cell death, apoptosis versus necrosis, induced by 7β-hydroxycholesterol and 7-ketocholesterol in the cells of the vascular wall. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1190–1200. [Google Scholar] [CrossRef] [PubMed]
- Shibata, N.; Glass, C.K. Macrophages, oxysterols and atherosclerosis. Circulation 2010, 74, 2045–2051. [Google Scholar] [CrossRef]
- Nishio, E.; Watanabe, Y. Oxysterols induced apoptosis in cultured smooth muscle cells through CPP32 protease activation and bcl-2 protein downregulation. Biochem. Biophys. Res. Commun. 1996, 226, 928–934. [Google Scholar] [CrossRef] [PubMed]
- Lemaire, S.; Lizard, G.; Monier, S.; Miguet, C.; Gueldry, S.; Volot, F.; Gambert, P.; Néel, D. Different patterns of IL-1beta secretion, adhesion molecule expression and apoptosis induction in human endothelial cells treated with 7α-, 7β-hydroxycholesterol, or 7-ketocholesterol. FEBS Lett. 1998, 440, 434–439. [Google Scholar] [CrossRef]
- O’Callaghan, Y.C.; Woods, J.A.; O’Brien, N.M. Oxysterol-induced cell death in U937 and HepG2 cells at reduced and normal serum concentrations. Eur. J. Nutr. 1999, 38, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Lizard, G.; Deckert, V.; Dubrez, L.; Moisant, M.; Gambert, P.; Lagrost, L. Induction of apoptosis in endothelial cells treated with cholesterol oxides. Am. J. Pathol. 1996, 148, 1625–1638. [Google Scholar] [PubMed]
- Maxfield, F.R.; Tabas, I. Role of cholesterol and lipid organization in disease. Nature 2005, 438, 612–621. [Google Scholar] [CrossRef] [PubMed]
- Prunet, C.; Montange, T.; Véjux, A.; Laubriet, A.; Rohmer, J.F.; Riedinger, J.M.; Athias, A.; Lemaire Ewing, S.; Néel, D.; Petit, J.M.; et al. Multiplexed flow cytometric analyses of pro- and anti-inflammatory cytokines in the culture media of oxysterol-treated human monocytic cells and in the sera of atherosclerotic patients. Cytom. Part A 2006, 69A, 359–373. [Google Scholar] [CrossRef] [PubMed]
- Lemaire-Ewing, S.; Prunet, C.; Montange, T.; Vejux, A.; Berthier, A.; Bessède, G.; Corcos, L.; Gambert, P.; Néel, D.; Lizard, G. Comparison of the cytotoxic, pro-oxidant and pro-inflammatory characteristics of different oxysterols. Cell. Biol. Toxicol. 2005, 21, 97–114. [Google Scholar] [CrossRef] [PubMed]
- Rosenblat, M.; Aviram, M. Oxysterol-induced activation of macrophage NADPH-oxidase enhances cell-mediated oxidation of LDL in the atherosclerotic apolipoprotein E deficient mouse: Inhibitory role for vitamin E. Atherosclerosis 2002, 160, 69–80. [Google Scholar] [CrossRef]
- Selley, M.L.; McGuiness, J.A.; Ardlie, N.G. The effect of cholesterol oxidation products on human platelet aggregation. Thromb. Res. 1996, 83, 449–461. [Google Scholar] [CrossRef]
- Olkkonen, V.M.; Lehto, M. Oxysterols and oxysterol binding proteins: Role in lipid metabolism and atherosclerosis. Ann. Med. 2009, 36, 562–572. [Google Scholar] [CrossRef]
- Iuliano, L.; Micheletta, F.; Natoli, S.; Ginanni Corradini, S.; Iappelli, M.; Elisei, W.; Giovannelli, L.; Violi, F.; Diczfalusy, U. Measurement of oxysterols and alpha-tocopherol in plasma and tissue samples as indices of oxidant stress status. Anal. Biochem. 2003, 312, 217–223. [Google Scholar] [CrossRef]
- Bochkov, V.N.; Oskolkova, O.V.; Birukov, K.G.; Levonen, A.-L.; Binder, C.J.; Stöckl, J. Generation and biological activities of oxidized phospholipids. Antioxid. Redox Signal. 2010, 12, 1009–1059. [Google Scholar] [CrossRef] [PubMed]
- Radi, R.; Beckman, J.S.; Bush, K.M.; Freeman, B.A. Peroxynitrite-induced membrane lipid peroxidation: The cytotoxic potential of superoxide and nitric oxide. Arch. Biochem. Biophys. 1991, 288, 481–487. [Google Scholar] [CrossRef]
- Jerlich, A.; Pitt, A.R.; Schaur, R.J.; Spickett, C.M. Pathways of phospholipid oxidation by HOCl in human LDL detected by LC-MS. Free Radic. Biol. Med. 2000, 28, 673–682. [Google Scholar] [CrossRef]
- Carr, A.C.; Winterbourn, C.C.; van den Berg, J.J. Peroxidase-mediated bromination of unsaturated fatty acids to form bromohydrins. Arch. Biochem. Biophys. 1996, 327, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, H.; Belkner, J.; Wiesner, R.; Brash, A.R. Oxygenation of biological membranes by the pure reticulocyte lipoxygenase. J. Biol. Chem. 1990, 265, 18351–18361. [Google Scholar] [PubMed]
- Belkner, J.; Wiesner, R.; Rathman, J.; Barnett, J.; Sigal, E.; Kuhn, H. Oxygenation of lipoproteins by mammalian lipoxygenases. Eur. J. Biochem. 1993, 213, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Heydeck, D.; Upston, J.M.; Viita, H.; Ylä-Herttuala, S.; Stocker, R. Oxidation of LDL by rabbit and human 15-lipoxygenase: Prevalence of nonenzymatic reactions. J. Lipid Res. 2001, 42, 1082–1088. [Google Scholar] [PubMed]
- Berliner, J.A.; Leitinger, N.; Tsimikas, S. The role of oxidized phospholipids in atherosclerosis. J. Lipid Res. 2009, 50, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Cole, A.L.; Subbanagounder, G.; Mukhopadhyay, S.; Berliner, J.A.; Vora, D.K. Oxidized phospholipid-induced endothelial cell/monocyte interaction is mediated by a cAMP-dependent R-Ras/PI3-kinase pathway. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1384–1390. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Mouillesseaux, K.P.; Montoya, D.; Cruz, D.; Gharavi, N.; Dun, M.; Koroniak, L.; Berliner, J.A. Identification of prostaglandin E2 receptor subtype 2 as a receptor activated by OxPAPC. Circ. Res. 2006, 98, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Vora, D.K.; Fang, Z.T.; Liva, S.M.; Tyner, T.R.; Parhami, F.; Watson, A.D.; Drake, T.A.; Territo, M.C.; Berliner, J.A. Induction of P-selectin by oxidized lipoproteins. Separate effects on synthesis and surface expression. Circ. Res. 1997, 80, 810–818. [Google Scholar] [CrossRef] [PubMed]
- Dever, G.J.; Benson, R.; Wainwright, C.L.; Kennedy, S.; Spickett, C.M. Phospholipid chlorohydrin induces leukocyte adhesion to ApoE−/− mouse arteries via upregulation of P-selectin. Free Radic. Biol. Med. 2008, 44, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Rouhanizadeh, M.; Hwang, J.; Clempus, R.E.; Marcu, L.; Lassègue, B.; Sevanian, A.; Hsiai, T.K. Oxidized-1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine induces vascular endothelial superoxide production: Implication of NADPH oxidase. Free Radic. Biol. Med. 2005, 39, 1512–1522. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, E.A.; Li, H.; Lawson, J.A.; Rokach, J.; FitzGerald, G.A.; Axelsen, P.H. Prothrombinase acceleration by oxidatively damaged phospholipids. J. Biol. Chem. 2000, 275, 22925–22930. [Google Scholar] [CrossRef] [PubMed]
- Safa, O.; Hensley, K.; Smirnov, M.D.; Esmon, C.T.; Esmon, N.L. Lipid oxidation enhances the function of activated protein C. J. Biol. Chem. 2001, 276, 1829–1836. [Google Scholar] [CrossRef] [PubMed]
- Bochkov, V.N.; Mechtcheriakova, D.; Lucerna, M.; Huber, J.; Malli, R.; Graier, W.F.; Hofer, E.; Binder, B.R.; Leitinger, N. Oxidized phospholipids stimulate tissue factor expression in human endothelial cells via activation of ERK/EGR-1 and Ca++/NFAT. Blood 2002, 99, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Haserück, N.; Erl, W.; Pandey, D.; Tigyi, G.; Ohlmann, P.; Ravanat, C.; Gachet, C.; Siess, W. The plaque lipid lysophosphatidic acid stimulates platelet activation and platelet-monocyte aggregate formation in whole blood: Involvement of P2Y1 and P2Y12 receptors. Blood 2004, 103, 2585–2592. [Google Scholar] [CrossRef] [PubMed]
- Marathe, G.K.; Zimmerman, G.A.; Prescott, S.M.; McIntyre, T.M. Activation of vascular cells by PAF-like lipids in oxidized LDL. Vascul. Pharmacol. 2002, 38, 193–200. [Google Scholar] [CrossRef]
- Subbanagounder, G.; Leitinger, N.; Shih, P.T.; Faull, K.F.; Berliner, J.A. Evidence that phospholipid oxidation products and/or platelet-activating factor play an important role in early atherogenesis: In vitro and In vivo inhibition by WEB 2086. Circ. Res. 1999, 85, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Tokumura, A.; Sumida, T.; Toujima, M.; Kogure, K.; Fukuzawa, K. Platelet-activating factor (PAF)-like oxidized phospholipids: Relevance to atherosclerosis. Biofactors 2000, 13, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Deigner, H.-P.; Hermetter, A. Oxidized phospholipids: Emerging lipid mediators in pathophysiology. Curr. Opin. Lipidol. 2008, 19, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Podrez, E.A. Identification of a novel family of oxidized phospholipids that serve as ligands for the macrophage scavenger receptor CD36. J. Biol. Chem. 2002, 277, 38503–38516. [Google Scholar] [CrossRef] [PubMed]
- Hazen, S.L. Oxidized phospholipids as endogenous pattern recognition ligands in innate immunity. J. Biol. Chem. 2008, 283, 15527–15531. [Google Scholar] [CrossRef] [PubMed]
- Frostegård, J.; Svenungsson, E.; Wu, R.; Gunnarsson, I.; Lundberg, I.E.; Klareskog, L.; Hörkkö, S.; Witztum, J.L. Lipid peroxidation is enhanced in patients with systemic lupus erythematosus and is associated with arterial and renal disease manifestations. Arthritis Rheum. 2005, 52, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Marnett, L.J. Lipid peroxidation—DNA damage by malondialdehyde. Mutat. Res. 1999, 424, 83–95. [Google Scholar] [CrossRef]
- VanderVeen, L.A.; Hashim, M.F.; Shyr, Y.; Marnett, L.J. Induction of frameshift and base pair substitution mutations by the major DNA adduct of the endogenous carcinogen malondialdehyde. PNAS 2003, 100, 14247–14252. [Google Scholar] [CrossRef] [PubMed]
- Uchida, K. Role of reactive aldehyde in cardiovascular diseases. Free Radic. Biol. Med. 2000, 28, 1685–1696. [Google Scholar] [CrossRef]
- Palinski, W.; Ord, V.A.; Plump, A.S.; Breslow, J.L.; Steinberg, D.; Witztum, J.L. ApoE-deficient mice are a model of lipoprotein oxidation in atherogenesis. Demonstration of oxidation-specific epitopes in lesions and high titers of autoantibodies to malondialdehyde-lysine in serum. Arterioscler. Thromb. Vasc. Biol. 1994, 14, 605–616. [Google Scholar] [CrossRef]
- Del Rio, D.; Stewart, A.J.; Pellegrini, N. A review of recent studies on malondialdehyde as toxic molecule and biological marker of oxidative stress. Nutr. Metabol. Cardiovasc. Dis. 2005, 15, 316–328. [Google Scholar] [CrossRef] [PubMed]
- De Vecchi, A.F.; Bamonti, F.; Novembrino, C.; Ippolito, S.; Guerra, L.; Lonati, S.; Salini, S.; Aman, C.S.; Scurati-Manzoni, E.; Cighetti, G. Free and total plasma malondialdehyde in chronic renal insufficiency and in dialysis patients. Nephrol. Dial. Transplant. 2009, 24, 2524–2529. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.I.; Saglam, M.; Caglar, K.; Cakir, E.; Sonmez, A.; Ozgurtas, T.; Aydin, A.; Eyileten, T.; Ozcan, O.; Acikel, C.; et al. The determinants of endothelial dysfunction in CKD: Oxidative stress and asymmetric dimethylarginine. Am. J. Kidney Dis. 2006, 47, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Capusa, C.; Stoian, I.; Rus, E.; Lixandru, D.; Barbulescu, C.; Mircescu, G. Does dialysis modality influence the oxidative stress of uremic patients? Kidney Blood Press. Res. 2012, 35, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Caimi, G.; Carollo, C.; Montana, M.; Iatrino, R.; Bondì, B.; Lo Presti, R. Nitric oxide metabolites, leukocyte activation markers and oxidative status in dialyzed subjects. Blood Purif. 2009, 27, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Dirican, M.; Sarandol, E.; Serdar, Z.; Ocak, N.; Dilek, K. Oxidative status and prevalent cardiovascular disease in patients with chronic renal failure treated by hemodialysis. Clin. Nephrol. 2007, 68, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Bober, J.; Kedzierska, K.; Kwiatkowska, E.; Stachowska, E.; Gołembiewska, E.; Mazur, O.; Staniewicz, Z.; Ciechanowski, K.; Chlubek, D. Does oxidative stress affect the activity of the sodium-proton exchanger? Ann. Acad. Med. Stetin. 2010, 56, 5–12. [Google Scholar] [PubMed]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Petersen, D.R.; Doorn, J.A. Reactions of 4-hydroxynonenal with proteins and cellular targets. Free Radic. Biol. Med. 2004, 37, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Siems, W.G.; Hapner, S.J.; Van Kuijk, F.J. 4-Hydroxynonenal inhibits Na(+)-K(+)-ATPase. Free Radic. Biol. Med. 1996, 20, 215–223. [Google Scholar] [CrossRef]
- Kristal, B.S.; Park, B.K.; Yu, B.P. 4-Hydroxyhexenal is a potent inducer of the mitochondrial permeability transition. J. Biol. Chem. 1996, 271, 6033–6038. [Google Scholar] [PubMed]
- Chen, J.J.; Yu, B.P. Alterations in mitochondrial membrane fluidity by lipid peroxidation products. Free Radic. Biol. Med. 1994, 17, 411–418. [Google Scholar] [CrossRef]
- Uchida, K.; Itakura, K.; Kawakishi, S.; Hiai, H.; Toyokuni, S.; Stadtman, E.R. Characterization of Epitopes Recognized by 4-Hydroxy-2-nonenal Specific Antibodies. Arch. Biochem. Biophys. 1995, 324, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Hoff, H.F.; O’Neil, J.; Chisolm, G.M.; Cole, T.B.; Quehenberger, O.; Esterbauer, H.; Jürgens, G. Modification of low density lipoprotein with 4-hydroxynonenal induces uptake by macrophages. Arterioscler. Thromb. Vasc. Biol. 1989, 9, 538–549. [Google Scholar] [CrossRef]
- Ruef, J.; Rao, G.N.; Li, F.; Bode, C.; Patterson, C.; Bhatnagar, A.; Runge, M.S. Induction of Rat Aortic Smooth Muscle Cell Growth by the Lipid Peroxidation Product 4-Hydroxy-2-Nonenal. Circulation 1998, 97, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Herbst, U.; Toborek, M.; Kaiser, S.; Mattson, M.P.; Hennig, B. 4-Hydroxynonenal induces dysfunction and apoptosis of cultured endothelial cells. J. Cell. Physiol. 1999, 181, 295–303. [Google Scholar] [CrossRef]
- Go, Y.-M.; Halvey, P.J.; Hansen, J.M.; Reed, M.; Pohl, J.; Jones, D.P. Reactive aldehyde modification of thioredoxin-1 activates early steps of inflammation and cell adhesion. Am. J. Pathol. 2007, 171, 1670–1681. [Google Scholar] [CrossRef] [PubMed]
- Piroddi, M.; Bartolini, D.; Ciffolilli, S.; Galli, F. Nondialyzable uremic toxins. Blood Purif. 2013, 35, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Sommerburg, O.; Grune, T.; Hampl, H.; Riedel, E.; van Kuijk, F.J.; Ehrich, J.H.; Siems, W.G. Does long-term treatment of renal anaemia with recombinant erythropoietin influence oxidative stress in haemodialysed patients? Nephrol. Dial. Transplant. 1998, 13, 2583–2587. [Google Scholar] [CrossRef] [PubMed]
- Alhamdani, M.S.S.; Al-Kassir, A.H.A.M.; Jaleel, N.A.; Hmood, A.M.; Ali, H.M. Elevated levels of Alkanals, alkenals and 4-ho-alkenals in plasma of hemodialysis patients. Am. J. Nephrol. 2006, 26, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Bae, E.H.; Cho, S.; Joo, S.Y.; Ma, S.K.; Kim, S.H.; Lee, J.; Kim, S.W. 4-Hydroxy-2-hexenal-induced apoptosis in human renal proximal tubular epithelial cells. Nephrol. Dial. Transplant. 2011, 26, 3866–3873. [Google Scholar] [CrossRef] [PubMed]
- Morrow, J.D.; Hill, K.E.; Burk, R.F.; Nammour, T.M.; Badr, K.F.; Roberts, L.J., 2nd. A series of prostaglandin F2-like compounds are produced in vivo in humans by a non-cyclooxygenase, free radical-catalyzed mechanism. Proc. Natl. Acad. Sci. USA 1990, 87, 9383–9387. [Google Scholar] [CrossRef] [PubMed]
- Roberts, L.J., II; Morrow, J.D. Measurement of F2-isoprostanes as an index of oxidative stress in vivo. Free Radic. Biol. Med. 2000, 28, 505–513. [Google Scholar] [CrossRef]
- Roberts, L.J., II; Fessel, J.P. The biochemistry of the isoprostane, neuroprostane, and isofuran pathways of lipid peroxidation. Chem. Phys. Lipids 2004, 128, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Morrow, J.D.; Minton, T.A.; Roberts, L.J. The F2-isoprostane, 8-epi-prostaglandin F2α, a potent agonist of the vascular thromboxane/endoperoxide receptor, is a platelet thromboxane/endoperoxide receptor antagonist. Prostaglandins 1992, 44, 155–163. [Google Scholar] [CrossRef]
- Yura, T.; Fukunaga, M.; Khan, R.; Nassar, G.N.; Badr, K.F. Free-radical-generated F2-isoprostane stimulates cell proliferation and endothelin-1 expression on endothelial cells. Kidney Int. 1999, 56, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Fukunaga, M.; Makita, N.; Roberts, L.J.; Morrow, J.D.; Takahashi, K.; Badr, K.F. Evidence for the existence of F2-isoprostane receptors on rat vascular smooth muscle cells. Am. J. Physiol. 1993, 264, 1619–1624. [Google Scholar]
- Takahashi, K.; Nammour, T.M.; Fukunaga, M.; Ebert, J.; Morrow, J.D.; Roberts, L.J., 2nd; Hoover, R.L.; Badr, K.F. Glomerular actions of a free radical-generated novel prostaglandin, 8-epi-prostaglandin F2 alpha, in the rat. Evidence for interaction with thromboxane A2 receptors. J. Clin. Investig. 1992, 90, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Reckelhoff, J.F.; Kanji, V.; Racusen, L.C.; Schmidt, A.M.; Yan, S.D.; Morrow, J.; Roberts, L.J.; Salahudeen, A.K. Vitamin E ameliorates enhanced renal lipid peroxidation and accumulation of F2-isoprostanes in aging kidneys. Am. J. Physiol. 1998, 274, 767–774. [Google Scholar]
- Cottone, S.; Lorito, M.C.; Riccobene, R.; Nardi, E. Oxidative stress, inflammation and cardiovascular disease in chronic renal failure. J. Nephrol. 2008, 21, 175–179. [Google Scholar] [PubMed]
- Lee, C.-Y.J.; Huang, S.H.; Jenner, A.M.; Halliwell, B. Measurement of F2-isoprostanes, hydroxyeicosatetraenoic products, and oxysterols from a single plasma sample. Free Radic. Biol. Med. 2008, 44, 1314–1322. [Google Scholar] [CrossRef] [PubMed]
- Ramezani, M.; Nazemian, F.; Shamsara, J.; Koohrokhi, R.; Mohammadpour, A.H. Effect of Ω-3 fatty acids on plasma level of 8-isoprostane in kidney transplant patients. J. Ren. Nutr. 2011, 21, 196–199. [Google Scholar] [CrossRef] [PubMed]
- Simmons, E.M.; Langone, A.; Sezer, M.T.; Vella, J.P.; Recupero, P.; Morrow, J.D.; Ikizler, T.A.; Himmelfarb, J. Effect of renal transplantation on biomarkers of inflammation and oxidative stress in end-stage renal disease patients. Transplantation 2005, 79, 914–919. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Normal | CKD | Clearance | HD Behavior | References | |
|---|---|---|---|---|---|
| Oxysterols (Total) | Liver metabolism | Generated during HD session | [113,114] | ||
| 7-ketocholesterol, nM | 32.3 ± 16.7 | 42.2 ± 30.1 δ | |||
| 7β-OH-cholesterol, nM | 14.4 ± 7.7 | 42.6 ± 24.1 δ | |||
| Oxidized Phospholipids | Enzymatic detoxification | Reduced after HD session | [56,103,115] | ||
| OxPL/ApoB ratio, AU | 0.068 ± 0.07 | 0.138 * ± 0.170 * | |||
| PUFAs Aldehydes | [7,9,23,116,117,118] [9,116,119,120,121] | ||||
| Malondialdehyde (MDA), μg/L | 257.7 ± 81.7 | 388.8 ± 21.6 δ | Enzymatic detoxification Renal excretion | Controversial (decrease, no change and increase) | |
| 4-hydroxy-decenal, μg/L | 10.3 ± 7.1 | 36.6 ± 22.3 δ | Enzymatic detoxification, rennal excretion | 4-HNE: Reduced after HD session | |
| 4-hydroxy-2-hexenal (4-HHE), μg/L | 25.1 ± 9.9 | 63.8 ± 25.3 δ | |||
| 4-hydroxy-2-nonenal (4-HNE), μg/L | 16.4 ± 9.0 | 117.3 ± 47.7 δ | |||
| 4-hydroxy-octenal, μg/L | 10.7 ± 3.6 | 27.8 ± 13.8 δ | |||
| Arachidonic Acid By-Products of Lipid Peroxidation | Renal excretion, Enzymatic detoxification | No change | [119,122,123,124,125,126,127,128] | ||
| Total F2-isoprostanes, pg/mL * | 162 ± 73 | 270 ± 10 δ | |||
| Unesterified F2-isoprostanes, pg/mL | 37.6 ± 17.2 | 96.2 ± 48.8 δ | |||
| Esterified F2-isoprostanes, pg/mL | 146.8 ± 58.4 | 220.4 ± 154.8 δ | |||
| Lipoprotein Products | |||||
| ApoB48 level, mg/L | 3.7 ± 2.3 | 19.3 ± 13.9 δ | [33] | ||
| Oxidized LDL, mg/L | 0.22 ± 0.05 | 1.92 ± 0.29 δ | Accumulation in atherosclerotic lesions | Increased after HD session | [53,54,55,56] |
| 3-chlorotyrosine, μmol/mol of tyrosine | <0.3 | 3.5 ± 0.5 δ | - | - | [64] |
| Lp(a) level, mg/dL | 18.4 ± 22.8 | 23.4 ± 34.6 δ | Renal and hepatic clearance | No changes or increased after HD session | [56,109,110,111] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Florens, N.; Calzada, C.; Lyasko, E.; Juillard, L.; Soulage, C.O. Modified Lipids and Lipoproteins in Chronic Kidney Disease: A New Class of Uremic Toxins. Toxins 2016, 8, 376. https://doi.org/10.3390/toxins8120376
Florens N, Calzada C, Lyasko E, Juillard L, Soulage CO. Modified Lipids and Lipoproteins in Chronic Kidney Disease: A New Class of Uremic Toxins. Toxins. 2016; 8(12):376. https://doi.org/10.3390/toxins8120376
Chicago/Turabian StyleFlorens, Nans, Catherine Calzada, Egor Lyasko, Laurent Juillard, and Christophe O. Soulage. 2016. "Modified Lipids and Lipoproteins in Chronic Kidney Disease: A New Class of Uremic Toxins" Toxins 8, no. 12: 376. https://doi.org/10.3390/toxins8120376