
Immunotoxin Therapies for the Treatment of Epidermal Growth Factor Receptor-Dependent Cancers

Abstract
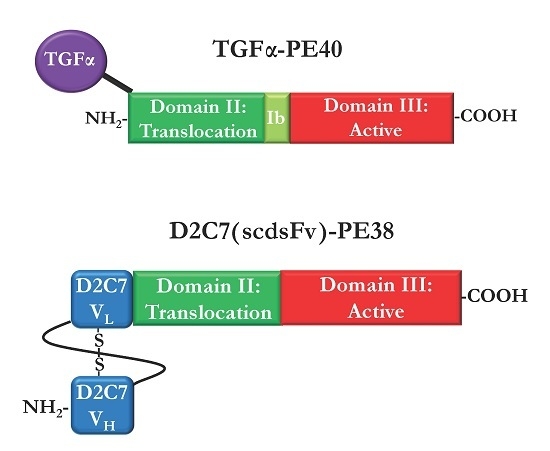
:
1. Introduction
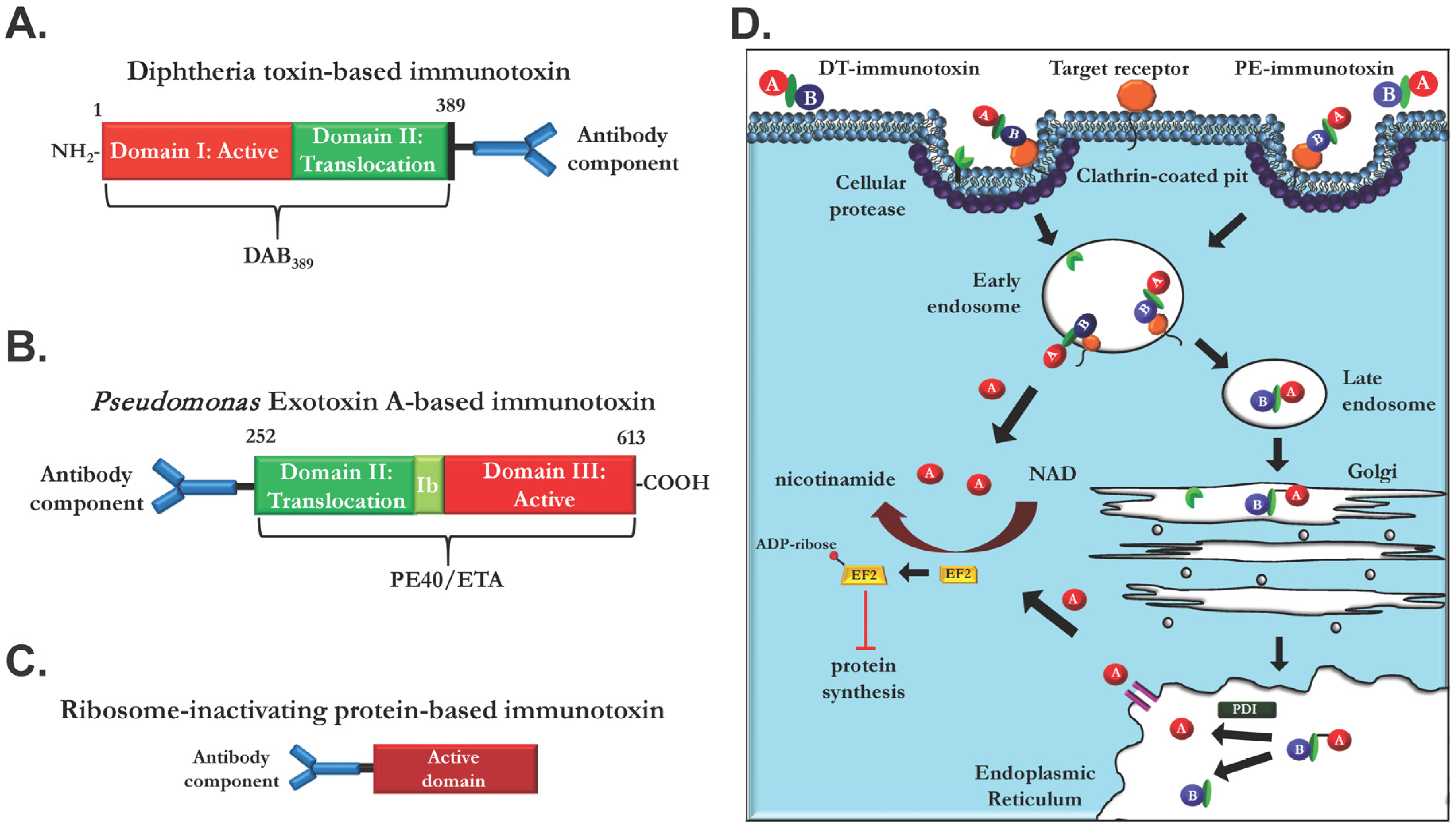
2. Literature
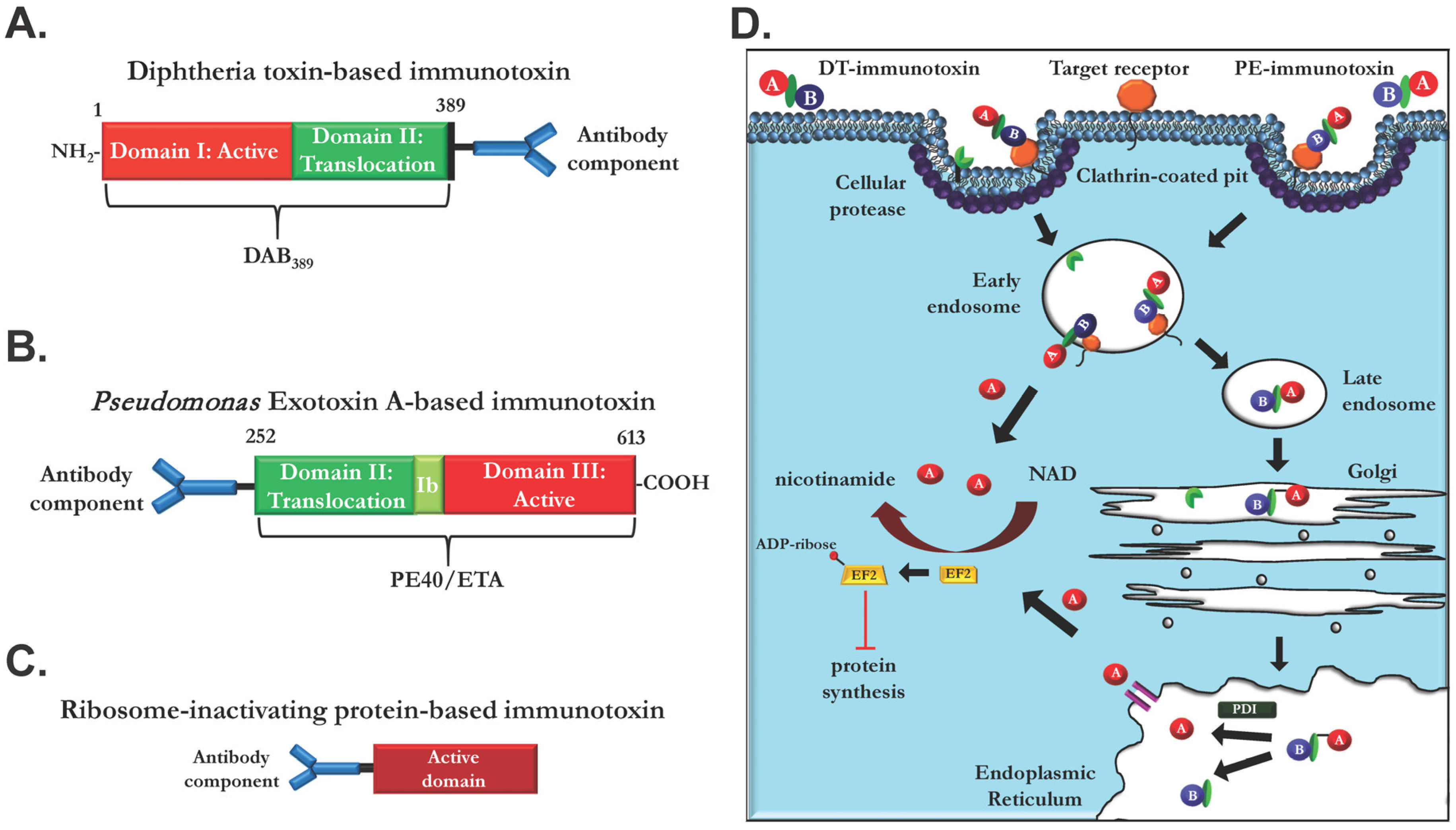
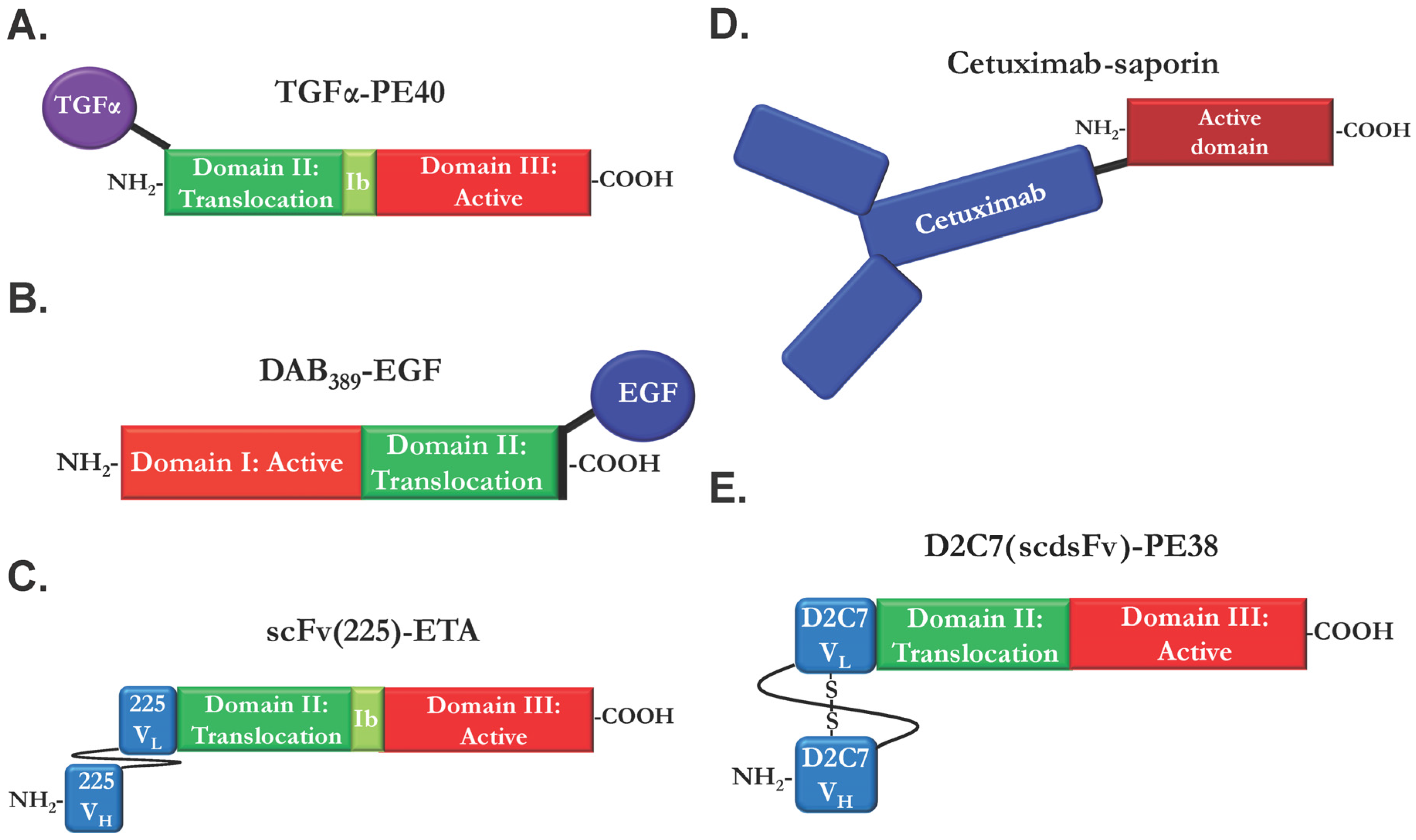
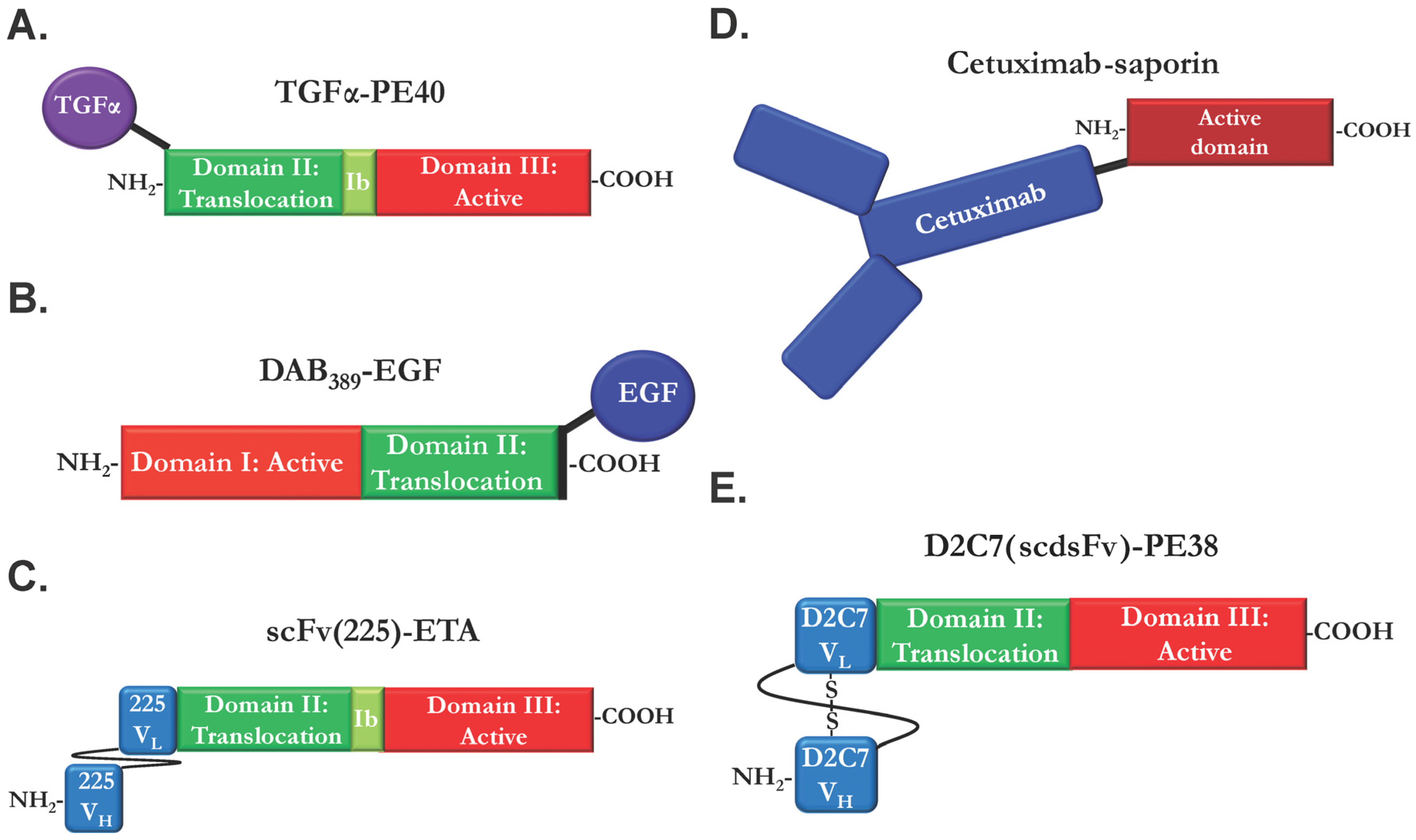
2.1. Ligand Immunotoxins
2.2. Immunotoxins Based on Monoclonal EGFR Antibodies
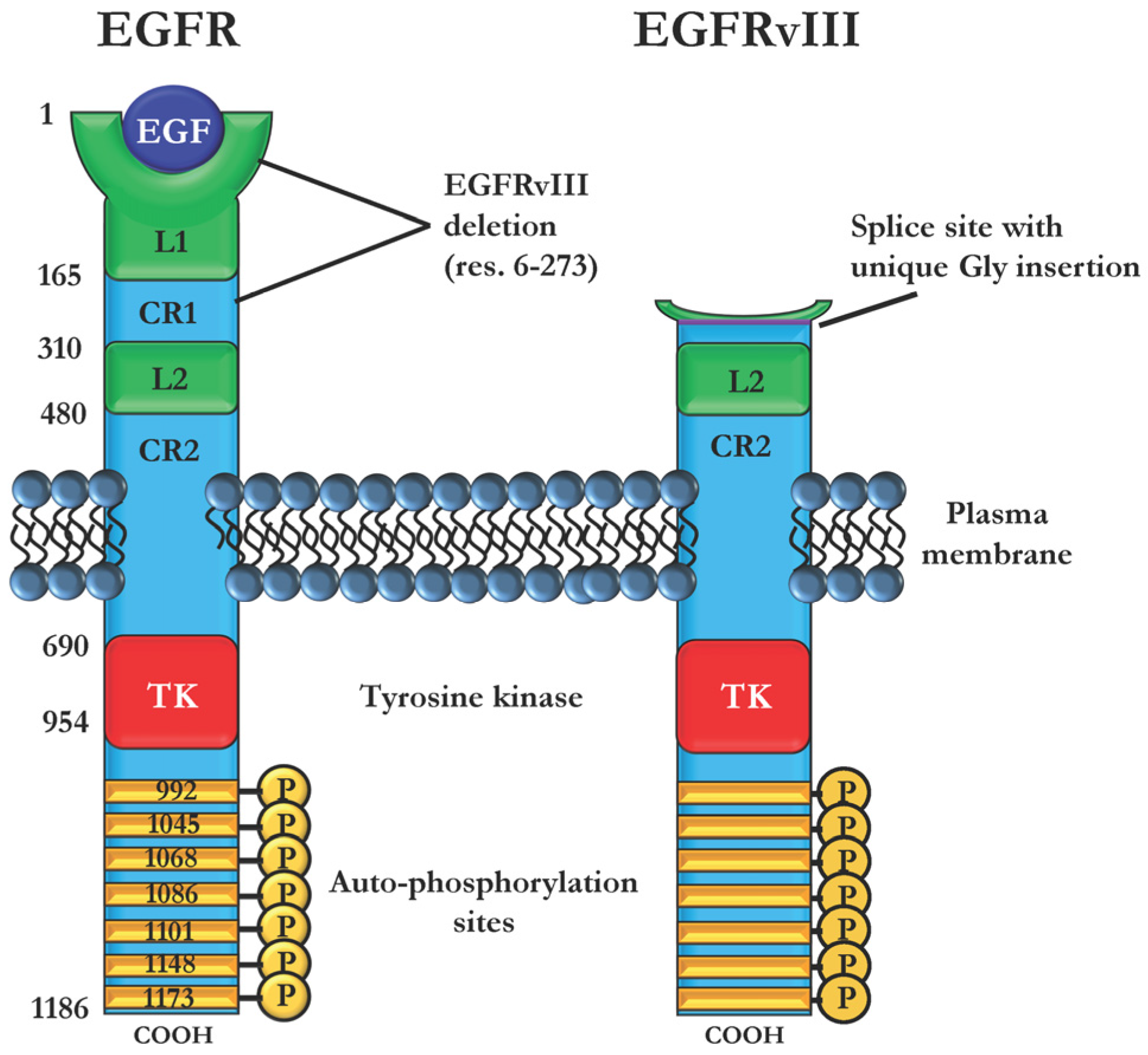
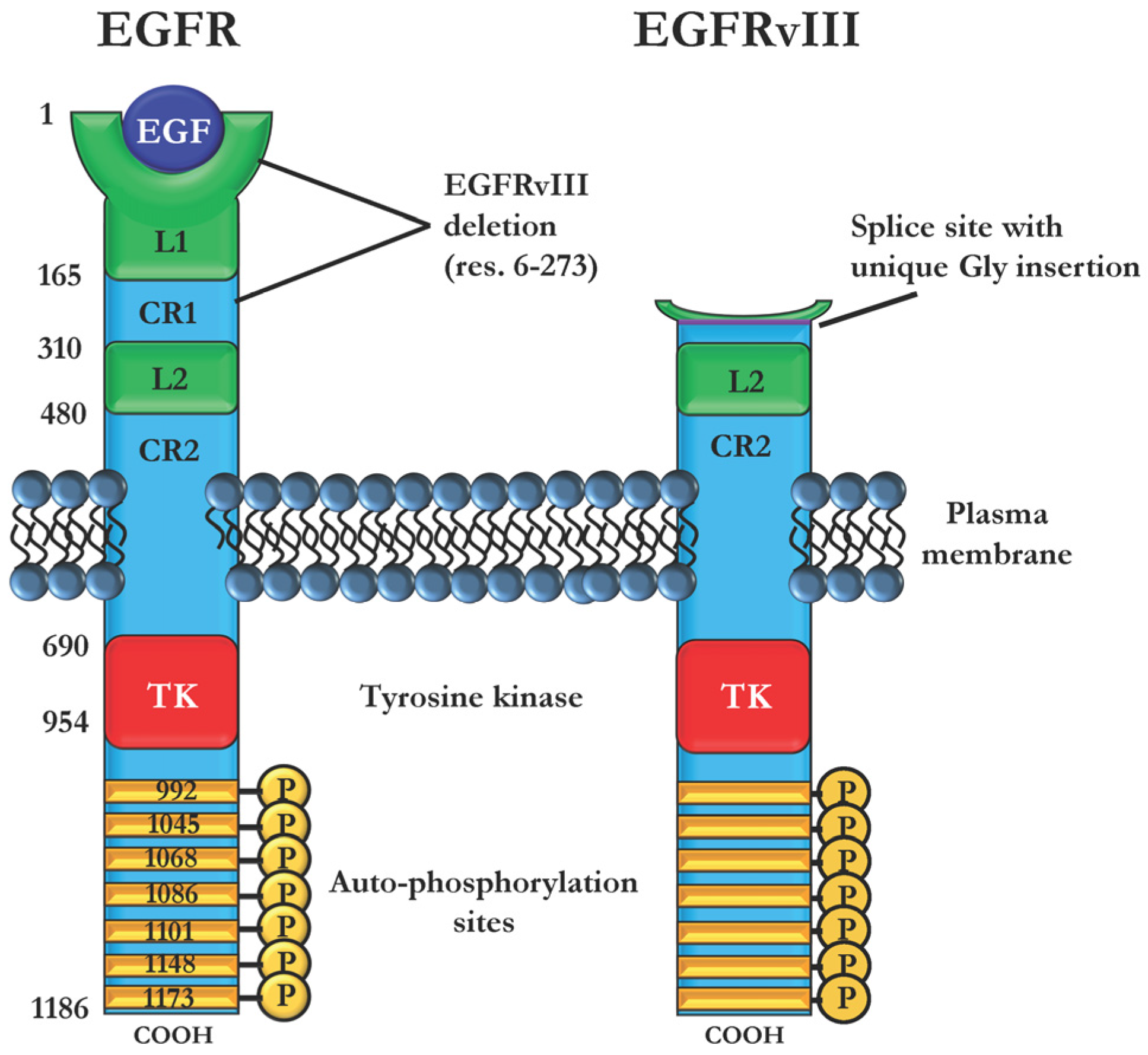
2.3. Immunotoxins Targeting Cancers Expressing Mutant EGFRvIII
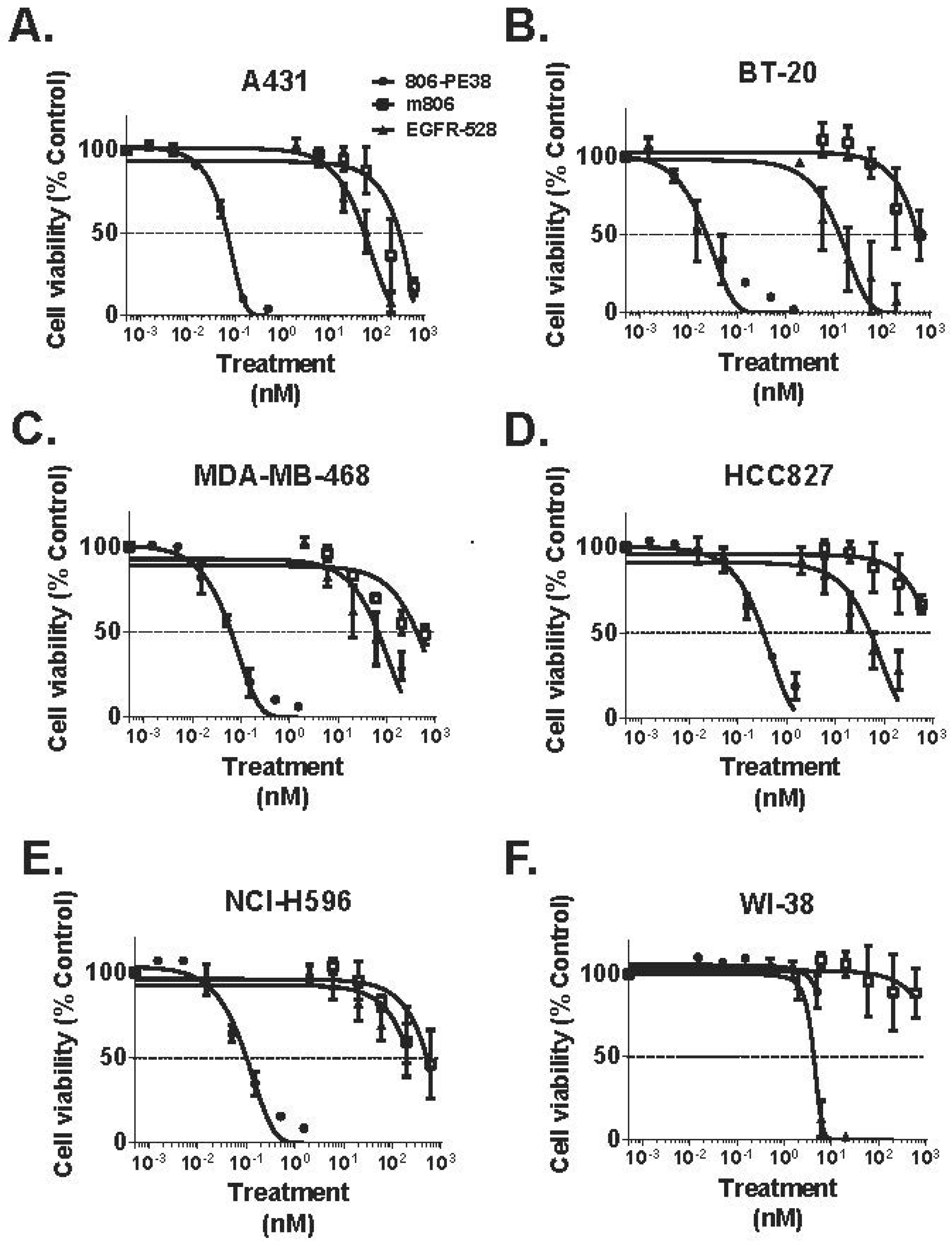
2.4. Bispecific and Conformation-Dependent Immunotoxins
2.5. Photochemical Delivery of Anti-EGFR Immunotoxins
3. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| EGFR | epidermal growth factor receptor |
| IT | immunotoxin |
| scFv | single-chain variable fragment |
| PE | Pseudomonas exotoxin |
| DT | diphtheria toxin |
| EGF | epidermal growth factor |
| TGFα | transforming growth factor alpha |
| HB-EGF | heparin binding-EGF |
| TKI | tyrosine kinase inhibitor |
| TNBC | triple-negative breast cancer |
| NSCLC | non-small cell lung cancer |
| EF2 | elongation factor 2 |
| RIP | ribosome-inactivating proteins |
| MAb | monoclonal antibody |
| NK cell | natural killer cell |
| CED | convection enhanced delivery |
| PCI | photochemical internalization |
References
- Chong, C.R.; Janne, P.A. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat. Med. 2013, 19, 1389–1400. [Google Scholar] [CrossRef] [PubMed]
- Hynes, N.E.; Lane, H.A. ERBB receptors and cancer: The complexity of targeted inhibitors. Nat. Rev. Cancer 2005, 5, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Scaltriti, M.; Baselga, J. The Epidermal Growth Factor Receptor Pathway: A Model for Targeted Therapy. Clin. Cancer Res. 2006, 12, 5268–5272. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Jänne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR Mutation and Resistance of Non–Small-Cell Lung Cancer to Gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating Mutations in the Epidermal Growth Factor Receptor Underlying Responsiveness of Non–Small-Cell Lung Cancer to Gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.K.; Cvrljevic, A.N.; Johns, T.G. The epidermal growth factor receptor variant III (EGFRvIII): Where wild things are altered. FEBS J. 2013, 280, 5350–5370. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, R.; Ji, X.D.; Harmon, R.C.; Lazar, C.S.; Gill, G.N.; Cavenee, W.K.; Huang, H.J. A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc. Natl. Acad. Sci. USA 1994, 91, 7727–7731. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Babic, I.; Nathanson, D.; Akhavan, D.; Guo, D.; Gini, B.; Dang, J.; Zhu, S.; Yang, H.; De Jesus, J.; et al. Oncogenic EGFR Signaling Activates an mTORC2–NF-κB Pathway That Promotes Chemotherapy Resistance. Cancer Discov. 2011, 1, 524–538. [Google Scholar] [CrossRef] [PubMed]
- Nagane, M.; Levitzki, A.; Gazit, A.; Cavenee, W.K.; Huang, H.-J.S. Drug resistance of human glioblastoma cells conferred by a tumor-specific mutant epidermal growth factor receptor through modulation of Bcl-XL and caspase-3-like proteases. Proc. Natl. Acad. Sci. USA 1998, 95, 5724–5729. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Jang, M.H.; Kim, E.J.; Kim, H.J.; Lee, H.J.; Kim, Y.J.; Kim, J.H.; Kang, E.; Kim, S.-W.; Kim, I.A.; et al. High EGFR gene copy number predicts poor outcome in triple-negative breast cancer. Mod. Pathol. 2014, 27, 1212–1222. [Google Scholar] [CrossRef] [PubMed]
- Lemoine, N.R.; Hughes, C.M.; Barton, C.M.; Poulsom, R.; Jeffery, R.E.; Klöppel, G.; Hall, P.A.; Gullick, W.J. The Epidermal Growth Factor Receptor in Human Pancreatic Cancer. J. Pathol. 1992, 166, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Korc, M.; Chandrasekar, B.; Yamanaka, Y.; Friess, H.; Buchier, M.; Beger, H.G. Overexpression of the epidermal growth factor receptor in human pancreatic cancer is associated with concomitant increases in the levels of epidermal growth factor and transforming growth factor alpha. J. Clin. Investig. 1992, 90, 1352–1360. [Google Scholar] [CrossRef] [PubMed]
- Grandis, J.R.; Melhem, M.F.; Barnes, E.L.; Tweardy, D.J. Quantitative immunohistochemical analysis of transforming growth factor-α and epidermal growth factor receptor in patients with squamous cell carcinoma of the head and neck. Cancer 1996, 78, 1284–1292. [Google Scholar] [CrossRef]
- Hirsch, F.R.; Scagliotti, G.V.; Langer, C.J.; Varella-Garcia, M.; Franklin, W.A. Epidermal growth factor family of receptors in preneoplasia and lung cancer: Perspectives for targeted therapies. Lung Cancer 2003, 41 (Suppl. 1), 29–42. [Google Scholar] [CrossRef]
- Rao, J.Y.; Hemstreet, G.P.; Hurst, R.E.; Bonner, R.B.; Jones, P.L.; Min, K.W.; Fradet, Y. Alterations in phenotypic biochemical markers in bladder epithelium during tumorigenesis. Proc. Natl. Acad. Sci. USA 1993, 90, 8287–8291. [Google Scholar] [CrossRef] [PubMed]
- Neal, D.; Bennett, M.; Hall, R.; Marsh, C.; Abel, P.; Sainsbury, J.R.C.; Harris, A. Epidermal-growth-factor receptors in human bladder cancer: Comparison of invasive and superficial tumours. Lancet 1985, 325, 366–368. [Google Scholar] [CrossRef]
- Wikstrand, C.J.; Hale, L.P.; Batra, S.K.; Hill, M.L.; Humphrey, P.A.; Kurpad, S.N.; McLendon, R.E.; Moscatello, D.; Pegram, C.N.; Reist, C.J.; et al. Monoclonal Antibodies against EGFRvIII Are Tumor Specific and React with Breast and Lung Carcinomas and Malignant Gliomas. Cancer Res. 1995, 55, 3140–3148. [Google Scholar] [PubMed]
- Wikstrand, C.J.; McLendon, R.E.; Friedman, A.H.; Bigner, D.D. Cell Surface Localization and Density of the Tumor-associated Variant of the Epidermal Growth Factor Receptor, EGFRvIII. Cancer Res. 1997, 57, 4130–4140. [Google Scholar] [PubMed]
- Arteaga, C.L.; Engelman, J.A. ERBB Receptors: From Oncogene Discovery to Basic Science to Mechanism-Based Cancer Therapeutics. Cancer Cell 2014, 25, 282–303. [Google Scholar]
- Salomon, D.S.; Brandt, R.; Ciardiello, F.; Normanno, N. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit. Rev. Oncol. Hematol. 1995, 19, 183–232. [Google Scholar] [CrossRef]
- Nicholson, R.I.; Gee, J.M.W.; Harper, M.E. EGFR and cancer prognosis. Eur. J. Cancer 2001, 37 (Suppl. 4), 9–15. [Google Scholar] [CrossRef]
- Martinelli, E.; De Palma, R.; Orditura, M.; De Vita, F.; Ciardiello, F. Anti-epidermal growth factor receptor monoclonal antibodies in cancer therapy. Clin. Exp. Immunol. 2009, 158, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Eriksen, J.G.; Maare, C.; Johansen, J.; Primdahl, H.; Evensen, J.F.; Kristensen, C.A.; Andersen, L.J.; Overgaard, J. Evaluation of the EGFR-Inhibitor Zalutumumab Given with Primary Curative (Chemo)radiation Therapy to Patients with Squamous Cell Carcinoma of the Head and Neck: Results of the DAHANCA 19 Randomized Phase 3 Trial. Int. J. Radiat. Oncol. Biol. Phys. 2014, 88, 465. [Google Scholar] [CrossRef]
- Westphal, M.; Heese, O.; Steinbach, J.P.; Schnell, O.; Schackert, G.; Mehdorn, M.; Schulz, D.; Simon, M.; Schlegel, U.; Senft, C.; et al. A randomised, open label phase III trial with nimotuzumab, an anti-epidermal growth factor receptor monoclonal antibody in the treatment of newly diagnosed adult glioblastoma. Eur. J. Cancer 2015, 51, 522–532. [Google Scholar] [CrossRef] [PubMed]
- Schiller, J.H.; von Pawel, J.; Schütt, P.; Ansari, R.H.; Thomas, M.; Saleh, M.; McCroskey, R.D.; Pfeifer, W.; Marsland, T.A.; Kloecker, G.H.; et al. Pemetrexed with or without Matuzumab as Second-Line Treatment for Patients with Stage IIIB/IV Non-small Cell Lung Cancer. J. Thoracic Oncol. 2010, 5, 1977–1985. [Google Scholar] [CrossRef] [PubMed]
- Seiden, M.V.; Burris, H.A.; Matulonis, U.; Hall, J.B.; Armstrong, D.K.; Speyer, J.; Weber, J.D.A.; Muggia, F. A phase II trial of EMD72000 (matuzumab), a humanized anti-EGFR monoclonal antibody, in patients with platinum-resistant ovarian and primary peritoneal malignancies. Gynecol. Oncol. 2007, 104, 727–731. [Google Scholar] [CrossRef] [PubMed]
- Beidler, C.B.; Petrovan, R.J.; Conner, E.M.; Boyles, J.S.; Yang, D.D.; Harlan, S.M.; Chu, S.; Ellis, B.; Datta-Mannan, A.; Johnson, R.L.; et al. Generation and Activity of a Humanized Monoclonal Antibody That Selectively Neutralizes the Epidermal Growth Factor Receptor Ligands Transforming Growth Factor-α and Epiregulin. J. Pharmacol. Exp. Ther. 2014, 349, 330–343. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Iwamoto, R.; Furuya, A.; Takahashi, K.; Sasaki, Y.; Ando, H.; Yotsumoto, F.; Yoneda, T.; Hamaoka, M.; Yagi, H.; et al. A Novel Anti-Human HB-EGF Monoclonal Antibody with Multiple Antitumor Mechanisms against Ovarian Cancer Cells. Clin. Cancer Res. 2011, 17, 6733–6741. [Google Scholar] [CrossRef] [PubMed]
- Lindzen, M.; Lavi, S.; Leitner, O.; Yarden, Y. Tailored cancer immunotherapy using combinations of chemotherapy and a mixture of antibodies against EGF-receptor ligands. Proc. Natl. Acad. Sci. USA 2010, 107, 12559–12563. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.; Wu, Y.-L.; Thongprasert, S.; Yang, C.-H.; Chu, D.-T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or Carboplatin–Paclitaxel in Pulmonary Adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, F.A.; Rodrigues Pereira, J.; Ciuleanu, T.; Tan, E.H.; Hirsh, V.; Thongprasert, S.; Campos, D.; Maoleekoonpiroj, S.; Smylie, M.; Martins, R.; et al. Erlotinib in Previously Treated Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2005, 353, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Goldstein, D.; Hamm, J.; Figer, A.; Hecht, J.R.; Gallinger, S.; Au, H.J.; Murawa, P.; Walde, D.; Wolff, R.A.; et al. Erlotinib Plus Gemcitabine Compared With Gemcitabine Alone in Patients With Advanced Pancreatic Cancer: A Phase III Trial of the National Cancer Institute of Canada Clinical Trials Group. Chin. Ger. J. Clin. Oncol. 2007, 25, 1960–1966. [Google Scholar] [CrossRef] [PubMed]
- Geyer, C.E.; Forster, J.; Lindquist, D.; Chan, S.; Romieu, C.G.; Pienkowski, T.; Jagiello-Gruszfeld, A.; Crown, J.; Chan, A.; Kaufman, B.; et al. Lapatinib plus Capecitabine for HER2-Positive Advanced Breast Cancer. N. Engl. J. Med. 2006, 355, 2733–2743. [Google Scholar] [CrossRef] [PubMed]
- Su, K.-Y.; Chen, H.-Y.; Li, K.-C.; Kuo, M.-L.; Yang, J.C.-H.; Chan, W.-K.; Ho, B.-C.; Chang, G.-C.; Shih, J.-Y.; Yu, S.-L.; et al. Pretreatment Epidermal Growth Factor Receptor (EGFR) T790M Mutation Predicts Shorter EGFR Tyrosine Kinase Inhibitor Response Duration in Patients With Non–Small-Cell Lung Cancer. Chin. Ger. J. Clin. Oncol. 2012, 30, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Cross, D.A.E.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.V.; Ward, R.A.; Mellor, M.J.; et al. AZD9291, an Irreversible EGFR TKI, Overcomes T790M-Mediated Resistance to EGFR Inhibitors in Lung Cancer. Cancer Discov. 2014, 4, 1046–1061. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.C.-H.; Wu, Y.-L.; Schuler, M.; Sebastian, M.; Popat, S.; Yamamoto, N.; Zhou, C.; Hu, C.-P.; O’Byrne, K.; Feng, J.; et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): Analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol. 2015, 16, 141–151. [Google Scholar] [CrossRef]
- Walter, A.O.; Sjin, R.T.T.; Haringsma, H.J.; Ohashi, K.; Sun, J.; Lee, K.; Dubrovskiy, A.; Labenski, M.; Zhu, Z.; Wang, Z.; et al. Discovery of a Mutant-Selective Covalent Inhibitor of EGFR that Overcomes T790M-Mediated Resistance in NSCLC. Cancer Discov. 2013, 3, 1404–1415. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Lee, J.-S.; Lee, K.H.; Kim, J.-H.; Min, Y.J.; Cho, J.Y.; Han, J.-Y.; Kim, B.-S.; Kim, J.-S.; Lee, D.H.; et al. Updated safety and efficacy results from phase I/II study of HM61713 in patients (pts) with EGFR mutation positive non-small cell lung cancer (NSCLC) who failed previous EGFR-tyrosine kinase inhibitor (TKI). In Proceedings of ASCO Annual Meeting, Chicago, IL, USA, 29 May–2 June 2015; p. 8084.
- Nathanson, D.A.; Gini, B.; Mottahedeh, J.; Visnyei, K.; Koga, T.; Gomez, G.; Eskin, A.; Hwang, K.; Wang, J.; Masui, K.; et al. Targeted Therapy Resistance Mediated by Dynamic Regulation of Extrachromosomal Mutant EGFR DNA. Science 2014, 343, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Widakowich, C.; de Castro, G.; de Azambuja, E.; Dinh, P.; Awada, A. Review: Side Effects of Approved Molecular Targeted Therapies in Solid Cancers. Oncologist 2007, 12, 1443–1455. [Google Scholar] [CrossRef] [PubMed]
- Blythman, H.E.; Casellas, P.; Gros, O.; Gros, P.; Jansen, F.K.; Paolucci, F.; Pau, B.; Vidal, H. Immunotoxins: Hybrid molecules of monoclonal antibodies and a toxin subunit specifically kill tumour cells. Nature 1981, 290, 145–146. [Google Scholar] [CrossRef] [PubMed]
- Antignani, A.; Fitzgerald, D. Immunotoxins: The role of the toxin. Toxins 2013, 5, 1486–1502. [Google Scholar] [CrossRef] [PubMed]
- Collier, R.J. Effect of diphtheria toxin on protein synthesis: Inactivation of one of the transfer factors. J. Mol. Biol. 1967, 25, 83–98. [Google Scholar] [CrossRef]
- Iglewski, B.H.; Liu, P.V.; Kabat, D. Mechanism of action of Pseudomonas aeruginosa exotoxin A: Adenosine diphosphate ribosylation of mammalian elongation factor 2 in vitro and in vivo. Infect. Immun. 1977, 15, 138–144. [Google Scholar] [PubMed]
- Morimoto, H.; Bonavida, B. Diphtheria toxin- and Pseudomonas A toxin-mediated apoptosis. ADP ribosylation of elongation factor-2 is required for DNA fragmentation and cell lysis and synergy with tumor necrosis factor-alpha. J. Immunol. 1992, 149, 2089–2094. [Google Scholar] [PubMed]
- Sandvig, K.; Olsnes, S. Diphtheria toxin entry into cells is facilitated by low pH. J. Cell Biol. 1980, 87, 828–832. [Google Scholar] [CrossRef] [PubMed]
- Weldon, J.E.; Pastan, I. A guide to taming a toxin--recombinant immunotoxins constructed from Pseudomonas exotoxin A for the treatment of cancer. FEBS J. 2011, 278, 4683–4700. [Google Scholar] [CrossRef] [PubMed]
- Yamaizumi, M.; Mekada, E.; Uchida, T.; Okada, Y. One molecule of diphtheria toxin fragment A introduced into a cell can kill the cell. Cell 1978, 15, 245–250. [Google Scholar] [CrossRef]
- Yokota, T.; Milenic, D.E.; Whitlow, M.; Schlom, J. Rapid Tumor Penetration of a Single-Chain Fv and Comparison with Other Immunoglobulin Forms. Cancer Res. 1992, 52, 3402–3408. [Google Scholar] [PubMed]
- Sandvig, K.; van Deurs, B. Delivery into cells: Lessons learned from plant and bacterial toxins. Gene Ther. 2005, 12, 865–872. [Google Scholar] [CrossRef] [PubMed]
- Banker, D.E.; Pastan, I.; Gottesman, M.M.; Herschman, H.R. An epidermal growth factor-ricin a chain (EGF-RTA)-resistant mutant and an epidermal growth factor-Pseudomonas endotoxin (EGF-PE)-resistant mutant have distinct phenotypes. J. Cell. Physiol. 1989, 139, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Lindstrom, A.L.; Erlandsen, S.L.; Kersey, J.H.; Pennell, C.A. An in vitro model for toxin-mediated vascular leak syndrome: ricin toxin A chain increases the permeability of human endothelial cell monolayers. Blood 1997, 90, 2323–2334. [Google Scholar] [PubMed]
- Hassan, R.; Bullock, S.; Premkumar, A.; Kreitman, R.J.; Kindler, H.; Willingham, M.C.; Pastan, I. Phase I study of SS1P, a recombinant anti-mesothelin immunotoxin given as a bolus I.V. infusion to patients with mesothelin-expressing mesothelioma, ovarian, and pancreatic cancers. Clin. Cancer Res. 2007, 13, 5144–5149. [Google Scholar] [CrossRef] [PubMed]
- Onda, M.; Ghoreschi, K.; Steward-Tharp, S.; Thomas, C.; O’Shea, J.J.; Pastan, I.H.; FitzGerald, D.J. Tofacitinib suppresses antibody responses to protein therapeutics in murine hosts. J. Immunol. 2014, 193, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Hassan, R.; Miller, A.C.; Sharon, E.; Thomas, A.; Reynolds, J.C.; Ling, A.; Kreitman, R.J.; Miettinen, M.M.; Steinberg, S.M.; Fowler, D.H.; et al. Major cancer regressions in mesothelioma after treatment with an anti-mesothelin immunotoxin and immune suppression. Sci. Transl. Med. 2013, 5, 208ra147. [Google Scholar] [CrossRef] [PubMed]
- Onda, M.; Beers, R.; Xiang, L.; Nagata, S.; Wang, Q.-C.; Pastan, I. An immunotoxin with greatly reduced immunogenicity by identification and removal of B cell epitopes. Proc. Natl. Acad. Sci. USA 2008, 105, 11311–11316. [Google Scholar] [CrossRef] [PubMed]
- Mazor, R.; Crown, D.; Addissie, S.; Jang, Y.; Kaplan, G.; Pastan, I. Elimination of murine and human T-cell epitopes in recombinant immunotoxin eliminates neutralizing and anti-drug antibodies in vivo. Cell Mol. Immunol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Olsen, E.; Duvic, M.; Frankel, A.; Kim, Y.; Martin, A.; Vonderheid, E.; Jegasothy, B.; Wood, G.; Gordon, M.; Heald, P.; et al. Pivotal phase III trial of two dose levels of denileukin diftitox for the treatment of cutaneous T-cell lymphoma. J. Clin. Oncol. 2001, 19, 376–388. [Google Scholar] [PubMed]
- Hassan, R.; Sharon, E.; Thomas, A.; Zhang, J.; Ling, A.; Miettinen, M.; Kreitman, R.J.; Steinberg, S.M.; Hollevoet, K.; Pastan, I. Phase 1 study of the antimesothelin immunotoxin SS1P in combination with pemetrexed and cisplatin for front-line therapy of pleural mesothelioma and correlation of tumor response with serum mesothelin, megakaryocyte potentiating factor, and cancer antigen 125. Cancer 2014, 120, 3311–3319. [Google Scholar] [PubMed]
- Kreitman, R.J.; Tallman, M.S.; Robak, T.; Coutre, S.; Wilson, W.H.; Stetler-Stevenson, M.; Fitzgerald, D.J.; Lechleider, R.; Pastan, I. Phase I trial of anti-CD22 recombinant immunotoxin moxetumomab pasudotox (CAT-8015 or HA22) in patients with hairy cell leukemia. J. Clin. Oncol. 2012, 30, 1822–1828. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, V.K.; FitzGerald, D.J.; Adhya, S.; Pastan, I. Activity of a recombinant fusion protein between transforming growth factor type alpha and Pseudomonas toxin. Proc. Natl. Acad. Sci. USA 1987, 84, 4538–4542. [Google Scholar] [CrossRef] [PubMed]
- Thakur, M.; Mergel, K.; Weng, A.; von Mallinckrodt, B.; Gilabert-Oriol, R.; Dürkop, H.; Melzig, M.F.; Fuchs, H. Targeted tumor therapy by epidermal growth factor appended toxin and purified saponin: An evaluation of toxicity and therapeutic potential in syngeneic tumor bearing mice. Mol. Oncol. 2013, 7, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Wels, W.; Beerli, R.; Hellmann, P.; Schmidt, M.; Marte, B.M.; Kornilova, E.S.; Hekele, A.; Mendelsohn, J.; Groner, B.; Hynes, N.E. EGF receptor and p185erbB-2-specific single-chain antibody toxins differ in their cell-killing activity on tumor cells expressing both receptor proteins. Int. J. Cancer 1995, 60, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Bruell, D.; Stöcker, M.; Huhn, M.; Redding, N.; Küpper, M.; Schumacher, P.; Paetz, A.; Bruns, C.J.; Haisma, H.J.; Fischer, R.; et al. The recombinant anti-EGF receptor immunotoxin 425(scFv)-ETA′ suppresses growth of a highly metastatic pancreatic carcinoma cell line. Int. J. Oncol. 2003, 23, 1179–1186. [Google Scholar] [PubMed]
- Niesen, J.; Stein, C.; Brehm, H.; Hehmann-Titt, G.; Fendel, R.; Melmer, G.; Fischer, R.; Barth, S. Novel EGFR-specific immunotoxins based on panitumumab and cetuximab show in vitro and ex vivo activity against different tumor entities. J. Cancer Res. Clin. Oncol. 2015, 141, 2079–2095. [Google Scholar] [CrossRef] [PubMed]
- Gilabert-Oriol, R.; Thakur, M.; von Mallinckrodt, B.; Hug, T.; Wiesner, B.; Eichhorst, J.; Melzig, M.F.; Fuchs, H.; Weng, A. Modified Trastuzumab and Cetuximab Mediate Efficient Toxin Delivery While Retaining Antibody-Dependent Cell-Mediated Cytotoxicity in Target Cells. Mol. Pharm. 2013, 10, 4347–4357. [Google Scholar] [CrossRef] [PubMed]
- Gilabert-Oriol, R.; Weng, A.; Trautner, A.; Weise, C.; Schmid, D.; Bhargava, C.; Niesler, N.; Wookey, P.J.; Fuchs, H.; Thakur, M. Combinatorial approach to increase efficacy of Cetuximab, Panitumumab and Trastuzumab by dianthin conjugation and co-application of SO1861. Biochem. Pharmacol. 2015, 97, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Lorimer, I.A.; Wikstrand, C.J.; Batra, S.K.; Bigner, D.D.; Pastan, I. Immunotoxins that target an oncogenic mutant epidermal growth factor receptor expressed in human tumors. Clin. Cancer Res. 1995, 1, 859–864. [Google Scholar] [PubMed]
- Lorimer, I.A.; Keppler-Hafkemeyer, A.; Beers, R.A.; Pegram, C.N.; Bigner, D.D.; Pastan, I. Recombinant immunotoxins specific for a mutant epidermal growth factor receptor: Targeting with a single chain antibody variable domain isolated by phage display. Proc. Natl. Acad. Sci. USA 1996, 93, 14815–14820. [Google Scholar] [CrossRef] [PubMed]
- Simon, N.; Antignani, A.; Sarnovsky, R.; Hewitt, S.M.; FitzGerald, D. Targeting a Cancer-Specific Epitope of the Epidermal Growth Factor Receptor in Triple-Negative Breast Cancer. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Liu, Y.; Gao, S.; Lin, S.; Gu, X.; Pomper, M.G.; Wang, P.C.; Shan, L. A bivalent recombinant immunotoxin with high potency against tumors with EGFR and EGFRvIII expression. Cancer Biol. Ther. 2015, 16, 1764–1774. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Vakalopoulou, E.; Schneider, D.W.; Wels, W. Construction and functional characterization of scFv(14E1)-ETA—A novel, highly potent antibody-toxin specific for the EGF receptor. Br. J. Cancer 1997, 75, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Sampson, J.H.; Akabani, G.; Archer, G.E.; Bigner, D.D.; Berger, M.S.; Friedman, A.H.; Friedman, H.S.; Herndon, J.E., 2nd; Kunwar, S.; Marcus, S.; et al. Progress report of a Phase I study of the intracerebral microinfusion of a recombinant chimeric protein composed of transforming growth factor (TGF)-alpha and a mutated form of the Pseudomonas exotoxin termed PE-38 (TP-38) for the treatment of malignant brain tumors. J. Neurooncol. 2003, 65, 27–35. [Google Scholar]
- Sampson, J.H.; Akabani, G.; Archer, G.E.; Berger, M.S.; Coleman, R.E.; Friedman, A.H.; Friedman, H.S.; Greer, K.; Herndon, J.E., 2nd; Kunwar, S.; et al. Intracerebral infusion of an EGFR-targeted toxin in recurrent malignant brain tumors. Neuro. Oncol. 2008, 10, 320–329. [Google Scholar]
- Theodoulou, M.; Baselga, J.; Scher, H.; Dantis, L.; Trainor, K.; Mendelsohn, J.; Howes, L.; Elledge, R.; Ravdin, P.; Bacha, P.; et al. Phase I dose-escalation study of the safety, tolerability, pharmacokinetics and biologic effects of DAB389EGF in patients with solid malignancies that express EGF receptors (EGFR). Proc. Am. Soc. Clin. Oncol. 1995, 14, 480. [Google Scholar]
- Bigner, D. Study of Immunotoxin, MR1–1 (MR1–1). Available online: https://clinicaltrials.gov/ct2/show/NCT01009866 (accessed on 22 March 2016).
- Bigner, D. D2C7 for Adult Patients With Recurrent Malignant Glioma. Available online: https://clinicaltrials.gov/ct2/show/NCT02303678 (accessed on 22 March 2016).
- Johannes, L.; Decaudin, D. Protein toxins: Intracellular trafficking for targeted therapy. Gene Ther. 2005, 12, 1360–1368. [Google Scholar] [CrossRef] [PubMed]
- Heimbrook, D.C.; Stirdivant, S.M.; Ahern, J.D.; Balishin, N.L.; Patrick, D.R.; Edwards, G.M.; Defeo-Jones, D.; FitzGerald, D.J.; Pastan, I.; Oliff, A. Transforming growth factor alpha-Pseudomonas exotoxin fusion protein prolongs survival of nude mice bearing tumor xenografts. Proc. Natl. Acad. Sci. USA 1990, 87, 4697–4701. [Google Scholar] [CrossRef] [PubMed]
- Pai, L.H.; Gallo, M.G.; FitzGerald, D.J.; Pastan, I. Antitumor Activity of a Transforming Growth Factor α-Pseudomonas Exotoxin Fusion Protein (TGF-α-PE40). Cancer Res. 1991, 51, 2808–2812. [Google Scholar] [PubMed]
- Kihara, A.; Pastan, I. Small Chimeric Toxins Containing Only Transforming Growth Factor α and Domain III of Pseudomonas Exotoxin with Good Antitumor Activity in Mice. Cancer Res. 1994, 54, 5154–5159. [Google Scholar] [PubMed]
- Theuer, C.P.; FitzGerald, D.J.; Pastan, I. A recombinant form of Pseudomonas exotoxin A containing transforming growth factor alpha near its carboxyl terminus for the treatment of bladder cancer. J. Urol. 1993, 149, 1626–1632. [Google Scholar] [PubMed]
- Phillips, P.C.; Levow, C.; Catterall, M.; Colvin, O.M.; Pastan, I.; Brem, H. Transforming growth factor-alpha-Pseudomonas exotoxin fusion protein (TGF-alpha-PE38) treatment of subcutaneous and intracranial human glioma and medulloblastoma xenografts in athymic mice. Cancer Res. 1994, 54, 1008–1015. [Google Scholar] [PubMed]
- Shaw, J.P.; Akiyoshi, D.E.; Arrigo, D.A.; Rhoad, A.E.; Sullivan, B.; Thomas, J.; Genbauffe, F.S.; Bacha, P.; Nichols, J.C. Cytotoxic properties of DAB486EGF and DAB389EGF, epidermal growth factor (EGF) receptor-targeted fusion toxins. J. Biol. Chem. 1991, 266, 21118–21124. [Google Scholar] [PubMed]
- Zalutsky, M.R.; Boskovitz, A.; Kuan, C.-T.; Pegram, C.N.; Ayriss, J.; Wikstrand, C.J.; Buckley, A.F.; Lipp, E.S.; Herndon, J.E., II; McLendon, R.E.; et al. Radioimmunotargeting of malignant glioma by monoclonal antibody D2C7 reactive against both wild-type and variant III mutant epidermal growth factor receptors. Nucl. Med. Biol. 2012, 39, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Mellon, J.K.; Cook, S.; Chambers, P.; Neal, D.E. Transforming growth factor alpha and epidermal growth factor levels in bladder cancer and their relationship to epidermal growth factor receptor. Br. J. Cancer 1996, 73, 654–658. [Google Scholar] [CrossRef] [PubMed]
- Sarosdy, M.F.; Hutzler, D.H.; Yee, D.; Von Hoff, D.D. In vitro sensitivity testing of human bladder cancers and cell lines to TP-40, a hybrid protein with selective targeting and cytotoxicity. J. Urol. 1993, 150, 1950–1955. [Google Scholar] [PubMed]
- Kameyama, S.; Kawamata, H.; Pastan, I.; Oyasu, R. Cytotoxic effect of a fusion protein from transforming growth factor alpha and Pseudomonas exotoxin on rat and human bladder carcinoma cells in vitro. J. Cancer Res. Clin. Oncol. 1994, 120, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, M.R.; Heimbrook, D.C.; Russo, P.; Sarosdy, M.F.; Greenberg, R.E.; Giantonio, B.J.; Linehan, W.M.; Walther, M.; Fisher, H.A.; Messing, E. Phase I clinical study of the recombinant oncotoxin TP40 in superficial bladder cancer. Clin. Cancer Res. 1995, 1, 57–61. [Google Scholar] [PubMed]
- Cawley, D.B.; Herschman, H.R.; Gary Gilliland, D.; John Collier, R. Epidermal growth factor-toxin A chain conjugates: EGF-Ricin A is a potent toxin while EGF-diphtheria fragment A is nontoxic. Cell 1980, 22, 563–570. [Google Scholar] [CrossRef]
- Yang, X.; Kessler, E.; Su, L.-J.; Thorburn, A.; Frankel, A.E.; Li, Y.; La Rosa, F.G.; Shen, J.; Li, C.-Y.; Varella-Garcia, M.; et al. Diphtheria Toxin-Epidermal Growth Factor Fusion Protein DAB(389)EGF for the Treatment of Bladder Cancer. Clin. Cance. Res. 2013, 19, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.F.; Cohen, K.A.; Ramage, J.G.; Willingham, M.C.; Thorburn, A.M.; Frankel, A.E. A Diphtheria Toxin-Epidermal Growth Factor Fusion Protein Is Cytotoxic to Human Glioblastoma Multiforme Cells. Cancer Res. 2003, 63, 1834–1837. [Google Scholar] [PubMed]
- Liu, T.F.; Hall, P.D.; Cohen, K.A.; Willingham, M.C.; Cai, J.; Thorburn, A.; Frankel, A.E. Interstitial Diphtheria Toxin-Epidermal Growth Factor Fusion Protein Therapy Produces Regressions of Subcutaneous Human Glioblastoma Multiforme Tumors in Athymic Nude Mice. Clin. Cancer Res. 2005, 11, 329–334. [Google Scholar] [PubMed]
- Huang, J.; Li, Y.M.; Massague, J.; Sicheneder, A.; Vallera, D.A.; Hall, W.A. Intracerebral infusion of the bispecific targeted toxin DTATEGF in a mouse xenograft model of a human metastatic non-small cell lung cancer. J. Neuro-Oncol. 2012, 109, 229–238. [Google Scholar] [CrossRef] [PubMed]
- FitzGerald, D.J.; Padmanabhan, R.; Pastan, I.; Willingham, M.C. Adenovirus-induced release of epidermal growth factor and pseudomonas toxin into the cytosol of KB cells during receptor-mediated endocytosis. Cell 1983, 32, 607–617. [Google Scholar] [CrossRef]
- Chandler, L.A.; Sosnowski, B.A.; McDonald, J.R.; Price, J.E.; Aukerman, S.L.; Baird, A.; Pierce, G.F.; Houston, L.L. Targeting tumor cells via EGF receptors: Selective toxicity of an HBEGF-toxin fusion protein. Int. J. Cancer 1998, 78, 106–111. [Google Scholar] [CrossRef]
- Chen, C.; Li, J.; Micko, C.J.; Pierce, G.F.; Cunningham, M.R.; Lumsden, A.B. Cytotoxic Effects of Basic FGF and Heparin Binding EGF Conjugated with Cytotoxin Saporin on Vascular Cell Cultures. J. Surg. Res. 1998, 75, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.M.; Wolchok, J.D.; Old, L.J. Antibody therapy of cancer. Nat. Rev. Cancer 2012, 12, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Van Gog, F.B.; Brakenhoff, R.H.; Stigter-van Walsum, M.; Snow, G.B.; van Dongen, G.A.M.S. Perspectives of combined radioimmunotherapy and anti-EGFR antibody therapy for the treatment of residual head and neck cancer. Int. J. Cancer 1998, 77, 13–18. [Google Scholar] [CrossRef]
- Perrotte, P.; Matsumoto, T.; Inoue, K.; Kuniyasu, H.; Eve, B.Y.; Hicklin, D.J.; Radinsky, R.; Dinney, C.P.N. Anti-epidermal Growth Factor Receptor Antibody C225 Inhibits Angiogenesis in Human Transitional Cell Carcinoma Growing Orthotopically in Nude Mice. Clin. Cancer Res. 1999, 5, 257–264. [Google Scholar] [PubMed]
- Baselga, J.; Norton, L.; Masui, H.; Pandiella, A.; Coplan, K.; Miller, W.H.; Mendelsohn, J. Antitumor Effects of Doxorubicin in Combination With Anti-epidermal Growth Factor Receptor Monoclonal Antibodies. J. Natl. Cancer Inst. 1993, 85, 1327–1333. [Google Scholar] [CrossRef]
- Fan, Z.; Baselga, J.; Masui, H.; Mendelsohn, J. Antitumor Effect of Anti-Epidermal Growth Factor Receptor Monoclonal Antibodies plus cis-Diamminedichloroplatinum on Well Established A431 Cell Xenografts. Cancer Res. 1993, 53, 4637–4642. [Google Scholar] [PubMed]
- Sato, J.D.; Kawamoto, T.; Le, A.D.; Mendelsohn, J.; Polikoff, J.; Sato, G.H. Biological effects in vitro of monoclonal antibodies to human epidermal growth factor receptors. Mol. Biol. Med. 1983, 1, 511–529. [Google Scholar] [PubMed]
- Sunada, H.; Magun, B.E.; Mendelsohn, J.; MacLeod, C.L. Monoclonal antibody against epidermal growth factor receptor is internalized without stimulating receptor phosphorylation. Proc. Natl. Acad. Sci. USA 1986, 83, 3825–3829. [Google Scholar] [CrossRef] [PubMed]
- Azemar, M.; Schmidt, M.; Arlt, F.; Kennel, P.; Brandt, B.; Papadimitriou, A.; Groner, B.; Wels, W. Recombinant antibody toxins specific for ErbB2 and EGF receptor inhibit the in vitro growth of human head and neck cancer cells and cause rapid tumor regression in vivo. Int. J. Cancer 2000, 86, 269–275. [Google Scholar] [CrossRef]
- Murthy, U.; Basu, A.; Rodeck, U.; Herlyn, M.; Ross, A.H.; Das, M. Binding of an antagonistic monoclonal antibody to an intact and fragmented EGF-receptor polypeptide. Arch. Biochem. Biophys. 1987, 252, 549–560. [Google Scholar] [CrossRef]
- Kamat, V.; Donaldson, J.M.; Kari, C.; Quadros, M.R.D.; Lelkes, P.I.; Chaiken, I.; Cocklin, S.; Williams, J.C.; Papazoglou, E.; Rodeck, U. Enhanced EGFR inhibition and distinct epitope recognition by EGFR antagonistic MABS C225 and 425. Cancer Biol. Ther. 2008, 7, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Rodeck, U.; Herlyn, M.; Herlyn, D.; Molthoff, C.; Atkinson, B.; Varello, M.; Steplewski, Z.; Koprowski, H. Tumor Growth Modulation by a Monoclonal Antibody to the Epidermal Growth Factor Receptor: Immunologically Mediated and Effector Cell-independent Effects. Cancer Res. 1987, 47, 3692–3696. [Google Scholar] [PubMed]
- Bruell, D.; Bruns, C.J.; Yezhelyev, M.; Huhn, M.; Müller, J.; Ischenko, I.; Fischer, R.; Finnern, R.; Jauch, K.; Barth, S. Recombinant anti-EGFR immunotoxin 425(scFv)-ETA′ demonstrates anti-tumor activity against disseminated human pancreatic cancer in nude mice. Int. J. Mol. Med. 2005, 15, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Niesen, J.; Brehm, H.; Stein, C.; Berges, N.; Pardo, A.; Fischer, R.; Haaf, A.; Gattenlöhner, S.; Tur, M.K.; Barth, S. In vitro effects and ex vivo binding of an EGFR-specific immunotoxin on rhabdomyosarcoma cells. J. Cancer Res. Clin. Oncol. 2014, 141, 1049–1061. [Google Scholar] [CrossRef] [PubMed]
- Galizia, G.; Lieto, E.; De Vita, F.; Orditura, M.; Castellano, P.; Troiani, T.; Imperatore, V.; Ciardiello, F. Cetuximab, a chimeric human mouse anti-epidermal growth factor receptor monoclonal antibody, in the treatment of human colorectal cancer. Oncogene 2007, 26, 3654–3660. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-D.; Jia, X.-C.; Corvalan, J.R.F.; Wang, P.; Davis, C.G.; Jakobovits, A. Eradication of Established Tumors by a Fully Human Monoclonal Antibody to the Epidermal Growth Factor Receptor without Concomitant hemotherapy. Cancer Res. 1999, 59, 1236–1243. [Google Scholar] [PubMed]
- Voigt, M.; Braig, F.; Göthel, M.; Schulte, A.; Lamszus, K.; Bokemeyer, C.; Binder, M. Functional Dissection of the Epidermal Growth Factor Receptor Epitopes Targeted by Panitumumab and Cetuximab. Neoplasia 2012, 14, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Erbe, A.K.; Hank, J.A.; Morris, Z.S.; Sondel, P.M. NK cell mediated antibody-dependent cellular cytotoxicity in cancer immunotherapy. Front. Immunol. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Clynes, R.A.; Towers, T.L.; Presta, L.G.; Ravetch, J.V. Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets. Nat. Med. 2000, 6, 443–446. [Google Scholar] [PubMed]
- Weng, A.; Thakur, M.; von Mallinckrodt, B.; Beceren-Braun, F.; Gilabert-Oriol, R.; Wiesner, B.; Eichhorst, J.; Böttger, S.; Melzig, M.F.; Fuchs, H. Saponins modulate the intracellular trafficking of protein toxins. J. Control. Release 2012, 164, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Heisler, I.; Sutherland, M.; Bachran, C.; Hebestreit, P.; Schnitger, A.; Melzig, M.F.; Fuchs, H. Combined application of saponin and chimeric toxins drastically enhances the targeted cytotoxicity on tumor cells. J. Control. Release 2005, 106, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Stirpe, F.; Williams, D.G.; Onyon, L.J.; Legg, R.F.; Stevens, W.A. Dianthins, ribosome-damaging proteins with anti-viral properties from Dianthus caryophyllus L. (carnation). Biochem. J. 1981, 195, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, P.A.; Wong, A.J.; Vogelstein, B.; Zalutsky, M.R.; Fuller, G.N.; Archer, G.E.; Friedman, H.S.; Kwatra, M.M.; Bigner, S.H.; Bigner, D.D. Anti-synthetic peptide antibody reacting at the fusion junction of deletion-mutant epidermal growth factor receptors in human glioblastoma. Proc. Natl. Acad. Sci. USA 1990, 87, 4207–4211. [Google Scholar] [CrossRef] [PubMed]
- Nagane, M.; Coufal, F.; Lin, H.; Bögler, O.; Cavenee, W.K.; Huang, H.-J.S. A Common Mutant Epidermal Growth Factor Receptor Confers Enhanced Tumorigenicity on Human Glioblastoma Cells by Increasing Proliferation and Reducing Apoptosis. Cancer Res. 1996, 56, 5079–5086. [Google Scholar] [PubMed]
- Huang, H.-J.S.; Nagane, M.; Klingbeil, C.K.; Lin, H.; Nishikawa, R.; Ji, X.-D.; Huang, C.-M.; Gill, G.N.; Wiley, H.S.; Cavenee, W.K. The Enhanced Tumorigenic Activity of a Mutant Epidermal Growth Factor Receptor Common in Human Cancers Is Mediated by Threshold Levels of Constitutive Tyrosine Phosphorylation and Unattenuated Signaling. J. Biol. Chem. 1997, 272, 2927–2935. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.K.; Kaye, A.H.; Luwor, R.B. The EGFRvIII variant in glioblastoma multiforme. J. Clin. Neurosci. 2009, 16, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Zhao, X.; Yuza, Y.; Shimamura, T.; Li, D.; Protopopov, A.; Jung, B.L.; McNamara, K.; Xia, H.; Glatt, K.A.; et al. Epidermal growth factor receptor variant III mutations in lung tumorigenesis and sensitivity to tyrosine kinase inhibitors. Proc. Natl. Acad. Sci. USA 2006, 103, 7817–7822. [Google Scholar] [CrossRef] [PubMed]
- Khattri, A.; Zuo, Z.; Brägelmann, J.; Keck, M.K.; El Dinali, M.; Brown, C.D.; Stricker, T.; Munagala, A.; Cohen, E.E.W.; Lingen, M.W.; et al. Rare occurrence of EGFRvIII deletion in head and neck squamous cell carcinoma. Oral Oncol. 2015, 51, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Rae, J.M.; Scheys, J.O.; Clark, K.M.; Chadwick, R.B.; Kiefer, M.C.; Lippman, M.E. EGFR and EGFRvIII expression in primary breast cancer and cell lines. Breast Cancer Res. Treat. 2004, 87, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Grossman, S.A.; Batara, J.F. Current management of glioblastoma multiforme. Semin. Oncol. 2004, 31, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Shinojima, N.; Tada, K.; Shiraishi, S.; Kamiryo, T.; Kochi, M.; Nakamura, H.; Makino, K.; Saya, H.; Hirano, H.; Kuratsu, J.-I.; et al. Prognostic Value of Epidermal Growth Factor Receptor in Patients with Glioblastoma Multiforme. Cancer Res. 2003, 63, 6962–6970. [Google Scholar] [PubMed]
- Bigner, S.H.; Humphrey, P.A.; Wong, A.J.; Vogelstein, B.; Mark, J.; Friedman, H.S.; Bigner, D.D. Characterization of the Epidermal Growth Factor Receptor in Human Glioma Cell Lines and Xenografts. Cancer Res. 1990, 50, 8017–8022. [Google Scholar] [PubMed]
- Archer, G.E.; Sampson, J.H.; Lorimer, I.A.; McLendon, R.E.; Kuan, C.T.; Friedman, A.H.; Friedman, H.S.; Pastan, I.H.; Bigner, D.D. Regional treatment of epidermal growth factor receptor vIII-expressing neoplastic meningitis with a single-chain immunotoxin, MR-1. Clin. Cancer Res. 1999, 5, 2646–2652. [Google Scholar] [PubMed]
- Beers, R.; Chowdhury, P.; Bigner, D.; Pastan, I. Immunotoxins with increased activity against epidermal growth factor receptor vIII-expressing cells produced by antibody phage display. Clin. Cancer Res. 2000, 6, 2835–2843. [Google Scholar] [PubMed]
- Kuan, C.T.; Wikstrand, C.J.; Archer, G.; Beers, R.; Pastan, I.; Zalutsky, M.R.; Bigner, D.D. Increased binding affinity enhances targeting of glioma xenografts by EGFRvIII-specific scFv. Int. J. Cancer 2000, 88, 962–969. [Google Scholar] [CrossRef]
- Bobo, R.H.; Laske, D.W.; Akbasak, A.; Morrison, P.F.; Dedrick, R.L.; Oldfield, E.H. Convection-enhanced delivery of macromolecules in the brain. Proc. Natl. Acad. Sci. USA 1994, 91, 2076–2080. [Google Scholar] [CrossRef] [PubMed]
- Ding, D.; Kanaly, C.W.; Bigner, D.D.; Cummings, T.J.; Herndon, J.E.; Pastan, I.; Raghavan, R.; Sampson, J.H. Convection-enhanced delivery of free gadolinium with the recombinant immunotoxin MR1–1. J. Neuro-Oncol. 2009, 98, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Chandramohan, V.; Bao, X.; Keir, S.T.; Pegram, C.N.; Szafranski, S.E.; Piao, H.; Wikstrand, C.J.; McLendon, R.E.; Kuan, C.-T.; Pastan, I.H.; et al. Construction of an Immunotoxin, D2C7-(scdsFv)-PE38KDEL, Targeting EGFRwt and EGFRvIII for Brain Tumor Therapy. Clin. Cancer Res. 2013, 19, 4717–4727. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Chandramohan, V.; Reynolds, R.P.; Norton, J.N.; Wetsel, W.C.; Rodriguiz, R.M.; Aryal, D.K.; McLendon, R.E.; Levin, E.D.; Petry, N.A.; et al. Preclinical toxicity evaluation of a novel immunotoxin, D2C7-(scdsFv)-PE38KDEL, administered via intracerebral convection-enhanced delivery in rats. Investig. New Drugs 2016, 34, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Luwor, R.B.; Johns, T.G.; Murone, C.; Huang, H.-J.S.; Cavenee, W.K.; Ritter, G.; Old, L.J.; Burgess, A.W.; Scott, A.M. Monoclonal Antibody 806 Inhibits the Growth of Tumor Xenografts Expressing Either the de2–7 or Amplified Epidermal Growth Factor Receptor (EGFR) but not Wild-Type EGFR. Cancer Res. 2001, 61, 5355–5361. [Google Scholar] [PubMed]
- Johns, T.G.; Stockert, E.; Ritter, G.; Jungbluth, A.A.; Huang, H.J.; Cavenee, W.K.; Smyth, F.E.; Hall, C.M.; Watson, N.; Nice, E.C.; et al. Novel monoclonal antibody specific for the de2–7 epidermal growth factor receptor (EGFR) that also recognizes the EGFR expressed in cells containing amplification of the EGFR gene. Int. J. Cancer 2002, 98, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Johns, T.G.; Adams, T.E.; Cochran, J.R.; Hall, N.E.; Hoyne, P.A.; Olsen, M.J.; Kim, Y.-S.; Rothacker, J.; Nice, E.C.; Walker, F.; et al. Identification of the Epitope for the Epidermal Growth Factor Receptor-specific Monoclonal Antibody 806 Reveals That It Preferentially Recognizes an Untethered Form of the Receptor. J. Biol. Chem. 2004, 279, 30375–30384. [Google Scholar] [CrossRef] [PubMed]
- Garrett, T.P.J.; Burgess, A.W.; Gan, H.K.; Luwor, R.B.; Cartwright, G.; Walker, F.; Orchard, S.G.; Clayton, A.H.A.; Nice, E.C.; Rothacker, J.; et al. Antibodies specifically targeting a locally misfolded region of tumor associated EGFR. Proc. Natl. Acad. Sci. USA 2009, 106, 5082–5087. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.M.; Lee, F.-T.; Tebbutt, N.; Herbertson, R.; Gill, S.S.; Liu, Z.; Skrinos, E.; Murone, C.; Saunder, T.H.; Chappell, B.; et al. A phase I clinical trial with monoclonal antibody ch806 targeting transitional state and mutant epidermal growth factor receptors. Proc. Natl. Acad. Sci. USA 2007, 104, 4071–4076. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Reiser, P.; Hills, D.; Gullick, W.J.; Wels, W. Expression of an oncogenic mutant EGF receptor markedly increases the sensitivity of cells to an EGF-receptor-specific antibody-toxin. Int. J. Cancer 1998, 75, 878–884. [Google Scholar] [CrossRef]
- Michaelis, M.; Bliss, J.; Arnold, S.C.; Hinsch, N.; Rothweiler, F.; Deubzer, H.E.; Witt, O.; Langer, K.; Doerr, H.W.; Wels, W.S.; et al. Cisplatin-Resistant Neuroblastoma Cells Express Enhanced Levels of Epidermal Growth Factor Receptor (EGFR) and Are Sensitive to Treatment with EGFR-Specific Toxins. Clin. Cancer Res. 2008, 14, 6531–6537. [Google Scholar] [CrossRef] [PubMed]
- Spyridonidis, A.; Schmidt, M.; Bernhardt, W.; Papadimitriou, A.; Azemar, M.; Wels, W.; Groner, B.; Henschler, R. Purging of Mammary Carcinoma Cells During Ex Vivo Culture of CD34+ Hematopoietic Progenitor Cells With Recombinant Immunotoxins. Blood 1998, 91, 1820–1827. [Google Scholar] [PubMed]
- Schmidt, M.; Maurer-Gebhard, M.; Groner, B.; Köhler, G.; Brochmann-Santos, G.; Wels, W. Suppression of metastasis formation by a recombinant single chain antibody-toxin targeted to full-length and oncogenic variant EGF receptors. Oncogene 1999, 18, 1711–1721. [Google Scholar] [CrossRef] [PubMed]
- Berg, K.; Weyergang, A.; Prasmickaite, L.; Bonsted, A.; Høgset, A.; Strand, M.-T.R.; Wagner, E.; Selbo, P.K. Photochemical Internalization (PCI): A Technology for Drug Delivery. In Photodynamic Therapy: Methods and Protocols; Gomer, J.C., Ed.; Humana Press: Totowa, NJ, USA, 2010; pp. 133–145. [Google Scholar]
- Weyergang, A.; Selbo, P.K.; Berg, K. Photochemically stimulated drug delivery increases the cytotoxicity and specificity of EGF–saporin. J. Control. Release 2006, 111, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Yip, W.L.; Weyergang, A.; Berg, K.; Tønnesen, H.H.; Selbo, P.K. Targeted Delivery and Enhanced Cytotoxicity of Cetuximab−Saporin by Photochemical Internalization in EGFR-Positive Cancer Cells. Mol. Pharm. 2007, 4, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Selbo, P.K.; Rosenblum, M.G.; Cheung, L.H.; Zhang, W.; Berg, K. Multi-Modality Therapeutics with Potent Anti-Tumor Effects: Photochemical Internalization Enhances Delivery of the Fusion Toxin scFvMEL/rGel. PLoS ONE 2009, 4, e6691. [Google Scholar] [CrossRef] [PubMed]
- Weyergang, A.; Selbo, P.K.; Berstad, M.E.B.; Bostad, M.; Berg, K. Photochemical internalization of tumor-targeted protein toxins. Lasers Surg. Med. 2011, 43, 721–733. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Immunotoxin | Targeting Moiety | Toxin Moiety | Susceptible Cancer cell Types | Ref. |
|---|---|---|---|---|
| TGFα-PE40 | TGFα; chemical conjugation | PE40 | Epidermoid; prostate | [61] |
| EGF-saporin | EGF; chemical | saporin | Colon, prostate,epithelial | [62] |
| scFv(225)-ETA | Anti-EGFR MAb225 scFv; fusion protein | PE40 | Breast; epidermoid; oral | [63] |
| 425(scFv)-ETA | Anti-EGFR MAb425 scFv; fusion | PE40 | Pancreatic; RMS | [64] |
| scFv2112-ETA | Cetuximab scFv; fusion | PE40 | Epidermoid; breast; prostate | [65] |
| scFv1711-ETA | Panitumumab scFv; fusion | PE40 | RMS; pancreatic | [65] |
| Cetuximab-saporin | Cetuximab IgG; chemical | saporin | - | [66] |
| Cetuximab-dianthin | Cetuximab IgG; chemical | dianthin | - | [67] |
| Panitumumab-dianthin | Panitumumab IgG; chemical | dianthin | - | [67] |
| L8A4-PE35 | Anti-EGFRvIII L8A4 IgG; chemical | PE35 | EGFRvIII-transfected N6M | [68] |
| MR1(Fv)-PE38 | Anti-EGFRvIII MR1 scFv; fusion | PE38 | Glioblastoma | [69] |
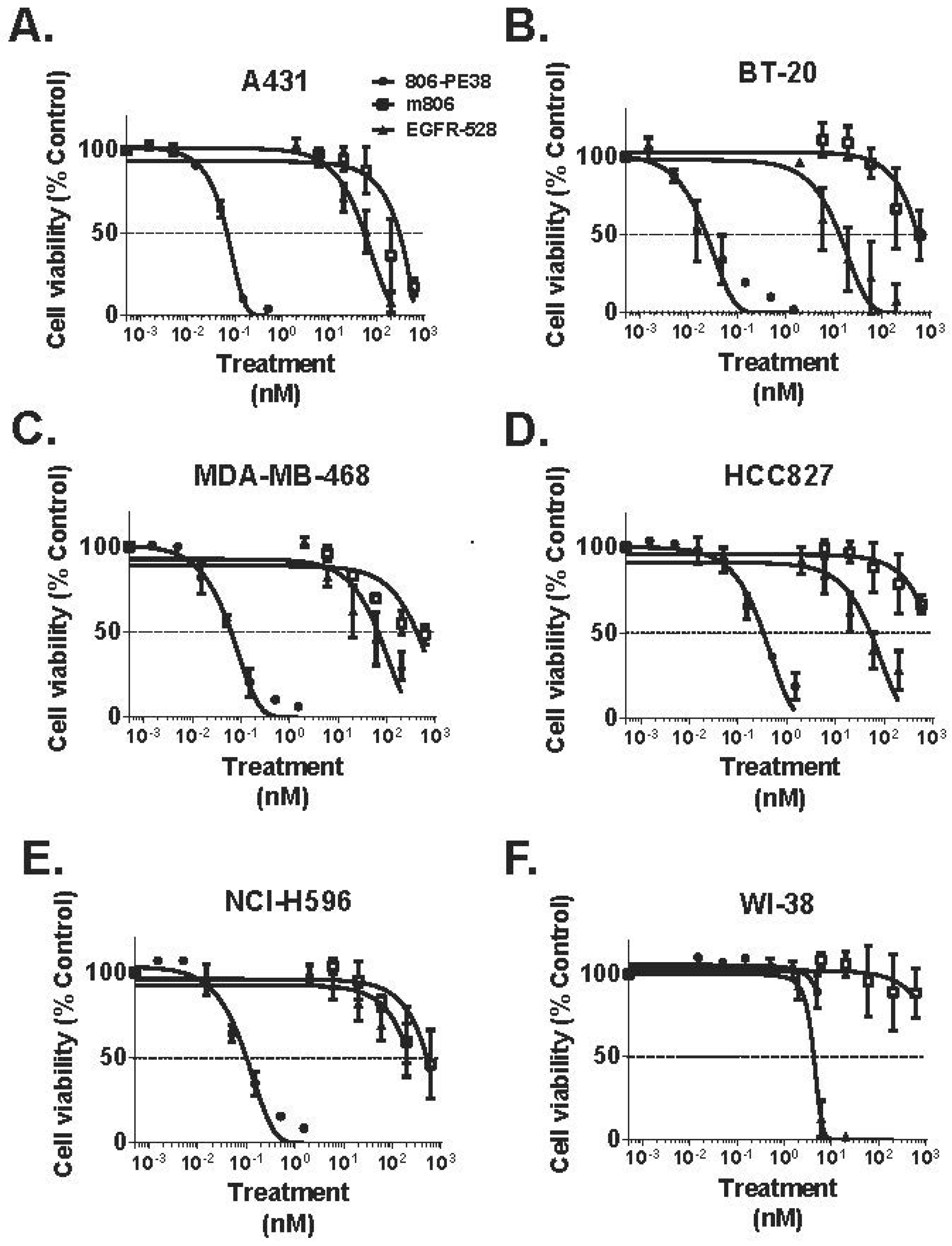
| 806-PE38 | Anti-EGFR/EGFRvIII MAb806 scFv; fusion | PE38 | TNBC; NSCLC; Epidermoid | [70] |
| DT390-BiscFv806 | Anti-EGFR/EGFRvIII MAb806 scFv; fusion | DT390 | Head-and-neck; EGFRvIII-transfected U87MG | [71] |
| scFv(14E1)-ETA | Anti-EGFR/EGFRvIII MAb14E1 scFv; fusion | PE40 | Glioblastoma; breast; renal | [72] |
| Immunotoxin | Targeting Moiety | Toxin Moiety | Clinical Trials: Indicated Cancer(s) | Clinical Trial Status | Ref. |
|---|---|---|---|---|---|
| TP38 | TGFα; chemical conjugation | PE38 | Malignant brain tumors | Phase I; discontinued | [73,74] |
| DAB389-EGF | EGF; chemical | DT389 | Solid tumors with EGFR overexpression | Phase I/II; discontinued | [75] |
| MR1-1(Fv)-PE38 | Anti-EGFRvIII MR1 scFv; fusion protein | PE38 | Malignant brain tumors | Phase I; discontinued | [76] |
| D2C7(scdsFv)-PE38 | Anti-EGFR/EGFRvIII MAbD2C7 scFv; fusion | PE38 | Malignant glioma | Phase I/II | [77] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simon, N.; FitzGerald, D. Immunotoxin Therapies for the Treatment of Epidermal Growth Factor Receptor-Dependent Cancers. Toxins 2016, 8, 137. https://doi.org/10.3390/toxins8050137
Simon N, FitzGerald D. Immunotoxin Therapies for the Treatment of Epidermal Growth Factor Receptor-Dependent Cancers. Toxins. 2016; 8(5):137. https://doi.org/10.3390/toxins8050137
Chicago/Turabian StyleSimon, Nathan, and David FitzGerald. 2016. "Immunotoxin Therapies for the Treatment of Epidermal Growth Factor Receptor-Dependent Cancers" Toxins 8, no. 5: 137. https://doi.org/10.3390/toxins8050137