
VacA’s Induction of VacA-Containing Vacuoles (VCVs) and Their Immunomodulatory Activities on Human T Cells


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Production of Functionally Distinct VacA Toxin Types by H. pylori
3. The Effects of VacA on Eukaryotic Cells
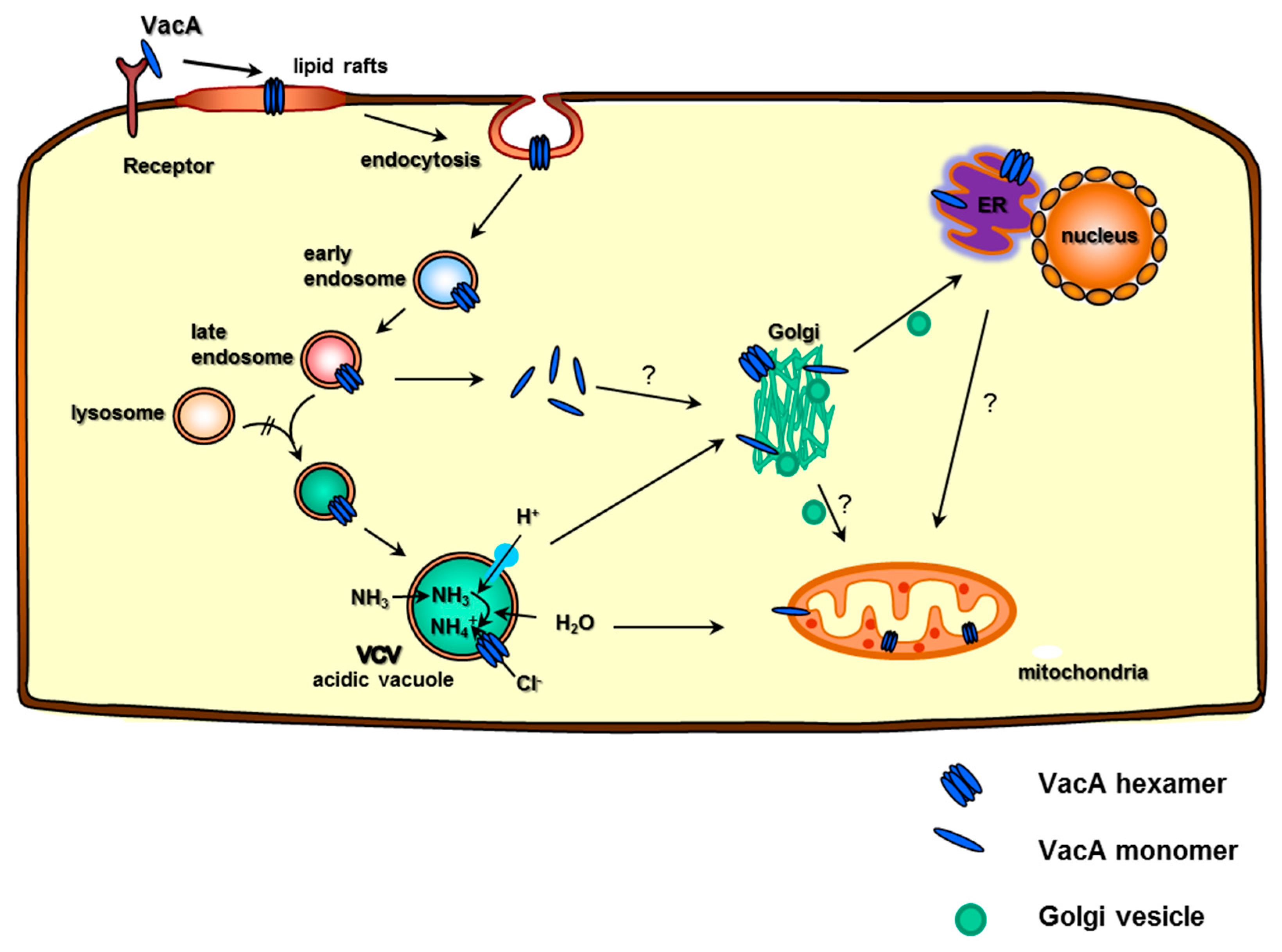
4. VacA—T Cell Interaction, Internalization, and Formation of Acidic Vacuoles
5. VCV Intracellular Transport, Toxin–Host Protein Interaction, and the Targeting of Cellular Organelles
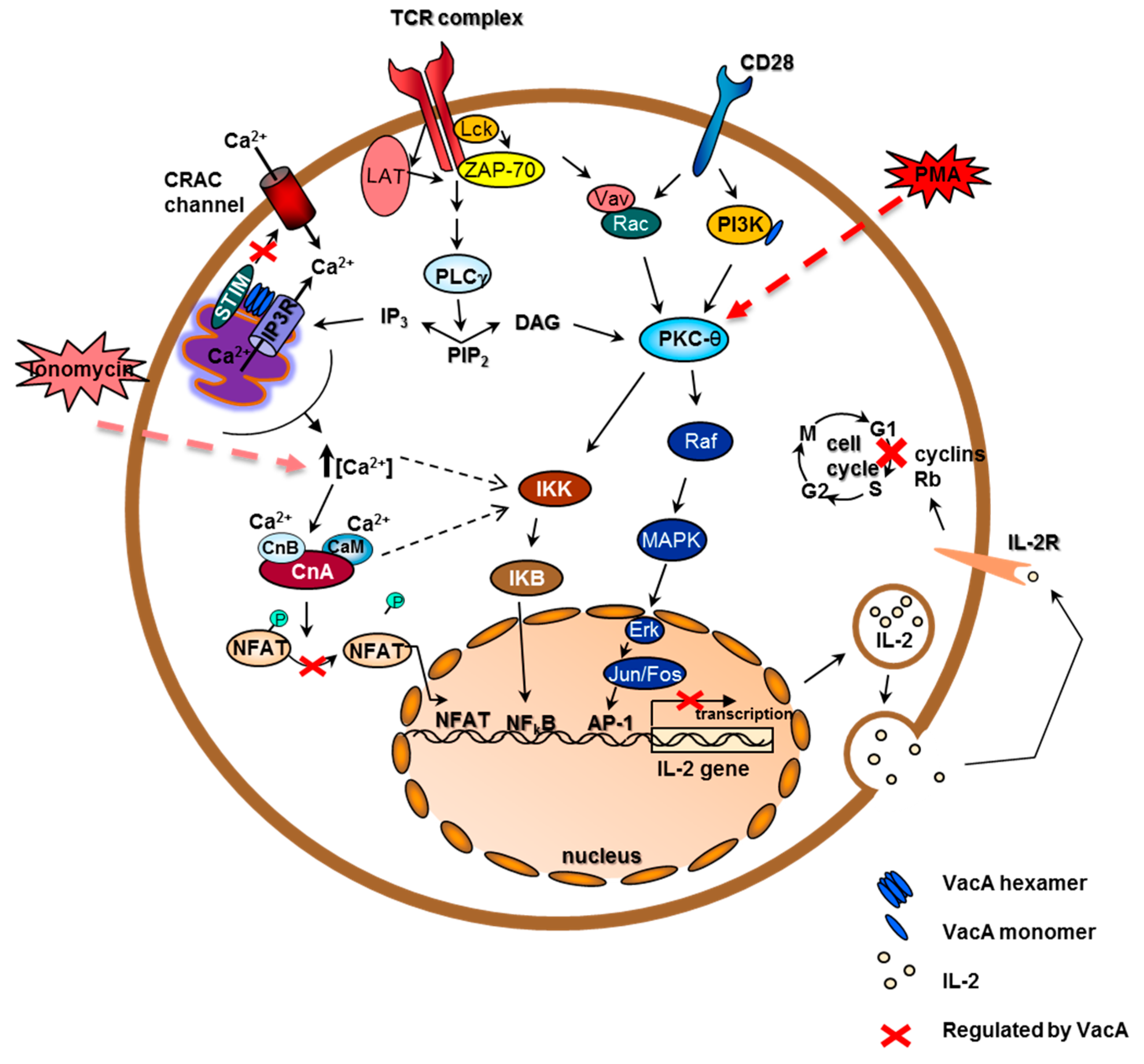
6. Cellular Immune Response and Immunoregulatory Activities of VacA in Human T Cells
7. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Cover, T.L.; Blaser, M.J. Helicobacter pylori in health and disease. Gastroenterology 2009, 136, 1863–1873. [Google Scholar] [CrossRef] [PubMed]
- Kusters, J.G.; van Vliet, A.H.; Kuipers, E.J. Pathogenesis of Helicobacter pylori infection. Clin. Microbiol. Rev. 2006, 19, 449–490. [Google Scholar] [CrossRef] [PubMed]
- Cover, T.L.; Blanke, S.R. Helicobacter pylori VacA, a paradigm for toxin multifunctionality. Nat. Rev. Microbiol. 2005, 3, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Boquet, P.; Ricci, V. Intoxication strategy of Helicobacter pylori VacA toxin. Trends Microbiol. 2012, 20, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Kern, B.; Jain, U.; Utsch, C.; Otto, A.; Busch, B.; Jimenez-Soto, L.; Becher, D.; Haas, R. Characterization of Helicobacter pylori VacA-containing vacuoles (VCVs), VacA intracellular trafficking and interference with calcium signalling in T lymphocytes. Cell. Microbiol. 2015, 12, 1811–1832. [Google Scholar] [CrossRef] [PubMed]
- Cover, T.L.; Blaser, M.J. Purification and characterization of the vacuolating toxin from Helicobacter pylori. J. Biol. Chem. 1992, 267, 10570–10575. [Google Scholar] [PubMed]
- Sewald, X.; Fischer, W.; Haas, R. Sticky socks: Helicobacter pylori VacA takes shape. Trends Microbiol. 2008, 16, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Atherton, J.C.; Peek, R.M., Jr.; Tummuru, M.K.R.; Blaser, M.J.; Cover, T.L. Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori. J. Biol. Chem. 1995, 270, 17771–17777. [Google Scholar] [PubMed]
- Algood, H.M.; Torres, V.J.; Unutmaz, D.; Cover, T.L. Resistance of primary murine CD4+ T cells to Helicobacter pylori vacuolating cytotoxin. Infect. Immun. 2007, 75, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rivera, C.; Algood, H.M.; Radin, J.N.; McClain, M.S.; Cover, T.L. The intermediate region of Helicobacter pylori VacA is a determinant of toxin potency in a Jurkat T cells assay. Infect. Immun. 2012, 80, 2578–2588. [Google Scholar] [CrossRef] [PubMed]
- Winter, J.A.; Letley, D.P.; Cook, K.W.; Rhead, J.L.; Zaitoun, A.A.; Ingram, R.J.; Amilon, K.R.; Croxall, N.J.; Kaye, P.V.; Robinson, K.; et al. A role for the vacuolating cytotoxin, VacA, in colonization and Helicobacter pylori-induced metaplasia in the stomach. J. Infect. Dis. 2014, 210, 954–963. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, W.; Haas, R. Genetic analysis of the Helicobacter pylori vacuolating cytotoxin: Structural similarities with the IgA protease type of exported protein. Mol. Microbiol. 1994, 12, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Chambers, M.G.; Pyburn, T.M.; Gonzalez-Rivera, C.; Collier, S.E.; Eli, I.; Yip, C.K.; Takizawa, Y.; Lacy, D.B.; Cover, T.L.; Ohi, M.D. Structural analysis of the oligomeric states of Helicobacter pylori VacA toxin. J. Mol. Biol. 2013, 425, 524–535. [Google Scholar] [CrossRef] [PubMed]
- Vinion-Dubiel, A.D.; McClain, M.S.; Czajkowsky, D.M.; Iwamoto, H.; Ye, D.; Cao, P.; Schraw, W.; Szabo, G.; Blanke, S.R.; Shao, Z.; et al. A dominant negative mutant of Helicobacter pylori vacuolating toxin (VacA) inhibits VacA-induced cell vacuolation. J. Biol. Chem. 1999, 274, 37736–37742. [Google Scholar] [CrossRef] [PubMed]
- Torres, V.J.; Ivie, S.E.; McClain, M.S.; Cover, T.L. Functional properties of the p33 and p55 domains of the Helicobacter pylori vacuolating cytotoxin. J. Biol. Chem. 2005, 280, 21107–21114. [Google Scholar] [CrossRef] [PubMed]
- Genisset, C.; Galeotti, C.L.; Lupetti, P.; Mercati, D.; Skibinski, D.A.; Barone, S.; Battistutta, R.; de Bernard, M.; Telford, J.L. A Helicobacter pylori vacuolating toxin mutant that fails to oligomerize has a dominant negative phenotype. Infect. Immun. 2006, 74, 1786–1794. [Google Scholar] [CrossRef] [PubMed]
- Marshall, B. Helicobacter pylori 20 years on. Clin. Med. 2002, 2, 147–152. [Google Scholar] [CrossRef]
- Szabo, I.; Brutsche, S.; Tombola, F.; Moschioni, M.; Satin, B.; Telford, J.; Rappuoli, R.; Montecucco, C.; Papini, E.; Zoratti, M. Formation of anion-selective channels in the cell plasma membrane by the toxin VacA of Helicobacter pylori is required for its biological activity. EMBO J. 1999, 18, 5517–5527. [Google Scholar] [CrossRef] [PubMed]
- Domanska, G.; Motz, C.; Meinecke, M.; Harsman, A.; Papatheodorou, P.; Reljic, B.; Dian-Lothrop, E.A.; Galmiche, A.; Kepp, O.; Becker, L.; et al. Helicobacter pylori VacA toxin/subunit p34: Targeting of an anion channel to the inner mitochondrial membrane. PLoS Pathog. 2010, 6, e1000878. [Google Scholar] [CrossRef] [PubMed]
- Willhite, D.C.; Blanke, S.R. Helicobacter pylori vacuolating cytotoxin enters cells, localizes to the mitochondria, and induces mitochondrial membrane permeability changes correlated to toxin channel activity. Cell. Microbiol. 2004, 6, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Galmiche, A.; Rassow, J.; Doye, A.; Cagnol, S.; Chambard, J.C.; Contamin, S.; de Thillot, V.; Just, I.; Ricci, V.; Solcia, E.; et al. The N-terminal 34 kDa fragment of Helicobacter pylori vacuolating cytotoxin targets mitochondria and induces cytochrome C release. EMBO J. 2000, 19, 6361–6370. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.; Luo, Z.-Q.; Blanke, S.R. Helicobacter pylori vacuolating cytotoxin a (VacA) engages the mitochondrial fission machinery to induce host cell death. Proc. Natl. Acad. Sci. USA 2011, 108, 16032–16037. [Google Scholar] [CrossRef] [PubMed]
- Cover, T.L.; Krishna, U.S.; Israel, D.A.; Peek, R.M. Induction of gastric epithelial cell apoptosis by Helicobacter pylori vacuolating cytotoxin. Cancer Res. 2003, 63, 951–957. [Google Scholar] [PubMed]
- Akazawa, Y.; Isomoto, H.; Matsushima, K.; Kanda, T.; Minami, H.; Yamaghchi, N.; Taura, N.; Shiozawa, K.; Ohnita, K.; Takeshima, F.; et al. Endoplasmic reticulum stress contributes to Helicobacter pylori VacA-induced apoptosis. PLoS ONE 2013, 8, e82322. [Google Scholar] [CrossRef] [PubMed]
- Radin, J.N.; Gonzalez-Rivera, C.; Ivie, S.E.; McClain, M.S.; Cover, T.L. Helicobacter pylori VacA induces programmed necrosis in gastric epithelial cells. Infect. Immun. 2011, 79, 2535–2543. [Google Scholar] [CrossRef] [PubMed]
- Amieva, M.R.; Vogelmann, R.; Covacci, A.; Tompkins, L.S.; Nelson, W.J.; Falkow, S. Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science 2003, 300, 1430–1434. [Google Scholar] [CrossRef] [PubMed]
- Gebert, B.; Fischer, W.; Weiss, E.; Hoffmann, R.; Haas, R. Helicobacter pylori vacuolating cytotoxin inhibits T lymphocyte activation. Science 2003, 301, 1099–1102. [Google Scholar] [CrossRef] [PubMed]
- Sundrud, M.S.; Torres, V.J.; Unutmaz, D.; Cover, T.L. Inhibition of primary human T cells proliferation by Helicobacter pylori vacuolating toxin (VacA) is independent of VacA effects on IL-2 secretion. Proc. Natl. Acad. Sci. USA 2004, 101, 7727–7732. [Google Scholar] [CrossRef] [PubMed]
- Ohta, A.; Sitkovsky, M. Role of g-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature 2001, 414, 916–920. [Google Scholar] [CrossRef] [PubMed]
- Robinson, K.; Argent, R.H.; Atherton, J.C. The inflammatory and immune response to Helicobacter pylori infection. Best Pract. Res. Clin. Gastroenterol. 2007, 21, 237–259. [Google Scholar] [CrossRef] [PubMed]
- Di Tommaso, A.; Xiang, Z.; Bugnoli, M.; Pileri, P.; Figura, N.; Bayeli, P.F.; Rappuoli, R.; Abrignani, S.; de Magistris, M.T. Helicobacter pylori-specific CD4+ T-cells clones from peripheral blood and gastric biopsies. Infect. Immun. 1995, 63, 1102–1106. [Google Scholar] [PubMed]
- McClain, M.S.; Schraw, W.; Ricci, V.; Boquet, P.; Cover, T.L. Acid activation of Helicobacter pylori vacuolating cytotoxin (VacA) results in toxin internalization by eukaryotic cells. Mol. Microbiol. 2000, 37, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Yahiro, K.; Niidome, T.; Kimura, M.; Hatakeyama, T.; Aoyagi, H.; Kurazono, H.; Imagawa, K.; Wada, A.; Moss, J.; Hirayama, T. Activation of Helicobacter pylori VacA toxin by alkaline or acid conditions increases its binding to a 250-kda receptor protein-tyrosine phosphatase beta. J. Biol. Chem. 1999, 274, 36693–36699. [Google Scholar] [CrossRef] [PubMed]
- Yahiro, K.; Wada, A.; Nakayama, M.; Kimura, T.; Ogushi, K.; Niidome, T.; Aoyagi, H.; Yoshino, K.; Yonezawa, K.; Moss, J.; et al. Protein-tyrosine phosphatase alpha, RPTP alpha, is a Helicobacter pylori VacA receptor. J. Biol. Chem. 2003, 278, 19183–19189. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.R.; Patel, H.K.; Kostolansky, S.S.; Ballivian, R.A.; Eichberg, J.; Blanke, S.R. Sphingomyelin functions as a novel receptor for Helicobacter pylori VacA. PLoS Pathog. 2008, 4, e1000073. [Google Scholar] [CrossRef] [PubMed]
- Ricci, V.; Galmiche, A.; Doye, A.; Necchi, V.; Solcia, E.; Boquet, P. High cell sensitivity to Helicobacter pylori VacA toxin depends on a GPI-anchored protein and is not blocked by inhibition of the clathrin-mediated pathway of endocytosis. Mol. Biol. Cell 2000, 11, 3897–3909. [Google Scholar] [CrossRef] [PubMed]
- Sewald, X.; Jimenez-Soto, L.; Haas, R. PKC-dependent endocytosis of the Helicobacter pylori vacuolating cytotoxin in primary T lymphocytes. Cell. Microbiol. 2011, 13, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Sewald, X.; Gebert-Vogl, B.; Prassl, S.; Barwig, I.; Weiss, E.; Fabbri, M.; Osicka, R.; Schiemann, M.; Busch, D.H.; Semmrich, M.; et al. Integrin subunit CD18 is the T-lymphocyte receptor for the Helicobacter pylori vacuolating cytotoxin. Cell Host Microbe 2008, 3, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Sommi, P.; Ricci, V.; Fiocca, R.; Necchi, V.; Romano, M.; Telford, J.L.; Solcia, E.; Ventura, U. Persistence of Helicobacter pylori VacA toxin and vacuolating potential in cultured gastric epithelial cells. Am. J. Physiol. 1998, 275, 681–688. [Google Scholar]
- Molinari, M.; Galli, C.; Norais, N.; Telford, J.L.; Rappuoli, R.; Luzio, J.P.; Montecucco, C. Vacuoles induced by Helicobacter pylori toxin contain both late endosomal and lysosomal markers. J. Biol. Chem. 1997, 272, 25339–25344. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, J.; Ohnishi, H.; Wada, A.; Hirayama, T.; Ohno, H.; Ueda, N.; Yasuda, H.; Iiri, T.; Wada, Y.; Futai, M.; et al. Involvement of syntaxin 7 in human gastric epithelial cell vacuolation induced by the Helicobacter pylori-produced cytotoxin VacA. J. Biol. Chem. 2003, 278, 25585–25590. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, N.C.; Monzo, P.; Gonzalez, T.; Doye, A.; Oldani, A.; Gounon, P.; Ricci, V.; Cormont, M.; Boquet, P. Early endosomes associated with dynamic F-actin structures are required for late trafficking of H. pylori VacA toxin. J. Cell Biol. 2007, 177, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Calore, F.; Genisset, C.; Casellato, A.; Rossato, M.; Codolo, G.; Esposti, M.D.; Scorrano, L.; de Bernard, M. Endosome-mitochondria juxtaposition during apoptosis induced by H. pylori VacA. Cell Death Differ. 2010, 17, 1707–1716. [Google Scholar] [CrossRef] [PubMed]
- Papini, E.; Satin, B.; de Bernard, M.; Molinari, M.; Arico, B.; Galli, C.; Telford, J.R.; Rappuoli, R.; Montecucco, C. Action site and cellular effects of cytotoxin VacA produced by Helicobacter pylori. Folia Microbiol. 1998, 43, 279–284. [Google Scholar] [CrossRef]
- Molinari, M.; Salio, M.; Galli, C.; Norais, N.; Rappuoli, R.; Lanzavecchia, A.; Montecucco, C. Selective inhibition of ii-dependent antigen presentation by Helicobacter pylori toxin VacA. J. Exp. Med. 1998, 187, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wandinger-Ness, A.; Goldenring, J.R.; Cover, T.L. Clustering and redistribution of late endocytic compartments in response to Helicobacter pylori vacuolating toxin. Mol. Biol. Cell 2004, 15, 1946–1959. [Google Scholar] [CrossRef] [PubMed]
- Larussa, T.; Leone, I.; Suraci, E.; Imeneo, M.; Luzza, F. Helicobacter pylori and T helper cells: Mechanisms of immune escape and tolerance. J. Immunol. Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Fassi Fehri, L.; Koch, M.; Belogolova, E.; Khalil, H.; Bolz, C.; Kalali, B.; Mollenkopf, H.J.; Beigier-Bompadre, M.; Karlas, A.; Schneider, T.; et al. Helicobacter pylori induces mir-155 in T cells in a cAMP-Foxp3-dependent manner. PLoS ONE 2010, 5, e9500. [Google Scholar] [CrossRef] [PubMed]
- Lundgren, A.; Suri-Payer, E.; Enarsson, K.; Svennerholm, A.M.; Lundin, B.S. Helicobacter pylori-specific CD4+ CD25 high regulatory T cells suppress memory T-cell responses to H. pylori in infected individuals. Infect. Immun. 2003, 71, 1755–1762. [Google Scholar] [CrossRef] [PubMed]
- Del Giudice, G.; Covacci, A.; Telford, J.L.; Montecucco, C.; Rappuoli, R. The design of vaccines against Helicobacter pylori and their development. Annu. Rev. Immunol. 2001, 19, 523–563. [Google Scholar] [CrossRef] [PubMed]
- Garhart, C.A.; Nedrud, J.G.; Heinzel, F.P.; Sigmund, N.E.; Czinn, S.J. Vaccine-induced protection against Helicobacter pylori in mice lacking both antibodies and interleukin-4. Infect. Immun. 2003, 71, 3628–3633. [Google Scholar] [CrossRef] [PubMed]
- Oertli, M.; Noben, M.; Engler, D.B.; Semper, R.P.; Reuter, S.; Maxeiner, J.; Gerhard, M.; Taube, C.; Muller, A. Helicobacter pylori gamma-glutamyl transpeptidase and vacuolating cytotoxin promote gastric persistence and immune tolerance. Proc. Natl. Acad. Sci. USA 2013, 110, 3047–3052. [Google Scholar] [CrossRef] [PubMed]
- Torres, V.J.; VanCompernolle, S.E.; Sundrud, M.S.; Unutmaz, D.; Cover, T.L. Helicobacter pylori vacuolating cytotoxin inhibits activation-induced proliferation of human T and B lymphocyte subsets. J. Immunol. 2007, 179, 5433–5440. [Google Scholar] [CrossRef] [PubMed]


© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Utsch, C.; Haas, R. VacA’s Induction of VacA-Containing Vacuoles (VCVs) and Their Immunomodulatory Activities on Human T Cells. Toxins 2016, 8, 190. https://doi.org/10.3390/toxins8060190
Utsch C, Haas R. VacA’s Induction of VacA-Containing Vacuoles (VCVs) and Their Immunomodulatory Activities on Human T Cells. Toxins. 2016; 8(6):190. https://doi.org/10.3390/toxins8060190
Chicago/Turabian StyleUtsch, Ciara, and Rainer Haas. 2016. "VacA’s Induction of VacA-Containing Vacuoles (VCVs) and Their Immunomodulatory Activities on Human T Cells" Toxins 8, no. 6: 190. https://doi.org/10.3390/toxins8060190