
Invasion of Dendritic Cells, Macrophages and Neutrophils by the Bordetella Adenylate Cyclase Toxin: A Subversive Move to Fool Host Immunity



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Immunity to B. pertussis
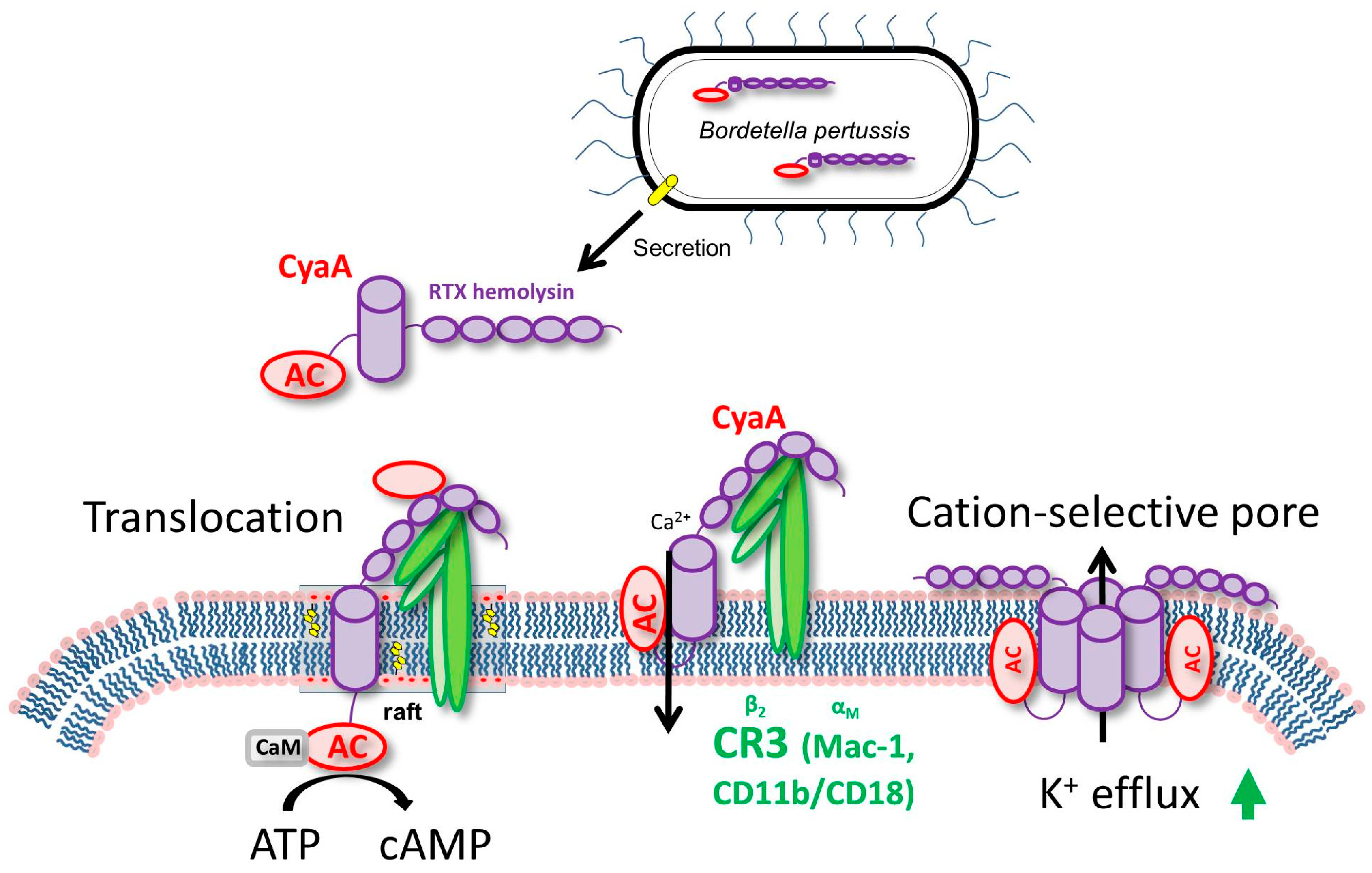
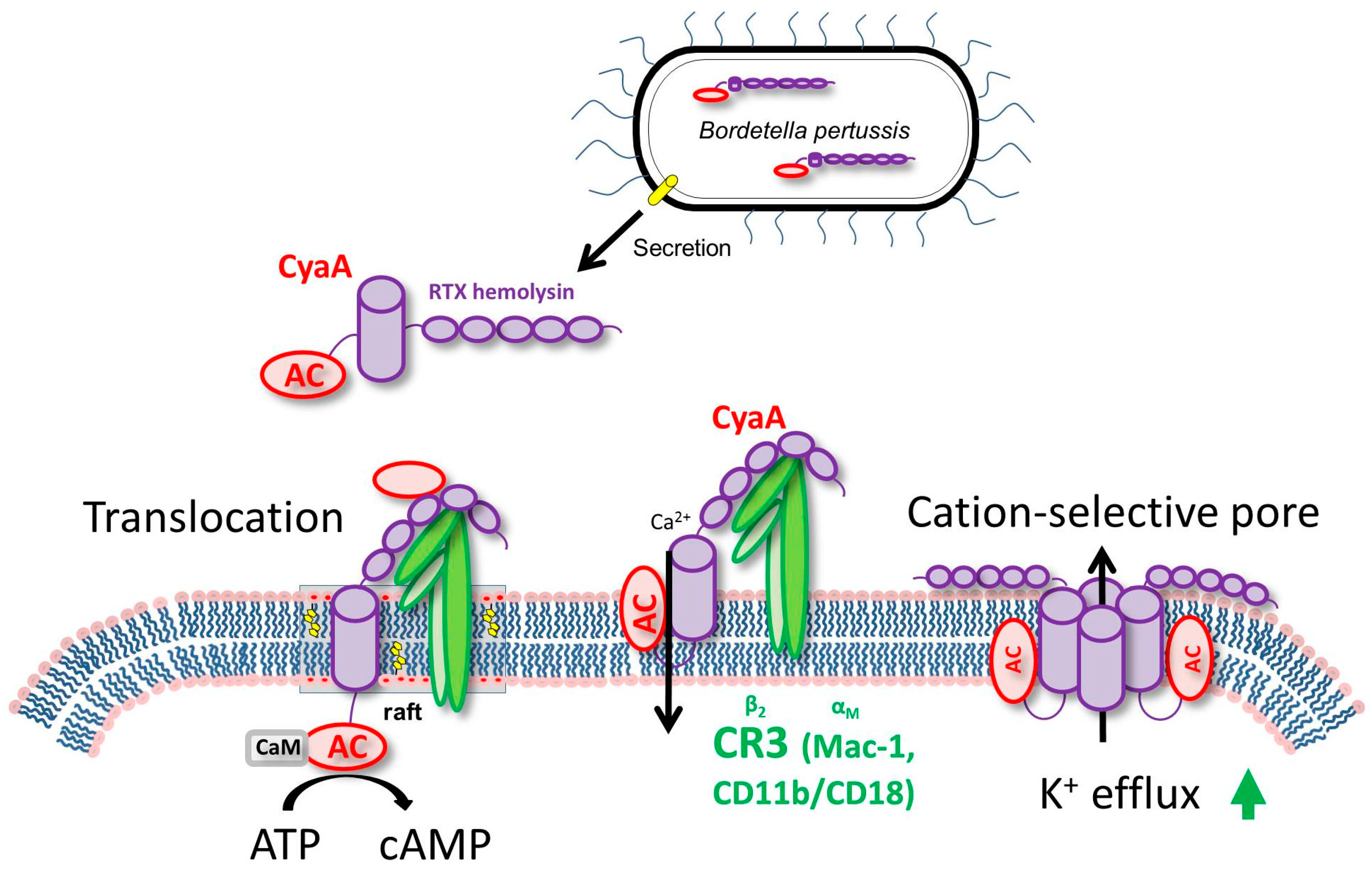
3. CyaA Mode of Action on Host Cells
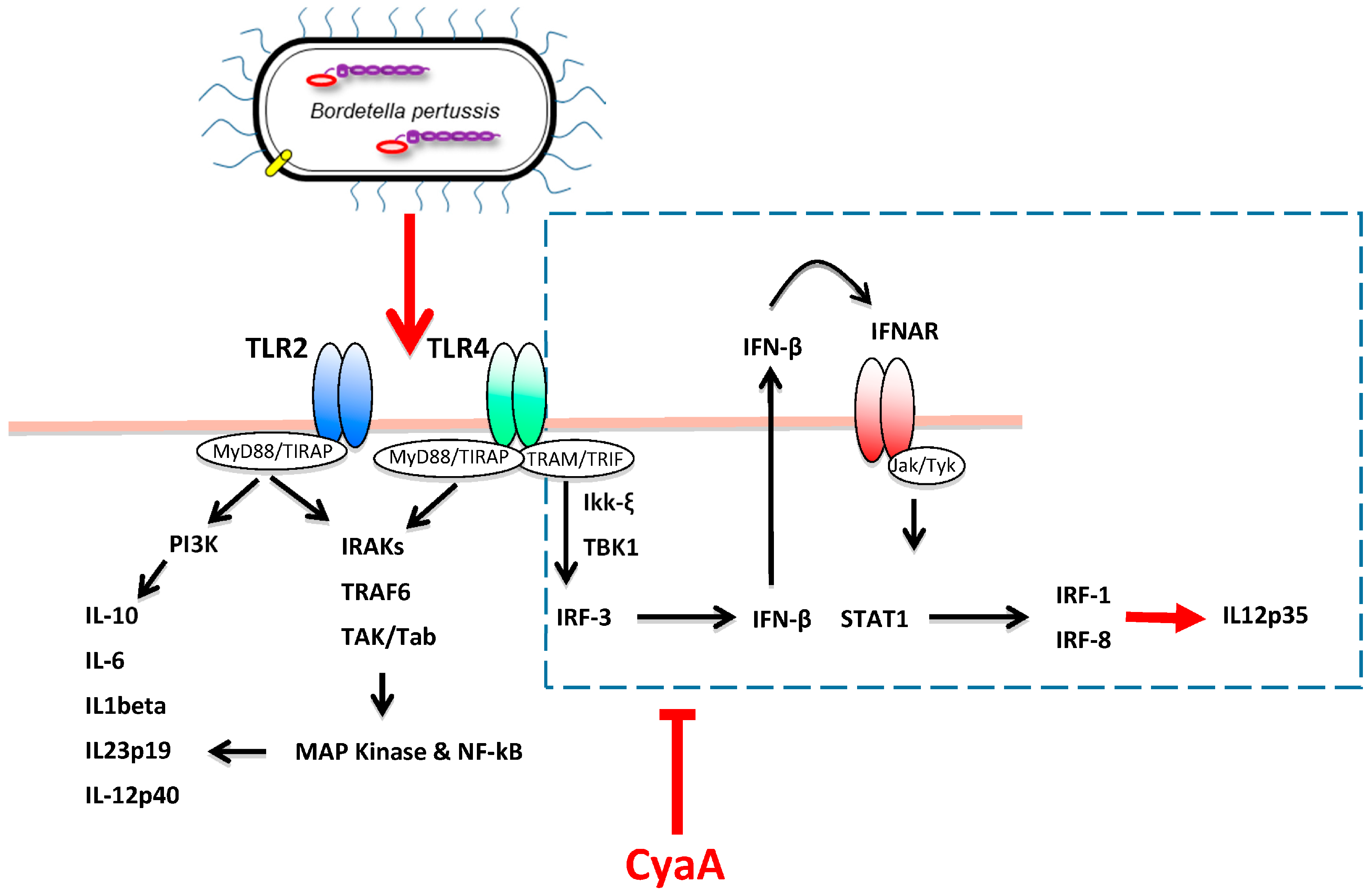
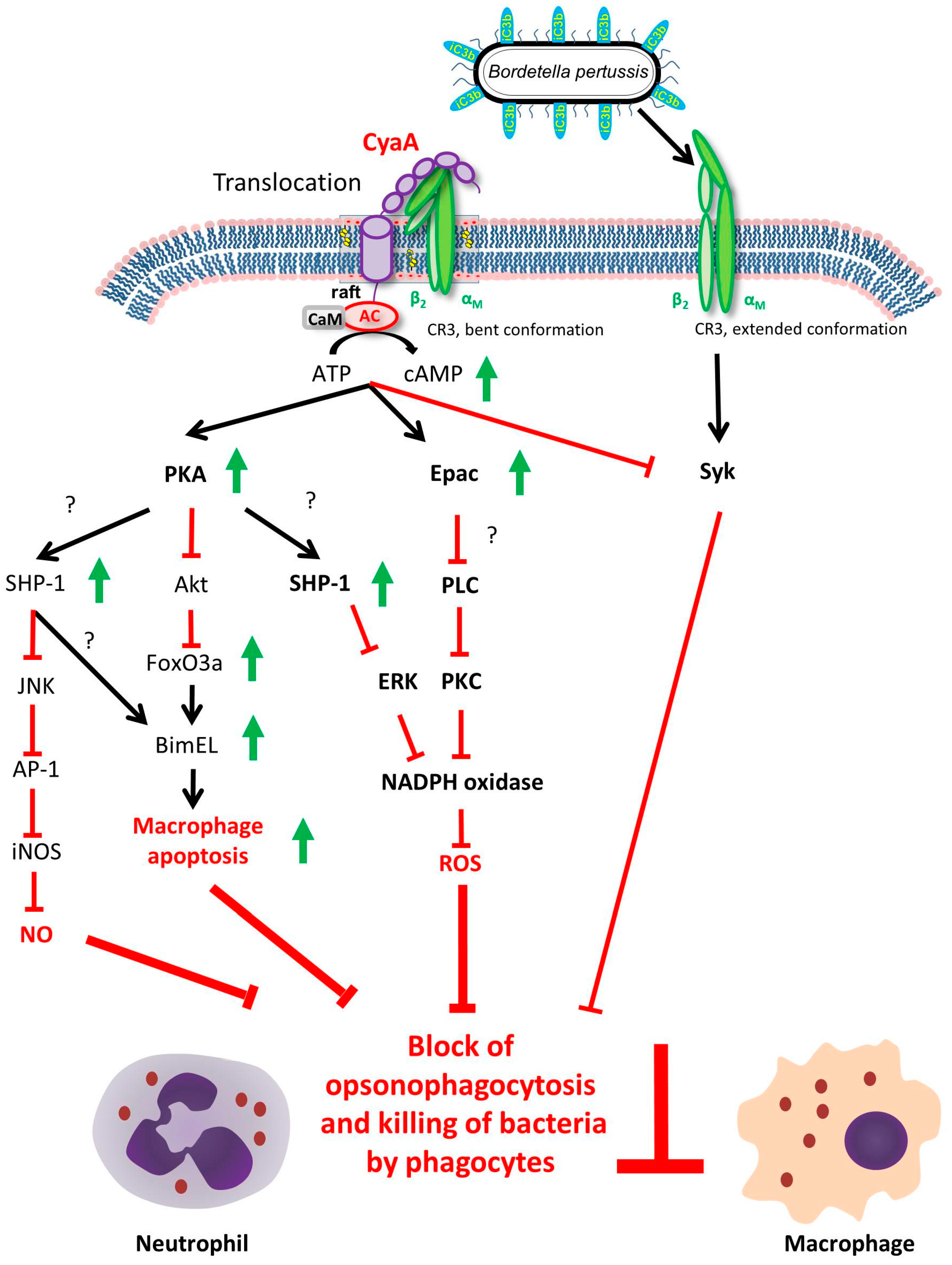
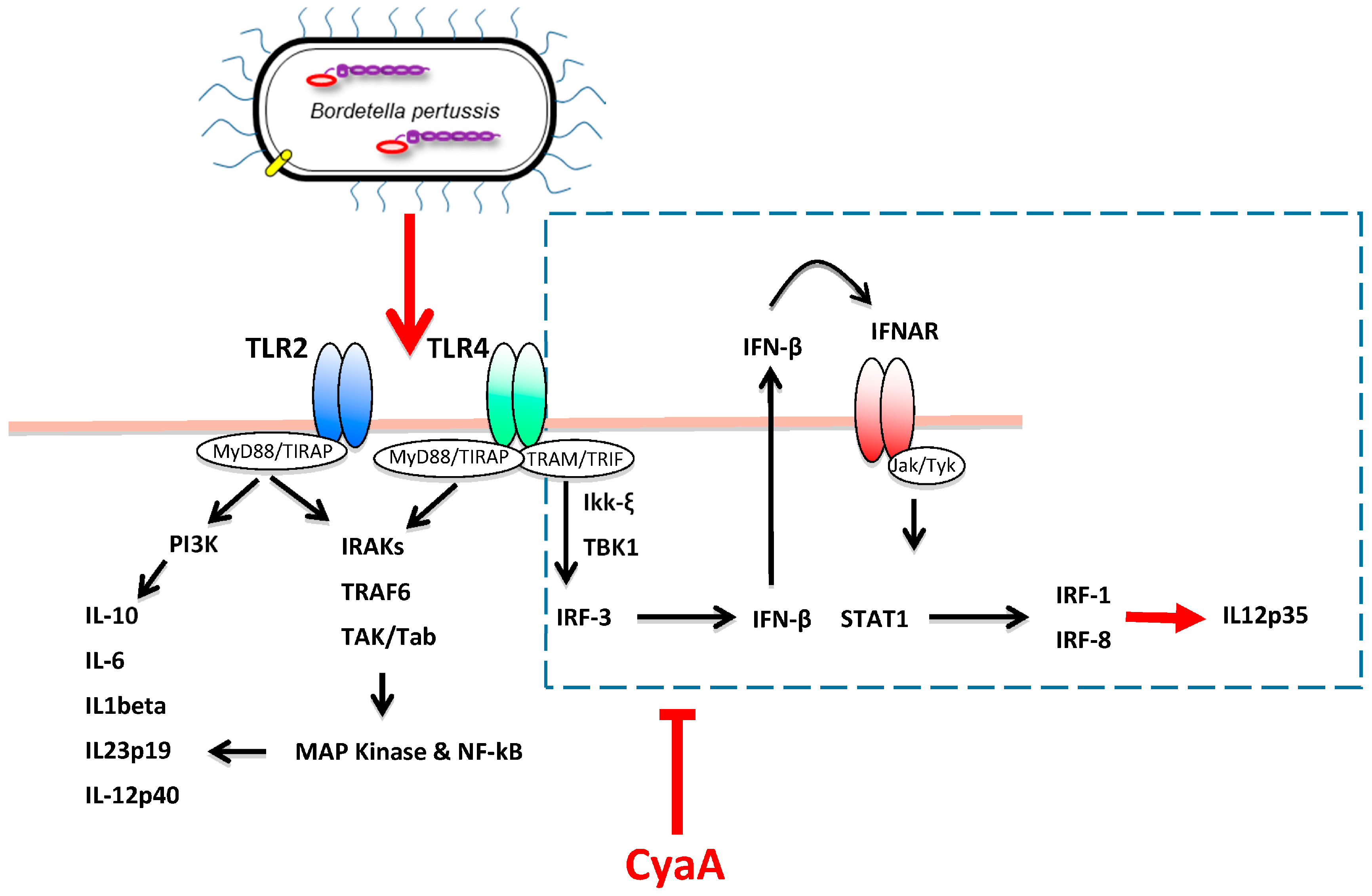
4. CyaA Interference with Innate Immune Defense
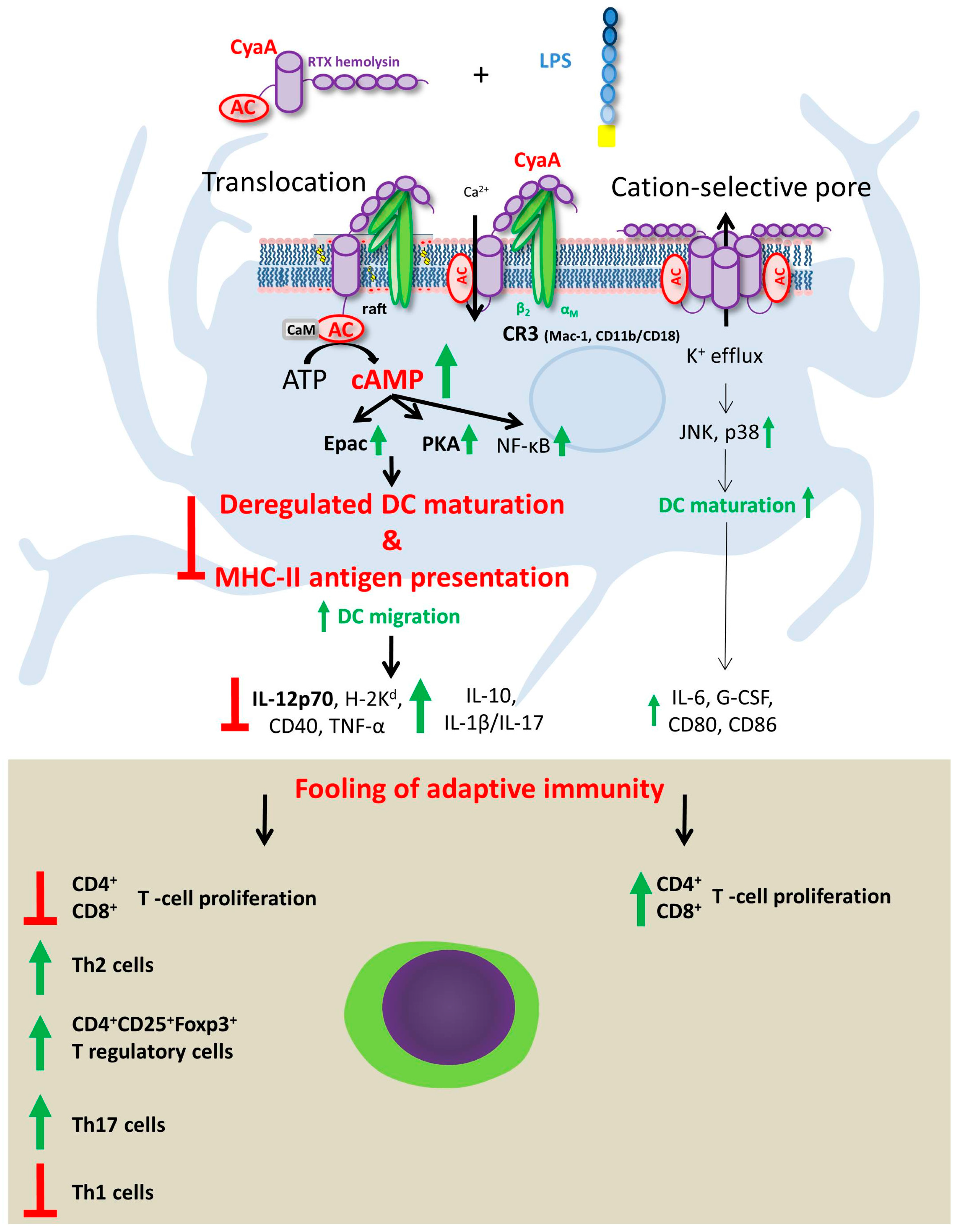
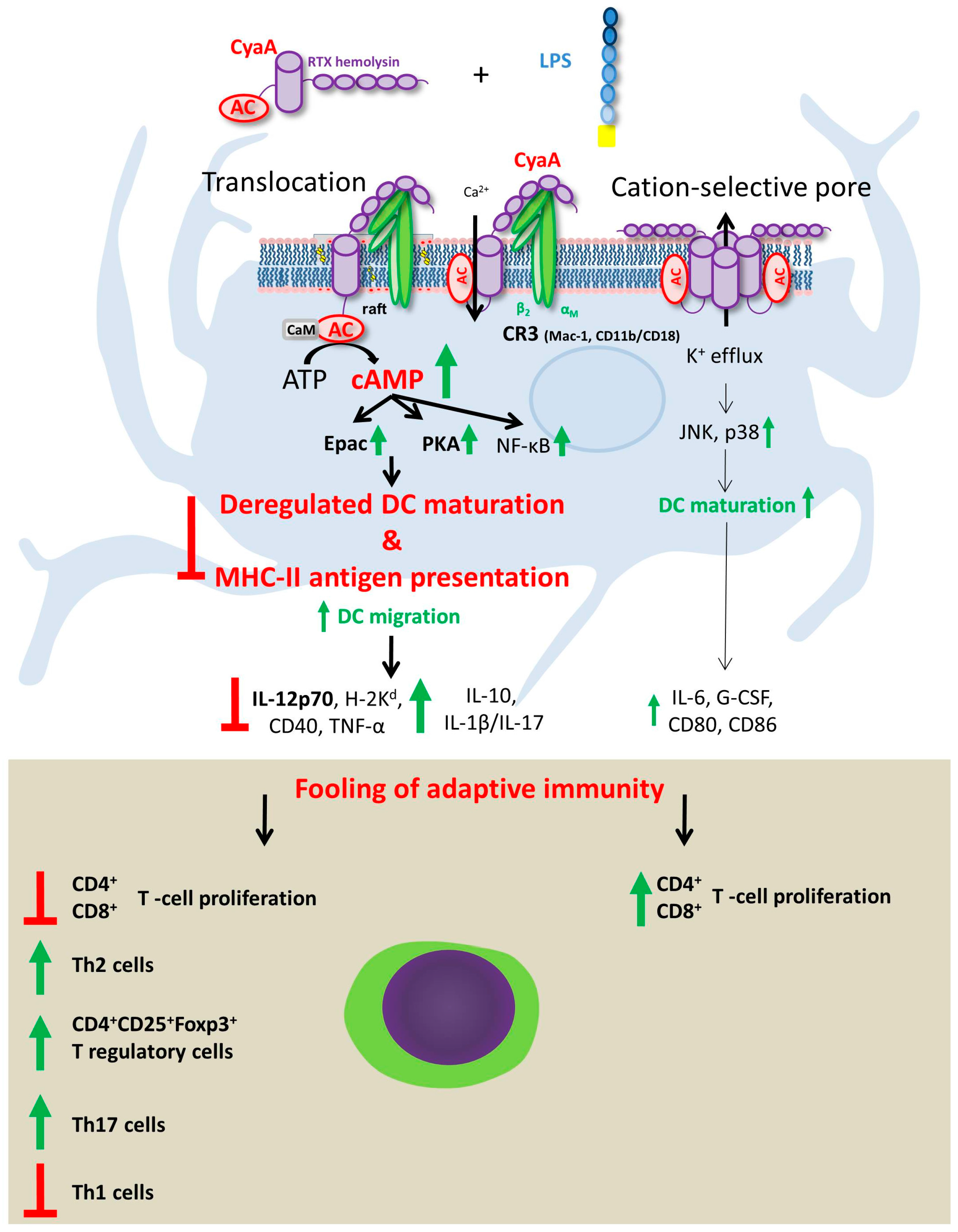
5. Effects of CyaA-Driven Intracellular cAMP Intoxication on DC Functions
6. Effects of Pore-Forming Activity of CyaA on DC Functions
7. Future Perspective
Acknowledgments
Conflicts of Interest
References
- Melvin, J.A.; Scheller, E.V.; Miller, J.F.; Cotter, P.A. Bordetella pertussis pathogenesis: Current and future challenges. Nat. Rev. Microbiol. 2014, 12, 274–288. [Google Scholar] [CrossRef] [PubMed]
- Confer, D.L.; Eaton, J.W. Phagocyte impotence caused by an invasive bacterial adenylate cyclase. Science 1982, 217, 948–950. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, M.S.; Weiss, A.A. Adenylate cyclase toxin is critical for colonization and pertussis toxin is critical for lethal infection by Bordetella pertussis in infant mice. Infect. Immun. 1990, 58, 3445–3447. [Google Scholar] [PubMed]
- Khelef, N.; Sakamoto, H.; Guiso, N. Both adenylate cyclase and hemolytic activities are required by Bordetella pertussis to initiate infection. Microb. Pathog. 1992, 12, 227–235. [Google Scholar] [CrossRef]
- Harvill, E.T.; Cotter, P.A.; Yuk, M.H.; Miller, J.F. Probing the function of Bordetella bronchiseptica adenylate cyclase toxin by manipulating host immunity. Infect. Immun. 1999, 67, 1493–1500. [Google Scholar] [PubMed]
- Cerny, O.; Anderson, K.E.; Stephens, L.R.; Hawkins, P.T.; Sebo, P. cAMP Signaling of Adenylate Cyclase Toxin Blocks the Oxidative Burst of Neutrophils through Epac-Mediated Inhibition of Phospholipase C Activity. J. Immunol. 2017, 198, 1285–1296. [Google Scholar] [CrossRef] [PubMed]
- Hewlett, E.; Wolff, J. Soluble adenylate cyclase from the culture medium of Bordetella pertussis: Purification and characterization. J. Bacteriol. 1976, 127, 890–898. [Google Scholar] [PubMed]
- Hewlett, E.L.; Manclark, C.R.; Wolff, J. Adenyl cyclase in Bordetella pertussis vaccines. J. Infect. Dis. 1977, 136, S216–S219. [Google Scholar] [CrossRef] [PubMed]
- Sebo, P.; Osicka, R.; Masin, J. Adenylate cyclase toxin-hemolysin relevance for pertussis vaccines. Expert Rev. Vaccines 2014, 13, 1215–1227. [Google Scholar] [CrossRef] [PubMed]
- O’Garra, A.; Robinson, D. Development and function of T helper 1 cells. Adv. Immunol. 2004, 83, 133–162. [Google Scholar] [PubMed]
- Mowen, K.A.; Glimcher, L.H. Signaling pathways in Th2 development. Immunol. Rev. 2004, 202, 203–222. [Google Scholar] [CrossRef] [PubMed]
- Harrington, L.E.; Mangan, P.R.; Weaver, C.T. Expanding the effector CD4 T-cell repertoire: The Th17 lineage. Curr. Opin. Immunol. 2006, 18, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.C.; Velichko, S.; Hung, L.Y.; Wu, R. IL-17A and Th17 cells in lung inflammation: An update on the role of Th17 cell differentiation and IL-17R signaling in host defense against infection. Clin. Dev. Immunol. 2013, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Liew, F.Y.; McInnes, I.B. The role of innate mediators in inflammatory response. Mol. Immunol. 2002, 38, 887–890. [Google Scholar] [CrossRef]
- Belkaid, Y.; Oldenhove, G. Tuning microenvironments: Induction of regulatory T cells by dendritic cells. Immunity 2008, 29, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Rudensky, A.Y. Foxp3 in control of the regulatory T cell lineage. Nat. Immunol. 2007, 8, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Wu, C. Differentiation, regulation and function of Th9 cells. Adv. Exp. Med. Biol. 2014, 841, 181–207. [Google Scholar] [CrossRef] [PubMed]
- Kagami, S.; Rizzo, H.L.; Lee, J.J.; Koguchi, Y.; Blauvelt, A. Circulating Th17, Th22, and Th1 cells are increased in psoriasis. J. Investig. Dermatol. 2010, 130, 1373–1383. [Google Scholar] [CrossRef] [PubMed]
- Vinuesa, C.G.; Linterman, M.A.; Yu, D.; MacLennan, I.C. Follicular Helper T Cells. Annu. Rev. Immunol. 2016, 34, 335–368. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Botran, R.; Sanders, V.M.; Mosmann, T.R.; Vitetta, E.S. Lymphokine-mediated regulation of the proliferative response of clones of T helper 1 and T helper 2 cells. J. Exp. Med. 1988, 168, 543–558. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, T.F.; Fitch, F.W. Anti-proliferative effect of IFN-gamma in immune regulation. I. IFN-gamma inhibits the proliferation of Th2 but not Th1 murine helper T lymphocyte clones. J. Immunol. 1988, 140, 4245–4252. [Google Scholar] [PubMed]
- Veldhoen, M.; Hocking, R.J.; Flavell, R.A.; Stockinger, B. Signals mediated by transforming growth factor-beta initiate autoimmune encephalomyelitis, but chronic inflammation is needed to sustain disease. Nat. Immunol. 2006, 7, 1151–1156. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Rodriguez, E.V.; Napolitani, G.; Lanzavecchia, A.; Sallusto, F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat. Immunol. 2007, 8, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Annunziato, F.; Cosmi, L.; Santarlasci, V.; Maggi, L.; Liotta, F.; Mazzinghi, B.; Parente, E.; Filì, L.; Ferri, S.; et al. Phenotypic and functional features of human Th17 cells. J. Exp. Med. 2007, 204, 1849–1861. [Google Scholar] [CrossRef] [PubMed]
- Koenen, H.J.; Smeets, R.L.; Vink, P.M.; van Rijssen, E.; Boots, A.M.; Joosten, I. Human CD25highFoxp3pos regulatory T cells differentiate into IL-17-producing cells. Blood 2008, 112, 2340–2352. [Google Scholar] [CrossRef] [PubMed]
- Mills, K.H.; Barnard, A.; Watkins, J.; Redhead, K. Cell-mediated immunity to Bordetella pertussis: Role of Th1 cells in bacterial clearance in a murine respiratory infection model. Infect. Immun. 1993, 61, 399–410. [Google Scholar] [PubMed]
- Leef, M.; Elkins, K.L.; Barbic, J.; Shahin, R.D. Protective immunity to Bordetella pertussis requires both B cells and CD4(+) T cells for key functions other than specific antibody production. J. Exp. Med. 2000, 191, 841–852. [Google Scholar] [CrossRef]
- Dirix, V.; Verscheure, V.; Vermeulen, F.; De Schutter, I.; Goetghebuer, T.; Locht, C.; Mascart, F. Both CD4+ and CD8+ lymphocytes participate in the IFN-γ response to filamentous hemagglutinin from Bordetella pertussis in infants, children, and adults. Clin. Dev. Immunol. 2012, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ausiello, C.M.; Urbani, F.; la Sala, A.; Lande, R.; Cassone, A. Vaccine- and antigen-dependent type 1 and type 2 cytokine induction after primary vaccination of infants with whole-cell or acellular pertussis vaccines. Infect. Immun. 1997, 65, 2168–2174. [Google Scholar] [PubMed]
- Ausiello, C.M.; Lande, R.; Urbani, F.; Di Carlo, B.; Stefanelli, P.; Salmaso, S.; Mastrantonio, P.; Cassone, A. Cell-mediated immunity and antibody responses to Bordetella pertussis antigens in children with a history of pertussis infection and in recipients of an acellular pertussis vaccine. J. Infect. Dis. 2000, 181, 1989–1995. [Google Scholar] [CrossRef] [PubMed]
- Mascart, F.; Verscheure, V.; Malfroot, A.; Hainaut, M.; Piérard, D.; Temerman, S.; Peltier, A.; Debrie, A.S.; Levy, J.; Del Giudice, G.; et al. Bordetella pertussis infection in 2-month-old infants promotes type 1 T cell responses. J. Immunol. 2003, 170, 1504–1509. [Google Scholar] [CrossRef] [PubMed]
- Higgins, S.C.; Jarnicki, A.G.; Lavelle, E.C.; Mills, K.H. TLR4 mediates vaccine-induced protective cellular immunity to Bordetella pertussis: Role of IL-17-producing T cells. J. Immunol. 2006, 177, 7980–7989. [Google Scholar] [CrossRef] [PubMed]
- Andreasen, C.; Powell, D.A.; Carbonetti, N.H. Pertussis toxin stimulates IL-17 production in response to Bordetella pertussis infection in mice. PLoS ONE 2009, 4, e7079. [Google Scholar] [CrossRef] [PubMed]
- Dunne, A.; Ross, P.J.; Pospisilova, E.; Masin, J.; Meaney, A.; Sutton, C.E.; Iwakura, Y.; Tschopp, J.; Sebo, P.; Mills, K.H. Inflammasome activation by adenylate cyclase toxin directs Th17 responses and protection against Bordetella pertussis. J. Immunol. 2010, 185, 1711–1719. [Google Scholar] [CrossRef] [PubMed]
- Feunou, P.F.; Bertout, J.; Locht, C. T- and B-cell-mediated protection induced by novel, live attenuated pertussis vaccine in mice. Cross protection against parapertussis. PLoS ONE 2010, 5, e10178. [Google Scholar] [CrossRef] [PubMed]
- Fedele, G.; Spensieri, F.; Palazzo, R.; Nasso, M.; Cheung, G.Y.; Coote, J.G.; Ausiello, C.M. Bordetella pertussis commits human dendritic cells to promote a Th1/Th17 response through the activity of adenylate cyclase toxin and MAPK-pathways. PLoS ONE 2010, 5, e8734. [Google Scholar] [CrossRef] [PubMed]
- Fedele, G.; Bianco, M.; Debrie, A.S.; Locht, C.; Ausiello, C.M. Attenuated Bordetella pertussis vaccine candidate BPZE1 promotes human dendritic cell CCL21-induced migration and drives a Th1/Th17 response. J. Immunol. 2011, 186, 5388–5396. [Google Scholar] [CrossRef] [PubMed]
- Sansonetti, P.J.; Di Santo, J.P. Debugging how bacteria manipulate the immune response. Immunity 2007, 26, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Fedele, G.; Nasso, M.; Spensieri, F.; Palazzo, R.; Frasca, L.; Watanabe, M.; Ausiello, C.M. Lipopolysaccharides from Bordetella pertussis and Bordetella parapertussis differently modulate human dendritic cell functions resulting in divergent prevalence of Th17-polarized responses. J. Immunol. 2008, 181, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Jongerius, I.; Schuijt, T.J.; Mooi, F.R.; Pinelli, E. Complement evasion by Bordetella pertussis: Implications for improving current vaccines. J. Mol. Med. 2015, 93, 395–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hovingh, E.S.; van den Broek, B.; Kuipers, B.; Pinelli, E.; Rooijakkers, S.H.M.; Jongerius, I. Acquisition of C1 inhibitor by Bordetella pertussis virulence associated gene 8 results in C2 and C4 consumption away from the bacterial surface. PLoS Pathog. 2017, 13, e1006531. [Google Scholar] [CrossRef] [PubMed]
- Coutte, L.; Locht, C. Investigating pertussis toxin and its impact on vaccination. Future Microbiol. 2015, 10, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Carbonetti, N.H. Bordetella pertussis: New concepts in pathogenesis and treatment. Curr. Opin. Infect. Dis. 2016, 29, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Carbonetti, N.H. Pertussis leukocytosis: Mechanisms, clinical relevance and treatment. Pathog. Dis. 2016, 74, pii:ftw087. [Google Scholar] [CrossRef] [PubMed]
- Lamberti, Y.A.; Hayes, J.A.; Perez Vidakovics, M.L.; Harvill, E.T.; Rodriguez, M.E. Intracellular trafficking of Bordetella pertussis in human macrophages. Infect. Immun. 2010, 78, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Lamberti, Y.; Gorgojo, J.; Massillo, C.; Rodriguez, M.E. Bordetella pertussis entry into respiratory epithelial cells and intracellular survival. Pathog. Dis. 2013, 69, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Nagamatsu, K.; Kuwae, A.; Konaka, T.; Nagai, S.; Yoshida, S.; Eguchi, M.; Watanabe, M.; Mimuro, H.; Koyasu, S.; Abe, A. Bordetella evades the host immune system by inducing IL-10 through a type III effector, BopN. J. Exp. Med. 2009, 206, 3073–3088. [Google Scholar] [CrossRef] [PubMed]
- Yuk, M.H.; Harvill, E.T.; Cotter, P.A.; Miller, J.F. Modulation of host immune responses, induction of apoptosis and inhibition of NF-kappaB activation by the Bordetella type III secretion system. Mol. Microbiol. 2000, 35, 991–1004. [Google Scholar] [CrossRef] [PubMed]
- Guermonprez, P.; Khelef, N.; Blouin, E.; Rieu, P.; Ricciardi-Castagnoli, P.; Guiso, N.; Ladant, D.; Leclerc, C. The adenylate cyclase toxin of Bordetella pertussis binds to target cells via the alpha(M)beta(2) integrin (CD11b/CD18). J. Exp. Med. 2001, 193, 1035–1044. [Google Scholar] [CrossRef] [PubMed]
- Vojtova, J.; Kamanova, J.; Sebo, P. Bordetella adenylate cyclase toxin: A swift saboteur of host defense. Curr. Opin. Microbiol. 2006, 9, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Linhartová, I.; Bumba, L.; Mašín, J.; Basler, M.; Osička, R.; Kamanová, J.; Procházková, K.; Adkins, I.; Hejnová-Holubová, J.; Sadílková, L.; et al. RTX proteins: A highly diverse family secreted by a common mechanism. FEMS Microbiol. Rev. 2010, 34, 1076–1112. [Google Scholar] [CrossRef] [PubMed]
- Fiser, R.; Masín, J.; Basler, M.; Krusek, J.; Spuláková, V.; Konopásek, I.; Sebo, P. Third activity of Bordetella adenylate cyclase (AC) toxin-hemolysin. Membrane translocation of AC domain polypeptide promotes calcium influx into CD11b+ monocytes independently of the catalytic and hemolytic activities. J. Biol. Chem. 2007, 282, 2808–2820. [Google Scholar] [CrossRef] [PubMed]
- Bumba, L.; Masin, J.; Fiser, R.; Sebo, P. Bordetella adenylate cyclase toxin mobilizes its beta2 integrin receptor into lipid rafts to accomplish translocation across target cell membrane in two steps. PLoS Pathog. 2010, 6, e1000901. [Google Scholar] [CrossRef] [PubMed]
- Fiser, R.; Masin, J.; Bumba, L.; Pospisilova, E.; Fayolle, C.; Basler, M.; Sadilkova, L.; Adkins, I.; Kamanova, J.; Cerny, J.; et al. Calcium influx rescues adenylate cyclase-hemolysin from rapid cell membrane removal and enables phagocyte permeabilization by toxin pores. PLoS Pathog. 2012, 8, e1002580. [Google Scholar] [CrossRef] [PubMed]
- Masin, J.; Osicka, R.; Bumba, L.; Sebo, P. Bordetella adenylate cyclase toxin: A unique combination of a pore-forming moiety with a cell-invading adenylate cyclase enzyme. Pathog. Dis. 2015, 73, ftv075. [Google Scholar] [CrossRef] [PubMed]
- Pearson, R.D.; Symes, P.; Conboy, M.; Weiss, A.A.; Hewlett, E.L. Inhibition of monocyte oxidative responses by Bordetella pertussis adenylate cyclase toxin. J. Immunol. 1987, 139, 2749–2754. [Google Scholar] [PubMed]
- Kamanova, J.; Kofronova, O.; Masin, J.; Genth, H.; Vojtova, J.; Linhartova, I.; Benada, O.; Just, I.; Sebo, P. Adenylate cyclase toxin subverts phagocyte function by RhoA inhibition and unproductive ruffling. J. Immunol. 2008, 181, 5587–5597. [Google Scholar] [CrossRef] [PubMed]
- Weingart, C.L.; Weiss, A.A. Bordetella pertussis virulence factors affect phagocytosis by human neutrophils. Infect. Immun. 2000, 68, 1735–1739. [Google Scholar] [CrossRef] [PubMed]
- Osicka, R.; Osickova, A.; Hasan, S.; Bumba, L.; Cerny, J.; Sebo, P. Bordetella adenylate cyclase toxin is a unique ligand of the integrin complement receptor 3. eLife 2015, 4, e10766. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.L.; Fiederlein, R.L.; Glasser, L.; Galgiani, J.N. Bordetella pertussis adenylate cyclase: Effects of affinity-purified adenylate cyclase on human polymorphonuclear leukocyte functions. Infect. Immun. 1987, 55, 135–140. [Google Scholar] [PubMed]
- Eby, J.C.; Gray, M.C.; Hewlett, E.L. Cyclic AMP-mediated suppression of neutrophil extracellular trap formation and apoptosis by the Bordetella pertussis adenylate cyclase toxin. Infect. Immun. 2014, 5256–5269. [Google Scholar] [CrossRef] [PubMed]
- Gorgojo, J.; Scharrig, E.; Gómez, R.M.; Harvill, E.T.; Rodríguez, M.E. Bordetella parapertussis Circumvents Neutrophil Extracellular Bactericidal Mechanisms. PLoS ONE 2017, 12, e0169936. [Google Scholar] [CrossRef] [PubMed]
- Cerny, O.; Kamanova, J.; Masin, J.; Bibova, I.; Skopova, K.; Sebo, P. Bordetella pertussis Adenylate Cyclase Toxin Blocks Induction of Bactericidal Nitric Oxide in Macrophages through cAMP-Dependent Activation of the SHP-1 Phosphatase. J. Immunol. 2015, 194, 4901–4913. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, J.N.; Cerny, O.; Linhartova, I.; Masin, J.; Osicka, R.; Sebo, P. cAMP signalling of Bordetella adenylate cyclase toxin through the SHP-1 phosphatase activates the BimEL-Bax pro-apoptotic cascade in phagocytes. Cell Microbiol. 2016, 18, 384–398. [Google Scholar] [CrossRef] [PubMed]
- Masure, H.R. The adenylate cyclase toxin contributes to the survival of Bordetella pertussis within human macrophages. Microb. Pathog. 1993, 14, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Khelef, N.; Zychlinsky, A.; Guiso, N. Bordetella pertussis induces apoptosis in macrophages: Role of adenylate cyclase-hemolysin. Infect. Immun. 1993, 61, 4064–4071. [Google Scholar] [PubMed]
- Gueirard, P.; Druilhe, A.; Pretolani, M.; Guiso, N. Role of adenylate cyclase-hemolysin in alveolar macrophage apoptosis during Bordetella pertussis infection in vivo. Infect. Immun. 1998, 66, 1718–1725. [Google Scholar] [PubMed]
- Skopova, K.; Tomalova, B.; Kanchev, I.; Rossmann, P.; Svedova, M.; Adkins, I.; Bibova, I.; Tomala, J.; Masin, J.; Guiso, N.; et al. Cyclic AMP-Elevating Capacity of Adenylate Cyclase Toxin-Hemolysin Is Sufficient for Lung Infection but Not for Full Virulence of Bordetella pertussis. Infect. Immun. 2017, 85, pii:e00937-16. [Google Scholar] [CrossRef] [PubMed]
- Eby, J.C.; Gray, M.C.; Warfel, J.M.; Paddock, C.D.; Jones, T.F.; Day, S.R.; Bowden, J.; Poulter, M.D.; Donato, G.M.; Merkel, T.J.; et al. Quantification of the adenylate cyclase toxin of Bordetella pertussis in vitro and during respiratory infection. Infect. Immun. 2013, 81, 1390–1398. [Google Scholar] [CrossRef] [PubMed]
- Gonyar, L.A.; Gray, M.C.; Christianson, G.J.; Mehrad, B.; Hewlett, E.L. Albumin, in the Presence of Calcium, Elicits a Massive Increase in Extracellular Bordetella Adenylate Cyclase Toxin. Infect. Immun. 2017, 85. [Google Scholar] [CrossRef] [PubMed]
- Bagley, K.C.; Abdelwahab, S.F.; Tuskan, R.G.; Fouts, T.R.; Lewis, G.K. Pertussis toxin and the adenylate cyclase toxin from Bordetella pertussis activate human monocyte-derived dendritic cells and dominantly inhibit cytokine production through a cAMP-dependent pathway. J. Leukoc. Biol. 2002, 72, 962–969. [Google Scholar] [PubMed]
- Ross, P.J.; Lavelle, E.C.; Mills, K.H.; Boyd, A.P. Adenylate cyclase toxin from Bordetella pertussis synergizes with lipopolysaccharide to promote innate interleukin-10 production and enhances the induction of Th2 and regulatory T cells. Infect. Immun. 2004, 72, 1568–1579. [Google Scholar] [CrossRef] [PubMed]
- Skinner, J.A.; Reissinger, A.; Shen, H.; Yuk, M.H. Bordetella type III secretion and adenylate cyclase toxin synergize to drive dendritic cells into a semimature state. J. Immunol. 2004, 173, 1934–1940. [Google Scholar] [CrossRef] [PubMed]
- Spensieri, F.; Fedele, G.; Fazio, C.; Nasso, M.; Stefanelli, P.; Mastrantonio, P.; Ausiello, C.M. Bordetella pertussis inhibition of interleukin-12 (IL-12) p70 in human monocyte-derived dendritic cells blocks IL-12 p35 through adenylate cyclase toxin-dependent cyclic AMP induction. Infect. Immun. 2006, 74, 2831–2838. [Google Scholar] [CrossRef] [PubMed]
- Hickey, F.B.; Brereton, C.F.; Mills, K.H. Adenylate cyclase toxin of Bordetella pertussis inhibits TLR-induced IRF-1 and IRF-8 activation and IL-12 production and enhances IL-10 through MAPK activation in dendritic cells. J. Leukoc. Biol. 2008, 84, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Adkins, I.; Kamanova, J.; Kocourkova, A.; Svedova, M.; Tomala, J.; Janova, H.; Masin, J.; Chladkova, B.; Bumba, L.; Kovar, M.; et al. Bordetella adenylate cyclase toxin differentially modulates toll-like receptor-stimulated activation, migration and T cell stimulatory capacity of dendritic cells. PLoS ONE 2014, 9, e104064. [Google Scholar] [CrossRef] [PubMed]
- Trinchieri, G.; Pflanz, S.; Kastelein, R.A. The IL-12 family of heterodimeric cytokines: New players in the regulation of T cell responses. Immunity 2003, 19, 641–644. [Google Scholar] [CrossRef]
- Gautier, G.; Humbert, M.; Deauvieau, F.; Scuiller, M.; Hiscott, J.; Bates, E.E.; Trinchieri, G.; Caux, C.; Garrone, P. A type I interferon autocrine-paracrine loop is involved in Toll-like receptor-induced interleukin-12p70 secretion by dendritic cells. J. Exp. Med. 2005, 201, 1435–1446. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Guan, X.; Tamura, T.; Ozato, K.; Ma, X. Synergistic activation of interleukin-12 p35 gene transcription by interferon regulatory factor-1 and interferon consensus sequence-binding protein. J. Biol. Chem. 2004, 279, 55609–55617. [Google Scholar] [CrossRef] [PubMed]
- Henderson, M.W.; Inatsuka, C.S.; Sheets, A.J.; Williams, C.L.; Benaron, D.J.; Donato, G.M.; Gray, M.C.; Hewlett, E.L.; Cotter, P.A. Contribution of Bordetella filamentous hemagglutinin and adenylate cyclase toxin to suppression and evasion of interleukin-17-mediated inflammation. Infect. Immun. 2012, 80, 2061–2075. [Google Scholar] [CrossRef] [PubMed]
- Macdonald-Fyall, J.; Xing, D.; Corbel, M.; Baillie, S.; Parton, R.; Coote, J. Adjuvanticity of native and detoxified adenylate cyclase toxin of Bordetella pertussis towards co-administered antigens. Vaccine 2004, 22, 4270–4281. [Google Scholar] [CrossRef] [PubMed]
- Orr, B.; Douce, G.; Baillie, S.; Parton, R.; Coote, J. Adjuvant effects of adenylate cyclase toxin of Bordetella pertussis after intranasal immunization of mice. Vaccine 2007, 25, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Cheung, G.Y.; Xing, D.; Prior, S.; Corbel, M.J.; Parton, R.; Coote, J.G. Effect of different forms of adenylate cyclase toxin of Bordetella pertussis on protection afforded by an acellular pertussis vaccine in a murine model. Infect. Immun. 2006, 74, 6797–6805. [Google Scholar] [CrossRef] [PubMed]
- Dadaglio, G.; Fayolle, C.; Zhang, X.; Ryffel, B.; Oberkampf, M.; Felix, T.; Hervas-Stubbs, S.; Osicka, R.; Sebo, P.; Ladant, D.; et al. Antigen targeting to CD11b+ dendritic cells in association with TLR4/TRIF signaling promotes strong CD8+ T cell responses. J. Immunol. 2014, 193, 1787–1798. [Google Scholar] [CrossRef] [PubMed]
- Svedova, M.; Masin, J.; Fiser, R.; Cerny, O.; Tomala, J.; Freudenberg, M.; Tuckova, L.; Kovar, M.; Dadaglio, G.; Adkins, I.; et al. Pore-formation by adenylate cyclase toxoid activates dendritic cells to prime CD8+ and CD4+ T cells. Immunol. Cell Biol. 2016, 94, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Simsova, M.; Sebo, P.; Leclerc, C. The adenylate cyclase toxin from Bordetella pertussis—A novel promising vehicle for antigen delivery to dendritic cells. Int. J. Med. Microbiol. 2004, 293, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Adkins, I.; Holubova, J.; Kosova, M.; Sadilkova, L. Bacteria and their toxins tamed for immunotherapy. Curr. Pharm. Biotechnol. 2012, 13, 1446–1473. [Google Scholar] [CrossRef] [PubMed]
- Sebo, P.; Fayolle, C.; d’Andria, O.; Ladant, D.; Leclerc, C.; Ullmann, A. Cell-invasive activity of epitope-tagged adenylate cyclase of Bordetella pertussis allows in vitro presentation of a foreign epitope to CD8+ cytotoxic T cells. Infect. Immun. 1995, 63, 3851–3857. [Google Scholar] [PubMed]
- Gmira, S.; Karimova, G.; Ladant, D. Characterization of recombinant Bordetella pertussis adenylate cyclase toxins carrying passenger proteins. Res. Microbiol. 2001, 152, 889–900. [Google Scholar] [CrossRef]
- Préville, X.; Ladant, D.; Timmerman, B.; Leclerc, C. Eradication of established tumors by vaccination with recombinant Bordetella pertussis adenylate cyclase carrying the human papillomavirus 16 E7 oncoprotein. Cancer Res. 2005, 65, 641–649. [Google Scholar] [PubMed]
- Holubova, J.; Kamanova, J.; Jelinek, J.; Tomala, J.; Masin, J.; Kosova, M.; Stanek, O.; Bumba, L.; Michalek, J.; Kovar, M.; et al. Delivery of large heterologous polypeptides across the cytoplasmic membrane of antigen-presenting cells by the Bordetella RTX hemolysin moiety lacking the adenylyl cyclase domain. Infect. Immun. 2012, 80, 1181–1192. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, P.; Bouillette-Marussig, M.; Hens, A.; De Coster, I.; Depuydt, C.; Goubier, A.; Van Tendeloo, V.; Cools, N.; Goossens, H.; Hercend, T.; et al. GTL001, A Therapeutic Vaccine for Women Infected with Human Papillomavirus 16 or 18 and Normal Cervical Cytology: Results of a Phase I Clinical Trial. Clin. Cancer Res. 2016, 22, 3238–3248. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fedele, G.; Schiavoni, I.; Adkins, I.; Klimova, N.; Sebo, P. Invasion of Dendritic Cells, Macrophages and Neutrophils by the Bordetella Adenylate Cyclase Toxin: A Subversive Move to Fool Host Immunity. Toxins 2017, 9, 293. https://doi.org/10.3390/toxins9100293
Fedele G, Schiavoni I, Adkins I, Klimova N, Sebo P. Invasion of Dendritic Cells, Macrophages and Neutrophils by the Bordetella Adenylate Cyclase Toxin: A Subversive Move to Fool Host Immunity. Toxins. 2017; 9(10):293. https://doi.org/10.3390/toxins9100293
Chicago/Turabian StyleFedele, Giorgio, Ilaria Schiavoni, Irena Adkins, Nela Klimova, and Peter Sebo. 2017. "Invasion of Dendritic Cells, Macrophages and Neutrophils by the Bordetella Adenylate Cyclase Toxin: A Subversive Move to Fool Host Immunity" Toxins 9, no. 10: 293. https://doi.org/10.3390/toxins9100293