
Determination of Ochratoxin A in Rye and Rye-Based Products by Fluorescence Polarization Immunoassay

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
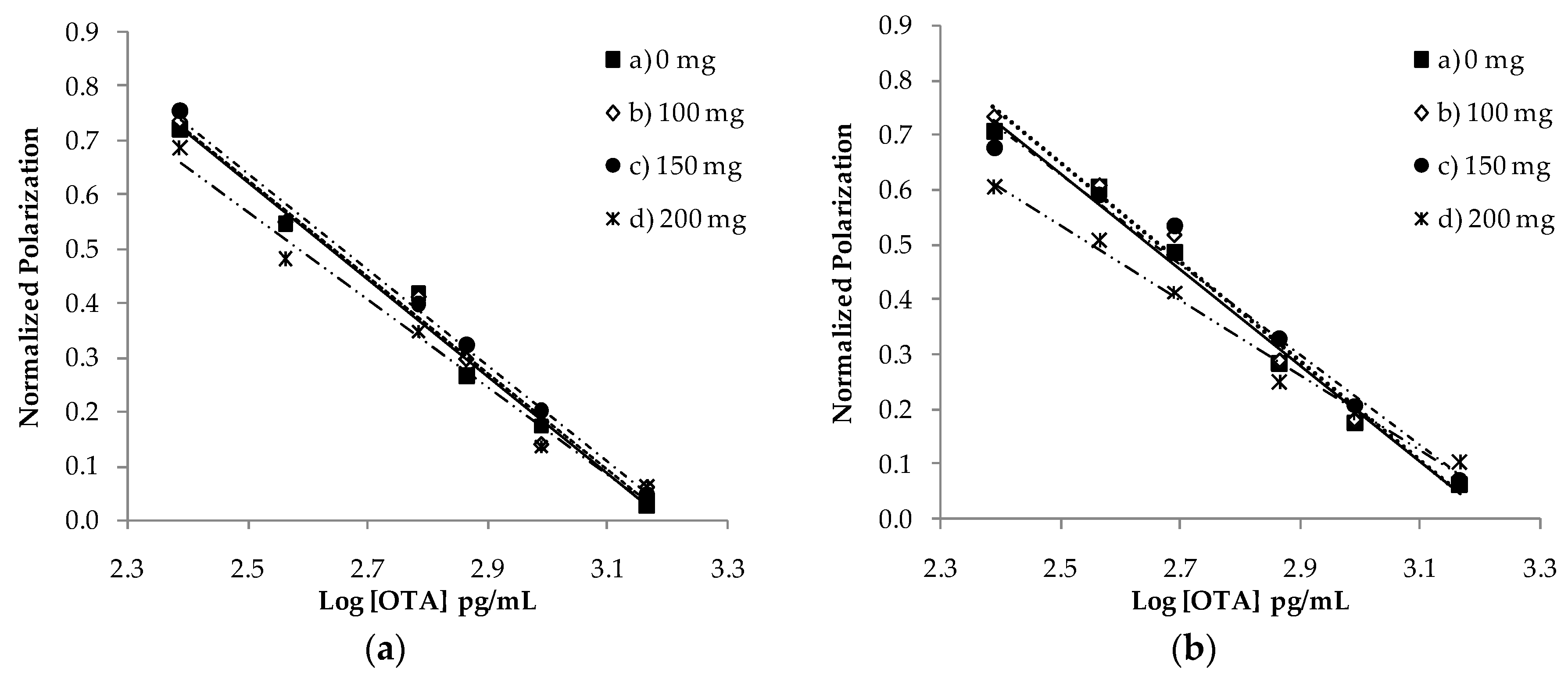
2.1. Matrix Effect and SPE Cleanup
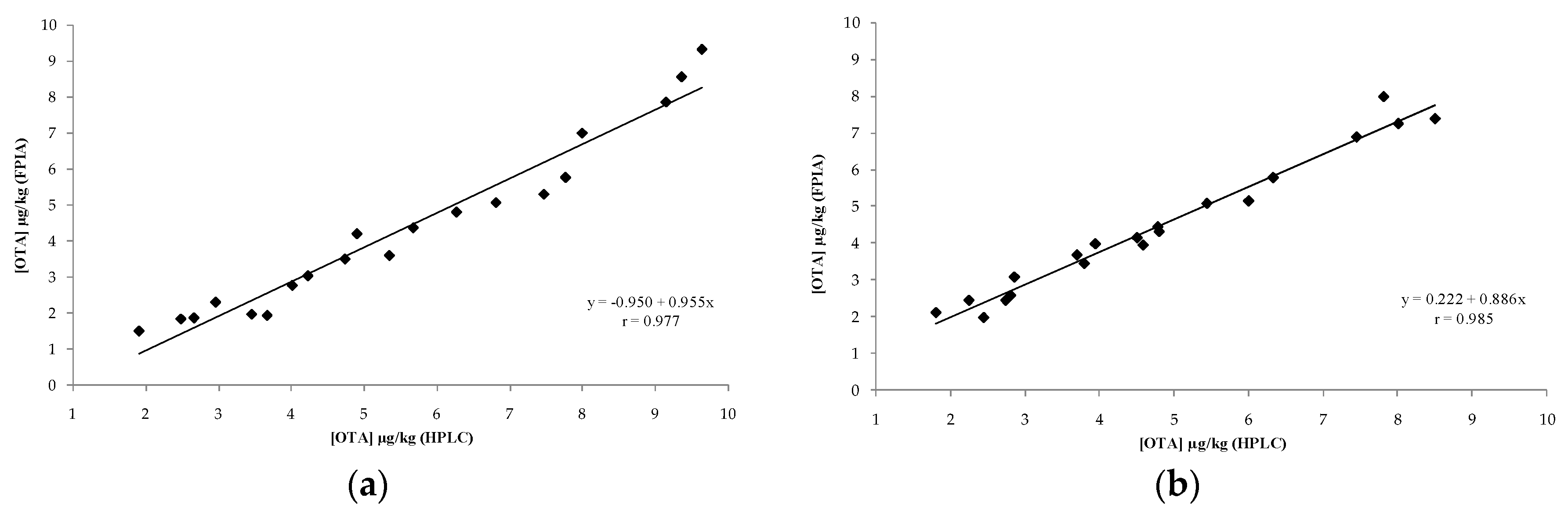
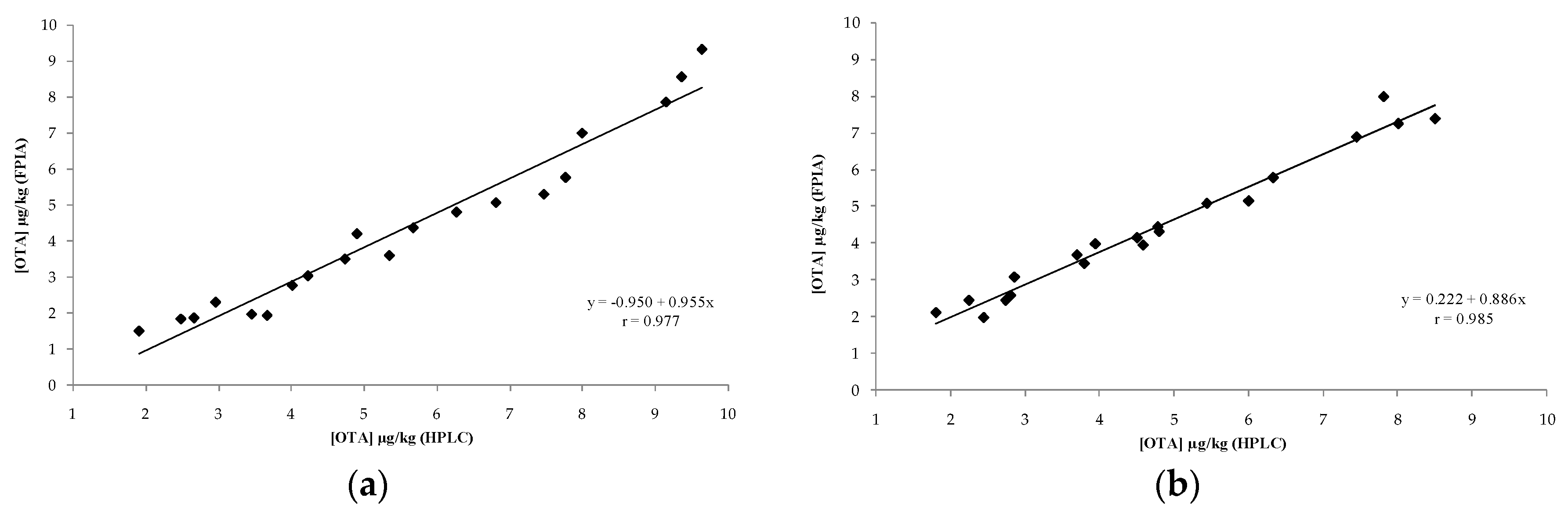
2.2. In-House Validation of the Optimized FPIA as Quantitative Method
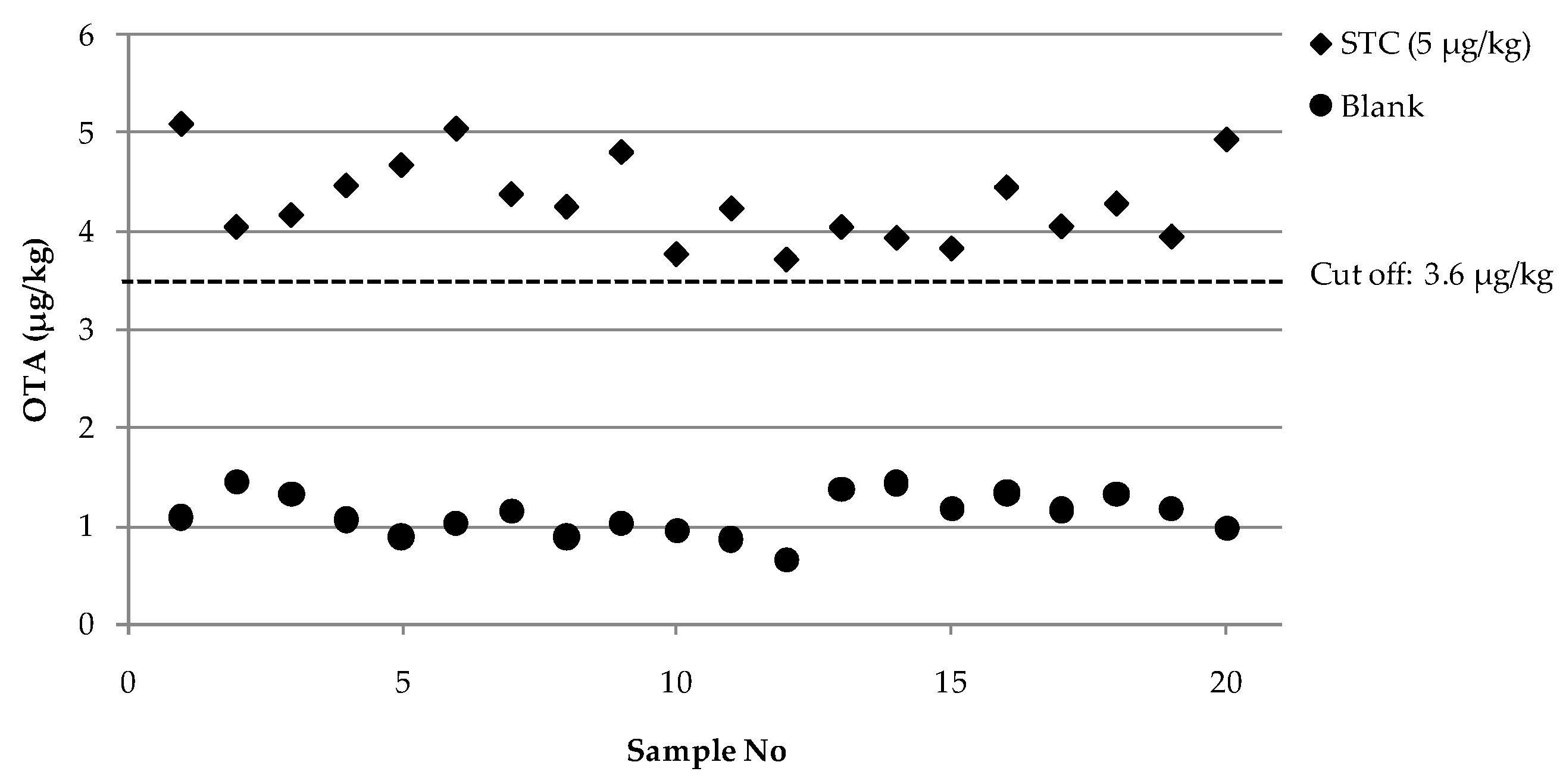
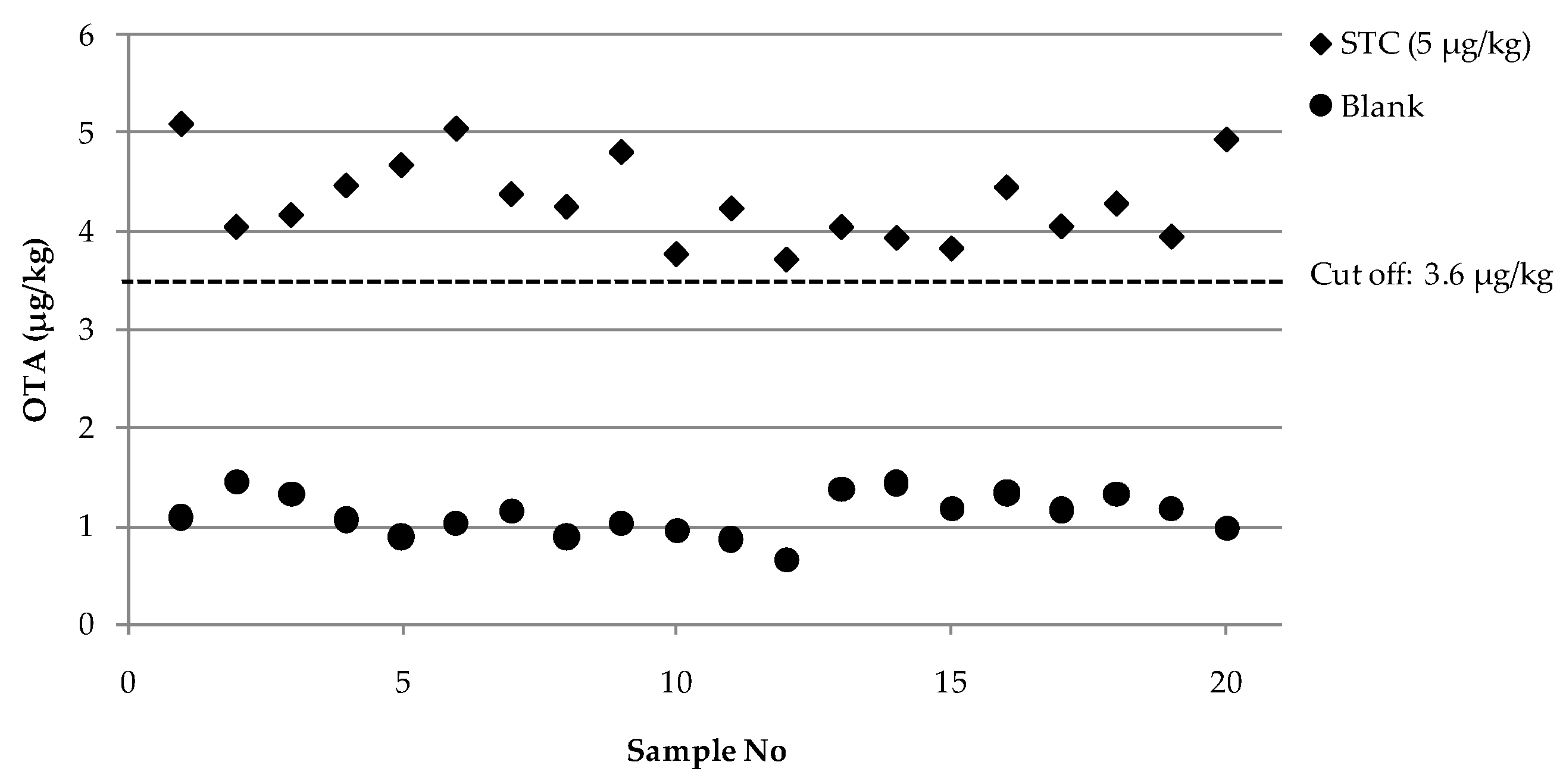
2.3. Evaluation of Analytical Performances of the Optimized FPIA According to Regulation 519/2014/EU
3. Conclusions
4. Materials and Methods
4.1. Reagents and Chemicals
4.2. OTA Standard and Immunoreagent Solutions
4.3. Sample Preparation
4.4. SPE Cleanup
4.5. FPIA Analysis
4.6. HPLC Analysis
4.7. In-House Validation of the Optimized FPIAs as Quantitative Method
4.8. In-House Validation of the Optimized FPIAs as Screening Method According to Regulation 519/2014/EU
4.9. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References and Note
- Wang, Y.; Wang, L.; Liu, F.; Wang, Q.; Selvaraj, J.N.; Xing, F.; Zhao, Y.; Liu, Y. Ochratoxin A Producing Fungi, Biosynthetic Pathway and Regulatory Mechanisms. Toxins 2016, 8, 83. [Google Scholar] [CrossRef] [PubMed]
- Malir, F.; Ostry, V.; Pfohl-Leszkowicz, A.; Malir, J.; Toman, J. Ochratoxin A: 50 Years of Research. Toxins 2016, 8, 191. [Google Scholar] [CrossRef] [PubMed]
- Bui-Klimke, T.R.; Wu, F. Ochratoxin A and Human Health Risk: A Review of the Evidence. Crit. Rev. Food Sci. Nutr. 2015, 55, 1860–1869. [Google Scholar] [CrossRef] [PubMed]
- International Agency for Research on Cancer (IARC). Monographs on the Evaluation of Carcinogenic Risks to Humans: Some Naturally Occuring Substances: Food Items and Costituents, Heterocyclic Aromatic Amines and Mycotoxins; IARC: Lyon, France, 1993; Volume 56, pp. 489–524. [Google Scholar]
- Kevin, J. Occurrence of ochratoxin A in commodities and processed food—A review of EU occurrence data. Food Addit. Contam. 2005, 22, 26–30. [Google Scholar] [CrossRef]
- Mateo, R.; Medina, A.; Mateo, E.M.; Mateo, F.; Jiménez, M. An overview of ochratoxin a in beer and wine. Int. J. Food Microbiol. 2007, 119, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Duarte, S.C.; Pena, A.; Lino, C.M. A review on OTA occurrence and effects of processing of cereal and cereal derived food products. Food Microbiol. 2010, 27, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Kabak, B.; Dobson, W. Mycotoxins in spices and herbs—An update. Crit. Rev. Food Sci. Nutr. 2017, 57, 18–34. [Google Scholar] [CrossRef] [PubMed]
- SCOOP (2002). Task Report 3.2.7 Assessment of Dietary Intake of Ochratoxin A by the Population of EU Member States. Available online: https://ec.europa.eu/food/sites/food/files/safety/docs/cs_contaminants_catalogue_ochratoxin_task_3-2-7_en.pdf (accessed on 25 September 2017).
- RASFF Portal. Available online: https://webgate.ec.europa.eu/rasff-window/portal/?event=SearchForm&cleanSearch=1 (accessed on 25 September 2017).
- FAO. Food and Agriculture Organization of the United Nations Statistics Division. 2014. Available online: http://faostat3.fao.org/download/Q/QC/E (accessed on 25 September 2017).
- Bondia-Pons, I.; Aura, A.M.; Vuorela, S.; Kolehmainen, M.; Mykkänen, H.; Poutanen, K. Rye phenolics in nutrition and health. J. Cereal Sci. 2009, 49, 323–336. [Google Scholar] [CrossRef]
- Mellen, P.B.; Walsh, T.F.; Herrington, D.M. Whole grain intake and cardiovascular diseases: A meta-analysis. Nutr. Metab. Cardiovasc. Dis. 2008, 18, 283–290. [Google Scholar] [CrossRef] [PubMed]
- European Commission. Commission Regulation (EC) No 1881/2006 of 19 December 2006 Setting Maximum Levels for Certain Contaminants in Foodstuffs. Off. J. Eur. Union 2006, L364, 5–24. [Google Scholar]
- Berthiller, F.; Brera, C.; Iha, M.H.; Krska, R.; Lattanzio, V.M.T.; MacDonald, S.; Malone, R.J.; Maragos, C.; Solfrizzo, M.; Stranska-Zachariasova, M.; et al. Developments in mycotoxin analysis: An update for 2015–2016. World Mycotoxin J. 2017, 10, 5–29. [Google Scholar] [CrossRef]
- Turner, N.W.; Bramhmbhatt, H.; Szabo-Vezse, M.; Poma, A.; Coker, R.; Piletsky, S.A. Analytical methods for determination of mycotoxins: An update (2009–2014). Anal. Chim. Acta 2015, 901, 12–33. [Google Scholar] [CrossRef] [PubMed]
- Visconti, A.; De Girolamo, A. Fitness for purpose ochratoxin a analytical developments. Food Addit. Contam. 2005, 22, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Entwisle, A.C.; Williams, A.C.; Mann, P.J.; Slack, P.T.; Gilbert, J. Liquid chromatographic method with immunoaffinity column cleanup for determination of ochratoxin A in barley: Collaborative study. J. AOAC Int. 2000, 83, 1377–1383. [Google Scholar] [PubMed]
- European Committee for Standardization (CEN). Foodstuffs—Determination of Ochratoxin A in Barley and Roasted Coffee: HPLC Method with Immunoaffinity Column Clean-Up; BSI: London, UK, 2009. [Google Scholar]
- European Committee for Standardization (CEN). Foodstuffs—Determination of Ochratoxin A in Cereal Based Foods for Infants and Young Children: HPLC Method with Immunoaffinity Column Clean-Up and Fluorescence Detection; BSI: London, UK, 2010. [Google Scholar]
- Lattanzio, V.M.T.; Ciasca, B.; Powers, S.; Visconti, A. Improved method for the simultaneous determination of aflatoxins, ochratoxin A and Fusarium toxins in cereals and derived products by liquid chromatography-tandem mass spectrometry after multi-toxin immunoaffinity clean up. J. Chromatogr. A 2014, 1354, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Venkataramana, M.; Rashmi, R.; Uppalapati, S.R.; Chandranayaka, S.; Balakrishna, K.; Radhika, M.; Gupta, V.K.; Batra, H.V. Development of sandwich dot-ELISA for specific detection of Ochratoxin A and its application on to contaminated cereal grains originating from India. Front. Microbiol. 2015, 6, 511. [Google Scholar] [CrossRef] [PubMed]
- Anfossi, L.; D’Arco, G.; Baggiani, C.; Giovannoli, C.; Giraudi, G. A lateral flow immunoassay for measuring ochratoxin A: Development of a single system for maize, wheat and durum wheat. Food Control 2011, 22, 1965–1970. [Google Scholar] [CrossRef]
- Saucedo-Friebe, J.C.; Karsunkea, X.Y.Z.; Vazaca, S.; Biselli, S.; Niessnera, R.; Knoppa, D. Regenerable immuno-biochip for screening ochratoxin A in green coffee extract using an automated microarray chip reader with chemiluminescence detection. Anal. Chim. Acta 2011, 689, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, J.G.; Castro, M.; Machado, S.; Barroso, M.F.; Nouws, H.P.A.; Delerue-Matos, C. Molecularly imprinted electrochemical sensor for ochratoxin A detection in food samples. Sens. Actuators B Chem. 2015, 215, 107–112. [Google Scholar] [CrossRef]
- Urusov, A.E.; Kostenko, S.N.; Sveshnikov, P.G.; Zherdev, A.V.; Dzantiev, B.B. Ochratoxin A immunoassay with surface plasmon resonance registration: Lowering limit of detection by the use of colloidal gold immunoconjugates. Sens. Actuators B Chem. 2011, 156, 343–349. [Google Scholar] [CrossRef]
- Ha, T.H. Recent Advances for the Detection of Ochratoxin A. Toxins 2015, 7, 5276–5300. [Google Scholar] [CrossRef] [PubMed]
- Lippolis, V.; Maragos, C. Fluorescence polarisation immunoassays for rapid, accurate and sensitive determination of mycotoxins. World Mycotoxin J. 2014, 7, 479–489. [Google Scholar] [CrossRef]
- Lippolis, V.; Pascale, M.; Visconti, A. Optimization of a fluorescence polarization immunoassay for rapid quantification of deoxynivalenol in durum wheat-based products. J. Food Prot. 2006, 69, 2712–2719. [Google Scholar] [CrossRef] [PubMed]
- Zezza, F.; Longobardi, F.; Pascale, M.; Eremin, S.A.; Visconti, A. Fluorescence polarization immunoassay for rapid screening of ochratoxin A in red wine. Anal. Bioanal. Chem. 2009, 395, 1317–1323. [Google Scholar] [CrossRef] [PubMed]
- Chun, H.S.; Choi, E.H.; Chang, H.J.; Choi, S.W.; Eremin, S.A. A fluorescence polarization immunoassay for the detection of zearalenone in corn. Anal. Chim. Acta 2009, 639, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Lippolis, V.; Pascale, M.; Valenzano, S.; Pluchinotta, V.; Baumgartner, S.; Krska, R.; Visconti, A. A rapid fluorescence polarization immunoassay for the determination of T-2 and HT-2 toxins in wheat. Anal. Bioanal. Chem. 2011, 401, 2561–2571. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Y.J.; Eremin, S.; Mi, T.J.; Zhang, S.X.; Shen, J.Z.; Wang, Z.H. The development of a fluorescence polarization immunoassay for aflatoxin detection. Biomed. Environ. Sci. 2014, 27, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Valenzano, S.; Lippolis, V.; Pascale, M.; De Marco, A.; Maragos, C.M.; Suman, M.; Visconti, A. Determination of Deoxynivalenol in Wheat Bran and Whole-Wheat Flour by Fluorescence Polarization Immunoassay. Food Anal. Methods 2014, 7, 806–813. [Google Scholar] [CrossRef]
- Beloglazova, N.V.; Eremin, S.A. Rapid screening of aflatoxin B1 in beer by fluorescence polarization immunoassay. Talanta 2015, 142, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Mi, T.; Conti, G.O.; Yu, Q.; Wen, K.; Shen, J.; Ferrante, M.; Wang, Z. Development of a screening fluorescence polarization immunoassay for the simultaneous detection of fumonisins B1 and B2 in maize. J. Agric. Food Chem. 2015, 63, 4940–4946. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wen, K.; Mi, T.; Zhang, X.; Zhang, H.; Zhang, S.; Shen, J.; Wang, Z. A universal multi-wavelength fluorescence polarization immunoassay for multiplexed detection of mycotoxins in maize. Biosens. Bioelectron. 2016, 79, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Porricelli, A.C.R.; Lippolis, L.; Valenzano, S.; Cortese, M.; Suman, M.; Zanardi, S.; Pascale, M. Optimization and Validation of a Fluorescence Polarization Immunoassay for Rapid Detection of T-2 and HT-2 Toxins in Cereals and Cereal-Based Products. Food Anal. Methods 2016, 9, 3310–3318. [Google Scholar] [CrossRef]
- Zhang, X.; Eremin, S.A.; Wen, K.; Yu, X.; Li, C.; Ke, Y.; Jiang, H.; Shen, J.; Wang, Z. Fluorescence Polarization Immunoassay Based on a New Monoclonal Antibody for the Detection of the Zearalenone Class of Mycotoxins in Maize. J. Agric. Food Chem. 2017, 65, 2240–2247. [Google Scholar] [CrossRef] [PubMed]
- Shim, W.B.; Kolosova, A.Y.; Kim, Y.J.; Yang, Z.Y.; Park, S.J.; Eremin, S.A.; Lee, I.S.; Chung, D.H. Fluorescence polarization immunoassay based on a monoclonal antibody for the detection of ochratoxin A. Int. J. Food Sci. Technol. 2004, 39, 829–837. [Google Scholar] [CrossRef]
- Park, J.H.; Chung, D.H.; Lee, I.S. Application of fluorescence polarization immunoassay for the screening of ochratoxin A in unpolished rice. J. Life Sci. 2006, 16, 1006–1013. [Google Scholar] [CrossRef]
- Bondarenko, A.P.; Eremin, S.A. Determination of zearalenone and ochratoxin A mycotoxins in grain by fluorescence polarization immunoassay. J. Anal. Chem. 2012, 67, 790–794. [Google Scholar] [CrossRef]
- Lippolis, V.; Pascale, M.; Valenzano, S.; Porricelli, A.C.R.; Suman, M.; Visconti, A. Fluorescence Polarization Immunoassay for Rapid, Accurate and Sensitive Determination of Ochratoxin A in Wheat. Food Anal. Methods 2014, 7, 298–307. [Google Scholar] [CrossRef]
- European Commission. Commission Regulation (EU) No 519/2014 of 16 May 2014 amending Regulation (EC) No 401/2006 as regards methods of sampling of large lots, spices and food supplements, performance criteria for T-2, HT-2 toxin and citrinin and screening methods of analysis. Off. J. Eur. Union 2014, L147, 29–43. [Google Scholar]
- Lattanzio, V.M.T.; Ciasca, B.; Powers, S.; von Holst, C. Validation of screening methods according to Regulation 519/2014/EU. Determination of deoxynivalenol in wheat by lateral flow immunoassay: A case study. Trends Anal. Chem. 2016, 76, 137–144. [Google Scholar] [CrossRef]
- Sanders, M.; McPartlin, D.; Moran, K.; Guo, Y.; Eeckhout, M.; O’Kennedy, R.; De Saeger, S.; Maragos, C. Comparison of Enzyme-Linked Immunosorbent Assay, Surface Plasmon Resonance and Biolayer Interferometry for Screening of Deoxynivalenol in Wheat and Wheat Dust. Toxins 2016, 8, 103. [Google Scholar] [CrossRef] [PubMed]
- Lattanzio, V.M.T. Toward Harmonization of Performance Criteria for Mycotoxin Screening Methods: The EU Perspective. J. AOAC Int. 2016, 99, 906–913. [Google Scholar] [CrossRef] [PubMed]
- European Committee for Standardization (CEN). CEN/TR 16059:2010. Food analysis—Performance Criteria for Single Laboratory Validation Methods of Analysis for the Determination of Mycotoxins.
- European Commission. Commission Regulation (EC) No 401/2006 of 23 February 2006 laying down the methods of sampling and analysis for the official control of the levels of mycotoxins in foodstuffs. Off. J. Eur. Union 2006, L70, 12–34. [Google Scholar]
- Miller, J.C.; Miller, J.N. Statistics for Analytical Chemistry; Ellis Horwood: Chichester, UK, 1984. [Google Scholar]
- Guidelines for Collaborative Study Procedures to Validate Characteristics of A Method of Analysis (Appendix D). AOAC Official Methods of Analysis, 2002, AOAC International. Available online: http://www.aoac.org/aoac_prod_imis/AOAC_Docs/StandardsDevelopment/Collaborative_Study_Validation_Guidelines.pdf (accessed on 25 September 2017).
- Soliani, L. Statistica Applicata per la Ricerca e Professioni Scientifiche. Manuale di Statistica Univariata e Bivariata Parametrica e Non-Parametrica; Uninova-Gruppo Pegaso: Parma, Italy, 2007. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
| Matrix | Spiking Level (μg/kg) | FPIA | HPLC | ||
|---|---|---|---|---|---|
| Recovery | RSD a (%) | Recovery | RSD a (%) | ||
| Rye | 2 | 86 | 2 | 99 | 3 |
| 5 | 84 | 2 | 98 | 2 | |
| 8 | 89 | 4 | 100 | 3 | |
| Overall average | 86 | 3 | 99 | 3 | |
| Rye crispbread | 2 | 99 | 6 | 91 | 1 |
| 5 | 86 | 2 | 96 | 6 | |
| 8 | 100 | 2 | 97 | 1 | |
| Overall average | 95 | 3 | 94 | 3 | |
| Validation | Statistical Assessment | Blank | STC a (5 μg/kg) |
|---|---|---|---|
| Single laboratory validation | Mean value b (μg/kg) | 1.1 | 4.3 |
| RSDr c (%) | 13 | 10 | |
| RSDRI d (%) | 19 | 10 | |
| Cut-off level | 3.6 | ||
| Rate of false suspect results (%) | <0.1 | ||
| Small scale collaborative trial | Mean value b (μg/kg) | 0.9 | 4.8 |
| RSDr e (%) | 16 | 9 | |
| RSDR f (%) | 22 | 10 | |
| Cut-off level | 4.0 | ||
| Rate of false suspect results (%) | <0.1 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lippolis, V.; Porricelli, A.C.R.; Cortese, M.; Suman, M.; Zanardi, S.; Pascale, M. Determination of Ochratoxin A in Rye and Rye-Based Products by Fluorescence Polarization Immunoassay. Toxins 2017, 9, 305. https://doi.org/10.3390/toxins9100305
Lippolis V, Porricelli ACR, Cortese M, Suman M, Zanardi S, Pascale M. Determination of Ochratoxin A in Rye and Rye-Based Products by Fluorescence Polarization Immunoassay. Toxins. 2017; 9(10):305. https://doi.org/10.3390/toxins9100305
Chicago/Turabian StyleLippolis, Vincenzo, Anna C. R. Porricelli, Marina Cortese, Michele Suman, Sandro Zanardi, and Michelangelo Pascale. 2017. "Determination of Ochratoxin A in Rye and Rye-Based Products by Fluorescence Polarization Immunoassay" Toxins 9, no. 10: 305. https://doi.org/10.3390/toxins9100305