
Shiga Toxins Induce Apoptosis and ER Stress in Human Retinal Pigment Epithelial Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
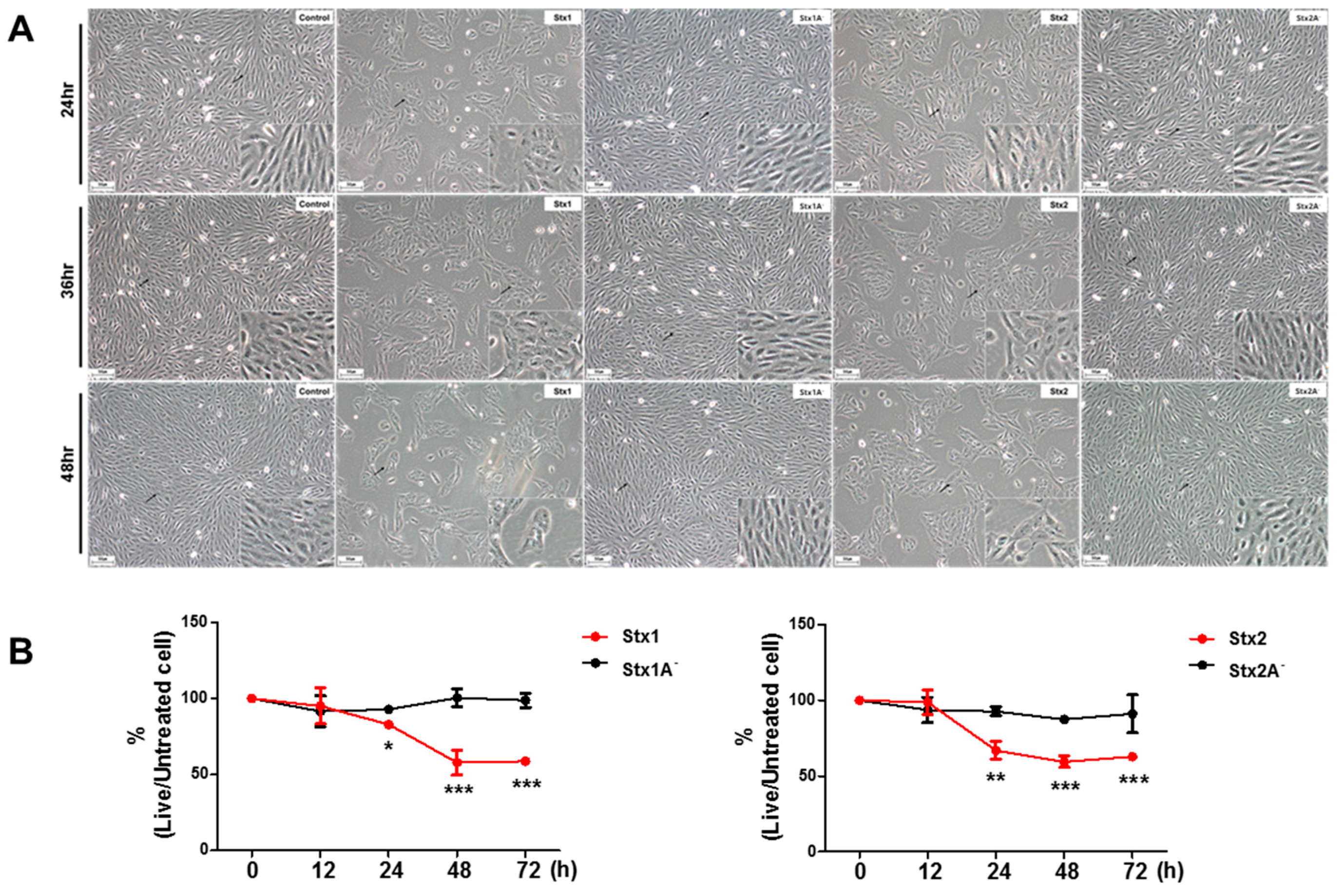
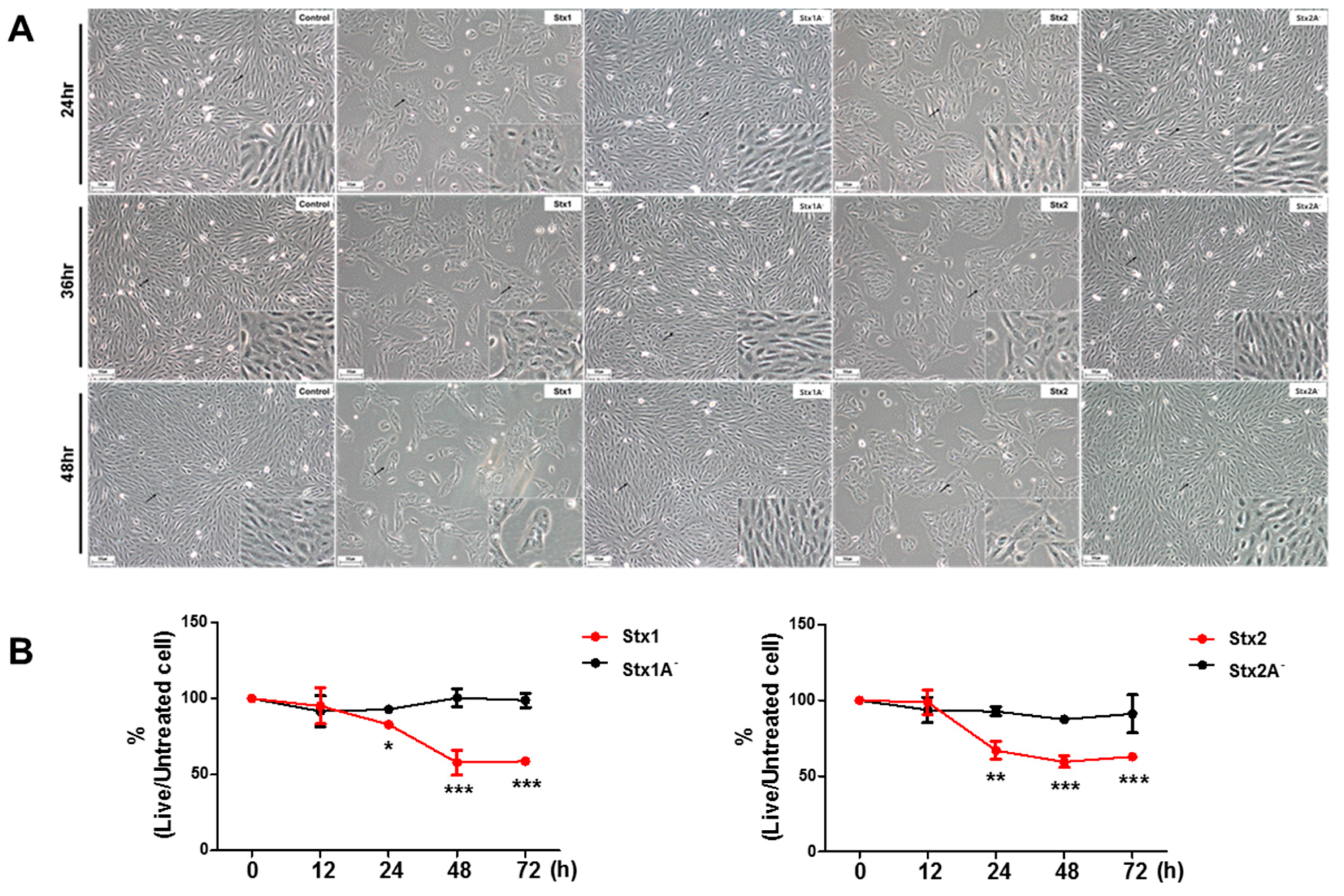
2.1. ARPE-19 Cells Are Sensitive to the Cytotoxic Effects of Stx1 and Stx2
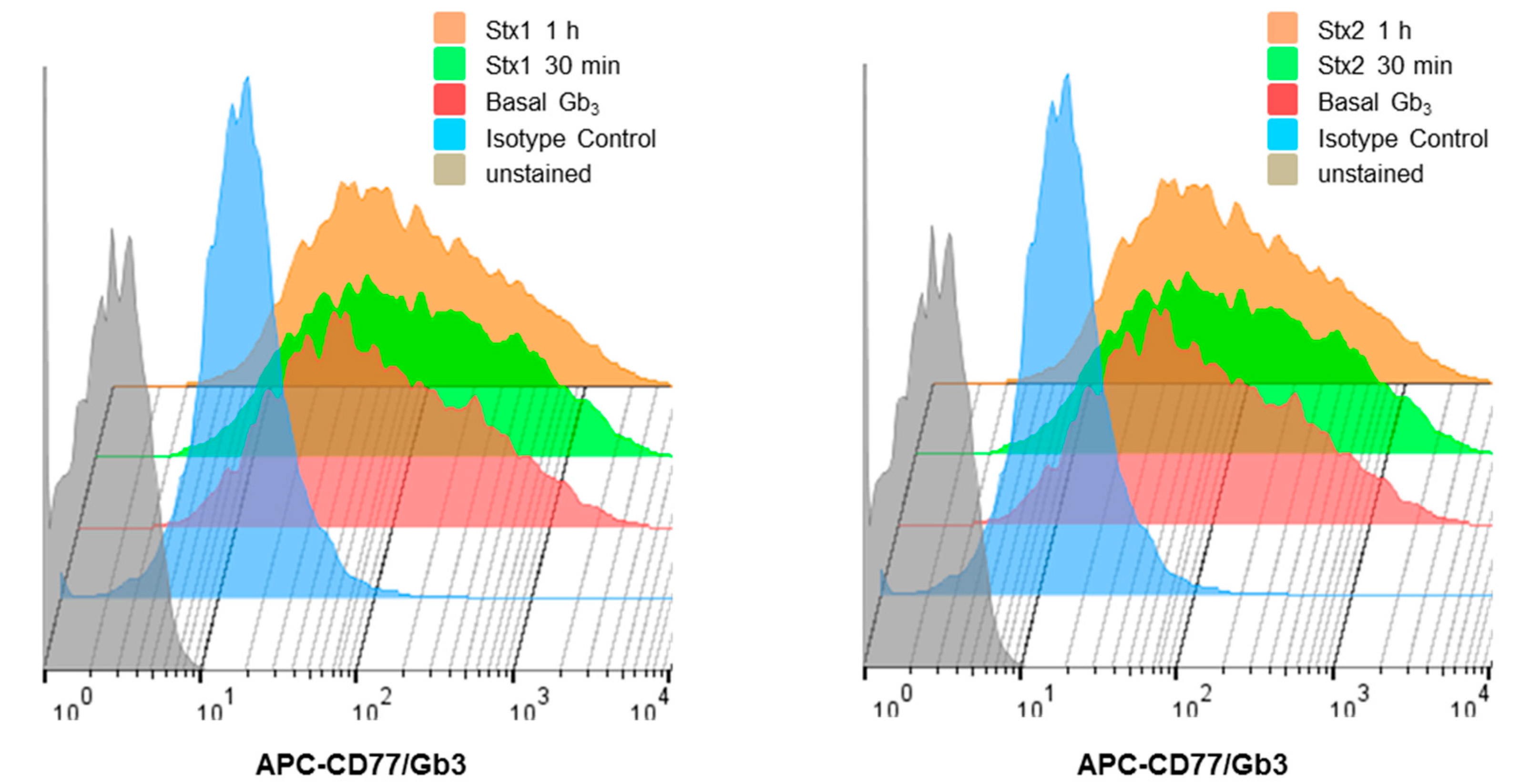
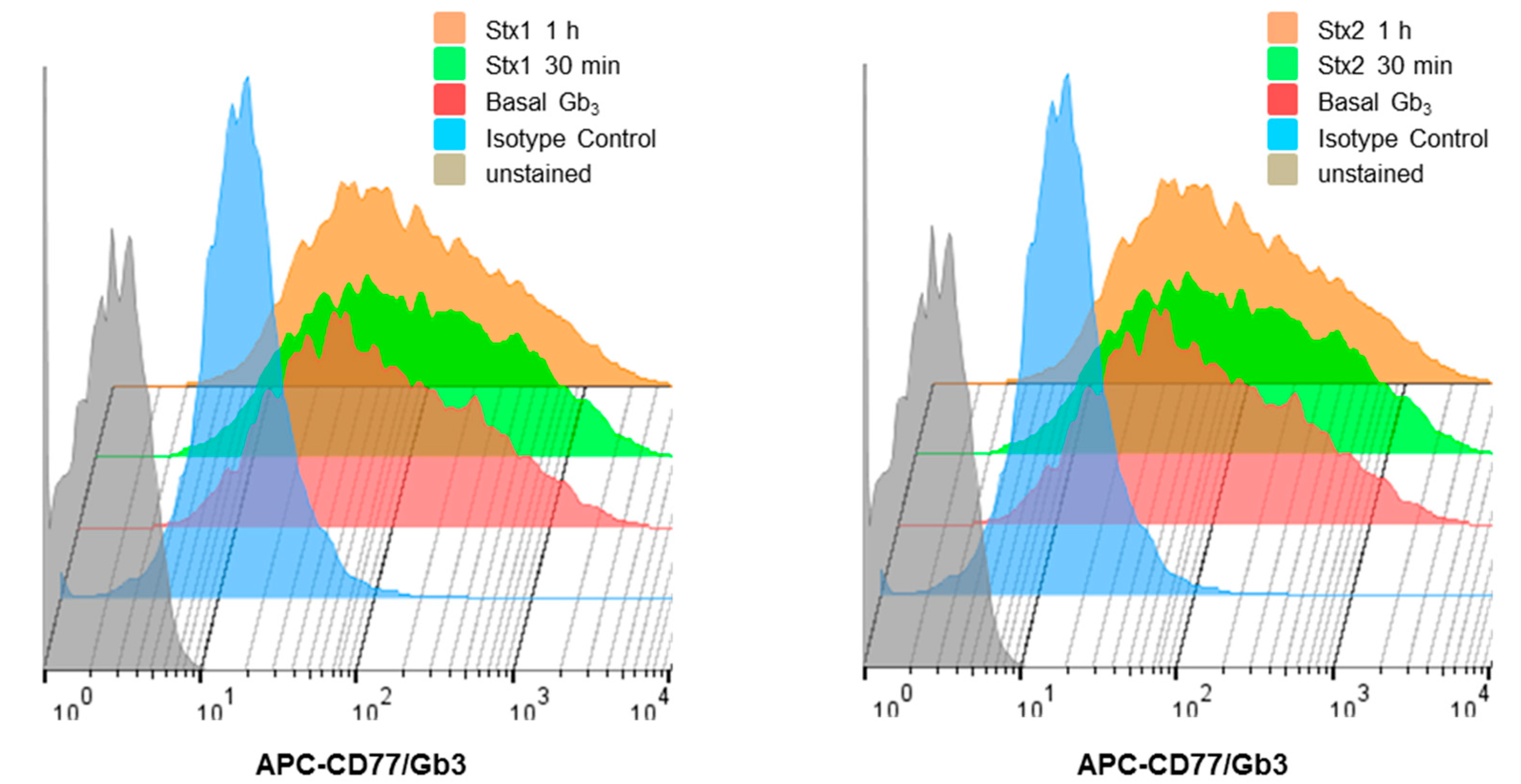
2.2. ARPE-19 Cells Express Membrane-Associated Stx-Receptor Gb3 Expression at the Cell Surface
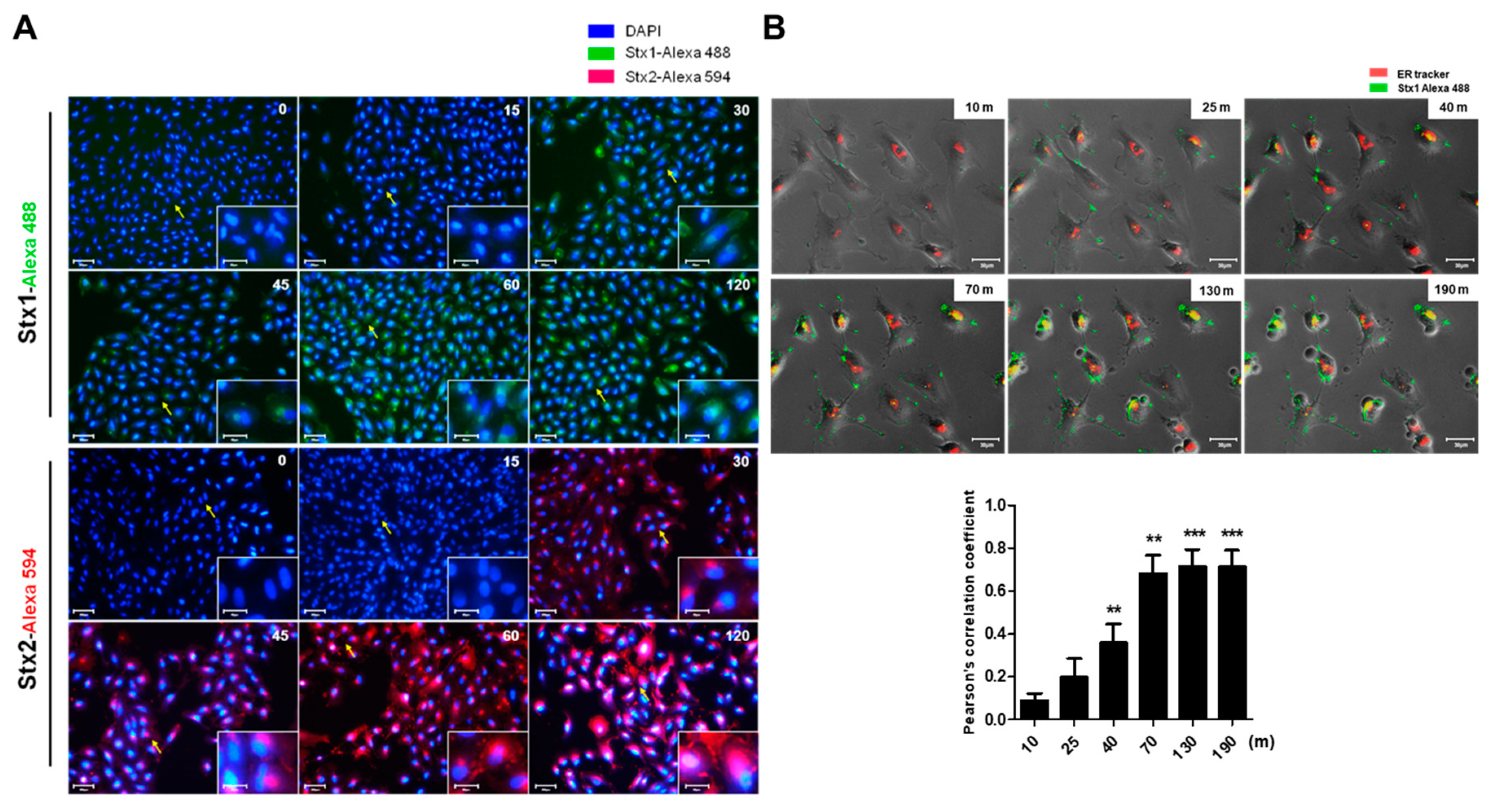
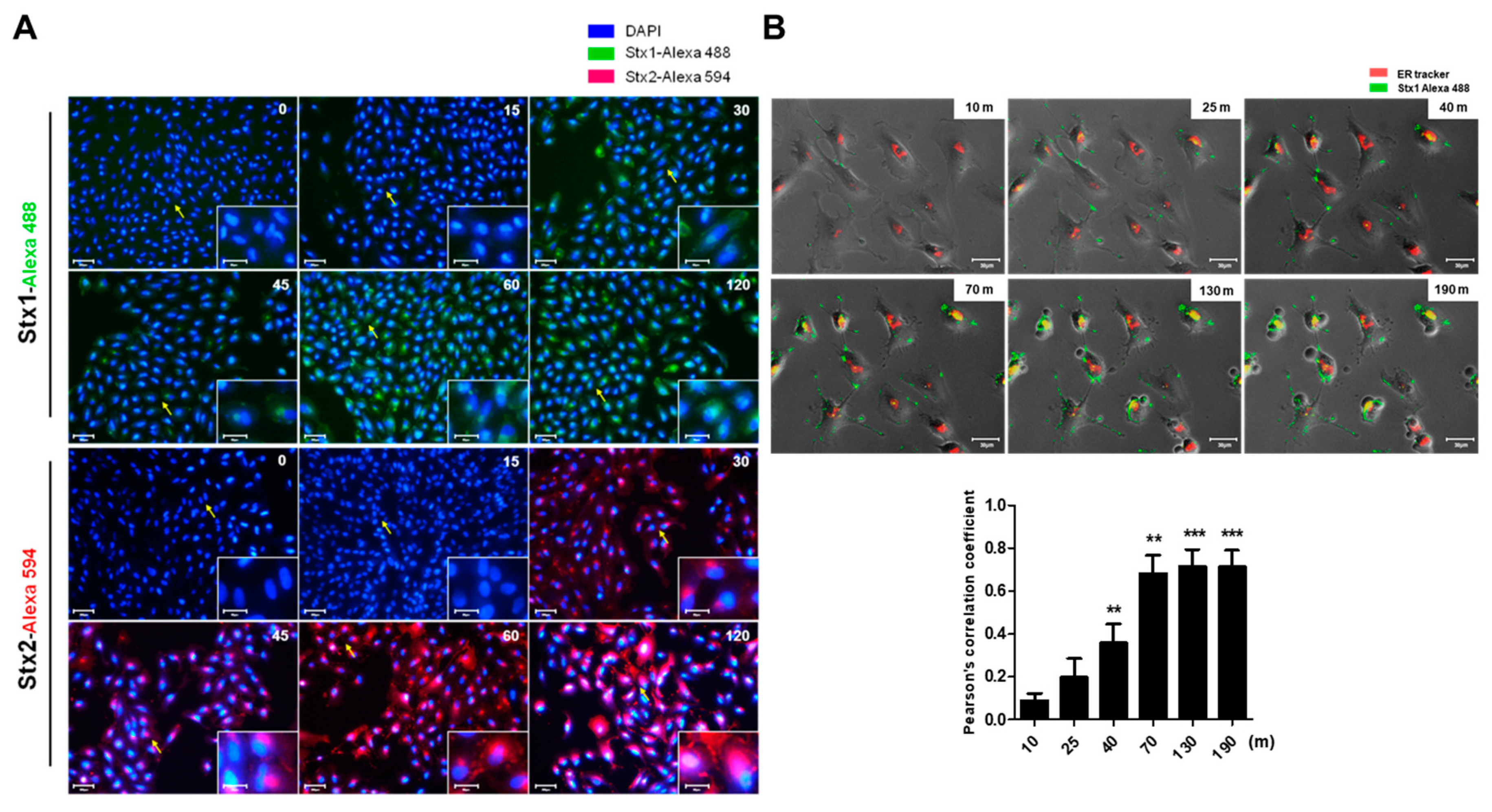
2.3. Intracellular Trafficking of Stxs to the ER in ARPE-19 Cells
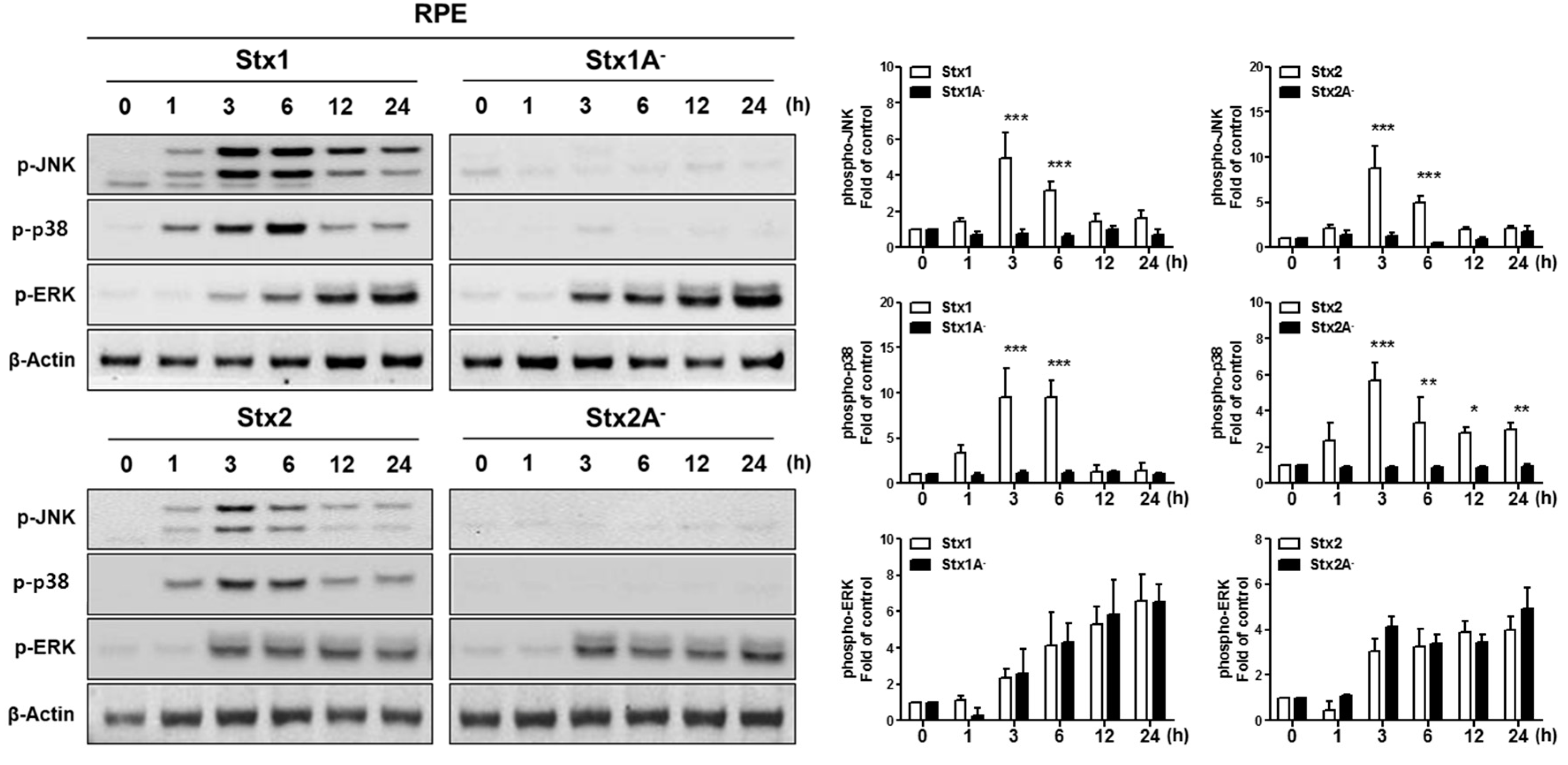
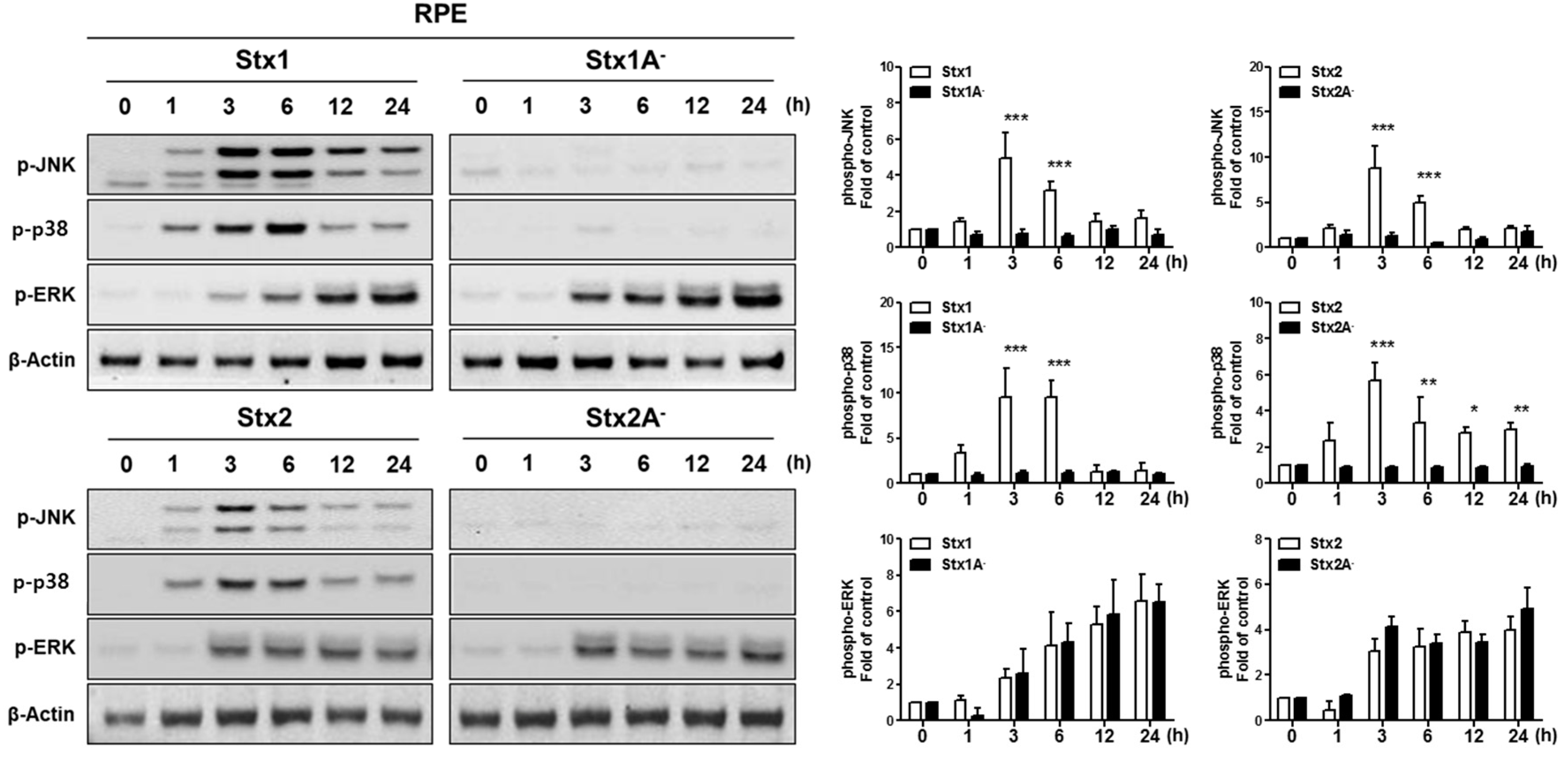
2.4. Stx1 and Stx2 Activate Stress-Associated MAPKs in ARPE-19 Cells
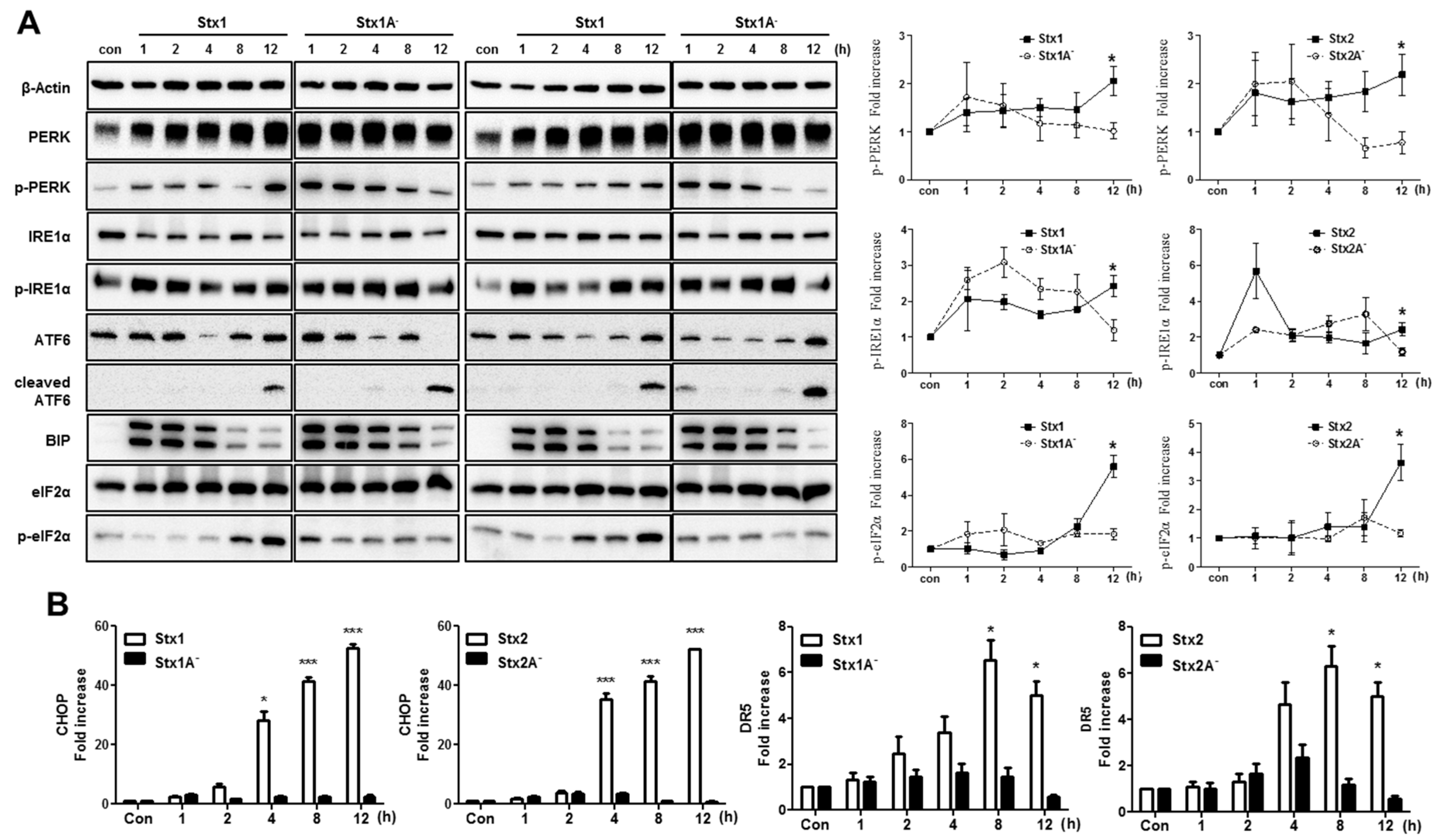
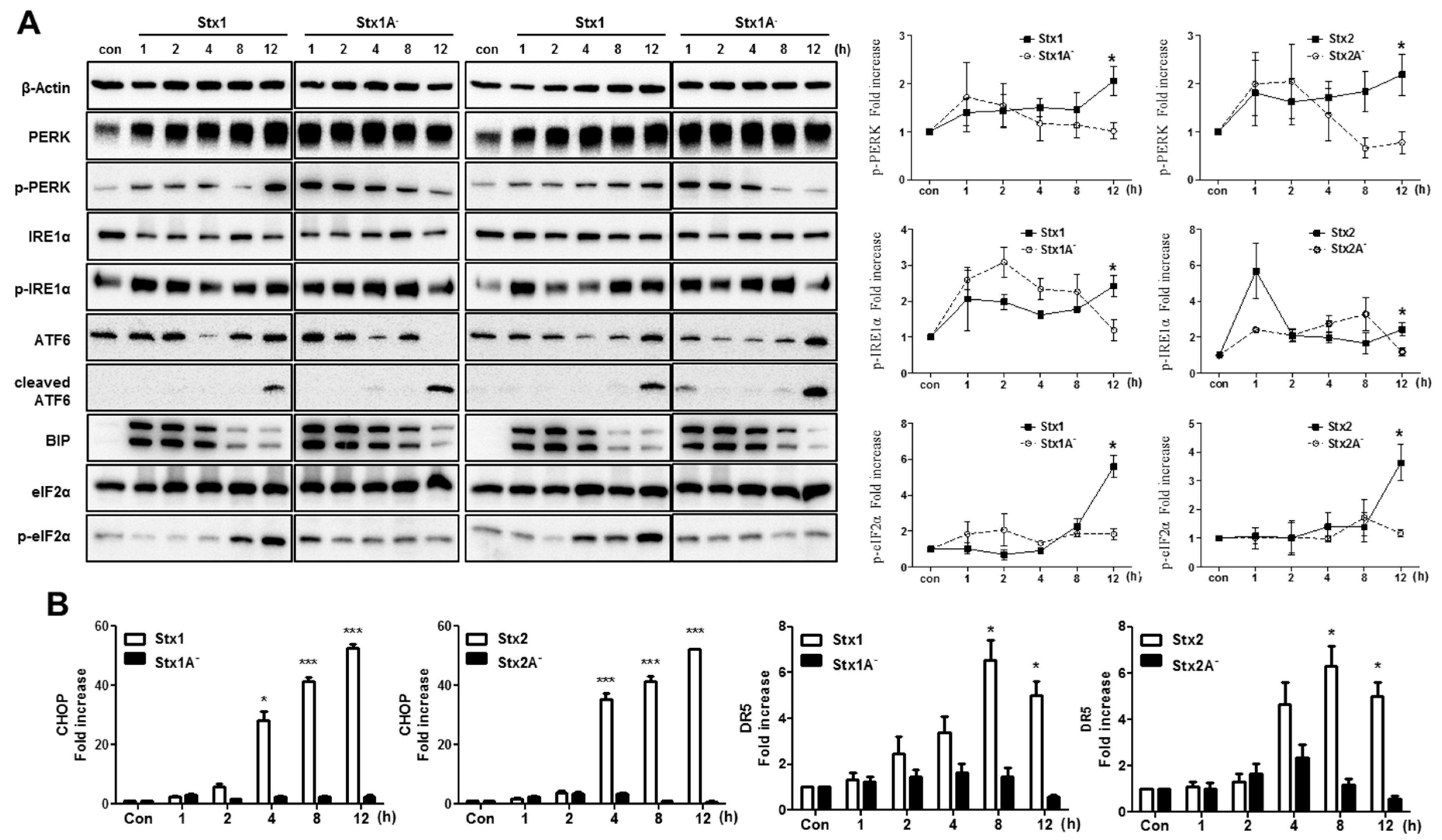
2.5. The ER Stress Response is Induced after Stx1 or Stx2 Exposure in ARPE-19 Cells
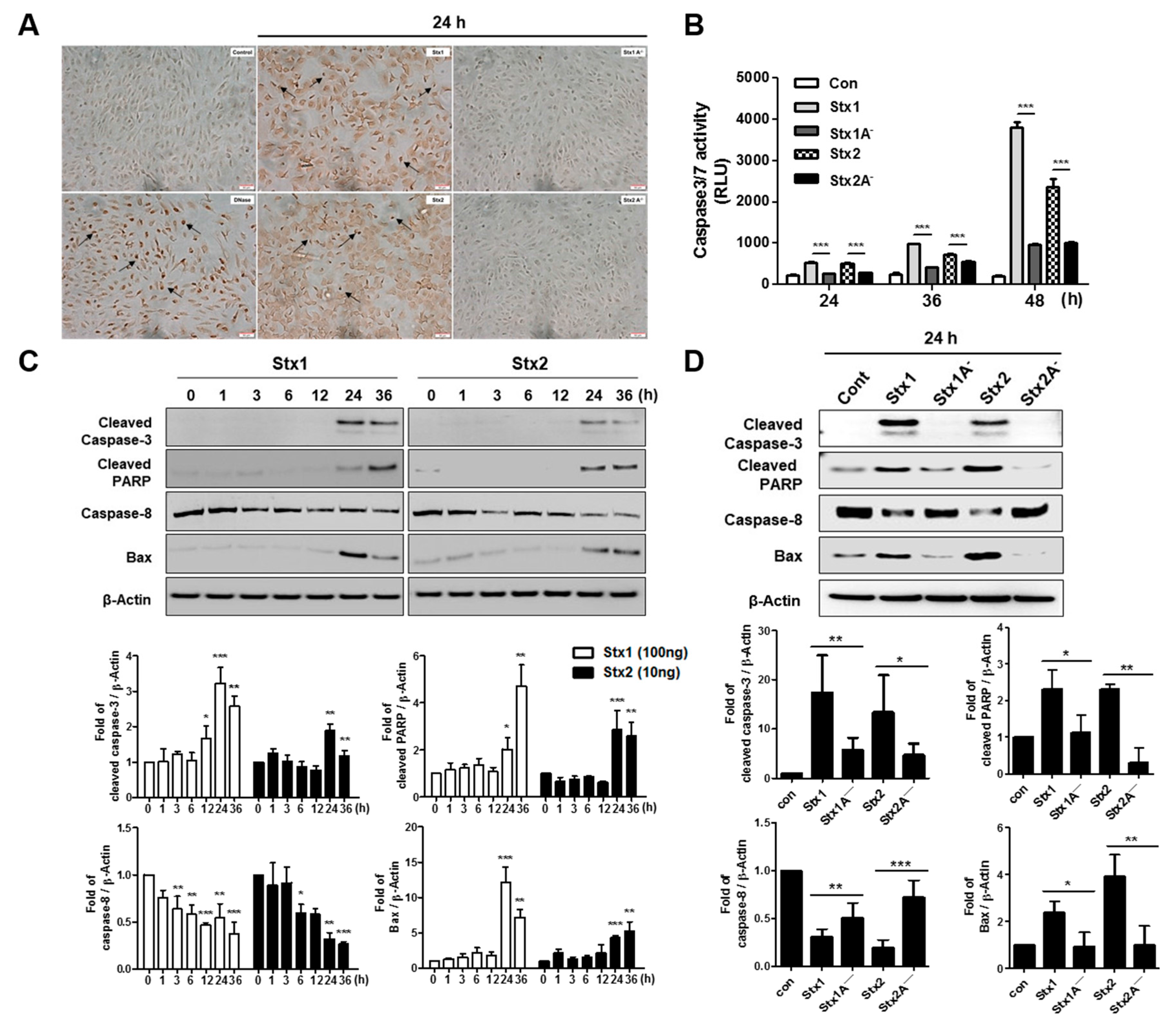
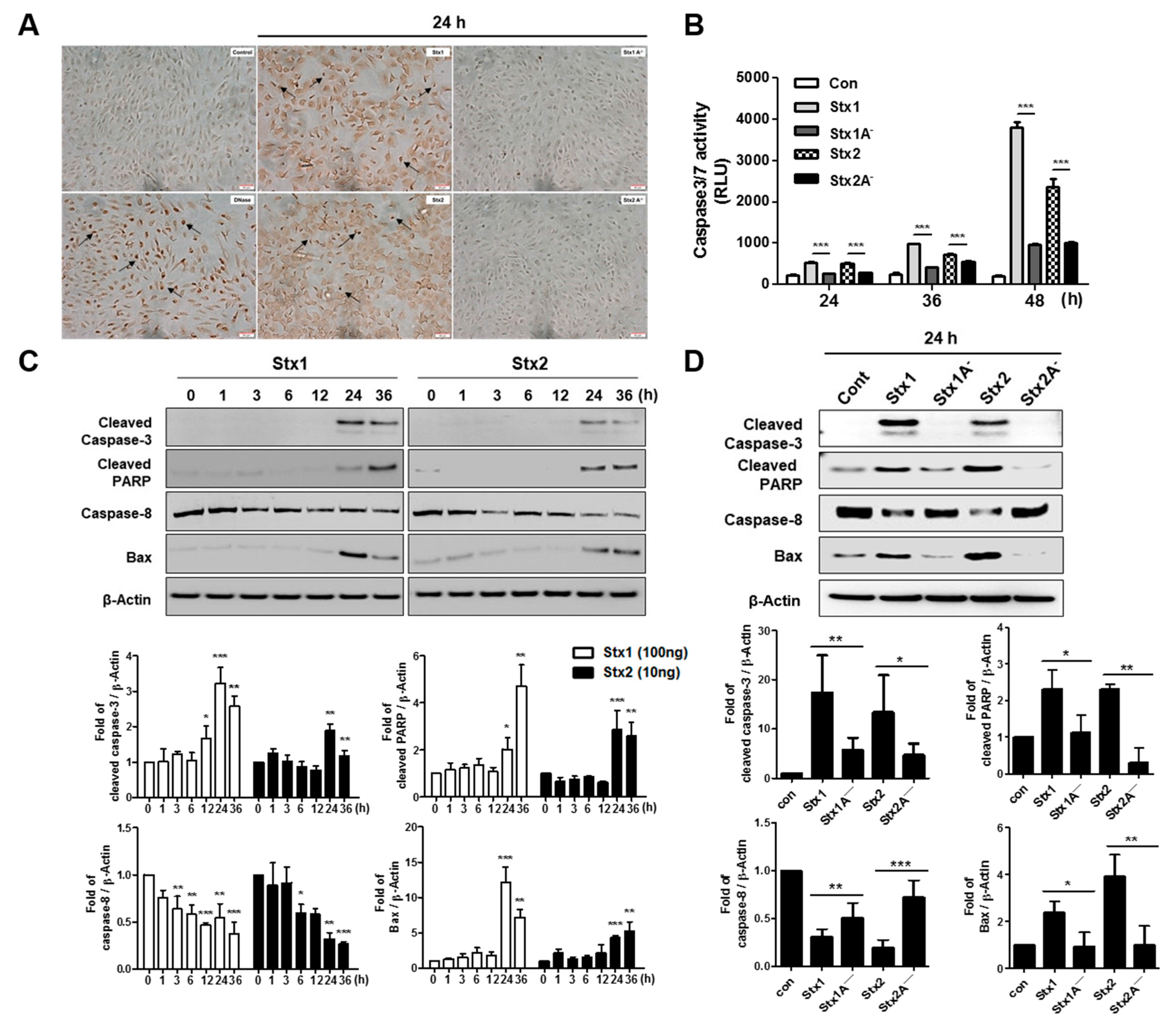
2.6. Stxs Activate Apoptotic Signaling Pathways in ARPE-19 Cells and Toxin Enzymatic Activity is Required for Apoptosis
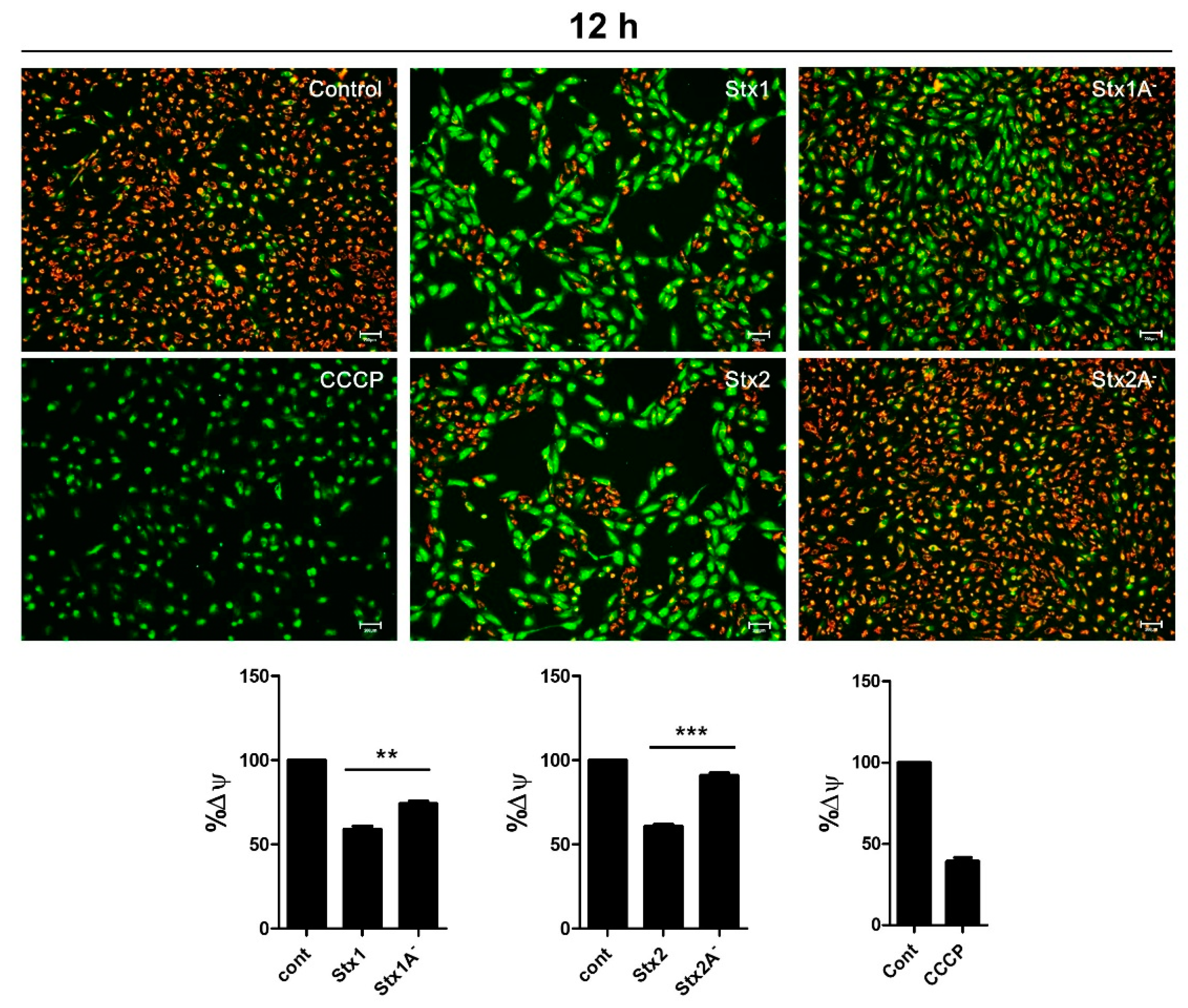
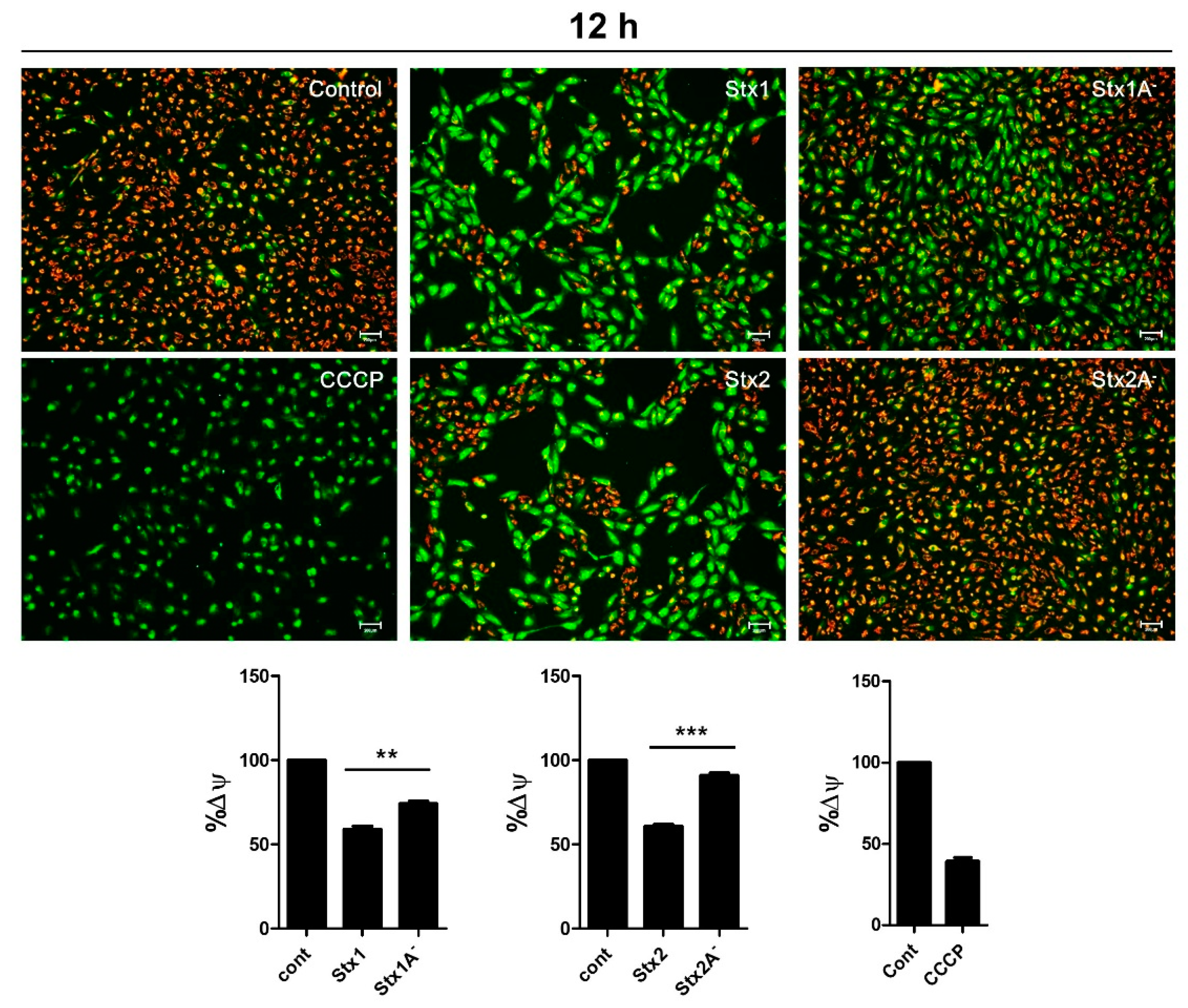
2.7. Stxs Induce Loss of Mitochondrial-Membrane Potential
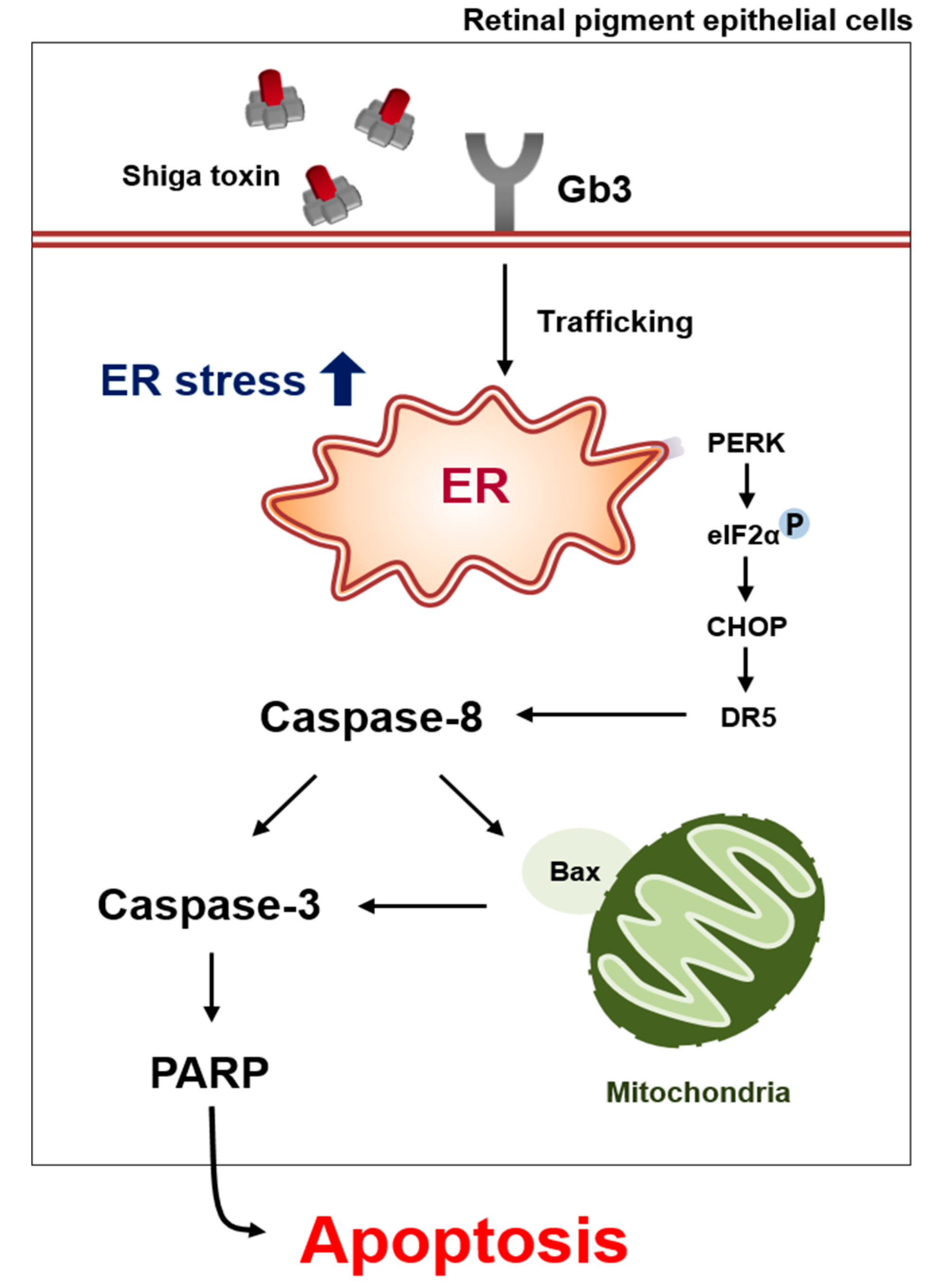
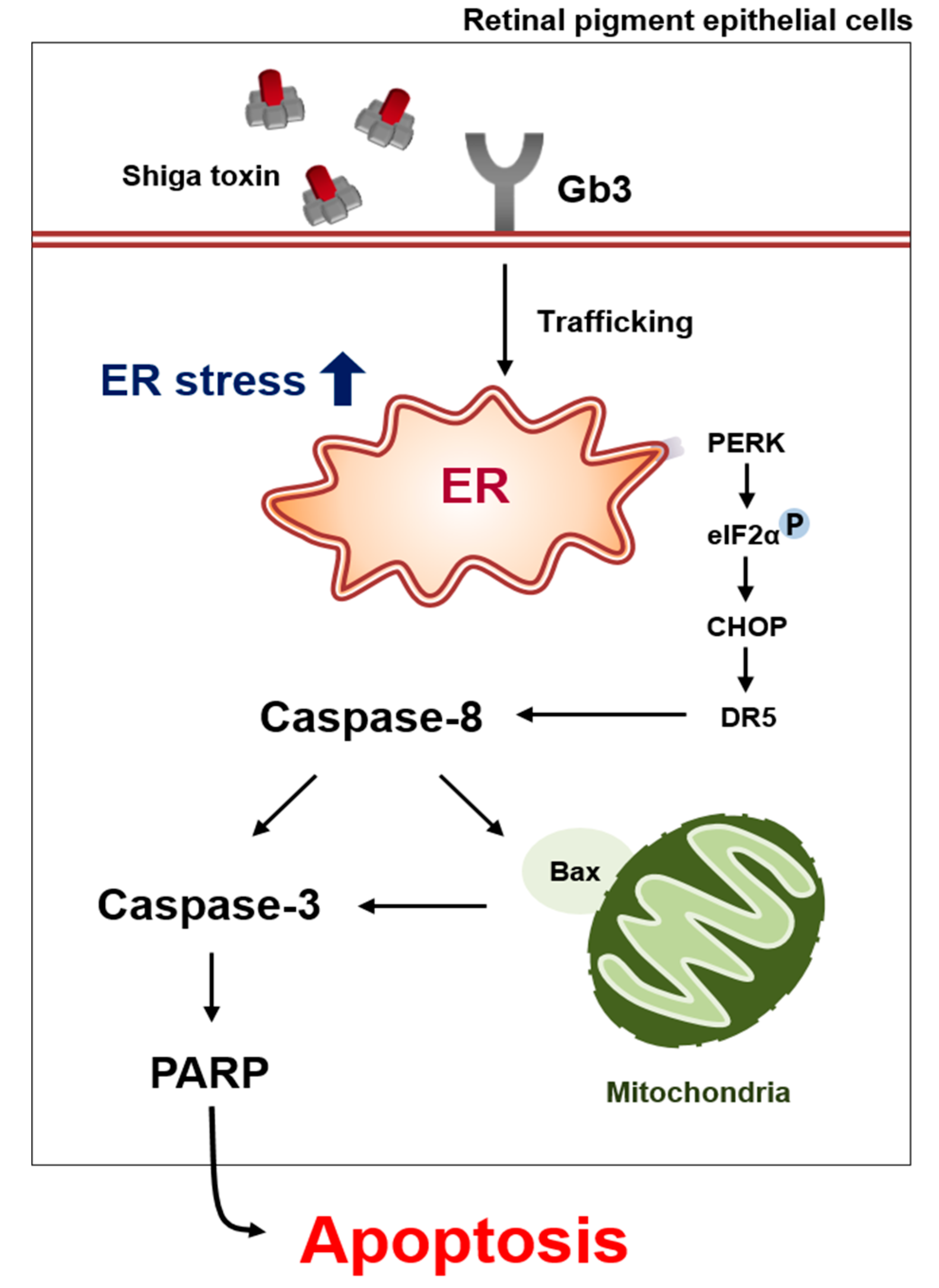
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Toxins
4.3. Cytotoxicity Assay
4.4. Flow Cytometric Analysis of Gb3 (CD77) Membrane Expression
4.5. Intracellular Trafficking Assay
4.6. Western Blotting
4.7. Real-Time Quantitative RT-PCR
4.8. TUNEL Assay
4.9. Caspase 3/7 Activity
4.10. Mitochondrial Membrane Potential Assay
4.11. Quantitative Analysis
4.12. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tarr, P.I.; Gordon, C.A.; Chandler, W.L. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet 2005, 365, 1073–1086. [Google Scholar] [CrossRef]
- Jacewicz, M.; Clausen, H.; Nudelman, E.; Donohue-Rolfe, A.; Keusch, G.T. Pathogenesis of shigella diarrhea. Xi. Isolation of a shigella toxin-binding glycolipid from rabbit jejunum and HeLa cells and its identification as globotriaosylceramide. J. Exp. Med. 1986, 163, 1391–1404. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, A.A.; Brown, J.E.; Stromberg, N.; Westling-Ryd, M.; Schultz, J.E.; Karlsson, K.A. Identification of the carbohydrate receptor for Shiga toxin produced by Shigella dysenteriae type 1. J. Biol. Chem. 1987, 262, 1779–1785. [Google Scholar] [PubMed]
- Lingwood, C.A.; Law, H.; Richardson, S.; Petric, M.; Brunton, J.L.; De Grandis, S.; Karmali, M. Glycolipid binding of purified and recombinant Escherichia coli produced verotoxin in vitro. J. Biol. Chem. 1987, 262, 8834–8839. [Google Scholar] [PubMed]
- Lingwood, C.A.; Binnington, B.; Manis, A.; Branch, D.R. Globotriaosyl ceramide receptor function—Where membrane structure and pathology intersect. FEBS Lett. 2010, 584, 1879–1886. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K.; Garred, O.; Prydz, K.; Kozlov, J.V.; Hansen, S.H.; van Deurs, B. Retrograde transport of endocytosed Shiga toxin to the endoplasmic reticulum. Nature 1992, 358, 510–512. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Cherla, R.P.; Tesh, V.L. Shiga toxins: Intracellular trafficking to the ER leading to activation of host cell stress responses. Toxins 2010, 2, 1515–1535. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K.; Bergan, J.; Kavaliauskiene, S.; Skotland, T. Lipid requirements for entry of protein toxins into cells. Prog. Lipid Res. 2014, 54, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Fraser, M.E.; Chernaia, M.M.; Kozlov, Y.V.; James, M.N. Crystal structure of the holotoxin from Shigella dysenteriae at 2.5 Å resolution. Nat. Struct. Biol. 1994, 1, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Fraser, M.E.; Fujinaga, M.; Cherney, M.M.; Melton-Celsa, A.R.; Twiddy, E.M.; O’Brien, A.D.; James, M.N. Structure of Shiga toxin type 2 (Stx2) from Escherichia coli O157:H7. J. Biol. Chem. 2004, 279, 27511–27517. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Lee, M.S.; Cherla, R.P.; Tesh, V.L. Shiga toxin 1 induces apoptosis through the endoplasmic reticulum stress response in human monocytic cells. Cell. Microbiol. 2008, 10, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Tsurugi, K.; Yutsudo, T.; Takeda, Y.; Ogasawara, T.; Igarashi, K. Site of action of a Vero toxin (VT2) from Escherichia coli O157:H7 and of Shiga toxin on eukaryotic ribosomes. RNA N-glycosidase activity of the toxins. Eur. J. Biochem. 1988, 171, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Saxena, S.K.; O’Brien, A.D.; Ackerman, E.J. Shiga toxin, Shiga-like toxin II variant, and ricin are all single-site RNA N-glycosidases of 28S rRNA when microinjected into Xenopus oocytes. J. Biol. Chem. 1989, 264, 596–601. [Google Scholar] [PubMed]
- Iordanov, M.S.; Pribnow, D.; Magun, J.L.; Dinh, T.H.; Pearson, J.A.; Chen, S.L.; Magun, B.E. Ribotoxic stress response: Activation of the stress-activated protein kinase JNK1 by inhibitors of the peptidyl transferase reaction and by sequence-specific RNA damage to the alpha-sarcin/ricin loop in the 28S rRNA. Mol. Cell. Biol. 1997, 17, 3373–3381. [Google Scholar] [CrossRef] [PubMed]
- Tesh, V.L. Induction of apoptosis by Shiga toxins. Future Microbiol. 2010, 5, 431–453. [Google Scholar] [CrossRef] [PubMed]
- Tesh, V.L.; Burris, J.A.; Owens, J.W.; Gordon, V.M.; Wadolkowski, E.A.; O’Brien, A.D.; Samuel, J.E. Comparison of the relative toxicities of Shiga-like toxins type I and type II for mice. Infect. Immun. 1993, 61, 3392–3402. [Google Scholar] [PubMed]
- Rutjes, N.W.; Binnington, B.A.; Smith, C.R.; Maloney, M.D.; Lingwood, C.A. Differential tissue targeting and pathogenesis of Verotoxins 1 and 2 in the mouse animal model. Kidney Int. 2002, 62, 832–845. [Google Scholar] [CrossRef] [PubMed]
- Sauter, K.A.; Melton-Celsa, A.R.; Larkin, K.; Troxell, M.L.; O’Brien, A.D.; Magun, B.E. Mouse model of hemolytic-uremic syndrome caused by endotoxin-free Shiga toxin 2 (Stx2) and protection from lethal outcome by anti-Stx2 antibody. Infect. Immun. 2008, 76, 4469–4478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohawk, K.L.; O’Brien, A.D. Mouse models of Escherichia coli O157:H7 infection and Shiga toxin injection. J. Biomed. Biotechnol. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Taylor, F.B., Jr.; Tesh, V.L.; DeBault, L.; Li, A.; Chang, A.C.; Kosanke, S.D.; Pysher, T.J.; Siegler, R.L. Characterization of the baboon responses to Shiga-like toxin: Descriptive study of a new primate model of toxic responses to Stx-1. Am. J. Pathol. 1999, 154, 1285–1299. [Google Scholar] [CrossRef]
- Stearns-Kurosawa, D.J.; Oh, S.Y.; Cherla, R.P.; Lee, M.S.; Tesh, V.L.; Papin, J.; Henderson, J.; Kurosawa, S. Distinct renal pathology and a chemotactic phenotype after enterohemorrhagic Escherichia coli Shiga toxins in non-human primate models of hemolytic uremic syndrome. Am. J. Pathol. 2013, 182, 1227–1238. [Google Scholar] [CrossRef] [PubMed]
- Murray, K.F.; Patterson, K. Escherichia coli O157:H7-induced hemolytic-uremic syndrome: Histopathologic changes in the colon over time. Pediatr. Dev. Pathol. 2000, 3, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Richardson, S.E.; Karmali, M.A.; Becker, L.E.; Smith, C.R. The histopathology of the hemolytic uremic syndrome associated with verocytotoxin-producing Escherichia coli infections. Hum. Pathol. 1988, 19, 1102–1108. [Google Scholar] [CrossRef]
- Tzipori, S.; Chow, C.W.; Powell, H.R. Cerebral infection with Escherichia coli O157:H7 in humans and gnotobiotic piglets. J. Clin. Pathol. 1988, 41, 1099–1103. [Google Scholar] [CrossRef] [PubMed]
- Theobald, I.; Kuwertz-Broking, E.; Schiborr, M.; Heindel, W. Central nervous system involvement in hemolytic uremic syndrome (HUS)—A retrospective analysis of cerebral CT and MRI studies. Clin. Nephrol. 2001, 56, S3–S8. [Google Scholar] [PubMed]
- Sheth, K.J.; Swick, H.M.; Haworth, N. Neurological involvement in hemolytic-uremic syndrome. Ann. Neurol. 1986, 19, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Loudon, S.E.; Dorresteijn, E.M.; Catsman-Berrevoets, C.E.; Verdijk, R.M.; Simonsz, H.J.; Jansen, A.J. Blinded by Shiga toxin-producing O104 Escherichia coli and hemolytic uremic syndrome. J. Pediatr. 2014, 165, 410–410.e1. [Google Scholar] [CrossRef] [PubMed]
- Bhutto, I.; Lutty, G. Understanding age-related macular between the photoreceptdegeneration (AMD): Relationships or/retinal pigment epithelium/Bruch's membrane/choriocapillaris complex. Mol. Asp. Med. 2012, 33, 295–317. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.E.; Kane, A.V.; Campbell, S.T.; Acheson, D.W.; Cochran, B.H.; Thorpe, C.M. Shiga toxin 1 triggers a ribotoxic stress response leading to p38 and JNK activation and induction of apoptosis in intestinal epithelial cells. Infect. Immun. 2003, 71, 1497–1504. [Google Scholar] [CrossRef] [PubMed]
- Lentz, E.K.; Leyva-Illades, D.; Lee, M.S.; Cherla, R.P.; Tesh, V.L. Differential response of the human renal proximal tubular epithelial cell line HK-2 to Shiga toxin types 1 and 2. Infect. Immun. 2011, 79, 3527–3540. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.K.; Ergonul, Z.; Stricklett, P.K.; Kohan, D.E. Molecular basis for high renal cell sensitivity to the cytotoxic effects of shigatoxin-1: Upregulation of globotriaosylceramide expression. J. Am. Soc. Nephrol. 2002, 13, 2239–2245. [Google Scholar] [CrossRef] [PubMed]
- Ramegowda, B.; Samuel, J.E.; Tesh, V.L. Interaction of Shiga toxins with human brain microvascular endothelial cells: Cytokines as sensitizing agents. J. Infect. Dis. 1999, 180, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K.; Grimmer, S.; Lauvrak, S.U.; Torgersen, M.L.; Skretting, G.; van Deurs, B.; Iversen, T.G. Pathways followed by ricin and Shiga toxin into cells. Histochem. Cell Biol. 2002, 117, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.L.; Lapadat, R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 2002, 298, 1911–1912. [Google Scholar] [CrossRef] [PubMed]
- Tesh, V.L. Activation of cell stress response pathways by Shiga toxins. Cell. Microbiol. 2012, 14, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Kwon, H.; Lee, E.Y.; Kim, D.J.; Park, J.H.; Tesh, V.L.; Oh, T.K.; Kim, M.H. Shiga toxins activate the NLRP3 inflammasome pathway to promote both production of the proinflammatory cytokine interleukin-1β and apoptotic cell death. Infect. Immun. 2016, 84, 172–186. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Cherla, R.P.; Leyva-Illades, D.; Tesh, V.L. Bcl-2 regulates the onset of Shiga toxin 1-induced apoptosis in THP-1 cells. Infect. Immun. 2009, 77, 5233–5244. [Google Scholar] [CrossRef] [PubMed]
- Otero, J.H.; Lizak, B.; Hendershot, L.M. Life and death of a BiP substrate. Semin. Cell Dev. Biol. 2010, 21, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed]
- Pincus, D.; Chevalier, M.W.; Aragon, T.; van Anken, E.; Vidal, S.E.; El-Samad, H.; Walter, P. BiP binding to the ER-stress sensor IRE1 tunes the homeostatic behavior of the unfolded protein response. PLoS Biol. 2010, 8, e1000415. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Haze, K.; Yanagi, H.; Yura, T.; Mori, K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J. Biol. Chem. 1998, 273, 33741–33749. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Z.; Lawson, B.; Brewer, J.W.; Zinszner, H.; Sanjay, A.; Mi, L.J.; Boorstein, R.; Kreibich, G.; Hendershot, L.M.; Ron, D. Signals from the stressed endoplasmic reticulum induce C/EBP-homologous protein (CHOP/GADD153). Mol. Cell. Biol. 1996, 16, 4273–4280. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Cherla, R.P.; Lentz, E.K.; Leyva-Illades, D.; Tesh, V.L. Signaling through C/EBP homologous protein and death receptor 5 and calpain activation differentially regulate THP-1 cell maturation-dependent apoptosis induced by Shiga toxin type 1. Infect. Immun. 2010, 78, 3378–3391. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Wang, H.G. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J. Biol. Chem. 2004, 279, 45495–45502. [Google Scholar] [CrossRef] [PubMed]
- Cherla, R.P.; Lee, S.Y.; Tesh, V.L. Shiga toxins and apoptosis. FEMS Microbiol. Lett. 2003, 228, 159–166. [Google Scholar] [CrossRef]
- Nishitoh, H.; Matsuzawa, A.; Tobiume, K.; Saegusa, K.; Takeda, K.; Inoue, K.; Hori, S.; Kakizuka, A.; Ichijo, H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002, 16, 1345–1355. [Google Scholar] [CrossRef] [PubMed]
- Chandra, D.; Choy, G.; Deng, X.; Bhatia, B.; Daniel, P.; Tang, D.G. Association of active caspase 8 with the mitochondrial membrane during apoptosis: Potential roles in cleaving BAP31 and caspase 3 and mediating mitochondrion-endoplasmic reticulum cross talk in etoposide-induced cell death. Mol. Cell. Biol. 2004, 24, 6592–6607. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhu, H.; Xu, C.J.; Yuan, J. Cleavage of bid by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998, 94, 491–501. [Google Scholar] [CrossRef]
- Wang, C.; Youle, R.J. The role of mitochondria in apoptosis. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef] [PubMed]
- Tesh, V.L. The induction of apoptosis by Shiga toxins and ricin. Curr. Top. Microbiol. Immunol. 2012, 357, 137–178. [Google Scholar] [PubMed]
- Lee, M.S.; Koo, S.; Jeong, D.G.; Tesh, V.L. Shiga toxins as multi-functional proteins: Induction of host cellular stress responses, role in pathogenesis and therapeutic applications. Toxins 2016, 8, 77. [Google Scholar] [CrossRef] [PubMed]
- Mele, C.; Remuzzi, G.; Noris, M. Hemolytic uremic syndrome. Semin. Immunopathol. 2014, 36, 399–420. [Google Scholar] [CrossRef] [PubMed]
- Sturm, V.; Menke, M.N.; Landau, K.; Laube, G.F.; Neuhaus, T.J. Ocular involvement in paediatric haemolytic uraemic syndrome. Acta Ophthalmol. 2010, 88, 804–807. [Google Scholar] [CrossRef] [PubMed]
- Siegler, R.L.; Brewer, E.D.; Swartz, M. Ocular involvement in hemolytic-uremic syndrome. J. Pediatr. 1988, 112, 594–597. [Google Scholar] [CrossRef]
- Falguières, T.; Mallard, F.; Baron, C.; Hanau, D.; Lingwood, C.; Goud, B.; Salamero, J.; Johannes, L. Targeting of Shiga toxin B-subunit to retrograde transport route in association with detergent-resistant membranes. Mol. Biol. Cell 2001, 12, 2453–2468. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K.; Torgersen, M.L.; Engedal, N.; Skotland, T.; Iversen, T.G. Protein toxins from plants and bacteria: Probes for intracellular transport and tools in medicine. FEBS Lett. 2010, 584, 2626–2634. [Google Scholar] [CrossRef] [PubMed]
- Jandhyala, D.M.; Ahluwalia, A.; Obrig, T.; Thorpe, C.M. ZAK: A MAP3Kinase that transduces Shiga toxin- and ricin-induced proinflammatory cytokine expression. Cell. Microbiol. 2008, 10, 1468–1477. [Google Scholar] [CrossRef] [PubMed]
- Karpman, D.; Hakansson, A.; Perez, M.T.; Isaksson, C.; Carlemalm, E.; Caprioli, A.; Svanborg, C. Apoptosis of renal cortical cells in the hemolytic-uremic syndrome: In vivo and in vitro studies. Infect. Immun. 1998, 66, 636–644. [Google Scholar] [PubMed]
- Te Loo, D.M.; Monnens, L.A.; van Der Velden, T.J.; Vermeer, M.A.; Preyers, F.; Demacker, P.N.; van Den Heuvel, L.P.; van Hinsbergh, V.W. Binding and transfer of verocytotoxin by polymorphonuclear leukocytes in hemolytic uremic syndrome. Blood 2000, 95, 3396–3402. [Google Scholar] [PubMed]
- Lee, S.Y.; Cherla, R.P.; Tesh, V.L. Simultaneous induction of apoptotic and survival signaling pathways in macrophage-like THP-1 cells by Shiga toxin 1. Infect. Immun. 2007, 75, 1291–1302. [Google Scholar] [CrossRef] [PubMed]
- Fujii, J.; Wood, K.; Matsuda, F.; Carneiro-Filho, B.A.; Schlegel, K.H.; Yutsudo, T.; Binnington-Boyd, B.; Lingwood, C.A.; Obata, F.; Kim, K.S.; et al. Shiga toxin 2 causes apoptosis in human brain microvascular endothelial cells via C/EBP homologous protein. Infect. Immun. 2008, 76, 3679–3689. [Google Scholar] [CrossRef] [PubMed]
- Landoni, V.I.; Schierloh, P.; de Campos Nebel, M.; Fernandez, G.C.; Calatayud, C.; Lapponi, M.J.; Isturiz, M.A. Shiga toxin 1 induces on lipopolysaccharide-treated astrocytes the release of tumor necrosis factor-alpha that alter brain-like endothelium integrity. PLoS Pathog. 2012, 8, e1002632. [Google Scholar] [CrossRef] [PubMed]
- Richardson, S.E.; Rotman, T.A.; Jay, V.; Smith, C.R.; Becker, L.E.; Petric, M.; Olivieri, N.F.; Karmali, M.A. Experimental verocytotoxemia in rabbits. Infect. Immun. 1992, 60, 4154–4167. [Google Scholar] [PubMed]
- Zoja, C.; Corna, D.; Farina, C.; Sacchi, G.; Lingwood, C.; Doyle, M.P.; Padhye, V.V.; Abbate, M.; Remuzzi, G. Verotoxin glycolipid receptors determine the localization of microangiopathic process in rabbits given verotoxin-1. J. Lab. Clin. Med. 1992, 120, 229–238. [Google Scholar] [PubMed]
- Ren, J.; Utsunomiya, I.; Taguchi, K.; Ariga, T.; Tai, T.; Ihara, Y.; Miyatake, T. Localization of verotoxin receptors in nervous system. Brain Res. 1999, 825, 183–188. [Google Scholar] [CrossRef]
- Obata, F.; Tohyama, K.; Bonev, A.D.; Kolling, G.L.; Keepers, T.R.; Gross, L.K.; Nelson, M.T.; Sato, S.; Obrig, T.G. Shiga toxin 2 affects the central nervous system through receptor globotriaosylceramide localized to neurons. J. Infect. Dis. 2008, 198, 1398–1406. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Funata, N.; Ikuta, F.; Sato, S. Neuronal apoptosis and inflammatory responses in the central nervous system of a rabbit treated with Shiga toxin-2. J. Neuroinflamm. 2008, 5, 11. [Google Scholar] [CrossRef] [PubMed]
- Ohmura, M.; Yamasaki, S.; Kurazono, H.; Kashiwagi, K.; Igarashi, K.; Takeda, Y. Characterization of non-toxic mutant toxins of Vero toxin 1 that were constructed by replacing amino acids in the A subunit. Microb. Pathog. 1993, 15, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Cory, A.H.; Owen, T.C.; Barltrop, J.A.; Cory, J.G. Use of an aqueous soluble tetrazolium/formazan assay for cell growth assays in culture. Cancer Commun. 1991, 3, 207–212. [Google Scholar] [PubMed]
- Salvioli, S.; Ardizzoni, A.; Franceschi, C.; Cossarizza, A. JC-1, but not DiOC6(3) or rhodamine 123, is a reliable fluorescent probe to assess delta psi changes in intact cells: Implications for studies on mitochondrial functionality during apoptosis. FEBS Lett. 1997, 411, 77–82. [Google Scholar] [CrossRef]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, J.-Y.; Jeong, Y.-J.; Park, S.-K.; Yoon, S.-J.; Choi, S.; Jeong, D.G.; Chung, S.W.; Lee, B.J.; Kim, J.H.; Tesh, V.L.; et al. Shiga Toxins Induce Apoptosis and ER Stress in Human Retinal Pigment Epithelial Cells. Toxins 2017, 9, 319. https://doi.org/10.3390/toxins9100319
Park J-Y, Jeong Y-J, Park S-K, Yoon S-J, Choi S, Jeong DG, Chung SW, Lee BJ, Kim JH, Tesh VL, et al. Shiga Toxins Induce Apoptosis and ER Stress in Human Retinal Pigment Epithelial Cells. Toxins. 2017; 9(10):319. https://doi.org/10.3390/toxins9100319
Chicago/Turabian StylePark, Jun-Young, Yu-Jin Jeong, Sung-Kyun Park, Sung-Jin Yoon, Song Choi, Dae Gwin Jeong, Su Wol Chung, Byung Joo Lee, Jeong Hun Kim, Vernon L. Tesh, and et al. 2017. "Shiga Toxins Induce Apoptosis and ER Stress in Human Retinal Pigment Epithelial Cells" Toxins 9, no. 10: 319. https://doi.org/10.3390/toxins9100319