
The Use of Plant-Derived Ribosome Inactivating Proteins in Immunotoxin Development: Past, Present and Future Generations


{kind=link}
Abstract
:1. Introduction
2. Ribosome Inactivating Proteins with Ribosomal RNA N-Glycosidase Activity
3. The Development of RIP-Based Immunotoxins
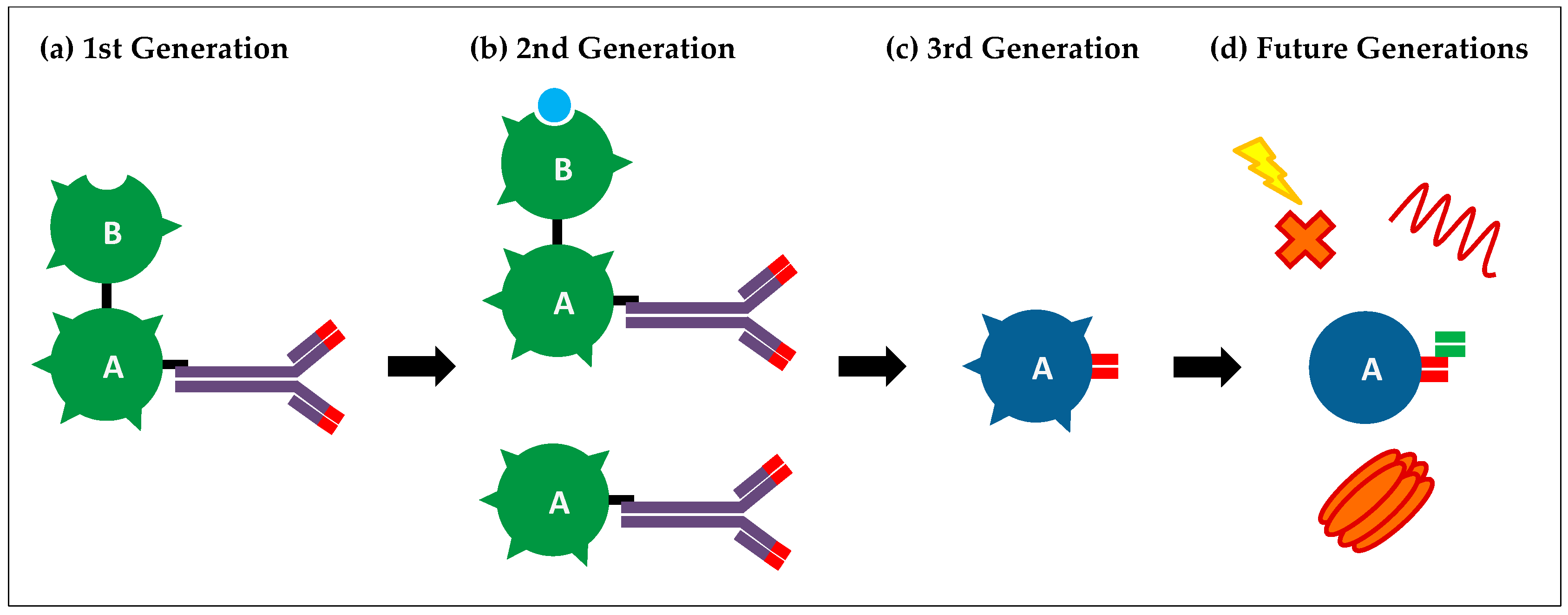
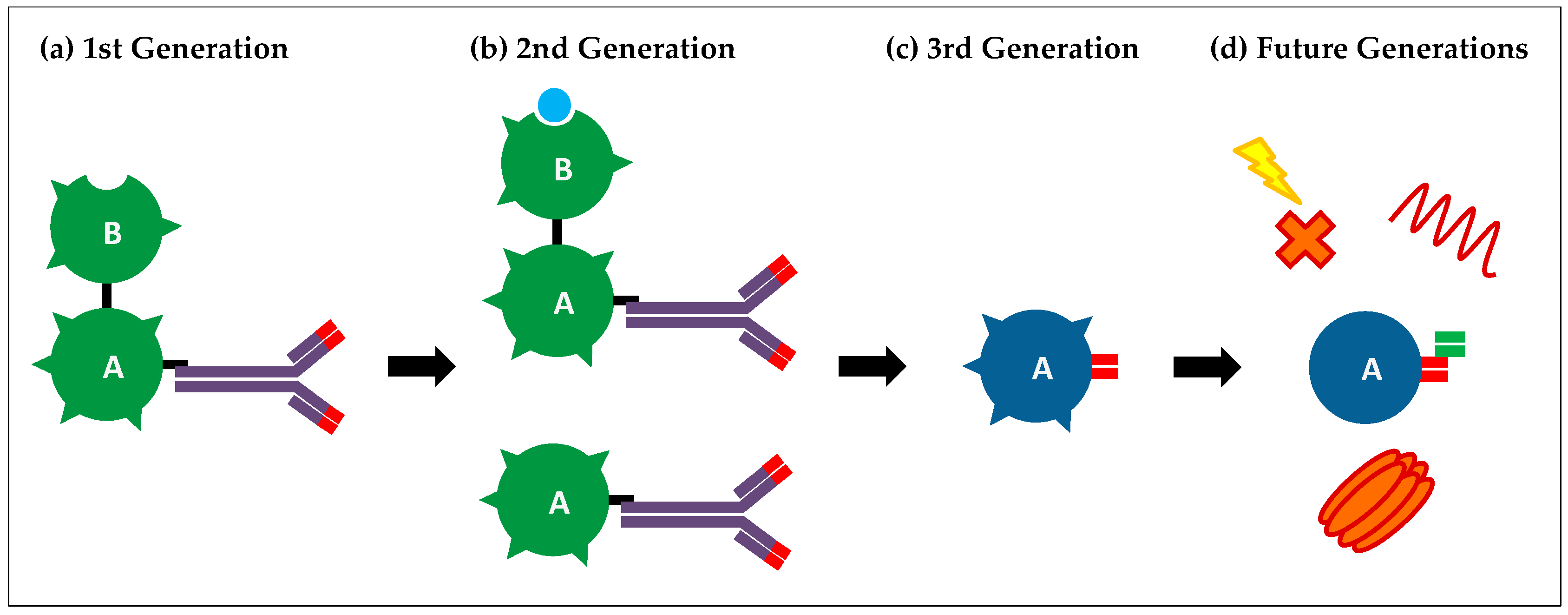
3.1. First-Generation Immunotoxins
3.2. Second-Generation Immunotoxins
3.3. Third-Generation Immunotoxins
4. Current Limitations and Future Generations of Immunotoxins
5. Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Schrot, J.; Weng, A.; Melzig, M.F. Ribosome-inactivating and related proteins. Toxins 2015, 7, 1556–1615. [Google Scholar] [CrossRef] [PubMed]
- Bolognesi, A.; Bortolotti, M.; Maiello, S.; Battelli, M.; Polito, L. Ribosome-inactivating proteins from plants: A historical overview. Molecules 2016, 21, 1627. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, M.S.; Katayama, M.; Nakase, I.; Vago, R. Plant ribosome-inactivating proteins: Progesses, challenges and biotechnological applications (and a few digressions). Toxins 2017, 9, 314. [Google Scholar] [CrossRef] [PubMed]
- Lapadula, W.J.; Sanchez Puerta, M.V.; Juri Ayub, M. Revising the taxonomic distribution, origin and evolution of ribosome inactivating protein genes. PLoS ONE 2013, 8, e72825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, M.J.; Dodd, J.E.; Hautbergue, G.M. Ribosome-inactivating proteins: Potent poisons and molecular tools. In Virulence; Taylor & Francis Group: Philadelphia, PA, USA, 2013; Volume 4, pp. 774–784. [Google Scholar]
- Antignani, A.; Fitzgerald, D. Immunotoxins: The role of the toxin. Toxins 2013, 5, 1486–1502. [Google Scholar] [CrossRef] [PubMed]
- Moolten, F.L.; Cooperband, S.R. Selective destruction of target cells by diphtheria toxin conjugated to antibody directed against antigens on the cells. Science 1970, 169, 68–70. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.N.S.; Mak, N.K.; Choi, W.T.; Law, P.T.W. Increased accumulation of trichosanthin in trichosanthes kirilowii induced by microorganisms. J. Exp. Bot. 1995, 46, 355–358. [Google Scholar] [CrossRef]
- Rippmann, J.F.; Michalowski, C.B.; Nelson, D.E.; Bohnert, H.J. Induction of a ribosome-inactivating protein upon environmental stress. Plant Mol. Biol. 1997, 35, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.H.; Wang, Y.; Yusufali, A.H.; Ashby, F.; Zhang, D.; Yin, Z.F.; Aslanidi, G.V.; Srivastava, A.; Ling, C.Q.; Ling, C. Cytotoxic genes from traditional chinese medicine inhibit tumor growth both in vitro and in vivo. J. Integr. Med. 2014, 12, 483–494. [Google Scholar] [CrossRef]
- Stirpe, F.; Battelli, M.G. Ribosome-inactivating proteins: Progress and problems. Cell. Mol. Life Sci. 2006, 63, 1850–1866. [Google Scholar] [CrossRef] [PubMed]
- Ferreras, J.M.; Citores, L.; Iglesias, R.; Jimenez, P.; Girbes, T. Use of ribosome-inactivating proteins from sambucus for the construction of immunotoxins and conjugates for cancer therapy. Toxins 2011, 3, 420–441. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, P.; Tejero, J.; Cordoba-Diaz, D.; Quinto, E.J.; Garrosa, M.; Gayoso, M.J.; Girbes, T. Ebulin from dwarf elder (sambucus ebulus L.): A mini-review. Toxins 2015, 7, 648–658. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K.; van Deurs, B. Endocytosis, intracellular transport, and cytotoxic action of shiga toxin and ricin. Physiol. Rev. 1996, 76, 949–966. [Google Scholar] [PubMed]
- Roberts, L.M.; Lord, J.M. Ribosome-inactivating proteins: Entry into mammalian cells and intracellular routing. Mini Rev. Med. Chem. 2004, 4, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Spooner, R.A.; Watson, P.D.; Marsden, C.J.; Smith, D.C.; Moore, K.A.; Cook, J.P.; Lord, J.M.; Roberts, L.M. Protein disulphide-isomerase reduces ricin to its a and b chains in the endoplasmic reticulum. Biochem. J. 2004, 383, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Spooner, R.A.; Hart, P.J.; Cook, J.P.; Pietroni, P.; Rogon, C.; Hohfeld, J.; Roberts, L.M.; Lord, J.M. Cytosolic chaperones influence the fate of a toxin dislocated from the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2008, 105, 17408–17413. [Google Scholar] [CrossRef] [PubMed]
- Polito, L.; Bortolotti, M.; Mercatelli, D.; Battelli, M.G.; Bolognesi, A. Saporin-s6: A useful tool in cancer therapy. Toxins 2013, 5, 1698–1722. [Google Scholar] [CrossRef] [PubMed]
- Cavallaro, U.; Nykjaer, A.; Nielsen, M.; Soria, M.R. Alpha 2-macroglobulin receptor mediates binding and cytotoxicity of plant ribosome-inactivating proteins. Eur. J. Biochem. 1995, 232, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Bagga, S.; Hosur, M.V.; Batra, J.K. Cytotoxicity of ribosome-inactivating protein saporin is not mediated through alpha2-macroglobulin receptor. FEBS Lett. 2003, 541, 16–20. [Google Scholar] [CrossRef]
- Vago, R.; Marsden, C.J.; Lord, J.M.; Ippoliti, R.; Flavell, D.J.; Flavell, S.U.; Ceriotti, A.; Fabbrini, M.S. Saporin and ricin a chain follow different intracellular routes to enter the cytosol of intoxicated cells. FEBS J. 2005, 272, 4983–4995. [Google Scholar] [CrossRef] [PubMed]
- Rust, A.; Hassan, H.H.; Sedelnikova, S.; Niranjan, D.; Hautbergue, G.; Abbas, S.A.; Partridge, L.; Rice, D.; Binz, T.; Davletov, B. Two complementary approaches for intracellular delivery of exogenous enzymes. Sci. Rep. 2015, 5, 12444. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Tsurugi, K. Rna n-glycosidase activity of ricin a-chain. Mechanism of action of the toxic lectin ricin on eukaryotic ribosomes. J. Biol. Chem. 1987, 262, 8128–8130. [Google Scholar] [PubMed]
- Endo, Y.; Mitsui, K.; Motizuki, M.; Tsurugi, K. The mechanism of action of ricin and related toxic lectins on eukaryotic ribosomes. The site and the characteristics of the modification in 28 s ribosomal rna caused by the toxins. J. Biol. Chem. 1987, 262, 5908–5912. [Google Scholar] [PubMed]
- Endo, Y.; Tsurugi, K. The rna n-glycosidase activity of ricin a-chain. The characteristics of the enzymatic activity of ricin a-chain with ribosomes and with rrna. J. Biol. Chem. 1988, 263, 8735–8739. [Google Scholar] [PubMed]
- Montanaro, L.; Sperti, S.; Mattioli, A.; Testoni, G.; Stirpe, F. Inhibition by ricin of protein synthesis in vitro. Inhibition of the binding of elongation factor 2 and of adenosine diphosphate-ribosylated elongation factor 2 to ribosomes. Biochem. J. 1975, 146, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, L.; Valbonesi, P.; Bonora, E.; Gorini, P.; Bolognesi, A.; Stirpe, F. Polynucleotide:Adenosine glycosidase activity of ribosome-inactivating proteins: Effect on dna, rna and poly(a). Nucleic Acids Res. 1997, 25, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Brigotti, M.; Alfieri, R.; Sestili, P.; Bonelli, M.; Petronini, P.G.; Guidarelli, A.; Barbieri, L.; Stirpe, F.; Sperti, S. Damage to nuclear dna induced by shiga toxin 1 and ricin in human endothelial cells. FASEB J. 2002, 16, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Sikriwal, D.; Ghosh, P.; Batra, J.K. Ribosome inactivating protein saporin induces apoptosis through mitochondrial cascade, independent of translation inhibition. Int. J. Biochem. Cell Biol. 2008, 40, 2880–2888. [Google Scholar] [CrossRef] [PubMed]
- Das, M.K.; Sharma, R.S.; Mishra, V. Induction of apoptosis by ribosome inactivating proteins: Importance of n-glycosidase activity. Appl. Biochem. Biotechnol. 2012, 166, 1552–1561. [Google Scholar] [CrossRef] [PubMed]
- Gilabert-Oriol, R.; Weng, A.; Mallinckrodt, B.; Melzig, M.F.; Fuchs, H.; Thakur, M. Immunotoxins constructed with ribosome-inactivating proteins and their enhancers: A lethal cocktail with tumor specific efficacy. Curr. Pharm. Des. 2014, 20, 6584–6643. [Google Scholar] [CrossRef] [PubMed]
- Alewine, C.; Hassan, R.; Pastan, I. Advances in anticancer immunotoxin therapy. Oncologist 2015, 20, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Madhumathi, J.; Verma, R.S. Therapeutic targets and recent advances in protein immunotoxins. Curr. Opin. Microbiol. 2012, 15, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Moolten, F.L.; Capparell, N.J.; Cooperband, S.R. Antitumor effects of antibody-diphtheria toxin conjugates: Use of hapten-coated tumor cells as an antigenic target. J. Natl. Cancer Inst. 1972, 49, 1057–1062. [Google Scholar] [PubMed]
- Samagh, B.S.; Gregory, K.F. Antibody to lactate dehydrogenase. V. Use as a carrier for introducing diphtheria toxin into mouse tumor cells. Biochim. Biophys. Acta 1972, 273, 188–198. [Google Scholar] [CrossRef]
- Lin, J.Y.; Tserng, K.Y.; Chen, C.C.; Lin, L.T.; Tung, T.C. Abrin and ricin: New anti-tumour substances. Nature 1970, 227, 292–293. [Google Scholar] [CrossRef] [PubMed]
- Fodstad, O.; Olsnes, S.; Pihl, A. Toxicity, distribution and elimination of the cancerostatic lectins abrin and ricin after parenteral injection into mice. Br. J. Cancer 1976, 34, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Moolten, F.; Zajdel, S.; Cooperband, S. Immunotherapy of experimental animal tumors with antitumor antibodies conjugated to diphtheria toxin or ricin. Ann. N. Y. Acad. Sci. 1976, 277, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Spitler, L.E. Immunotoxin therapy of malignant melanoma. Med. Oncol. Tumor Pharmacother. 1986, 3, 147–152. [Google Scholar] [PubMed]
- Polito, L.; Djemil, A.; Bortolotti, M. Plant toxin-based immunotoxins for cancer therapy: A short overview. Biomedicines 2016, 4, 12. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Neville, D.M., Jr. Anti-thy 1.2 monoclonal antibody linked to ricin is a potent cell-type-specific toxin. Proc. Natl. Acad. Sci. USA 1980, 77, 5483–5486. [Google Scholar] [CrossRef] [PubMed]
- Wawrzynczak, E.J.; Watson, G.J.; Cumber, A.J.; Henry, R.V.; Parnell, G.D.; Rieber, E.P.; Thorpe, P.E. Blocked and non-blocked ricin immunotoxins against the cd4 antigen exhibit higher cytotoxic potency than a ricin a chain immunotoxin potentiated with ricin b chain or with a ricin b chain immunotoxin. Cancer Immunol. Immunother. 1991, 32, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, P.E.; Brown, A.N.; Ross, W.C.; Cumber, A.J.; Detre, S.I.; Edwards, D.C.; Davies, A.J.; Stirpe, F. Cytotoxicity acquired by conjugation of an anti-thy1.1 monoclonal antibody and the ribosome-inactivating protein, gelonin. Eur. J. Biochem. 1981, 116, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, S.; Houston, L.L. Comparison of the selective cytotoxic effects of immunotoxins containing ricin a chain or pokeweed antiviral protein and anti-thy 1.1 monoclonal antibodies. Cancer Res. 1984, 44, 201–208. [Google Scholar] [PubMed]
- O’Hare, M.; Roberts, L.M.; Thorpe, P.E.; Watson, G.J.; Prior, B.; Lord, J.M. Expression of ricin a chain in escherichia coli. FEBS Lett. 1987, 216, 73–78. [Google Scholar] [CrossRef]
- Prieto, I.; Lappi, D.A.; Ong, M.; Matsunami, R.; Benatti, L.; Villares, R.; Soria, M.; Sarmientos, P.; Baird, A. Expression and characterization of a basic fibroblast growth factor-saporin fusion protein in escherichia coli. Ann. N. Y. Acad. Sci. 1991, 638, 434–437. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, M.G.; Kohr, W.A.; Beattie, K.L.; Beattie, W.G.; Marks, W.; Toman, P.D.; Cheung, L. Amino acid sequence analysis, gene construction, cloning, and expression of gelonin, a toxin derived from gelonium multiflorum. J. Interferon Cytokine Res. 1995, 15, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Laurent, G.; Pris, J.; Farcet, J.P.; Carayon, P.; Blythman, H.; Casellas, P.; Poncelet, P.; Jansen, F.K. Effects of therapy with t101 ricin a-chain immunotoxin in two leukemia patients. Blood 1986, 67, 1680–1687. [Google Scholar] [PubMed]
- Spitler, L.E.; del Rio, M.; Khentigan, A.; Wedel, N.I.; Brophy, N.A.; Miller, L.L.; Harkonen, W.S.; Rosendorf, L.L.; Lee, H.M.; Mischak, R.P.; et al. Therapy of patients with malignant melanoma using a monoclonal antimelanoma antibody-ricin a chain immunotoxin. Cancer Res. 1987, 47, 1717–1723. [Google Scholar] [PubMed]
- Falini, B.; Bolognesi, A.; Flenghi, L.; Tazzari, P.L.; Broe, M.K.; Stein, H.; Durkop, H.; Aversa, F.; Corneli, P.; Pizzolo, G.; et al. Response of refractory hodgkin’s disease to monoclonal anti-cd30 immunotoxin. Lancet 1992, 339, 1195–1196. [Google Scholar] [CrossRef]
- LeMaistre, C.F.; Rosen, S.; Frankel, A.; Kornfeld, S.; Saria, E.; Meneghetti, C.; Drajesk, J.; Fishwild, D.; Scannon, P.; Byers, V. Phase i trial of h65-rta immunoconjugate in patients with cutaneous t-cell lymphoma. Blood 1991, 78, 1173–1182. [Google Scholar] [PubMed]
- Selvaggi, K.; Saria, E.A.; Schwartz, R.; Vlock, D.R.; Ackerman, S.; Wedel, N.; Kirkwood, J.M.; Jones, H.; Ernstoff, M.S. Phase I/II study of murine monoclonal antibody-ricin a chain (xomazyme-mel) immunoconjugate plus cyclosporine a in patients with metastatic melanoma. J. Immunother. Emphasis Tumor Immunol. 1993, 13, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Baluna, R.; Vitetta, E.S. Vascular leak syndrome: A side effect of immunotherapy. Immunopharmacology 1997, 37, 117–132. [Google Scholar] [CrossRef]
- Vitetta, E.S. Immunotoxins and vascular leak syndrome. Cancer J. 2000, 6 (Suppl. 3), S218–S224. [Google Scholar] [PubMed]
- Ahmad, A.; Law, K. Strategies for designing antibody-toxin conjugates. Trends Biotechnol. 1988, 6, 246–251. [Google Scholar] [CrossRef]
- Shan, L.; Liu, Y.; Wang, P. Recombinant immunotoxin therapy of solid tumors: Challenges and strategies. J. Basic Clin. Med. 2013, 2, 1–6. [Google Scholar] [PubMed]
- Hertler, A.A.; Frankel, A.E. Immunotoxins: A clinical review of their use in the treatment of malignancies. J. Clin. Oncol. 1989, 7, 1932–1942. [Google Scholar] [CrossRef] [PubMed]
- Harkonen, S.; Stoudemire, J.; Mischak, R.; Spitler, L.E.; Lopez, H.; Scannon, P. Toxicity and immunogenicity of monoclonal antimelanoma antibody-ricin a chain immunotoxin in rats. Cancer Res. 1987, 47, 1377–1382. [Google Scholar] [PubMed]
- Li, M.; Liu, Z.S.; Liu, X.L.; Hui, Q.; Lu, S.Y.; Qu, L.L.; Li, Y.S.; Zhou, Y.; Ren, H.L.; Hu, P. Clinical targeting recombinant immunotoxins for cancer therapy. OncoTargets Ther. 2017, 10, 3645–3665. [Google Scholar] [CrossRef] [PubMed]
- Pastan, I.H.; Pai, L.H.; Brinkmann, U.; Fitzgerald, D.J. Recombinant toxins: New therapeutic agents for cancer. Ann. N. Y. Acad. Sci. 1995, 758, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Weldon, J.E.; Skarzynski, M.; Therres, J.A.; Ostovitz, J.R.; Zhou, H.; Kreitman, R.J.; Pastan, I. Designing the furin-cleavable linker in recombinant immunotoxins based on pseudomonas exotoxin a. Bioconj. Chem. 2015, 26, 1120–1128. [Google Scholar] [CrossRef] [PubMed]
- Vallera, D.A.; Panoskaltsis-Mortari, A.; Blazar, B.R. Renal dysfunction accounts for the dose limiting toxicity of dt390anti-cd3sfv, a potential new recombinant anti-gvhd immunotoxin. Protein Eng. 1997, 10, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Pennell, C.A.; Erickson, H.A. Designing immunotoxins for cancer therapy. Immunol. Res. 2002, 25, 177–191. [Google Scholar] [CrossRef]
- Pastan, I.; Hassan, R.; FitzGerald, D.J.; Kreitman, R.J. Immunotoxin treatment of cancer. Annu. Rev. Med. 2007, 58, 221–237. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, U.; Pai, L.H.; FitzGerald, D.J.; Willingham, M.; Pastan, I. B3(fv)-pe38kdel, a single-chain immunotoxin that causes complete regression of a human carcinoma in mice. Proc. Natl. Acad. Sci. USA 1991, 88, 8616–8620. [Google Scholar] [CrossRef] [PubMed]
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hamers, C.; Songa, E.B.; Bendahman, N.; Hamers, R. Naturally occurring antibodies devoid of light chains. Nature 1993, 363, 446–448. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Qi, S.; Unger, M.; Hou, Y.N.; Deng, Q.W.; Liu, J.; Lam, C.M.; Wang, X.W.; Xin, D.; Zhang, P.; et al. Immuno-targeting the multifunctional cd38 using nanobody. Sci. Rep. 2016, 6, 27055. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Li, J.; Zhu, X.; Tang, X.; Bao, Y.; Sun, X.; Huang, Y.; Tian, F.; Liu, X.; Yang, L. Humanized cd7 nanobody-based immunotoxins exhibit promising anti-t-cell acute lymphoblastic leukemia potential. Int. J. Nanomed. 2017, 12, 1969–1983. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Li, J.; Zhu, X.; Yu, Y.; Chen, D.; Yuan, L.; Gu, Z.; Zhang, X.; Qi, L.; Gong, Z.; et al. Novel cd7-specific nanobody-based immunotoxins potently enhanced apoptosis of cd7-positive malignant cells. Oncotarget 2016, 7, 34070–34083. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, M.G.; Cheung, L.H.; Liu, Y.; Marks, J.W., 3rd. Design, expression, purification, and characterization, in vitro and in vivo, of an antimelanoma single-chain fv antibody fused to the toxin gelonin. Cancer Res. 2003, 63, 3995–4002. [Google Scholar] [PubMed]
- Zhou, H.; Ekmekcioglu, S.; Marks, J.W.; Mohamedali, K.A.; Asrani, K.; Phillips, K.K.; Brown, S.A.; Cheng, E.; Weiss, M.B.; Hittelman, W.N.; et al. The tweak receptor fn14 is a novel therapeutic target in melanoma: Immunotoxins targeting fn14 receptor for malignant melanoma treatment. J. Investig. Dermatol. 2013, 133, 1052–1062. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.H.; Liu, Y.Y.; Mathias, A.; Stavrou, S.; Wang, Z.; Thompson, J.; Neville, D.M., Jr. Gene optimization is necessary to express a bivalent anti-human anti-t cell immunotoxin in pichia pastoris. Protein Expr. Purif. 2002, 25, 270–282. [Google Scholar] [CrossRef]
- Della Cristina, P.; Castagna, M.; Lombardi, A.; Barison, E.; Tagliabue, G.; Ceriotti, A.; Koutris, I.; Di Leandro, L.; Giansanti, F.; Vago, R.; et al. Systematic comparison of single-chain fv antibody-fusion toxin constructs containing pseudomonas exotoxin a or saporin produced in different microbial expression systems. Microb. Cell Fact. 2015, 14, 19. [Google Scholar] [CrossRef] [PubMed]
- Schindler, J.; Gajavelli, S.; Ravandi, F.; Shen, Y.; Parekh, S.; Braunchweig, I.; Barta, S.; Ghetie, V.; Vitetta, E.; Verma, A. A phase i study of a combination of anti-cd19 and anti-cd22 immunotoxins (combotox) in adult patients with refractory b-lineage acute lymphoblastic leukaemia. Br. J. Haematol. 2011, 154, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Herrera, L.; Bostrom, B.; Gore, L.; Sandler, E.; Lew, G.; Schlegel, P.G.; Aquino, V.; Ghetie, V.; Vitetta, E.S.; Schindler, J. A phase 1 study of combotox in pediatric patients with refractory b-lineage acute lymphoblastic leukemia. J. Pediatr. Hematol. Oncol. 2009, 31, 936–941. [Google Scholar] [CrossRef] [PubMed]
- Furman, R.R.; Grossbard, M.L.; Johnson, J.L.; Pecora, A.L.; Cassileth, P.A.; Jung, S.H.; Peterson, B.A.; Nadler, L.M.; Freedman, A.; Bayer, R.L.; et al. A phase iii study of anti-b4-blocked ricin as adjuvant therapy post-autologous bone marrow transplant: Calgb 9254. Leuk. Lymphoma 2011, 52, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Gould, B.J.; Borowitz, M.J.; Groves, E.S.; Carter, P.W.; Anthony, D.; Weiner, L.M.; Frankel, A.E. Phase i study of an anti-breast cancer immunotoxin by continuous infusion: Report of a targeted toxic effect not predicted by animal studies. J. Natl. Cancer Inst. 1989, 81, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Weiner, L.M.; O’Dwyer, J.; Kitson, J.; Comis, R.L.; Frankel, A.E.; Bauer, R.J.; Konrad, M.S.; Groves, E.S. Phase I evaluation of an anti-breast carcinoma monoclonal antibody 260f9-recombinant ricin a chain immunoconjugate. Cancer Res. 1989, 49, 4062–4067. [Google Scholar] [PubMed]
- Borthakur, G.; Rosenblum, M.G.; Talpaz, M.; Daver, N.; Ravandi, F.; Faderl, S.; Freireich, E.J.; Kadia, T.; Garcia-Manero, G.; Kantarjian, H.; et al. Phase 1 study of an anti-cd33 immunotoxin, humanized monoclonal antibody m195 conjugated to recombinant gelonin (hum-195/rgel), in patients with advanced myeloid malignancies. Haematologica 2013, 98, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Turturro, F. Denileukin diftitox: A biotherapeutic paradigm shift in the treatment of lymphoid-derived disorders. Expert Rev. Anticancer Ther. 2007, 7, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Hayashida, K. Substance p-saporin for bone cancer pain in dogs: Can man’s best friend solve the lost in translation problem in analgesic development? Anesthesiology 2013, 119, 999–1000. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.C.; Agnello, K. Intrathecal substance p-saporin in the dog: Efficacy in bone cancer pain. Anesthesiology 2013, 119, 1178–1185. [Google Scholar] [CrossRef] [PubMed]
- Higgins, S.C.; Fillmore, H.L.; Ashkan, K.; Butt, A.M.; Pilkington, G.J. Dual targeting ng2 and gd3a using mab-zap immunotoxin results in reduced glioma cell viability in vitro. Anticancer Res. 2015, 35, 77–84. [Google Scholar] [PubMed]
- Vallera, D.A.; Todhunter, D.A.; Kuroki, D.W.; Shu, Y.; Sicheneder, A.; Chen, H. A bispecific recombinant immunotoxin, dt2219, targeting human cd19 and cd22 receptors in a mouse xenograft model of b-cell leukemia/lymphoma. Clin. Cancer Res. 2005, 11, 3879–3888. [Google Scholar] [CrossRef] [PubMed]
- Vallera, D.A.; Oh, S.; Chen, H.; Shu, Y.; Frankel, A.E. Bioengineering a unique deimmunized bispecific targeted toxin that simultaneously recognizes human cd22 and cd19 receptors in a mouse model of b-cell metastases. Mol. Cancer Ther. 2010, 9, 1872–1883. [Google Scholar] [CrossRef] [PubMed]
- Fidias, P.; Grossbard, M.; Lynch, T.J., Jr. A phase ii study of the immunotoxin n901-blocked ricin in small-cell lung cancer. Clin. Lung Cancer 2002, 3, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Baluna, R.; Rizo, J.; Gordon, B.E.; Ghetie, V.; Vitetta, E.S. Evidence for a structural motif in toxins and interleukin-2 that may be responsible for binding to endothelial cells and initiating vascular leak syndrome. Proc. Natl. Acad. Sci. USA 1999, 96, 3957–3962. [Google Scholar] [CrossRef] [PubMed]
- Smallshaw, J.E.; Ghetie, V.; Rizo, J.; Fulmer, J.R.; Trahan, L.L.; Ghetie, M.A.; Vitetta, E.S. Genetic engineering of an immunotoxin to eliminate pulmonary vascular leak in mice. Nat. Biotechnol. 2003, 21, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Pastan, I.; Hassan, R.; Fitzgerald, D.J.; Kreitman, R.J. Immunotoxin therapy of cancer. Nat. Rev. Cancer 2006, 6, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Pirie, C.M.; Hackel, B.J.; Rosenblum, M.G.; Wittrup, K.D. Convergent potency of internalized gelonin immunotoxins across varied cell lines, antigens, and targeting moieties. J. Biol. Chem. 2011, 286, 4165–4172. [Google Scholar] [CrossRef] [PubMed]
- Pirie, C.M.; Liu, D.V.; Wittrup, K.D. Targeted cytolysins synergistically potentiate cytoplasmic delivery of gelonin immunotoxin. Mol. Cancer Ther. 2013, 12, 1774–1782. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, H.; Bachran, D.; Panjideh, H.; Schellmann, N.; Weng, A.; Melzig, M.F.; Sutherland, M.; Bachran, C. Saponins as tool for improved targeted tumor therapies. Curr. Drug Targets 2009, 10, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Nakase, I.; Kobayashi, S.; Futaki, S. Endosome-disruptive peptides for improving cytosolic delivery of bioactive macromolecules. Biopolymers 2010, 94, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Berg, K.; Folini, M.; Prasmickaite, L.; Selbo, P.K.; Bonsted, A.; Engesaeter, B.O.; Zaffaroni, N.; Weyergang, A.; Dietze, A.; Maelandsmo, G.M.; et al. Photochemical internalization: A new tool for drug delivery. Curr. Pharm. Biotechnol. 2007, 8, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Weyergang, A.; Selbo, P.K.; Berstad, M.E.; Bostad, M.; Berg, K. Photochemical internalization of tumor-targeted protein toxins. Lasers Surg. Med. 2011, 43, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Bostad, M.; Olsen, C.E.; Peng, Q.; Berg, K.; Hogset, A.; Selbo, P.K. Light-controlled endosomal escape of the novel cd133-targeting immunotoxin ac133-saporin by photochemical internalization—A minimally invasive cancer stem cell-targeting strategy. J. Control. Release 2015, 206, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Queen, C.; Schneider, W.P.; Selick, H.E.; Payne, P.W.; Landolfi, N.F.; Duncan, J.F.; Avdalovic, N.M.; Levitt, M.; Junghans, R.P.; Waldmann, T.A. A humanized antibody that binds to the interleukin 2 receptor. Proc. Natl. Acad. Sci. USA 1989, 86, 10029–10033. [Google Scholar] [CrossRef] [PubMed]
- Molineux, G. Pegylation: Engineering improved pharmaceuticals for enhanced therapy. Cancer Treat. Rev. 2002, 28, 13–16. [Google Scholar] [CrossRef]
- Tsutsumi, Y.; Onda, M.; Nagata, S.; Lee, B.; Kreitman, R.J.; Pastan, I. Site-specific chemical modification with polyethylene glycol of recombinant immunotoxin anti-tac(fv)-pe38 (lmb-2) improves antitumor activity and reduces animal toxicity and immunogenicity. Proc. Natl. Acad. Sci. USA 2000, 97, 8548–8553. [Google Scholar] [CrossRef] [PubMed]
- Cheung, L.H.; Marks, J.W.; Rosenblum, M.G. Development of “designer toxins” with reduced antigenicity and size. Proc. Amer. Assoc. Cancer Res. 2004, 64, 874–875. [Google Scholar]
- Cizeau, J.; Grenkow, D.M.; Brown, J.G.; Entwistle, J.; MacDonald, G.C. Engineering and biological characterization of vb6–845, an anti-epcam immunotoxin containing a t-cell epitope-depleted variant of the plant toxin bouganin. J. Immunother. 2009, 32, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Entwistle, J.; Brown, J.G.; Chooniedass, S.; Cizeau, J.; MacDonald, G.C. Preclinical evaluation of vb6–845: An anti-epcam immunotoxin with reduced immunogenic potential. Cancer Biother. Radiopharm. 2012, 27, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Onda, M.; Lee, B.; Kreitman, R.J.; Hassan, R.; Xiang, L.; Pastan, I. Recombinant immunotoxin engineered for low immunogenicity and antigenicity by identifying and silencing human b-cell epitopes. Proc. Natl. Acad. Sci. USA 2012, 109, 11782–11787. [Google Scholar] [CrossRef] [PubMed]
- Tachtsidis, A.; McInnes, L.M.; Jacobsen, N.; Thompson, E.W.; Saunders, C.M. Minimal residual disease in breast cancer: An overview of circulating and disseminated tumour cells. Clin. Exp. Metastasis 2016, 33, 521–550. [Google Scholar] [CrossRef] [PubMed]
- Sarosdy, M.F.; Hutzler, D.H.; Yee, D.; von Hoff, D.D. In vitro sensitivity testing of human bladder cancers and cell lines to tp-40, a hybrid protein with selective targeting and cytotoxicity. J. Urol. 1993, 150, 1950–1955. [Google Scholar] [CrossRef]
- Battelli, M.G.; Polito, L.; Bolognesi, A.; Lafleur, L.; Fradet, Y.; Stirpe, F. Toxicity of ribosome-inactivating proteins-containing immunotoxins to a human bladder carcinoma cell line. Int. J. Cancer 1996, 65, 485–490. [Google Scholar] [CrossRef]
- Li, C.; Yan, R.; Yang, Z.; Wang, H.; Zhang, R.; Chen, H.; Wang, J. Bcmab1-ra, a novel immunotoxin that bcmab1 antibody coupled to ricin a chain, can eliminate bladder tumor. Oncotarget 2017, 8, 46704–46705. [Google Scholar] [CrossRef] [PubMed]
- Dalken, B.; Giesubel, U.; Knauer, S.K.; Wels, W.S. Targeted induction of apoptosis by chimeric granzyme b fusion proteins carrying antibody and growth factor domains for cell recognition. Cell Death Differ. 2006, 13, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Rybak, S.M.; Arndt, M.A.; Schirrmann, T.; Dubel, S.; Krauss, J. Ribonucleases and immunornases as anticancer drugs. Curr. Pharm. Des. 2009, 15, 2665–2675. [Google Scholar] [CrossRef] [PubMed]
- Mathew, M.; Verma, R.S. Humanized immunotoxins: A new generation of immunotoxins for targeted cancer therapy. Cancer Sci. 2009, 100, 1359–1365. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rust, A.; Partridge, L.J.; Davletov, B.; Hautbergue, G.M. The Use of Plant-Derived Ribosome Inactivating Proteins in Immunotoxin Development: Past, Present and Future Generations. Toxins 2017, 9, 344. https://doi.org/10.3390/toxins9110344
Rust A, Partridge LJ, Davletov B, Hautbergue GM. The Use of Plant-Derived Ribosome Inactivating Proteins in Immunotoxin Development: Past, Present and Future Generations. Toxins. 2017; 9(11):344. https://doi.org/10.3390/toxins9110344
Chicago/Turabian StyleRust, Aleksander, Lynda J. Partridge, Bazbek Davletov, and Guillaume M. Hautbergue. 2017. "The Use of Plant-Derived Ribosome Inactivating Proteins in Immunotoxin Development: Past, Present and Future Generations" Toxins 9, no. 11: 344. https://doi.org/10.3390/toxins9110344