
Microvesicle Involvement in Shiga Toxin-Associated Infection


Department of Pediatrics, Clinical Sciences Lund, Lund University, 22184 Lund, Sweden
*
Author to whom correspondence should be addressed.
Toxins 2017, 9(11), 376; https://doi.org/10.3390/toxins9110376
Submission received: 31 October 2017
/
Revised: 15 November 2017
/
Accepted: 16 November 2017
/
Published: 19 November 2017
(This article belongs to the Special Issue Ribosome Inactivating Toxins)
Abstract
:Shiga toxin is the main virulence factor of enterohemorrhagic Escherichia coli, a non-invasive pathogen that releases virulence factors in the intestine, causing hemorrhagic colitis and, in severe cases, hemolytic uremic syndrome (HUS). HUS manifests with acute renal failure, hemolytic anemia and thrombocytopenia. Shiga toxin induces endothelial cell damage leading to platelet deposition in thrombi within the microvasculature and the development of thrombotic microangiopathy, mostly affecting the kidney. Red blood cells are destroyed in the occlusive capillary lesions. This review focuses on the importance of microvesicles shed from blood cells and their participation in the prothrombotic lesion, in hemolysis and in the transfer of toxin from the circulation into the kidney. Shiga toxin binds to blood cells and may undergo endocytosis and be released within microvesicles. Microvesicles normally contribute to intracellular communication and remove unwanted components from cells. Many microvesicles are prothrombotic as they are tissue factor- and phosphatidylserine-positive. Shiga toxin induces complement-mediated hemolysis and the release of complement-coated red blood cell-derived microvesicles. Toxin was demonstrated within blood cell-derived microvesicles that transported it to renal cells, where microvesicles were taken up and released their contents. Microvesicles are thereby involved in all cardinal aspects of Shiga toxin-associated HUS, thrombosis, hemolysis and renal failure.
1. Introduction
Shiga toxin-producing Escherichia coli (STEC) or enterohemorrhagic E. coli (EHEC) may cause disease in humans manifesting with diarrhea, bloody diarrhea (hemorrhagic colitis) and, in approximately 15% of cases, the severe systemic complication of hemolytic uremic syndrome (HUS) [1]. HUS is characterized by the post-diarrheal acute onset of non-immune hemolytic anemia, thrombocytopenia and renal failure. The most common clinical EHEC isolate is E. coli O157:H7 [2], although many other non-O157 serotypes have been described, notably the E. coli O104:H4 serotype that caused a huge outbreak in 2011 [3]. EHEC is a non-invasive pathogen [4] that colonizes the intestine where it expresses and also releases virulence factors. Some of these allow adherence to the intestinal mucosa by forming attaching and effacing lesions leading to colonization [5], while flagella are associated with bacterial motility [6]. EHEC interaction with commensal strains and host hormones enhances colonization and virulence by a genetically determined phenomenon known as quorum sensing [7]. The major and unique virulence factor strongly associated with EHEC-induced morbidity is Shiga toxin [8]. In addition, EHEC possesses lipopolysaccharide (LPS) and other factors capable of activating the host response [9]. A prerequisite for the strain to cause systemic and target organ damage, such as renal failure or brain damage [10], is the ability of virulence factors to gain access to the bloodstream and thereby reach target organ cells.
Shiga toxin may be capable of binding to intestine epithelial cells and thereafter translocate [11,12,13]. The intestinal inflammatory response is multifactorial depending on the interaction between the toxin, other virulence factors, and the host response [9]. Shiga toxin-producing EHEC strains are diarrheogenic. The diarrhea may become bloody leading to hemorrhagic colitis. This form of intestinal injury appears to be specifically associated with Shiga toxin production, as demonstrated in a monkey model of Shigella infection [14]. The massive erosion of the intestinal mucosal lining allows virulence factors released from EHEC to gain access to the circulation. Once within the bloodstream most of the toxin does not circulate in free form [15,16] but rather bound to blood cells such as leukocytes [17] and platelets as well as aggregates between these cells [18]. Red blood cells are also capable of binding the toxin [19,20]. Blood cells are activated by toxin binding and, thereafter, shed microvesicles which are pro-inflammatory, pro-thrombotic [18], and, importantly, transport the toxin to its target organ [21]. This does not exclude other mechanisms of toxin transfer from blood cells to affected cells [22], but has been suggested to be one of the main mechanisms of toxin-induced systemic and targeted organ injury [1].
Microvesicles are a subtype of extracellular vesicles shed directly from the plasma membrane of cells upon activation, stress and apoptosis [23]. Microvesicles can originate from blood cells [24,25,26] as well as non-circulating organ-specific cells [27,28]. Vesicles may be enriched in components of the parent cells such as proteins, receptors, RNAs (mRNA and miRNA) and lipids, enabling them to interact with cells in their immediate vicinity and at a distance [29]. Vesicle release may also maintain cellular integrity by ridding the cell of harmful substances [30]. Increasing evidence suggests that microvesicles are key players in several diseases, including cancer [31], renal diseases [32], cardiovascular disease [33] and inflammatory diseases [34]. In these diseases, the number of circulating microvesicles is significantly increased, indicating a disruption in physiological processes. In Shiga toxin-associated disease, Shiga toxin-bearing microvesicles have been found in the circulation of EHEC-infected patients as well as within the kidney [21], enabling toxin evasion of the immune system and thereby protection of the toxin from degradation. This review will mainly focus on the functions of microvesicles, in general and in the context of bacterial infections, particularly with respect to Shiga toxin-associated infection.
2. Shiga Toxin
Shiga toxin, encoded by a bacteriophage, is released from bacteria in the gut, most probably during bacterial lysis [35]. Shiga toxin is a ribosomal-inactivating protein. It is an AB5 toxin composed of two subunits, an A-subunit and a pentrameric B-subunit, linked together by non-covalent bonds [36]. The A-subunit accounts for the enzymatic cytotoxic activity whereas the pentameric B-subunit binds to glycosphingolipid receptors mainly the globotriaosylceramide (Gb3) receptor [37,38] and, to a lesser extent, the Gb4 receptor [39]. The density of Gb3 in the cell membrane and its association with lipid rafts affect toxin binding [40].
After Shiga toxin binds to its glycolipid receptor it can be taken up by endocytosis. Various endocytic routes have been described involving formation of membrane microtubular structures mainly in a clathrin-independent manner but also by a clathrin-dependent mechanism [41,42,43,44], as recently reviewed [45]. Uptake in intestinal cells by macropinocytosis, in a Gb3-independent manner, has also been reported [46,47]. Once within a cell, Shiga toxin is ultimately destined to reach ribosomes in the cytosol [48]. Shiga toxin is transported in a retrograded manner from early endosomes to the trans-Golgi network and further to the endoplasmic reticulum. Within the endoplasmic reticulum the A subunit is cleaved by furin into the A1 and A2 subunits [49]. From the endoplasmic reticulum, Shiga toxin is transported out to the cytosol, accessing the ribosomes [50].
2.1. Cytotoxicity of Shiga Toxin
The enzymatically active A1 subunit of Shiga toxin exerts a cytotoxic effect by N-glycosidic cleavage of a specific adenine base from 28S rRNA [51] leading to inhibition of protein synthesis followed by cell death. Moreover, Shiga toxin activates apoptotic pathways [52,53], most probably by inducing ribosomal damage and further activation of mitogen-activated protein (MAP) kinase pathways, the so-called ribosomal stress response [54,55]. Shiga toxin has been shown to induce intestinal cell apoptosis [56] and renal cell apoptosis in vivo and in vitro [52].
2.2. Inflammatory Effects of Shiga Toxin and Lipopolysaccharide in the Intestine
In addition to its cytotoxic effects, Shiga toxin is capable of activating an inflammatory response in the intestine, in its interaction with blood cells and after binding to target organ cells [9]. These effects occur simultaneously with the cytotoxic effects and are associated with the release of a wide range of pro-inflammatory cytokines and chemokines. In vitro studies have shown that Shiga toxin can trigger neutrophil influx into the intestine by inducing the release of interleukin (IL)-8 and other C-X-C chemokines [57,58,59]. The interaction with peritoneal macrophages also led to release of pro-inflammatory cytokines such as tumor necrosis factor-alpha (TNF-α) and IL-6 [60]. LPS may also contribute to the inflammatory response in the intestine [60]. Studies in mice have indicated that the initial host response to E. coli O157:H7 LPS is essential for bacterial elimination from the gut, thus mice lacking an adequate response to LPS were subject to more severe disease [61,62].
2.3. Interactions between Shiga Toxin and Blood Cells
During HUS, elevated neutrophil counts and decreased platelet counts suggest a worse prognosis [63]. Shiga toxin may circulate bound to neutrophils in vivo [17,64], and aggregates between platelets and neutrophils [18]. Both Shiga toxin and LPS induce the formation of these cell aggregates in which neutrophils are activated. Thus, the toxin may induce neutrophil activation and degranulation [9]. Similarly, the toxin may bind to monocytes [65] and has been detected on monocytes or platelet-monocyte aggregates in patient samples [18]. Shiga toxin induces the release of pro-inflammatory cytokines from monocytes including IL-6, IL-8, TNF-α, IL-1β and RANTES [65,66], as well as the expression of pro-thrombotic tissue factor activating the extrinsic pathway of coagulation leading to thrombin generation and blood clotting [18,67].
Platelets are activated during HUS and deposit in microthrombi. Their aggregation and consumption on injured endothelium leads to lowered platelet counts [68,69]. In the circulation, platelets are degranulated [70] with consequent elevated platelet-derived proteins such as platelet factor-4, β-thromboglobulin and P-selectin [71,72]. Shiga toxin binds to platelets via Gb3 and an alternative glycolipid receptor [73]. LPS binds to platelets via a toll-like receptor (TLR)4-CD62 receptor complex [74] and activates platelets. LPS derived from E. coli O157 was particularly potent in this respect [74]. Once activated, platelets further respond to Shiga toxin [75,76], may take up the toxin and exhibit excessive fibrinogen binding, enhancing their thrombotic potential. Platelet activation induced by Shiga toxin may be further exacerbated by toxin-mediated endothelial cell damage, exposing the subendothelium with release of von Willebrand factor, fibrinogen and collagen [68,69], as well as complement deposition. Furthermore, complement deposits on platelet and platelet–leukocyte aggregates in response to stimulation with Shiga toxin and E. coli O157 LPS [77]. Studies have shown that complement deposition on platelets initiates thrombin-mediated aggregation [78]. Moreover, once thrombin is formed [79], it can further propagate complement system activation [80,81] enhancing the inflammatory activity at the interface of platelets and the endothelium [82]. Once activated, platelets may contribute to the inflammatory state by the release of potent chemokines, as previously reviewed [9,69].
Patients with HUS exhibit acute hemolysis with fragmented red blood cells [1]. Hemolysis may be caused by the mechanical breakdown of red blood cells within occluded blood vessels, but complement activation on red blood cells may also contribute to this process, since it is known that complement deposition on red blood cells induces hemolysis [83]. Patients with HUS exhibit C3 deposition on red blood cells [19], suggesting that complement is activated on blood cell surfaces. Shiga toxin binds to the Gb3 receptor on red blood cells, also known as the Pk antigen of the P1Pk blood group system [20,84]. In vitro experiments demonstrated that Shiga toxin induced hemolysis by activating complement on human red blood cells in the presence of plasma, an effect abrogated by complement inactivation or by addition of the terminal complement pathway inhibitor eculizumab, directed against C5 [19].
Taken together, Shiga toxin is capable of binding to neutrophils, monocytes, platelets and red blood cells and thus it may be transported on, or within, blood cells in the circulation. Blood cells are activated thus potentiating the inflammatory and thrombotic process occurring during HUS. Furthermore, the toxin may thereby reach its target organs [85], mainly the kidney and the brain, although this mechanism of transfer does not fully clarify how the toxin is released from blood cells and taken up by recipient cells [86].
2.4. Thrombus Formation During HUS
Shiga toxin binds to endothelial and epithelial cells expressing the Gb3 receptor, such as the glomerular endothelium and the tubular epithelium in the human renal cortex [52,87]. Interestingly, cytokine release and exposure to bacterial LPS enhances Gb3 expression and augments toxin binding [88,89]. The mechanisms contributing to thrombus formation are multifactorial involving toxin-mediated endothelial cell damage [90], platelet activation [74,76], the formation of platelet-monocyte aggregates expressing tissue factor [18] as well as the release of inflammatory mediators from monocytes and endothelial cells that further activate platelets [66,91]. In addition, activation of coagulation and impaired fibrinolysis occur before HUS develops [79] promoting thrombus stabilization during HUS, after which fibrinolysis is enhanced [92,93,94]. Of note, the fibrinolytic system is activated in the murine kidney both in the endothelium and in tubular epithelial cells [95] but Shiga toxin may actually lower production of fibrinolytic parameters by inhibiting protein synthesis [94] and thereby tip the balance towards enhanced thrombus formation.
2.5. Shiga Toxin Induces the Release of Blood Cell-Derived Microvesicles
Binding of Shiga toxin to circulating blood cells may initiate cell activation and the release of blood cell-derived microvesicles, particularly in the presence of LPS [18,77]. During the acute phase of HUS, levels of circulating microvesicles are elevated [18,19,21,96] and Shiga toxin was detected within blood cell-derived microvesicles originating from neutrophils, monocytes, platelets and red blood cells [21]. In the following section, we will describe extracellular vesicles, their formation, shedding and uptake. We will also describe the function of microvesicles in normal cellular interactions and contribution to pathological processes, in particular during infectious diseases. Thereafter, we will focus on their contribution to HUS.
3. Characteristics of Extracellular Vesicles
Extracellular vesicles are membranous particles, released from cells, that are characterized based on their cell of origin and their size (Figure 1). Exosomes (30–100 nm in diameter) are formed by the release of the contents of intracellular endosomal multivesicular bodies containing intraluminal vesicles. Once extruded, these vesicles are termed exosomes [97]. Microvesicles (100–1000 nm) are released from cells by direct budding of the plasma membrane, while apoptotic bodies (1–5 μm) are released by the breakdown of cells during programmed cell death [98]. The latter are distinctly different from other vesicles because they contain larger cellular degradation products, such as organelles. The properties of extracellular vesicles are summarized in Table 1. Detection methods include flow cytometry, nanoparticle tracking and transmission electron microscopy, among others, as well as proteomic analysis of vesicular contents, as reviewed elsewhere [99,100,101,102]. Extracellular vesicles can be differentiated based on their mechanism of secretion and based on cellular markers [29,103], often allowing to determine the parent cell from which the vesicles were released. Web-based databases are available in which data regarding vesicle content and properties is summarized (see Vesiclepedia, ExoCarta or EVpedia, online).
3.1. Exosomes
Intracellular multivesicular bodies are late endosomes containing intraluminal vesicles. When they dock onto the plasma membrane and fuse, the vesicles are released as exosomes into the extracellular space [104]. The formation of intraluminal vesicles and their release as exosomes is mainly regulated by two pathways, either via the endosomal sorting complex required for transport (ESCRT) machinery [105,106] or by an ESCRT-independent process (including tetraspanins and ceramide generation) [107,108]. Exosomes are characteristically different from other vesicle populations due to their endosomal origin with a unique composition of lipids and proteins. Compared to the parent cell, exosomes may be enriched in cholesterol, glycosphingolipids, sphingomyelin and phosphatidylserine [109]. Proteins enriched in exosomes include tetraspanins [110], heat shock proteins, component of the ESCRT family [111] and MHC class I [112]. While these proteins are generally enriched in exosomal populations, other proteins may be specifically enriched in exosomes dependent on the cell of origin [111,113]. Comprehensive reviews regarding exosomal formation and their biological functions have been published elsewhere [103,104,114], and herein we will focus on microvesicles.
3.2. Microvesicles
Microvesicles are shed directly from the plasma membrane together with contents of the parent cell. The content may include proteins (cytokines, chemokines, enzymes, receptors) [32,115], RNAs (mRNAs and non-coding RNAs, particularly miRNAs) [23] and lipids [116]. The content will vary depending on the cell of origin and reflect the biological status of the cell and degree of activation [117]. The structure of microvesicles protects bioactive materials and enables the transfer of such substances from one cell to another as a means of cell-to-cell communication [29,118,119]. Microvesicles can be taken up by neighboring or distant cells and have the potential to phenotypically alter the recipient cell [120,121], as elaborated on below. Another important function of microvesicles is to discard the parent cell of unwanted substances and thus preserve cellular integrity [122,123,124].
3.2.1. Microvesicle Formation
The underlying mechanism of microvesicle formation involves numerous cellular events leading to local budding of the plasma membrane, followed by a fission event whereby the vesicle is pinched off and released into the extracellular space [125]. Shedding of microvesicles occurs spontaneously in resting cells [23], and is increased during cell activation in response to various stimuli such as hypoxia, oxidative stress, shear stress, pro-inflammatory mediators, cell damage or ligand-binding [126,127,128]. A common feature during activation is an increase in the cytosolic calcium level, which promotes microvesicle release [129]. Increasing evidence suggests that the inner content as well as the surface of microvesicles are selectively packaged, rather than randomly, by a controlled mechanism of cargo trafficking within microvesicles, as reviewed [121]. The regulatory protein, ADP-ribosylation factor 6, selectively recruits protein cargo onto microvesicles and has been shown to be an important component of this process [130]. The exact mechanisms by which proteins, lipids and RNAs are targeted into microvesicles remain to be elucidated. The process is affected by external stimuli triggering vesicle release, as well as by the cellular environment [126]. The same cell may thus release microvesicles with varied content [131]. Interestingly, microvesicles may incorporate membranous contents at a higher density than the parent cell due to enrichment in lipid rafts [132], thereby enhancing the procoagulant potency of platelet-derived microvesicles [133].
In resting cells, plasma membrane lipids are arranged in an asymmetric pattern whereby the outer leaflet contains phosphatidylcholine and sphingomyelin, and the inner membrane leaflet is enriched with aminophospholipids (phosphatidylserine and phosphatidylethanolamine) [134,135]. Phospholipid asymmetry is controlled by specific enzymes such as flippase (transfers phosphatidylserine to the inner leaflet), floppase (controls outward phospholipid translocation), and enzymes with scramblase activity (transport lipids in a bidirectional Ca++-dependent manner) [136,137]. During cell activation, concurrent with a rise in the intracellular calcium level, floppase and scramblase activities are activated, while flippase is inactivated leading to disruption of lipid asymmetry with exposure of phosphatidylserine on the outer leaflet. The increased calcium level also enhances the activity of cytosolic enzymes such as calpain, involved in cytoskeleton changes that facilitate microvesicle shedding [138]. Microvesicle membranes are characterized by a loss of lipid asymmetry, in comparison to the parent cell, and may expose phosphatidylserine, although this is not a feature of all microvesicles [126]. The mechanism associated with the release of phosphatidylserine-negative microvesicles is poorly understood. A possible mechanism involving cellular proteins able to alter the membrane curvature has been described [139].
3.2.2. Microvesicle Uptake
Uptake of microvesicles by recipient cells can be mediated by various endocytic pathways or fusion with the plasma membrane. Endocytosis may be facilitated by receptor-ligand interactions. Uptake may involve clathrin-dependent or -independent endocytosis, caveolin-dependent endocytosis, macropinocytosis, phagocytosis or lipid raft-mediated uptake, as reviewed [140]. Fusion of microvesicles with the membrane of the target cell is a highly dynamic process, resembling the process utilized by retroviruses, involving high affinity binding to the target cell, lipid reorganization and restructuring of proteins [141]. The fusion event enables the bioactive vesicular content to be inserted into the cytosol of the target cell [142]. Several factors have been shown to affect fusion; these include an acidic environment, the degree of ligand-receptor binding and the lipid composition of the microvesicle [143].
The uptake of a microvesicle may depend on the presence of receptors and/or proteins on the microvesicle surface and their interaction with counterparts on the recipient cell [140,144]. These interactions may lead to the formation of a microvesicle–cell complex, thereby coating the cell and possibly initiating intracellular signaling events due to receptor-ligand interactions. The microvesicle may be taken up by the cell and release its content into the cell [119]. Ligands specifically enriched on microvesicles such as glycoproteins (integrins, selectins and tissue factor) and phospholipids may favor cell-specific uptake [145,146,147,148]. For example, platelet-derived microvesicles transfer tissue factor to monocytes but not to neutrophils [149], demonstrating uptake by a defined cell type.
3.2.3. The role of Microvesicles in Intercellular Communication
The diversity of bioactive materials covering the surface of microvesicles and contained within microvesicles may affect the phenotype of the recipient cell by transfer of nucleic acids, proteins, receptors and lipids. RNAs may be translated in the target cell activating or silencing certain properties [150,151,152]. The physiological or pathological consequences may result in induction of angiogenesis, thrombosis, altered hemostasis, immune modulation, invasive potential, matrix alteration and tissue regeneration, as recently reviewed [32]. The transfer of functionally active receptors may activate pro-inflammatory or pro-thrombotic signaling pathways as well as proliferative capacity in the recipient cell, as has been exemplified regarding chemokine receptors [153,154], epidermal growth factor receptor [155], kinin B1 receptor [156] and platelet receptors [157]. The transfer of the platelet receptor GPIIb/IIIa to neutrophils will induce neutrophil binding to the endothelium via fibronectin and thereby have a pro-inflammatory effect [158]. Cancer cell-derived microvesicles have been shown to bear epidermal growth factor receptor, that could be transferred to neighboring cells and influence cell morphology and growth capacity [120]. The invasive capacity has been demonstrated in microvesicles derived from cancer cells which are enriched in metalloproteinases capable of breaking down extracellular matrix to promote tumor growth [159].
Microvesicles derived from blood and endothelial cells may induce an inflammatory response. As mentioned above, microvesicles can transfer chemokine receptors [153]. Furthermore, microvesicles from platelets or from endothelial cells can induce monocyte [160] or neutrophil chemotaxis [161]. The recruited monocytes will deposit on endothelial cells [162]. Likewise, platelet-derived microvesicles can recruit CD34-positive hematopoietic cells [157]. The immunomodulatory effects of microvesicles also encompass the transfer of anti-inflammatory mediators (RNAs or proteins), particularly studied in microvesicles derived from stem cells [163] or neutrophils [164], potentially contributing to a beneficial effect during tissue regeneration.
Importantly, microvesicles may possess pro-thrombotic potential. The exposure of phosphatidylserine on the outer leaflet of microvesicles generates a negatively-charged surface promoting binding of prothrombin, factor Va and factor Xa [165]. When scramblase activity is defective, such as in Scott syndrome, the number of phosphatidylserine-positive microvesicles is reduced and coagulation is impaired resulting in a bleeding disorder [166,167]. Phosphatidylserine exposure on monocytic microvesicles was shown to differ depending on the stimulus inducing microvesicle release suggesting that environmental factors can affect the composition of microvesicles and their pro-thrombotic properties [128]. In addition, certain microvesicles, mainly from monocytes and platelets, carry tissue factor on their surface contributing to thrombus formation [18,146,168,169]. Monocyte-derived microvesicles transfer tissue factor to platelets [170]. Red blood cell-derived microvesicles are also thrombogenic and may induce thrombin generation in the absence of tissue factor by activation of the intrinsic coagulation pathway on their surfaces [171]. Similarly, platelet-derived microvesicles containing arachidonic acid metabolized to thromboxane A2 induce platelet aggregation [116].
4. Microvesicles in Infectious Diseases
Infections, and sepsis in particular, are accompanied by a pro-inflammatory and pro-thrombotic host response. The properties of blood cell-derived microvesicles can contribute to these effects and to multi-organ failure. Patients with sepsis have high levels of platelet- and leukocyte-derived microvesicles [172,173,174,175]. Blood cell-derived microvesicles were tissue factor-positive in patients with meningococcal sepsis [172] and febrile urinary tract infection [176]. Patients with disseminated intravascular coagulopathy had elevated endothelial and leukocyte microvesicles [177]. An enhanced inflammatory response during sepsis, as reflected by microvesicle levels, may, however, predict a more favorable outcome [178], and circulatory microvesicle levels correlated negatively with the degree of acute kidney injury during sepsis, although this may actually reflect the degree of microvesicle deposition in damaged tissue [174].
When microvesicles released during sepsis were injected into mice they enhanced vascular reactivity and thromboxane A2 production [175]. Likewise, microvesicles derived from rats with peritonitis induced an inflammatory response when injected into healthy rats [179]. Thus, microvesicles may have damaging effects during sepsis [180,181]. Beneficial effects may also prevail as neutrophil-derived microvesicles, that were elevated during bacteremia, exhibited anti-microbial effects [182]. Similar effects were also demonstrated as blood cell-derived microvesicles prevented in vivo spreading of Streptococcus pyogenes [183].
Microvesicles May Transfer Infectious Agents or Their Virulence Factors
Bacteria and viruses have developed ways of utilizing extracellular vesicles for the transfer of their antigens and virulence factors. For example, Mycobacterium tuberculosis infected macrophages release microvesicles containing M. tuberculosis antigen in complex with MHC-II that can induce an antimicrobial T-cell response [184]. HIV budding from cells can utilize components of the ESCRT pathway and yet be shed directly from the cell membrane [185,186] suggesting that viral dissemination may occur via extracellular vesicles [187] possibly utilizing phosphatidylserine receptors [188]. Other viruses, such as herpes viruses, also utilize extracellular vesicles to transfer viral RNAs and proteins from cell to cell [189,190].
Pathogens can thereby exploit the microvesicle transport system for their own benefit to evade host response by covering themselves with the host membrane within a vesicle. The vesicle containing pathogen virulence factors is taken up by recipient cells thereby affecting target cells. Interestingly, pathogens can also release their own vesicles. Protozoan parasites, such as Trypanosoma cruzi, release vesicles capable of interacting with host cells [191,192]. Similarly, mycobacterium may release vesicles capable of inducing an inflammatory response in the host [193]. Outer membrane vesicles are released from gram-negative bacteria, such as EHEC, and may transfer toxins, such as Shiga toxin, into intestinal epithelial cells as well as renal or brain endothelial cells [194]. The major difference between pathogen-derived vesicles [194] and host blood cell-derived microvesicles [21], both containing virulence factors, such as Shiga toxin, is that the latter, host-derived extracellular vesicles, will be recognized as “self” and thereby avoid attack by the immune system whereas pathogen-derived vesicles are “non-self” and subject to immune attack.
5. Shiga Toxin-Induced Microvesicles in Laboratory Models
Shiga toxin binds to neutrophils, monocytes, platelets and red blood cells [20,22,64,65,73]. These blood cells are resistant to the cytotoxic effects of the toxin, in part due to minimal protein synthesis in platelets and red blood cells. The cells may instead become activated and release microvesicles [18,19,66,76,77]. In vitro experiments have shown that Shiga toxin induces the release of microvesicles from human monocytes, platelets [18,77] and red blood cells [19]. Co-stimulation with Shiga toxin and E. coli O157-LPS enhanced microvesicle release from platelets and leukocytes compared to each stimulant alone [18]. Microvesicles shed from platelets and monocytes carry tissue factor and phosphatidylserine [18], as well as complement C3 and C9 [77]. C5b-9 was also detected on microvesicles from red blood cells which also exposed phosphatidylserine after stimulation with Shiga toxin [19]. The toxin itself may be incorporated in blood cell-derived microvesicles originating from neutrophils, monocytes, platelets and red blood cells [21]. Permeabilization of microvesicles was required to detect Shiga toxin, suggesting that the toxin was mainly localized within the microvesicles and not on their outer membrane [21].
Toxin within microvesicles can be transferred to target organ cells and be taken up after endocytosis of the entire microvesicle [21] (Figure 2). In a mouse model of EHEC infection [62], blood cell-derived microvesicles carrying Shiga toxin were demonstrated to be taken up by renal glomerular endothelial cells and tubular cells [21]. One notable aspect of this finding is that, in contrast to the human glomerulus, mouse glomerular endothelial cells lack the toxin Gb3 receptor [87]. Thus, it is possible that this process is Gb3- or toxin receptor-dependent up until the toxin binds to its receptor on blood cells and is internalized in these cells. After the toxin is shed from the blood cells, within microvesicles, the toxin-containing microvesicles may be taken up even by cells that lack the Gb3 [1].
6. Microvesicles in the Pathogenesis of Hemolytic Uremic Syndrome
During EHEC-associated HUS, only minimal amounts of freely circulating Shiga toxin have been found in patient samples and Shiga toxin is mainly found bound to blood cells [15,18]. In acute phase HUS patients, the number of circulating blood cell-derived microvesicles is elevated [18,19,21,96], decreasing after recovery. Shiga toxin circulates within microvesicles originating from neutrophils, monocytes, platelets and red blood cells [21]. Shiga toxin-containing microvesicles from blood cells were further demonstrated to be taken up by glomerular endothelial cells in a patient sample [21]. Kidney cells, including glomerular endothelial cells, mesangial cells, podocytes and tubular cells are highly sensitive to Shiga toxin [52,195,196]. The effects of Shiga toxin and blood cell-derived microvesicles in the circulation of acute phase HUS patients are described in Figure 3.
The main features of HUS are thrombocytopenia due to platelet consumption in microthrombotic lesions on the damaged endothelium, hemolysis with fragmented red blood cells and renal failure. Microvesicles may contribute to and partake in each of these processes. The pro-thrombotic properties of microvesicles, bearing tissue factor and exposing phosphatidylserine on their outer leaflet, as described above, and reviewed [32], may contribute to thrombin generation and platelet activation on damaged microvasculature. Complement deposition on red blood cells will induce hemolysis and the release of complement-coated red blood cell-derived microvesicles during HUS [19] suggesting that hemolysis is induced not only by mechanical fragmentation of red blood cells in occluded blood vessels but also by complement activation. The pro-thrombotic effect may be further enhanced by circulating red blood cell-derived microvesicles capable of activating the intrinsic coagulation pathway [171].
The presence of complement C3 and C9 on platelet and monocyte-derived microvesicles in HUS patients [77] would reflect complement activation on the parent cells and could also contribute to the thrombotic process as complement activation on platelet membranes may promote their activation [197]. During HUS complement is deposited on the injured vascular wall [198], on platelets and microvesicles derived thereof [77], suggesting that complement contributes to the tissue injury during HUS [82]. Indeed, mouse models of EHEC infection or Shiga toxin injection in which the alternative, lectin or terminal complement pathways were blocked, exhibited reduced renal injury [198,199,200].
Shiga toxin can be transferred from the bloodstream to the kidney within blood cell-derived microvesicles and taken up in glomerular endothelial cells or peritubular capillary endothelial cells [21]. This mechanism could explain how the toxin enters target organ cells after circulating within activated blood cells that release microvesicles. In a mouse model, toxin-positive microvesicles were released from blood cells during the early stages of infection before symptoms develop [21]. They were further taken up by renal cells before clinical signs of disease were evident. Microvesicles were demonstrated to release their content of Shiga toxin within 12 h of entering the cell and the toxin reached ribosomes within 24 h, inhibiting protein synthesis, thus providing evidence that toxin within microvesiscles retains its cytotoxicity. An interesting finding was that not all microvesicles emptied their contents after endocytosis, certain microvesicles transcytosed the recipient cells, migrated through the corresponding glomerular or tubular basement membranes and passed into podocytes or tubular cells, respectively [21]. The capacity of microvesicles to navigate through cells and basement membranes could be secondary to tissue injury during HUS or possibly the toxic content of the microvesicles.
7. Conclusions
During Shiga toxin-associated HUS, microvesicles are released from blood cells capable of transferring the toxin to target organ cells inducing renal cell death and promoting thrombosis. Red blood cell-derived microvesicles are also involved in the hemolytic process. Microvesicles are thereby involved in all aspects of HUS.
Microvesicles transfer an array of nucleic acids, proteins and lipids, and are thus a potent mechanism for intercellular communication, which may have detrimental effects during infection and inflammation, but may also have beneficial effects in tissue regeneration. Furthermore, the shedding of microvesicles carrying unwanted cellular components may maintain cell integrity after a trigger of cell activation. Therefore, future studies should address the importance of microvesicles and possible effects of blocking their release in Shiga toxin-mediated infection and kidney damage.
Acknowledgments
D.K. is supported by The Swedish Research Council (K2013-64X-14008-13-5 and K2015-99X-22877-01-6), The Knut and Alice Wallenberg Foundation (Wallenberg Clinical Scholar 2015.0320), The Torsten Söderberg Foundation, Skåne Centre of Excellence in Health, Crown Princess Lovisa’s Society for Child Care, Region Skåne, the IngaBritt and Arne Lundberg Research Foundation and The Konung Gustaf V:s 80-års minnesfond.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Karpman, D.; Loos, S.; Tati, R.; Arvidsson, I. Haemolytic uraemic syndrome. J. Intern. Med. 2017, 281, 123–148. [Google Scholar] [CrossRef] [PubMed]
- Griffin, P.M.; Ostroff, S.M.; Tauxe, R.V.; Greene, K.D.; Wells, J.G.; Lewis, J.H.; Blake, P.A. Illnesses associated with Escherichia coli O157:H7 infections. A broad clinical spectrum. Ann. Intern. Med. 1988, 109, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Frank, C.; Werber, D.; Cramer, J.P.; Askar, M.; Faber, M.; an der Heiden, M.; Bernard, H.; Fruth, A.; Prager, R.; Spode, A.; et al. Epidemic profile of Shiga-toxin-producing Escherichia coli O104:H4 outbreak in Germany. N. Engl. J. Med. 2011, 365, 1771–1780. [Google Scholar] [CrossRef] [PubMed]
- McKee, M.L.; O’Brien, A.D. Investigation of enterohemorrhagic Escherichia coli o157:H7 adherence characteristics and invasion potential reveals a new attachment pattern shared by intestinal E. coli. Infect. Immun. 1995, 63, 2070–2074. [Google Scholar] [PubMed]
- Kaper, J.B. The locus of enterocyte effacement pathogenicity island of Shiga toxin-producing Escherichia coli O157:H7 and other attaching and effacing E. coli. Jpn. J. Med. Sci. Biol. 1998, 51 (Suppl. 1), S101–S107. [Google Scholar] [CrossRef] [PubMed]
- Rogers, T.J.; Paton, J.C.; Wang, H.; Talbot, U.M.; Paton, A.W. Reduced virulence of an flic mutant of Shiga-toxigenic Escherichia coli O113:H21. Infect. Immun. 2006, 74, 1962–1966. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, A.R.; Sperandio, V. Inter-kingdom signaling: Chemical language between bacteria and host. Curr. Opin. Microbiol. 2009, 12, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Karmali, M.A.; Petric, M.; Lim, C.; Fleming, P.C.; Arbus, G.S.; Lior, H. The association between idiopathic hemolytic uremic syndrome and infection by verotoxin-producing Escherichia coli. J. Infect. Dis. 1985, 151, 775–782. [Google Scholar] [CrossRef] [PubMed]
- Karpman, D.; Ståhl, A.L. Enterohemorrhagic Escherichia coli pathogenesis and the host response. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Loos, S.; Ahlenstiel, T.; Kranz, B.; Staude, H.; Pape, L.; Hartel, C.; Vester, U.; Buchtala, L.; Benz, K.; Hoppe, B.; et al. An outbreak of Shiga toxin-producing Escherichia coli O104:H4 hemolytic uremic syndrome in Germany: Presentation and short-term outcome in children. Clin. Infect. Dis. 2012, 55, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Schuller, S.; Frankel, G.; Phillips, A.D. Interaction of Shiga toxin from Escherichia coli with human intestinal epithelial cell lines and explants: Stx2 induces epithelial damage in organ culture. Cell Microbiol. 2004, 6, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Schuller, S.; Heuschkel, R.; Torrente, F.; Kaper, J.B.; Phillips, A.D. Shiga toxin binding in normal and inflamed human intestinal mucosa. Microbes Infect. 2007, 9, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Hurley, B.P.; Thorpe, C.M.; Acheson, D.W. Shiga toxin translocation across intestinal epithelial cells is enhanced by neutrophil transmigration. Infect. Immun. 2001, 69, 6148–6155. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, A.; Arondel, J.; Sansonetti, P.J. Role of Shiga toxin in the pathogenesis of bacillary dysentery, studied by using a tox- mutant of Shigella dysenteriae 1. Infect. Immun. 1988, 56, 3099–3109. [Google Scholar] [PubMed]
- Brigotti, M.; Tazzari, P.L.; Ravanelli, E.; Carnicelli, D.; Rocchi, L.; Arfilli, V.; Scavia, G.; Minelli, F.; Ricci, F.; Pagliaro, P.; et al. Clinical relevance of Shiga toxin concentrations in the blood of patients with hemolytic uremic syndrome. Pediatr. Infect. Dis. J. 2011, 30, 486–490. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Quinones, B.; Loo, M.T.; Loos, S.; Scavia, G.; Brigotti, M.; Levtchenko, E.; Monnens, L. Serum Shiga toxin 2 values in patients during acute phase of diarrhoea-associated haemolytic uraemic syndrome. Acta Paediatr. 2015, 104, e564–e568. [Google Scholar] [CrossRef] [PubMed]
- Te Loo, D.M.; van Hinsbergh, V.W.; van den Heuvel, L.P.; Monnens, L.A. Detection of verocytotoxin bound to circulating polymorphonuclear leukocytes of patients with hemolytic uremic syndrome. J. Am. Soc. Nephrol. 2001, 12, 800–806. [Google Scholar] [PubMed]
- Ståhl, A.L.; Sartz, L.; Nelsson, A.; Békássy, Z.D.; Karpman, D. Shiga toxin and lipopolysaccharide induce platelet-leukocyte aggregates and tissue factor release, a thrombotic mechanism in hemolytic uremic syndrome. PLoS ONE 2009, 4, e6990. [Google Scholar] [CrossRef] [PubMed]
- Arvidsson, I.; Ståhl, A.L.; Hedström, M.M.; Kristoffersson, A.C.; Rylander, C.; Westman, J.S.; Storry, J.R.; Olsson, M.L.; Karpman, D. Shiga toxin-induced complement-mediated hemolysis and release of complement-coated red blood cell-derived microvesicles in hemolytic uremic syndrome. J. Immunol. 2015, 194, 2309–2318. [Google Scholar] [CrossRef] [PubMed]
- Bitzan, M.; Richardson, S.; Huang, C.; Boyd, B.; Petric, M.; Karmali, M.A. Evidence that verotoxins (Shiga-like toxins) from Escherichia coli bind to p blood group antigens of human erythrocytes in vitro. Infect. Immun. 1994, 62, 3337–3347. [Google Scholar] [PubMed]
- Ståhl, A.L.; Arvidsson, I.; Johansson, K.E.; Chromek, M.; Rebetz, J.; Loos, S.; Kristoffersson, A.C.; Békássy, Z.D.; Mörgelin, M.; Karpman, D. A novel mechanism of bacterial toxin transfer within host blood cell-derived microvesicles. PLoS Pathog. 2015, 11, e1004619. [Google Scholar] [CrossRef] [PubMed]
- Te Loo, D.M.; Monnens, L.A.; van Der Velden, T.J.; Vermeer, M.A.; Preyers, F.; Demacker, P.N.; van Den Heuvel, L.P.; van Hinsbergh, V.W. Binding and transfer of verocytotoxin by polymorphonuclear leukocytes in hemolytic uremic syndrome. Blood 2000, 95, 3396–3402. [Google Scholar] [PubMed]
- Ratajczak, J.; Wysoczynski, M.; Hayek, F.; Janowska-Wieczorek, A.; Ratajczak, M.Z. Membrane-derived microvesicles: Important and underappreciated mediators of cell-to-cell communication. Leukemia 2006, 20, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Flaumenhaft, R.; Dilks, J.R.; Richardson, J.; Alden, E.; Patel-Hett, S.R.; Battinelli, E.; Klement, G.L.; Sola-Visner, M.; Italiano, J.E., Jr. Megakaryocyte-derived microparticles: Direct visualization and distinction from platelet-derived microparticles. Blood 2009, 113, 1112–1121. [Google Scholar] [CrossRef] [PubMed]
- Angelillo-Scherrer, A. Leukocyte-derived microparticles in vascular homeostasis. Circ. Res. 2012, 110, 356–369. [Google Scholar] [CrossRef] [PubMed]
- Rubin, O.; Canellini, G.; Delobel, J.; Lion, N.; Tissot, J.D. Red blood cell microparticles: Clinical relevance. Transfus. Med. Hemother. 2012, 39, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Chironi, G.N.; Boulanger, C.M.; Simon, A.; Dignat-George, F.; Freyssinet, J.M.; Tedgui, A. Endothelial microparticles in diseases. Cell Tissue Res. 2009, 335, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Turco, A.E.; Lam, W.; Rule, A.D.; Denic, A.; Lieske, J.C.; Miller, V.M.; Larson, J.J.; Kremers, W.K.; Jayachandran, M. Specific renal parenchymal-derived urinary extracellular vesicles identify age-associated structural changes in living donor kidneys. J. Extracell. Vesicles 2016, 5, 29642. [Google Scholar] [CrossRef] [PubMed]
- Camussi, G.; Deregibus, M.C.; Bruno, S.; Cantaluppi, V.; Biancone, L. Exosomes/microvesicles as a mechanism of cell-to-cell communication. Kidney Int. 2010, 78, 838–848. [Google Scholar] [CrossRef] [PubMed]
- Abid Hussein, M.N.; Boing, A.N.; Sturk, A.; Hau, C.M.; Nieuwland, R. Inhibition of microparticle release triggers endothelial cell apoptosis and detachment. Thromb. Haemost. 2007, 98, 1096–1107. [Google Scholar] [CrossRef] [PubMed]
- Pap, E. The role of microvesicles in malignancies. Adv. Exp. Med. Biol. 2011, 714, 183–199. [Google Scholar] [PubMed]
- Karpman, D.; Ståhl, A.L.; Arvidsson, I. Extracellular vesicles in renal disease. Nat. Rev. Nephrol. 2017, 13, 545–562. [Google Scholar] [CrossRef] [PubMed]
- Lawson, C.; Vicencio, J.M.; Yellon, D.M.; Davidson, S.M. Microvesicles and exosomes: New players in metabolic and cardiovascular disease. J. Endocrinol. 2016, 228, R57–R71. [Google Scholar] [CrossRef] [PubMed]
- Distler, J.H.; Huber, L.C.; Gay, S.; Distler, O.; Pisetsky, D.S. Microparticles as mediators of cellular cross-talk in inflammatory disease. Autoimmunity 2006, 39, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Waldor, M.K.; Friedman, D.I. Phage regulatory circuits and virulence gene expression. Curr. Opin. Microbiol. 2005, 8, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Stein, P.E.; Boodhoo, A.; Tyrrell, G.J.; Brunton, J.L.; Read, R.J. Crystal structure of the cell-binding b oligomer of verotoxin-1 from E. coli. Nature 1992, 355, 748–750. [Google Scholar] [CrossRef] [PubMed]
- Lingwood, C.A.; Law, H.; Richardson, S.; Petric, M.; Brunton, J.L.; De Grandis, S.; Karmali, M. Glycolipid binding of purified and recombinant Escherichia coli produced verotoxin in vitro. J. Biol. Chem. 1987, 262, 8834–8839. [Google Scholar] [PubMed]
- Lindberg, A.A.; Brown, J.E.; Strömberg, N.; Westling-Ryd, M.; Schultz, J.E.; Karlsson, K.A. Identification of the carbohydrate receptor for Shiga toxin produced by Shigella dysenteriae type 1. J. Biol. Chem. 1987, 262, 1779–1785. [Google Scholar] [PubMed]
- Nakajima, H.; Kiyokawa, N.; Katagiri, Y.U.; Taguchi, T.; Suzuki, T.; Sekino, T.; Mimori, K.; Ebata, T.; Saito, M.; Nakao, H.; et al. Kinetic analysis of binding between Shiga toxin and receptor glycolipid gb3cer by surface plasmon resonance. J. Biol. Chem. 2001, 276, 42915–42922. [Google Scholar] [CrossRef] [PubMed]
- Kovbasnjuk, O.; Edidin, M.; Donowitz, M. Role of lipid rafts in Shiga toxin 1 interaction with the apical surface of caco-2 cells. J. Cell Sci. 2001, 114, 4025–4031. [Google Scholar] [PubMed]
- Romer, W.; Berland, L.; Chambon, V.; Gaus, K.; Windschiegl, B.; Tenza, D.; Aly, M.R.; Fraisier, V.; Florent, J.C.; Perrais, D.; et al. Shiga toxin induces tubular membrane invaginations for its uptake into cells. Nature 2007, 450, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Torgersen, M.L.; Lauvrak, S.U.; Sandvig, K. The a-subunit of surface-bound Shiga toxin stimulates clathrin-dependent uptake of the toxin. FEBS J. 2005, 272, 4103–4113. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K.; Skotland, T.; van Deurs, B.; Klokk, T.I. Retrograde transport of protein toxins through the golgi apparatus. Histochem. Cell Biol. 2013, 140, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Romer, W.; Pontani, L.L.; Sorre, B.; Rentero, C.; Berland, L.; Chambon, V.; Lamaze, C.; Bassereau, P.; Sykes, C.; Gaus, K.; et al. Actin dynamics drive membrane reorganization and scission in clathrin-independent endocytosis. Cell 2010, 140, 540–553. [Google Scholar] [CrossRef] [PubMed]
- Johannes, L.; Parton, R.G.; Bassereau, P.; Mayor, S. Building endocytic pits without clathrin. Nat. Rev. Mol. Cell Biol. 2015, 16, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Malyukova, I.; Murray, K.F.; Zhu, C.; Boedeker, E.; Kane, A.; Patterson, K.; Peterson, J.R.; Donowitz, M.; Kovbasnjuk, O. Macropinocytosis in Shiga toxin 1 uptake by human intestinal epithelial cells and transcellular transcytosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G78–G92. [Google Scholar] [CrossRef] [PubMed]
- Schuller, S. Shiga toxin interaction with human intestinal epithelium. Toxins (Basel) 2011, 3, 626–639. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K.; Garred, O.; Prydz, K.; Kozlov, J.V.; Hansen, S.H.; van Deurs, B. Retrograde transport of endocytosed Shiga toxin to the endoplasmic reticulum. Nature 1992, 358, 510–512. [Google Scholar] [CrossRef] [PubMed]
- Garred, O.; van Deurs, B.; Sandvig, K. Furin-induced cleavage and activation of Shiga toxin. J Biol. Chem. 1995, 270, 10817–10821. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K. Shiga toxins. Toxicon 2001, 39, 1629–1635. [Google Scholar] [CrossRef]
- Endo, Y.; Tsurugi, K.; Yutsudo, T.; Takeda, Y.; Ogasawara, T.; Igarashi, K. Site of action of a vero toxin (vt2) from Escherichia coli O157:H7 and of Shiga toxin on eukaryotic ribosomes. RNA n-glycosidase activity of the toxins. Eur. J. Biochem. 1988, 171, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Karpman, D.; Håkansson, A.; Perez, M.T.; Isaksson, C.; Carlemalm, E.; Caprioli, A.; Svanborg, C. Apoptosis of renal cortical cells in the hemolytic-uremic syndrome: In vivo and in vitro studies. Infect. Immun. 1998, 66, 636–644. [Google Scholar] [PubMed]
- Burlaka, I.; Liu, X.L.; Rebetz, J.; Arvidsson, I.; Yang, L.; Brismar, H.; Karpman, D.; Aperia, A. Ouabain protects against Shiga toxin-triggered apoptosis by reversing the imbalance between bax and bcl-xl. J. Am. Soc. Nephrol. 2013, 24, 1413–1423. [Google Scholar] [CrossRef] [PubMed]
- Tesh, V.L. Activation of cell stress response pathways by Shiga toxins. Cell. Microbiol. 2012, 14, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Koo, S.; Jeong, D.G.; Tesh, V.L. Shiga toxins as multi-functional proteins: Induction of host cellular stress responses, role in pathogenesis and therapeutic applications. Toxins (Basel) 2016, 8, 77. [Google Scholar] [CrossRef] [PubMed]
- Békássy, Z.D.; Calderon Toledo, C.; Leoj, G.; Kristoffersson, A.; Leopold, S.R.; Perez, M.T.; Karpman, D. Intestinal damage in enterohemorrhagic Escherichia coli infection. Pediatr. Nephrol. 2011, 26, 2059–2071. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, C.M.; Hurley, B.P.; Lincicome, L.L.; Jacewicz, M.S.; Keusch, G.T.; Acheson, D.W. Shiga toxins stimulate secretion of interleukin-8 from intestinal epithelial cells. Infect. Immun. 1999, 67, 5985–5993. [Google Scholar] [PubMed]
- Thorpe, C.M.; Smith, W.E.; Hurley, B.P.; Acheson, D.W. Shiga toxins induce, superinduce, and stabilize a variety of c-x-c chemokine mRNAs in intestinal epithelial cells, resulting in increased chemokine expression. Infect. Immun. 2001, 69, 6140–6147. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, C.; Natori, Y.; Zeng, X.T.; Ohmura, M.; Yamasaki, S.; Takeda, Y.; Natori, Y. Induction of cytokines in a human colon epithelial cell line by Shiga toxin 1 (stx1) and stx2 but not by non-toxic mutant stx1 which lacks n-glycosidase activity. FEBS Lett. 1999, 442, 231–234. [Google Scholar] [CrossRef]
- Tesh, V.L.; Ramegowda, B.; Samuel, J.E. Purified Shiga-like toxins induce expression of proinflammatory cytokines from murine peritoneal macrophages. Infect. Immun. 1994, 62, 5085–5094. [Google Scholar] [PubMed]
- Karpman, D.; Connell, H.; Svensson, M.; Scheutz, F.; Alm, P.; Svanborg, C. The role of lipopolysaccharide and Shiga-like toxin in a mouse model of Escherichia coli O157:H7 infection. J. Infect. Dis. 1997, 175, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Calderon Toledo, C.; Rogers, T.J.; Svensson, M.; Tati, R.; Fischer, H.; Svanborg, C.; Karpman, D. Shiga toxin-mediated disease in myd88-deficient mice infected with Escherichia coli O157:H7. Am. J. Pathol. 2008, 173, 1428–1439. [Google Scholar] [CrossRef] [PubMed]
- Robson, W.L.; Fick, G.H.; Wilson, P.C. Prognostic factors in typical postdiarrhea hemolytic-uremic syndrome. Child Nephrol. Urol. 1988, 9, 203–207. [Google Scholar] [PubMed]
- Tazzari, P.L.; Ricci, F.; Carnicelli, D.; Caprioli, A.; Tozzi, A.E.; Rizzoni, G.; Conte, R.; Brigotti, M. Flow cytometry detection of Shiga toxins in the blood from children with hemolytic uremic syndrome. Cytometry B. Clin. Cytom. 2004, 61, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Van Setten, P.A.; Monnens, L.A.; Verstraten, R.G.; van den Heuvel, L.P.; van Hinsbergh, V.W. Effects of verocytotoxin-1 on nonadherent human monocytes: Binding characteristics, protein synthesis, and induction of cytokine release. Blood 1996, 88, 174–183. [Google Scholar] [PubMed]
- Guessous, F.; Marcinkiewicz, M.; Polanowska-Grabowska, R.; Keepers, T.R.; Obrig, T.; Gear, A.R. Shiga toxin 2 and lipopolysaccharide cause monocytic thp-1 cells to release factors which activate platelet function. Thromb. Haemost. 2005, 94, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Murata, K.; Higuchi, T.; Takada, K.; Oida, K.; Horie, S.; Ishii, H. Verotoxin-1 stimulation of macrophage-like THP-1 cells up-regulates tissue factor expression through activation of c-yes tyrosine kinase: Possible signal transduction in tissue factor up-regulation. Biochim. Biophys. Acta 2006, 1762, 835–843. [Google Scholar] [CrossRef] [PubMed]
- Zoja, C.; Buelli, S.; Morigi, M. Shiga toxin-associated hemolytic uremic syndrome: Pathophysiology of endothelial dysfunction. Pediatr. Nephrol. 2010, 25, 2231–2240. [Google Scholar] [CrossRef] [PubMed]
- Karpman, D.; Manea, M.; Vaziri-Sani, F.; Ståhl, A.L.; Kristoffersson, A.C. Platelet activation in hemolytic uremic syndrome. Semin. Thromb. Hemost. 2006, 32, 128–145. [Google Scholar] [CrossRef] [PubMed]
- Fong, J.S.; Kaplan, B.S. Impairment of platelet aggregation in hemolytic uremic syndrome: Evidence for platelet “exhaustion”. Blood 1982, 60, 564–570. [Google Scholar] [PubMed]
- Appiani, A.C.; Edefonti, A.; Bettinelli, A.; Cossu, M.M.; Paracchini, M.L.; Rossi, E. The relationship between plasma levels of the factor viii complex and platelet release products (beta-thromboglobulin and platelet factor 4) in children with the hemolytic-uremic syndrome. Clin. Nephrol. 1982, 17, 195–199. [Google Scholar] [PubMed]
- Katayama, M.; Handa, M.; Araki, Y.; Ambo, H.; Kawai, Y.; Watanabe, K.; Ikeda, Y. Soluble p-selectin is present in normal circulation and its plasma level is elevated in patients with thrombotic thrombocytopenic purpura and haemolytic uraemic syndrome. Br. J. Haematol. 1993, 84, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Cooling, L.L.; Walker, K.E.; Gille, T.; Koerner, T.A. Shiga toxin binds human platelets via globotriaosylceramide (pk antigen) and a novel platelet glycosphingolipid. Infect. Immun. 1998, 66, 4355–4366. [Google Scholar] [PubMed]
- Ståhl, A.L.; Svensson, M.; Mörgelin, M.; Svanborg, C.; Tarr, P.I.; Mooney, J.C.; Watkins, S.L.; Johnson, R.; Karpman, D. Lipopolysaccharide from enterohemorrhagic Escherichia coli binds to platelets through TLR4 and CD62 and is detected on circulating platelets in patients with hemolytic uremic syndrome. Blood 2006, 108, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.A.; Polanowska-Grabowska, R.K.; Fujii, J.; Obrig, T.; Gear, A.R. Shiga toxin binds to activated platelets. J. Thromb. Haemost. 2004, 2, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Karpman, D.; Papadopoulou, D.; Nilsson, K.; Sjögren, A.C.; Mikaelsson, C.; Lethagen, S. Platelet activation by Shiga toxin and circulatory factors as a pathogenetic mechanism in the hemolytic uremic syndrome. Blood 2001, 97, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Ståhl, A.L.; Sartz, L.; Karpman, D. Complement activation on platelet-leukocyte complexes and microparticles in enterohemorrhagic Escherichia coli-induced hemolytic uremic syndrome. Blood 2011, 117, 5503–5513. [Google Scholar] [CrossRef] [PubMed]
- Polley, M.J.; Nachman, R.L. Human complement in thrombin-mediated platelet function: Uptake of the C5b-9 complex. J. Exp. Med. 1979, 150, 633–645. [Google Scholar] [CrossRef] [PubMed]
- Chandler, W.L.; Jelacic, S.; Boster, D.R.; Ciol, M.A.; Williams, G.D.; Watkins, S.L.; Igarashi, T.; Tarr, P.I. Prothrombotic coagulation abnormalities preceding the hemolytic-uremic syndrome. N. Engl. J. Med. 2002, 346, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Polley, M.J.; Nachman, R. The human complement system in thrombin-mediated platelet function. J. Exp. Med. 1978, 147, 1713–1726. [Google Scholar] [CrossRef] [PubMed]
- Huber-Lang, M.; Sarma, J.V.; Zetoune, F.S.; Rittirsch, D.; Neff, T.A.; McGuire, S.R.; Lambris, J.D.; Warner, R.L.; Flierl, M.A.; Hoesel, L.M.; et al. Generation of C5a in the absence of C3: A new complement activation pathway. Nat. Med. 2006, 12, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Karpman, D.; Tati, R. Complement contributes to the pathogenesis of Shiga toxin-associated hemolytic uremic syndrome. Kidney Int. 2016, 90, 726–729. [Google Scholar] [CrossRef] [PubMed]
- Risitano, A.M.; Marotta, S. Therapeutic complement inhibition in complement-mediated hemolytic anemias: Past, present and future. Semin. Immunol. 2016, 28, 223–240. [Google Scholar] [CrossRef] [PubMed]
- Spitalnik, P.F.; Spitalnik, S.L. The P blood group system: Biochemical, serological, and clinical aspects. Transfus. Med. Rev. 1995, 9, 110–122. [Google Scholar] [CrossRef]
- Karpman, D. Management of Shiga toxin-associated Escherichia coli-induced haemolytic uraemic syndrome: Randomized clinical trials are needed. Nephrol. Dial. Transplant. 2012, 27, 3669–3674. [Google Scholar] [CrossRef] [PubMed]
- Geelen, J.M.; van der Velden, T.J.; van den Heuvel, L.P.; Monnens, L.A. Interactions of Shiga-like toxin with human peripheral blood monocytes. Pediatr. Nephrol. 2007, 22, 1181–1187. [Google Scholar] [CrossRef] [PubMed]
- Psotka, M.A.; Obata, F.; Kolling, G.L.; Gross, L.K.; Saleem, M.A.; Satchell, S.C.; Mathieson, P.W.; Obrig, T.G. Shiga toxin 2 targets the murine renal collecting duct epithelium. Infect. Immun. 2009, 77, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Van de Kar, N.C.; Monnens, L.A.; Karmali, M.A.; van Hinsbergh, V.W. Tumor necrosis factor and interleukin-1 induce expression of the verocytotoxin receptor globotriaosylceramide on human endothelial cells: Implications for the pathogenesis of the hemolytic uremic syndrome. Blood 1992, 80, 2755–2764. [Google Scholar] [PubMed]
- Stone, M.K.; Kolling, G.L.; Lindner, M.H.; Obrig, T.G. P38 mitogen-activated protein kinase mediates lipopolysaccharide and tumor necrosis factor alpha induction of Shiga toxin 2 sensitivity in human umbilical vein endothelial cells. Infect. Immun. 2008, 76, 1115–1121. [Google Scholar] [CrossRef] [PubMed]
- Louise, C.B.; Obrig, T.G. Specific interaction of Escherichia coli O157:H7-derived Shiga-like toxin ii with human renal endothelial cells. J. Infect. Dis. 1995, 172, 1397–1401. [Google Scholar] [CrossRef] [PubMed]
- Guessous, F.; Marcinkiewicz, M.; Polanowska-Grabowska, R.; Kongkhum, S.; Heatherly, D.; Obrig, T.; Gear, A.R. Shiga toxin 2 and lipopolysaccharide induce human microvascular endothelial cells to release chemokines and factors that stimulate platelet function. Infect. Immun. 2005, 73, 8306–8316. [Google Scholar] [CrossRef] [PubMed]
- Nevard, C.H.; Jurd, K.M.; Lane, D.A.; Philippou, H.; Haycock, G.B.; Hunt, B.J. Activation of coagulation and fibrinolysis in childhood diarrhoea-associated haemolytic uraemic syndrome. Thromb. Haemost. 1997, 78, 1450–1455. [Google Scholar] [PubMed]
- Van Geet, C.; Proesmans, W.; Arnout, J.; Vermylen, J.; Declerck, P.J. Activation of both coagulation and fibrinolysis in childhood hemolytic uremic syndrome. Kidney Int. 1998, 54, 1324–1330. [Google Scholar] [CrossRef] [PubMed]
- Van de Kar, N.C.; van Hinsbergh, V.W.; Brommer, E.J.; Monnens, L.A. The fibrinolytic system in the hemolytic uremic syndrome: In vivo and in vitro studies. Pediatr. Res. 1994, 36, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Sappino, A.P.; Huarte, J.; Vassalli, J.D.; Belin, D. Sites of synthesis of urokinase and tissue-type plasminogen activators in the murine kidney. J. Clin. Investig. 1991, 87, 962–970. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.; Hertel, B.; Emden, S.H.; Beneke, J.; Menne, J.; Haller, H.; von Vietinghoff, S. Microparticle generation and leucocyte death in Shiga toxin-mediated hus. Nephrol. Dial. Transplant. 2012, 27, 2768–2775. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Raposo, G.; Thery, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef] [PubMed]
- Gyorgy, B.; Szabo, T.G.; Pasztoi, M.; Pal, Z.; Misjak, P.; Aradi, B.; Laszlo, V.; Pallinger, E.; Pap, E.; Kittel, A.; et al. Membrane vesicles, current state-of-the-art: Emerging role of extracellular vesicles. Cell. Mol. Life. Sci. 2011, 68, 2667–2688. [Google Scholar] [CrossRef] [PubMed]
- Erdbrügger, U.; Lannigan, J. Analytical challenges of extracellular vesicle detection: A comparison of different techniques. Cytometry A. 2016, 89, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Witwer, K.W.; Buzas, E.I.; Bemis, L.T.; Bora, A.; Lasser, C.; Lotvall, J.; Nolte-’t Hoen, E.N.; Piper, M.G.; Sivaraman, S.; Skog, J.; et al. Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J. Extracell. Vesicles 2013, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, D.S.; Kim, D.K.; Kim, Y.K.; Gho, Y.S. Proteomics of extracellular vesicles: Exosomes and ectosomes. Mass Spectrom. Rev. 2015, 34, 474–490. [Google Scholar] [CrossRef] [PubMed]
- Van der Pol, E.; Coumans, F.A.; Grootemaat, A.E.; Gardiner, C.; Sargent, I.L.; Harrison, P.; Sturk, A.; van Leeuwen, T.G.; Nieuwland, R. Particle size distribution of exosomes and microvesicles determined by transmission electron microscopy, flow cytometry, nanoparticle tracking analysis, and resistive pulse sensing. J. Thromb. Haemost. 2014, 12, 1182–1192. [Google Scholar] [CrossRef] [PubMed]
- Thery, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [PubMed]
- Kowal, J.; Tkach, M.; Thery, C. Biogenesis and secretion of exosomes. Curr. Opin. Cell Biol. 2014, 29, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Moita, C.; van Niel, G.; Kowal, J.; Vigneron, J.; Benaroch, P.; Manel, N.; Moita, L.F.; Thery, C.; Raposo, G. Analysis of escrt functions in exosome biogenesis, composition and secretion highlights the heterogeneity of extracellular vesicles. J. Cell Sci. 2013, 126, 5553–5565. [Google Scholar] [CrossRef] [PubMed]
- Hanson, P.I.; Roth, R.; Lin, Y.; Heuser, J.E. Plasma membrane deformation by circular arrays of ESCRT-III protein filaments. J. Cell Biol. 2008, 180, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Van Niel, G.; Charrin, S.; Simoes, S.; Romao, M.; Rochin, L.; Saftig, P.; Marks, M.S.; Rubinstein, E.; Raposo, G. The tetraspanin CD63 regulates ESCRT-independent and -dependent endosomal sorting during melanogenesis. Dev. Cell 2011, 21, 708–721. [Google Scholar] [CrossRef] [PubMed]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brugger, B.; Simons, M. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef] [PubMed]
- Skotland, T.; Sandvig, K.; Llorente, A. Lipids in exosomes: Current knowledge and the way forward. Prog. Lipid Res. 2017, 66, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Escola, J.M.; Kleijmeer, M.J.; Stoorvogel, W.; Griffith, J.M.; Yoshie, O.; Geuze, H.J. Selective enrichment of tetraspan proteins on the internal vesicles of multivesicular endosomes and on exosomes secreted by human b-lymphocytes. J. Biol. Chem. 1998, 273, 20121–20127. [Google Scholar] [CrossRef] [PubMed]
- Thery, C.; Boussac, M.; Veron, P.; Ricciardi-Castagnoli, P.; Raposo, G.; Garin, J.; Amigorena, S. Proteomic analysis of dendritic cell-derived exosomes: A secreted subcellular compartment distinct from apoptotic vesicles. J. Immunol. 2001, 166, 7309–7318. [Google Scholar] [CrossRef] [PubMed]
- Lynch, S.; Santos, S.G.; Campbell, E.C.; Nimmo, A.M.; Botting, C.; Prescott, A.; Antoniou, A.N.; Powis, S.J. Novel MHC class I structures on exosomes. J. Immunol. 2009, 183, 1884–1891. [Google Scholar] [CrossRef] [PubMed]
- Haraszti, R.A.; Didiot, M.C.; Sapp, E.; Leszyk, J.; Shaffer, S.A.; Rockwell, H.E.; Gao, F.; Narain, N.R.; DiFiglia, M.; Kiebish, M.A.; et al. High-resolution proteomic and lipidomic analysis of exosomes and microvesicles from different cell sources. J. Extracell. Vesicles 2016, 5, 32570. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Xu, Q. Functions and application of exosomes. Acta Pol. Pharm. 2014, 71, 537–543. [Google Scholar] [PubMed]
- Little, K.M.; Smalley, D.M.; Harthun, N.L.; Ley, K. The plasma microparticle proteome. Semin. Thromb. Hemost. 2010, 36, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Barry, O.P.; Pratico, D.; Lawson, J.A.; FitzGerald, G.A. Transcellular activation of platelets and endothelial cells by bioactive lipids in platelet microparticles. J. Clin. Investig. 1997, 99, 2118–2127. [Google Scholar] [CrossRef] [PubMed]
- Sapet, C.; Simoncini, S.; Loriod, B.; Puthier, D.; Sampol, J.; Nguyen, C.; Dignat-George, F.; Anfosso, F. Thrombin-induced endothelial microparticle generation: Identification of a novel pathway involving rock-II activation by caspase-2. Blood 2006, 108, 1868–1876. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, J.; Miekus, K.; Kucia, M.; Zhang, J.; Reca, R.; Dvorak, P.; Ratajczak, M.Z. Embryonic stem cell-derived microvesicles reprogram hematopoietic progenitors: Evidence for horizontal transfer of mRNA and protein delivery. Leukemia 2006, 20, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Cocucci, E.; Racchetti, G.; Meldolesi, J. Shedding microvesicles: Artefacts no more. Trends Cell Biol. 2009, 19, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Al-Nedawi, K.; Meehan, B.; Micallef, J.; Lhotak, V.; May, L.; Guha, A.; Rak, J. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat. Cell Biol. 2008, 10, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Tricarico, C.; Clancy, J.; D’Souza-Schorey, C. Biology and biogenesis of shed microvesicles. Small GTPases 2016, 8, 220–232. [Google Scholar] [CrossRef] [PubMed]
- Pomatto, M.A.C.; Gai, C.; Bussolati, B.; Camussi, G. Extracellular vesicles in renal pathophysiology. Front. Mol. Biosci. 2017, 4, 37. [Google Scholar] [CrossRef] [PubMed]
- Iida, K.; Whitlow, M.B.; Nussenzweig, V. Membrane vesiculation protects erythrocytes from destruction by complement. J. Immunol. 1991, 147, 2638–2642. [Google Scholar] [PubMed]
- Willekens, F.L.; Werre, J.M.; Groenen-Dopp, Y.A.; Roerdinkholder-Stoelwinder, B.; de Pauw, B.; Bosman, G.J. Erythrocyte vesiculation: A self-protective mechanism? Br. J. Haematol. 2008, 141, 549–556. [Google Scholar] [CrossRef] [PubMed]
- McMahon, H.T.; Boucrot, E. Membrane curvature at a glance. J. Cell Sci. 2015, 128, 1065–1070. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, J.J.; Jy, W.; Mauro, L.M.; Soderland, C.; Horstman, L.L.; Ahn, Y.S. Endothelial cells release phenotypically and quantitatively distinct microparticles in activation and apoptosis. Thromb. Res. 2003, 109, 175–180. [Google Scholar] [CrossRef]
- Nomura, S.; Tandon, N.N.; Nakamura, T.; Cone, J.; Fukuhara, S.; Kambayashi, J. High-shear-stress-induced activation of platelets and microparticles enhances expression of cell adhesion molecules in THP-1 and endothelial cells. Atherosclerosis 2001, 158, 277–287. [Google Scholar] [CrossRef]
- Bernimoulin, M.; Waters, E.K.; Foy, M.; Steele, B.M.; Sullivan, M.; Falet, H.; Walsh, M.T.; Barteneva, N.; Geng, J.G.; Hartwig, J.H.; et al. Differential stimulation of monocytic cells results in distinct populations of microparticles. J. Thromb. Haemost. 2009, 7, 1019–1028. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, L.J.; Gibbons, E.; Bailey, R.W.; Fairbourn, J.; Nguyen, T.; Smith, S.K.; Best, K.B.; Nelson, J.; Judd, A.M.; Bell, J.D. The influence of membrane physical properties on microvesicle release in human erythrocytes. PMC Biophys. 2009, 2, 7. [Google Scholar] [CrossRef] [PubMed]
- Muralidharan-Chari, V.; Clancy, J.; Plou, C.; Romao, M.; Chavrier, P.; Raposo, G.; D’Souza-Schorey, C. ARF6-regulated shedding of tumor cell-derived plasma membrane microvesicles. Curr. Biol. 2009, 19, 1875–1885. [Google Scholar] [CrossRef] [PubMed]
- Lai, R.C.; Tan, S.S.; Yeo, R.W.; Choo, A.B.; Reiner, A.T.; Su, Y.; Shen, Y.; Fu, Z.; Alexander, L.; Sze, S.K.; et al. MSC secretes at least 3 EV types each with a unique permutation of membrane lipid, protein and RNA. J. Extracell. Vesicles 2016, 5, 29828. [Google Scholar] [CrossRef] [PubMed]
- Biro, E.; Akkerman, J.W.; Hoek, F.J.; Gorter, G.; Pronk, L.M.; Sturk, A.; Nieuwland, R. The phospholipid composition and cholesterol content of platelet-derived microparticles: A comparison with platelet membrane fractions. J. Thromb. Haemost. 2005, 3, 2754–2763. [Google Scholar] [CrossRef] [PubMed]
- Sinauridze, E.I.; Kireev, D.A.; Popenko, N.Y.; Pichugin, A.V.; Panteleev, M.A.; Krymskaya, O.V.; Ataullakhanov, F.I. Platelet microparticle membranes have 50- to 100-fold higher specific procoagulant activity than activated platelets. Thromb. Haemost. 2007, 97, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Seigneuret, M.; Zachowski, A.; Hermann, A.; Devaux, P.F. Asymmetric lipid fluidity in human erythrocyte membrane: New spin-label evidence. Biochemistry 1984, 23, 4271–4275. [Google Scholar] [CrossRef] [PubMed]
- Manno, S.; Takakuwa, Y.; Mohandas, N. Identification of a functional role for lipid asymmetry in biological membranes: Phosphatidylserine-skeletal protein interactions modulate membrane stability. Proc. Natl. Acad. Sci. USA 2002, 99, 1943–1948. [Google Scholar] [CrossRef] [PubMed]
- Daleke, D.L. Regulation of transbilayer plasma membrane phospholipid asymmetry. J. Lipid Res. 2003, 44, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Kim, A.; David, T.; Palmer, D.; Jin, T.; Tien, J.; Huang, F.; Cheng, T.; Coughlin, S.R.; Jan, Y.N.; et al. Tmem16f forms a Ca2+-activated cation channel required for lipid scrambling in platelets during blood coagulation. Cell 2012, 151, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Morel, O.; Jesel, L.; Freyssinet, J.M.; Toti, F. Cellular mechanisms underlying the formation of circulating microparticles. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Farsad, K.; De Camilli, P. Mechanisms of membrane deformation. Curr. Opin. Cell Biol. 2003, 15, 372–381. [Google Scholar] [CrossRef]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podbilewicz, B. Virus and cell fusion mechanisms. Annu. Rev. Cell Dev. Biol. 2014, 30, 111–139. [Google Scholar] [CrossRef] [PubMed]
- Prada, I.; Meldolesi, J. Binding and fusion of extracellular vesicles to the plasma membrane of their cell targets. Int. J. Mol. Sci. 2016, 17, 1296. [Google Scholar] [CrossRef] [PubMed]
- Maas, S.L.; Breakefield, X.O.; Weaver, A.M. Extracellular vesicles: Unique intercellular delivery vehicles. Trends Cell Biol. 2017, 27, 172–188. [Google Scholar] [CrossRef] [PubMed]
- French, K.C.; Antonyak, M.A.; Cerione, R.A. Extracellular vesicle docking at the cellular port: Extracellular vesicle binding and uptake. Semin. Cell Dev. Biol. 2017, 67, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Pluskota, E.; Woody, N.M.; Szpak, D.; Ballantyne, C.M.; Soloviev, D.A.; Simon, D.I.; Plow, E.F. Expression, activation, and function of integrin alphaMbeta2 (Mac-1) on neutrophil-derived microparticles. Blood 2008, 112, 2327–2335. [Google Scholar] [CrossRef] [PubMed]
- Falati, S.; Liu, Q.; Gross, P.; Merrill-Skoloff, G.; Chou, J.; Vandendries, E.; Celi, A.; Croce, K.; Furie, B.C.; Furie, B. Accumulation of tissue factor into developing thrombi in vivo is dependent upon microparticle p-selectin glycoprotein ligand 1 and platelet p-selectin. J. Exp. Med. 2003, 197, 1585–1598. [Google Scholar] [CrossRef] [PubMed]
- Piccin, A.; Murphy, W.G.; Smith, O.P. Circulating microparticles: Pathophysiology and clinical implications. Blood Rev. 2007, 21, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Rossaint, J.; Kuhne, K.; Skupski, J.; Van Aken, H.; Looney, M.R.; Hidalgo, A.; Zarbock, A. Directed transport of neutrophil-derived extracellular vesicles enables platelet-mediated innate immune response. Nat. Commun. 2016, 7, 13464. [Google Scholar] [CrossRef] [PubMed]
- Losche, W.; Scholz, T.; Temmler, U.; Oberle, V.; Claus, R.A. Platelet-derived microvesicles transfer tissue factor to monocytes but not to neutrophils. Platelets 2004, 15, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Deregibus, M.C.; Cantaluppi, V.; Calogero, R.; Lo Iacono, M.; Tetta, C.; Biancone, L.; Bruno, S.; Bussolati, B.; Camussi, G. Endothelial progenitor cell derived microvesicles activate an angiogenic program in endothelial cells by a horizontal transfer of mrna. Blood 2007, 110, 2440–2448. [Google Scholar] [CrossRef] [PubMed]
- Mause, S.F.; Weber, C. Microparticles: Protagonists of a novel communication network for intercellular information exchange. Circ. Res. 2010, 107, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Collino, F.; Bruno, S.; Incarnato, D.; Dettori, D.; Neri, F.; Provero, P.; Pomatto, M.; Oliviero, S.; Tetta, C.; Quesenberry, P.J.; et al. AKI recovery induced by mesenchymal stromal cell-derived extracellular vesicles carrying micrornas. J. Am. Soc. Nephrol. 2015, 26, 2349–2360. [Google Scholar] [CrossRef] [PubMed]
- Mack, M.; Kleinschmidt, A.; Bruhl, H.; Klier, C.; Nelson, P.J.; Cihak, J.; Plachy, J.; Stangassinger, M.; Erfle, V.; Schlondorff, D. Transfer of the chemokine receptor CCR5 between cells by membrane-derived microparticles: A mechanism for cellular human immunodeficiency virus 1 infection. Nat. Med. 2000, 6, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Rozmyslowicz, T.; Majka, M.; Kijowski, J.; Murphy, S.L.; Conover, D.O.; Poncz, M.; Ratajczak, J.; Gaulton, G.N.; Ratajczak, M.Z. Platelet- and megakaryocyte-derived microparticles transfer CXCR4 receptor to CXCR4-null cells and make them susceptible to infection by X4-HIV. AIDS 2003, 17, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Al-Nedawi, K.; Meehan, B.; Kerbel, R.S.; Allison, A.C.; Rak, J. Endothelial expression of autocrine VEGF upon the uptake of tumor-derived microvesicles containing oncogenic EGFR. Proc. Natl. Acad. Sci. USA 2009, 106, 3794–3799. [Google Scholar] [CrossRef] [PubMed]
- Kahn, R.; Mossberg, M.; Ståhl, A.L.; Johansson, K.; Lopatko Lindman, I.; Heijl, C.; Segelmark, M.; Mörgelin, M.; Leeb-Lundberg, L.M.; Karpman, D. Microvesicle transfer of kinin B1-receptors is a novel inflammatory mechanism in vasculitis. Kidney Int. 2017, 91, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Baj-Krzyworzeka, M.; Majka, M.; Pratico, D.; Ratajczak, J.; Vilaire, G.; Kijowski, J.; Reca, R.; Janowska-Wieczorek, A.; Ratajczak, M.Z. Platelet-derived microparticles stimulate proliferation, survival, adhesion, and chemotaxis of hematopoietic cells. Exp. Hematol. 2002, 30, 450–459. [Google Scholar] [CrossRef]
- Salanova, B.; Choi, M.; Rolle, S.; Wellner, M.; Luft, F.C.; Kettritz, R. Beta2-integrins and acquired glycoprotein IIb/IIIa (gpIIb/IIIa) receptors cooperate in NF-kappaB activation of human neutrophils. J. Biol. Chem. 2007, 282, 27960–27969. [Google Scholar] [CrossRef] [PubMed]
- Taraboletti, G.; D’Ascenzo, S.; Borsotti, P.; Giavazzi, R.; Pavan, A.; Dolo, V. Shedding of the matrix metalloproteinases mmp-2, mmp-9, and mt1-mmp as membrane vesicle-associated components by endothelial cells. Am. J. Pathol. 2002, 160, 673–680. [Google Scholar] [CrossRef]
- Barry, O.P.; Pratico, D.; Savani, R.C.; FitzGerald, G.A. Modulation of monocyte-endothelial cell interactions by platelet microparticles. J. Clin. Investig. 1998, 102, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Mossberg, M.; Ståhl, A.L.; Kahn, R.; Kristoffersson, A.C.; Tati, R.; Heijl, C.; Segelmark, M.; Leeb-Lundberg, L.M.F.; Karpman, D. C1-inhibitor decreases the release of vasculitis-like chemotactic endothelial microvesicles. J. Am. Soc. Nephrol. 2017, 28, 2472–2481. [Google Scholar] [CrossRef] [PubMed]
- Mause, S.F.; von Hundelshausen, P.; Zernecke, A.; Koenen, R.R.; Weber, C. Platelet microparticles: A transcellular delivery system for rantes promoting monocyte recruitment on endothelium. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1512–1518. [Google Scholar] [CrossRef] [PubMed]
- Biancone, L.; Bruno, S.; Deregibus, M.C.; Tetta, C.; Camussi, G. Therapeutic potential of mesenchymal stem cell-derived microvesicles. Nephrol. Dial. Transplant. 2012, 27, 3037–3042. [Google Scholar] [CrossRef] [PubMed]
- Gasser, O.; Schifferli, J.A. Activated polymorphonuclear neutrophils disseminate anti-inflammatory microparticles by ectocytosis. Blood 2004, 104, 2543–2548. [Google Scholar] [CrossRef] [PubMed]
- Bevers, E.M.; Williamson, P.L. Getting to the outer leaflet: Physiology of phosphatidylserine exposure at the plasma membrane. Physiol. Rev. 2016, 96, 605–645. [Google Scholar] [CrossRef] [PubMed]
- Sims, P.J.; Wiedmer, T.; Esmon, C.T.; Weiss, H.J.; Shattil, S.J. Assembly of the platelet prothrombinase complex is linked to vesiculation of the platelet plasma membrane. Studies in scott syndrome: An isolated defect in platelet procoagulant activity. J. Biol. Chem. 1989, 264, 17049–17057. [Google Scholar] [PubMed]
- Zwaal, R.F.; Comfurius, P.; Bevers, E.M. Surface exposure of phosphatidylserine in pathological cells. Cell. Mol. Life Sci. 2005, 62, 971–988. [Google Scholar] [CrossRef] [PubMed]
- Ståhl, A.L.; Vaziri-Sani, F.; Heinen, S.; Kristoffersson, A.C.; Gydell, K.H.; Raafat, R.; Gutierrez, A.; Beringer, O.; Zipfel, P.F.; Karpman, D. Factor H dysfunction in patients with atypical hemolytic uremic syndrome contributes to complement deposition on platelets and their activation. Blood 2008, 111, 5307–5315. [Google Scholar] [CrossRef] [PubMed]
- Diamant, M.; Nieuwland, R.; Pablo, R.F.; Sturk, A.; Smit, J.W.; Radder, J.K. Elevated numbers of tissue-factor exposing microparticles correlate with components of the metabolic syndrome in uncomplicated type 2 diabetes mellitus. Circulation 2002, 106, 2442–2447. [Google Scholar] [CrossRef] [PubMed]
- Del Conde, I.; Shrimpton, C.N.; Thiagarajan, P.; Lopez, J.A. Tissue-factor-bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood 2005, 106, 1604–1611. [Google Scholar] [CrossRef] [PubMed]
- Rubin, O.; Delobel, J.; Prudent, M.; Lion, N.; Kohl, K.; Tucker, E.I.; Tissot, J.D.; Angelillo-Scherrer, A. Red blood cell-derived microparticles isolated from blood units initiate and propagate thrombin generation. Transfusion 2013, 53, 1744–1754. [Google Scholar] [CrossRef] [PubMed]
- Nieuwland, R.; Berckmans, R.J.; McGregor, S.; Boing, A.N.; Romijn, F.P.; Westendorp, R.G.; Hack, C.E.; Sturk, A. Cellular origin and procoagulant properties of microparticles in meningococcal sepsis. Blood 2000, 95, 930–935. [Google Scholar] [PubMed]
- Fujimi, S.; Ogura, H.; Tanaka, H.; Koh, T.; Hosotsubo, H.; Nakamori, Y.; Kuwagata, Y.; Shimazu, T.; Sugimoto, H. Activated polymorphonuclear leukocytes enhance production of leukocyte microparticles with increased adhesion molecules in patients with sepsis. J. Trauma 2002, 52, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Tokes-Fuzesi, M.; Woth, G.; Ernyey, B.; Vermes, I.; Muhl, D.; Bogar, L.; Kovacs, G.L. Microparticles and acute renal dysfunction in septic patients. J. Crit. Care 2013, 28, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Mostefai, H.A.; Meziani, F.; Mastronardi, M.L.; Agouni, A.; Heymes, C.; Sargentini, C.; Asfar, P.; Martinez, M.C.; Andriantsitohaina, R. Circulating microparticles from patients with septic shock exert protective role in vascular function. Am. J. Respir. Crit. Care Med. 2008, 178, 1148–1155. [Google Scholar] [CrossRef] [PubMed]
- Woei, A.J.F.J.; van der Starre, W.E.; Tesselaar, M.E.; Garcia Rodriguez, P.; van Nieuwkoop, C.; Bertina, R.M.; van Dissel, J.T.; Osanto, S. Procoagulant tissue factor activity on microparticles is associated with disease severity and bacteremia in febrile urinary tract infections. Thromb. Res. 2014, 133, 799–803. [Google Scholar] [CrossRef] [PubMed]
- Delabranche, X.; Boisrame-Helms, J.; Asfar, P.; Berger, A.; Mootien, Y.; Lavigne, T.; Grunebaum, L.; Lanza, F.; Gachet, C.; Freyssinet, J.M.; et al. Microparticles are new biomarkers of septic shock-induced disseminated intravascular coagulopathy. Intensive Care Med. 2013, 39, 1695–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soriano, A.O.; Jy, W.; Chirinos, J.A.; Valdivia, M.A.; Velasquez, H.S.; Jimenez, J.J.; Horstman, L.L.; Kett, D.H.; Schein, R.M.; Ahn, Y.S. Levels of endothelial and platelet microparticles and their interactions with leukocytes negatively correlate with organ dysfunction and predict mortality in severe sepsis. Crit. Care Med. 2005, 33, 2540–2546. [Google Scholar] [CrossRef] [PubMed]
- Mortaza, S.; Martinez, M.C.; Baron-Menguy, C.; Burban, M.; de la Bourdonnaye, M.; Fizanne, L.; Pierrot, M.; Cales, P.; Henrion, D.; Andriantsitohaina, R.; et al. Detrimental hemodynamic and inflammatory effects of microparticles originating from septic rats. Crit. Care Med. 2009, 37, 2045–2050. [Google Scholar] [CrossRef] [PubMed]
- Meziani, F.; Delabranche, X.; Asfar, P.; Toti, F. Bench-to-bedside review: Circulating microparticles--a new player in sepsis? Crit. Care 2010, 14, 236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camussi, G.; Cantaluppi, V.; Deregibus, M.C.; Gatti, E.; Tetta, C. Role of microvesicles in acute kidney injury. Contrib. Nephrol. 2011, 174, 191–199. [Google Scholar] [PubMed]
- Timar, C.I.; Lorincz, A.M.; Csepanyi-Komi, R.; Valyi-Nagy, A.; Nagy, G.; Buzas, E.I.; Ivanyi, Z.; Kittel, A.; Powell, D.W.; McLeish, K.R.; et al. Antibacterial effect of microvesicles released from human neutrophilic granulocytes. Blood 2013, 121, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Oehmcke, S.; Westman, J.; Malmström, J.; Mörgelin, M.; Olin, A.I.; Kreikemeyer, B.; Herwald, H. A novel role for pro-coagulant microvesicles in the early host defense against streptococcus pyogenes. PLoS Pathog. 2013, 9, e1003529. [Google Scholar] [CrossRef] [PubMed]
- Ramachandra, L.; Qu, Y.; Wang, Y.; Lewis, C.J.; Cobb, B.A.; Takatsu, K.; Boom, W.H.; Dubyak, G.R.; Harding, C.V. Mycobacterium tuberculosis synergizes with ATP to induce release of microvesicles and exosomes containing major histocompatibility complex class ii molecules capable of antigen presentation. Infect. Immun. 2010, 78, 5116–5125. [Google Scholar] [CrossRef] [PubMed]
- Bieniasz, P.D. The cell biology of HIV-1 virion genesis. Cell Host Microbe 2009, 5, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Hurley, J.H. ESCRTs are everywhere. EMBO J. 2015, 34, 2398–2407. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo-Useros, N.; Naranjo-Gomez, M.; Archer, J.; Hatch, S.C.; Erkizia, I.; Blanco, J.; Borras, F.E.; Puertas, M.C.; Connor, J.H.; Fernandez-Figueras, M.T.; et al. Capture and transfer of HIV-1 particles by mature dendritic cells converges with the exosome-dissemination pathway. Blood 2009, 113, 2732–2741. [Google Scholar] [CrossRef] [PubMed]
- Sims, B.; Farrow, A.L.; Williams, S.D.; Bansal, A.; Krendelchtchikov, A.; Gu, L.; Matthews, Q.L. Role of tim-4 in exosome-dependent entry of HIV-1 into human immune cells. Int. J. Nanomed. 2017, 12, 4823–4833. [Google Scholar] [CrossRef] [PubMed]
- Meckes, D.G., Jr.; Raab-Traub, N. Microvesicles and viral infection. J. Virol. 2011, 85, 12844–12854. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.D.; Maier, C.L.; Pober, J.S. Cytomegalovirus-infected human endothelial cells can stimulate allogeneic CD4+ memory t cells by releasing antigenic exosomes. J. Immunol. 2009, 182, 1548–1559. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Silva, M.R.; Cabrera-Cabrera, F.; das Neves, R.F.; Souto-Padron, T.; de Souza, W.; Cayota, A. Gene expression changes induced by Trypanosoma cruzi shed microvesicles in mammalian host cells: Relevance of trna-derived halves. Biomed. Res. Int. 2014, 2014, 305239. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Silva, M.R.; das Neves, R.F.; Cabrera-Cabrera, F.; Sanguinetti, J.; Medeiros, L.C.; Robello, C.; Naya, H.; Fernandez-Calero, T.; Souto-Padron, T.; de Souza, W.; et al. Extracellular vesicles shed by Trypanosoma cruzi are linked to small RNA pathways, life cycle regulation, and susceptibility to infection of mammalian cells. Parasitol. Res. 2014, 113, 285–304. [Google Scholar] [CrossRef] [PubMed]
- Prados-Rosales, R.; Baena, A.; Martinez, L.R.; Luque-Garcia, J.; Kalscheuer, R.; Veeraraghavan, U.; Camara, C.; Nosanchuk, J.D.; Besra, G.S.; Chen, B.; et al. Mycobacteria release active membrane vesicles that modulate immune responses in a TLR2-dependent manner in mice. J. Clin. Investig. 2011, 121, 1471–1483. [Google Scholar] [CrossRef] [PubMed]
- Bielaszewska, M.; Ruter, C.; Bauwens, A.; Greune, L.; Jarosch, K.A.; Steil, D.; Zhang, W.; He, X.; Lloubes, R.; Fruth, A.; et al. Host cell interactions of outer membrane vesicle-associated virulence factors of enterohemorrhagic Escherichia coli O157: Intracellular delivery, trafficking and mechanisms of cell injury. PLoS Pathog. 2017, 13, e1006159. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.; Cleary, T.G.; Hernandez, J.D.; Abboud, H.E. Shiga toxin 1 elicits diverse biologic responses in mesangial cells. Kidney Int. 1998, 54, 1117–1127. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, M.; Buelli, S.; Pezzotta, A.; Corna, D.; Perico, L.; Tomasoni, S.; Rottoli, D.; Rizzo, P.; Conti, D.; Thurman, J.M.; et al. Shiga toxin promotes podocyte injury in experimental hemolytic uremic syndrome via activation of the alternative pathway of complement. J. Am. Soc. Nephrol. 2014, 25, 1786–1798. [Google Scholar] [CrossRef] [PubMed]
- Polley, M.J.; Nachman, R.L. Human platelet activation by C3a and C3a des-arg. J. Exp. Med. 1983, 158, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Arvidsson, I.; Rebetz, J.; Loos, S.; Herthelius, M.; Kristoffersson, A.C.; Englund, E.; Chromek, M.; Karpman, D. Early terminal complement blockade and C6 deficiency are protective in enterohemorrhagic Escherichia coli-infected mice. J. Immunol. 2016, 197, 1276–1286. [Google Scholar] [CrossRef] [PubMed]
- Morigi, M.; Galbusera, M.; Gastoldi, S.; Locatelli, M.; Buelli, S.; Pezzotta, A.; Pagani, C.; Noris, M.; Gobbi, M.; Stravalaci, M.; et al. Alternative pathway activation of complement by Shiga toxin promotes exuberant C3a formation that triggers microvascular thrombosis. J. Immunol. 2011, 187, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, M.; Kang, Y.; Tan, Y.S.; Pavlov, V.I.; Liu, B.; Boyle, D.C.; Kushak, R.I.; Skjoedt, M.O.; Grabowski, E.F.; Taira, Y.; et al. Human mannose-binding lectin inhibitor prevents Shiga toxin-induced renal injury. Kidney Int. 2016, 90, 774–782. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic presentation of the mechanisms of extracellular vesicle release. Exosomes, shown on the left, are derived from multivesicular bodies (MVB) that are late endosomes carrying intraluminal vesicles (ILVs). MVBs fuse with the plasma membrane to release the ILVs that are termed exosomes once shed into the extracellular space. Microvesicles, shown in the middle panel, originate from a direct budding of the plasma membrane. Apoptotic bodies, depicted on the right, are released during apoptosis and cellular degradation. ER: endoplasmic reticulum.
Figure 1.
Schematic presentation of the mechanisms of extracellular vesicle release. Exosomes, shown on the left, are derived from multivesicular bodies (MVB) that are late endosomes carrying intraluminal vesicles (ILVs). MVBs fuse with the plasma membrane to release the ILVs that are termed exosomes once shed into the extracellular space. Microvesicles, shown in the middle panel, originate from a direct budding of the plasma membrane. Apoptotic bodies, depicted on the right, are released during apoptosis and cellular degradation. ER: endoplasmic reticulum.

Figure 2.
Shiga toxin uptake and intracellular routing. Shiga toxin may be taken up directly after binding to its glycolipid receptor Gb3. Alternatively, the toxin may be endocytosed within a microvesicle. Either way the toxin will undergo retrograde transport via the Golgi to the endoplasmic reticulum and the ribosomes where it may inhibit protein synthesis or induce ribosomal stress and apoptosis.
Figure 2.
Shiga toxin uptake and intracellular routing. Shiga toxin may be taken up directly after binding to its glycolipid receptor Gb3. Alternatively, the toxin may be endocytosed within a microvesicle. Either way the toxin will undergo retrograde transport via the Golgi to the endoplasmic reticulum and the ribosomes where it may inhibit protein synthesis or induce ribosomal stress and apoptosis.
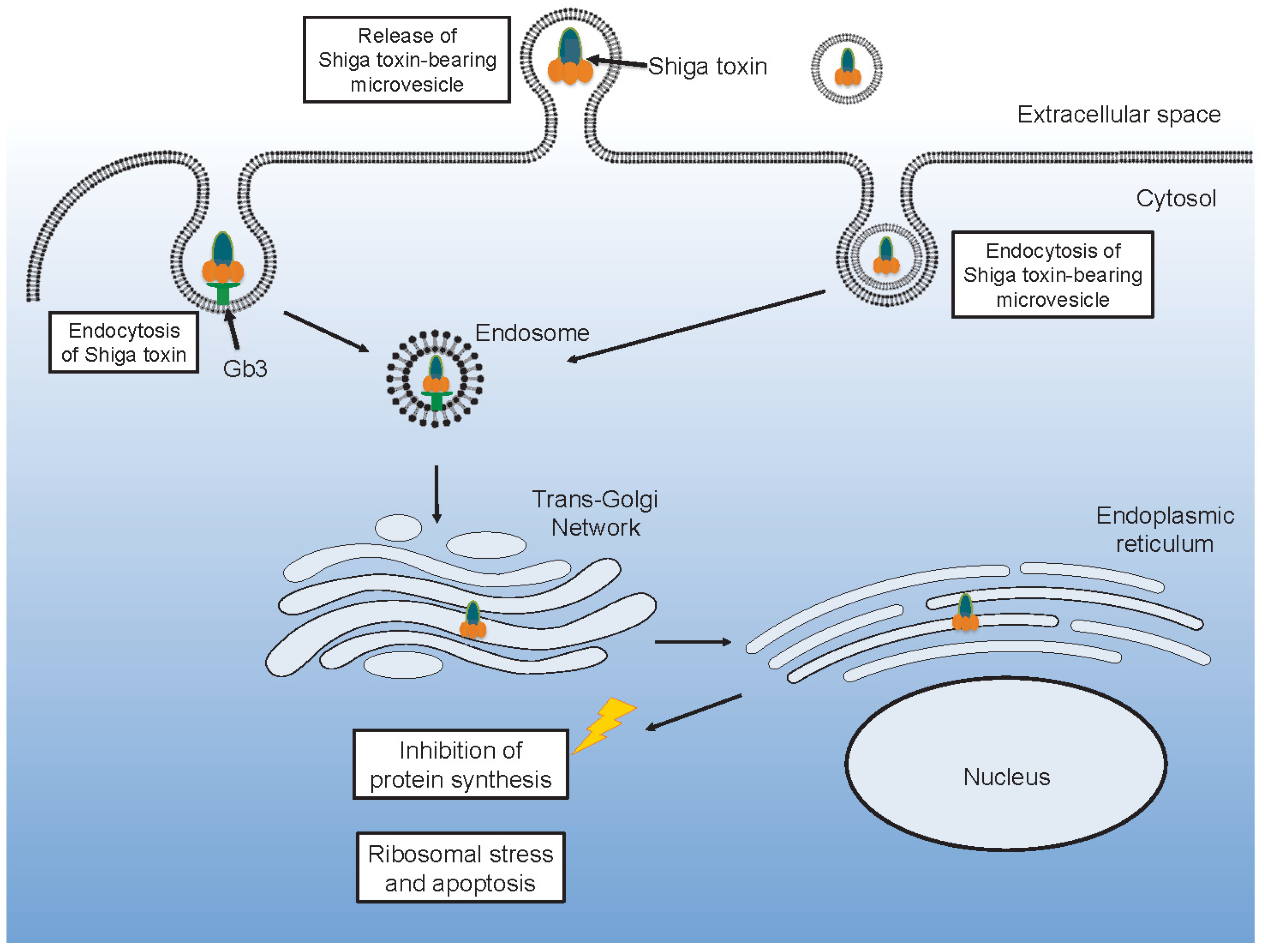
Figure 3.
Shiga toxin-induced activation of blood cells and damage to the glomerular endothelium. Within the bloodstream Shiga toxin can bind to monocytes, neutrophils, activated platelets and red blood cells. Aggregates are formed between platelets and leukocytes (depicted in the figure between a neutrophil and a platelet). Microvesicles are shed from blood cells (neutrophils, monocytes, platelets and red blood cells). Some of these contain the toxin. All these microvesicles may expose phosphatidylserine. Microvesicles from platelets and monocytes expose tissue factor. Complement may deposit on platelet, monocyte and red blood cell-derived microvesicles, as well as on the glomerular endothelium. Shiga toxin binds to glomerular endothelial cells via the Gb3 glycolipid receptor or undergoes endocytosis within blood cell-derived microvesicles. Shiga toxin-induced cytotoxicity and complement deposition lead to glomerular endothelial cell damage. Thrombosis is induced by toxin-mediated endothelial cell damage, platelet activation and the pro-thrombotic effects of microvesicles. Hemolysis is induced by mechanical breakdown of red blood cells in occluded capillaries as well as by complement deposition on red blood cells. Fragmented red blood cells are termed schistocytes. Gb3: globotriaosylceramide, MAC: membrane attack complex (C5b-9), RBC: red blood cell.
Figure 3.
Shiga toxin-induced activation of blood cells and damage to the glomerular endothelium. Within the bloodstream Shiga toxin can bind to monocytes, neutrophils, activated platelets and red blood cells. Aggregates are formed between platelets and leukocytes (depicted in the figure between a neutrophil and a platelet). Microvesicles are shed from blood cells (neutrophils, monocytes, platelets and red blood cells). Some of these contain the toxin. All these microvesicles may expose phosphatidylserine. Microvesicles from platelets and monocytes expose tissue factor. Complement may deposit on platelet, monocyte and red blood cell-derived microvesicles, as well as on the glomerular endothelium. Shiga toxin binds to glomerular endothelial cells via the Gb3 glycolipid receptor or undergoes endocytosis within blood cell-derived microvesicles. Shiga toxin-induced cytotoxicity and complement deposition lead to glomerular endothelial cell damage. Thrombosis is induced by toxin-mediated endothelial cell damage, platelet activation and the pro-thrombotic effects of microvesicles. Hemolysis is induced by mechanical breakdown of red blood cells in occluded capillaries as well as by complement deposition on red blood cells. Fragmented red blood cells are termed schistocytes. Gb3: globotriaosylceramide, MAC: membrane attack complex (C5b-9), RBC: red blood cell.
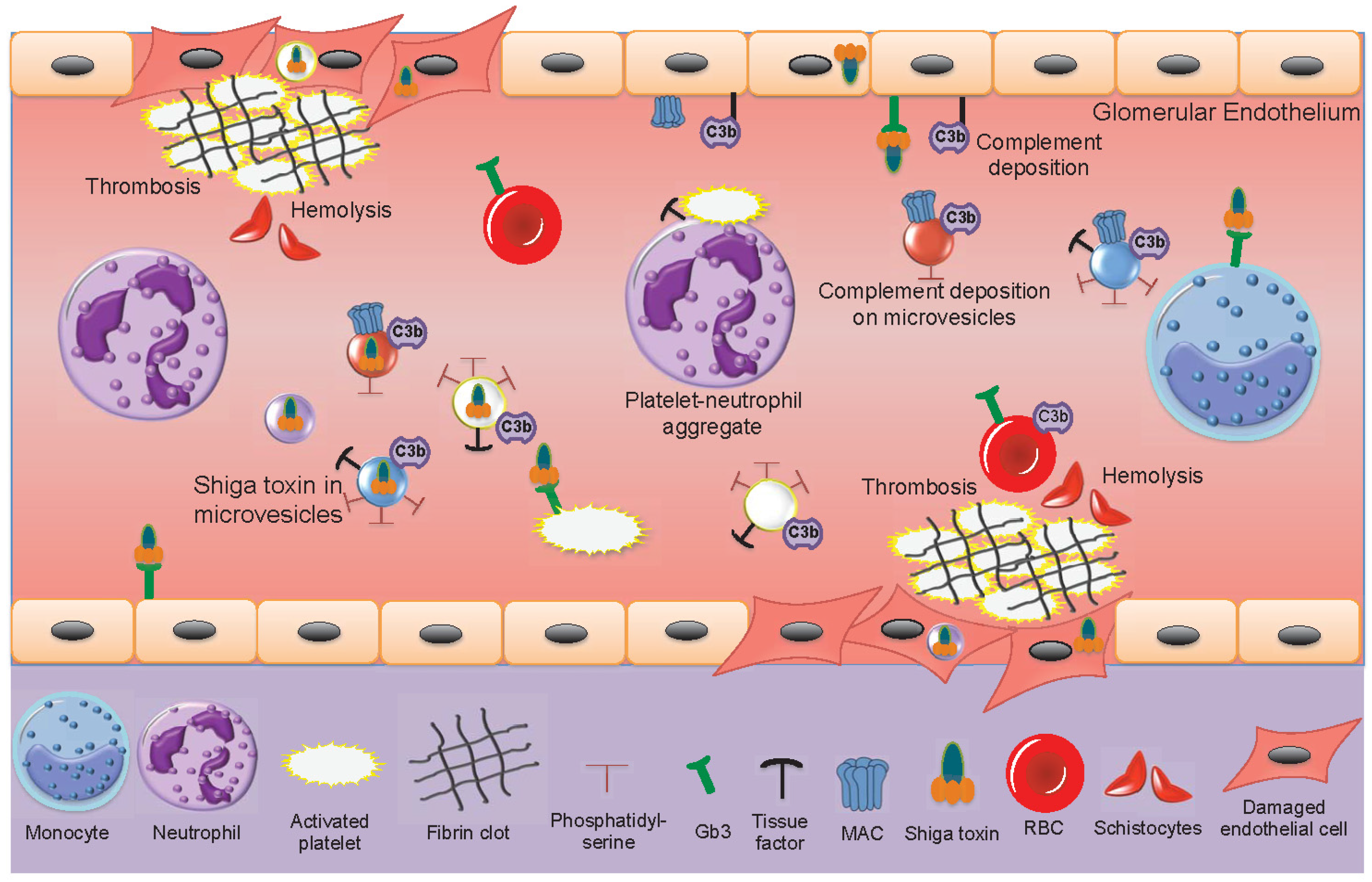

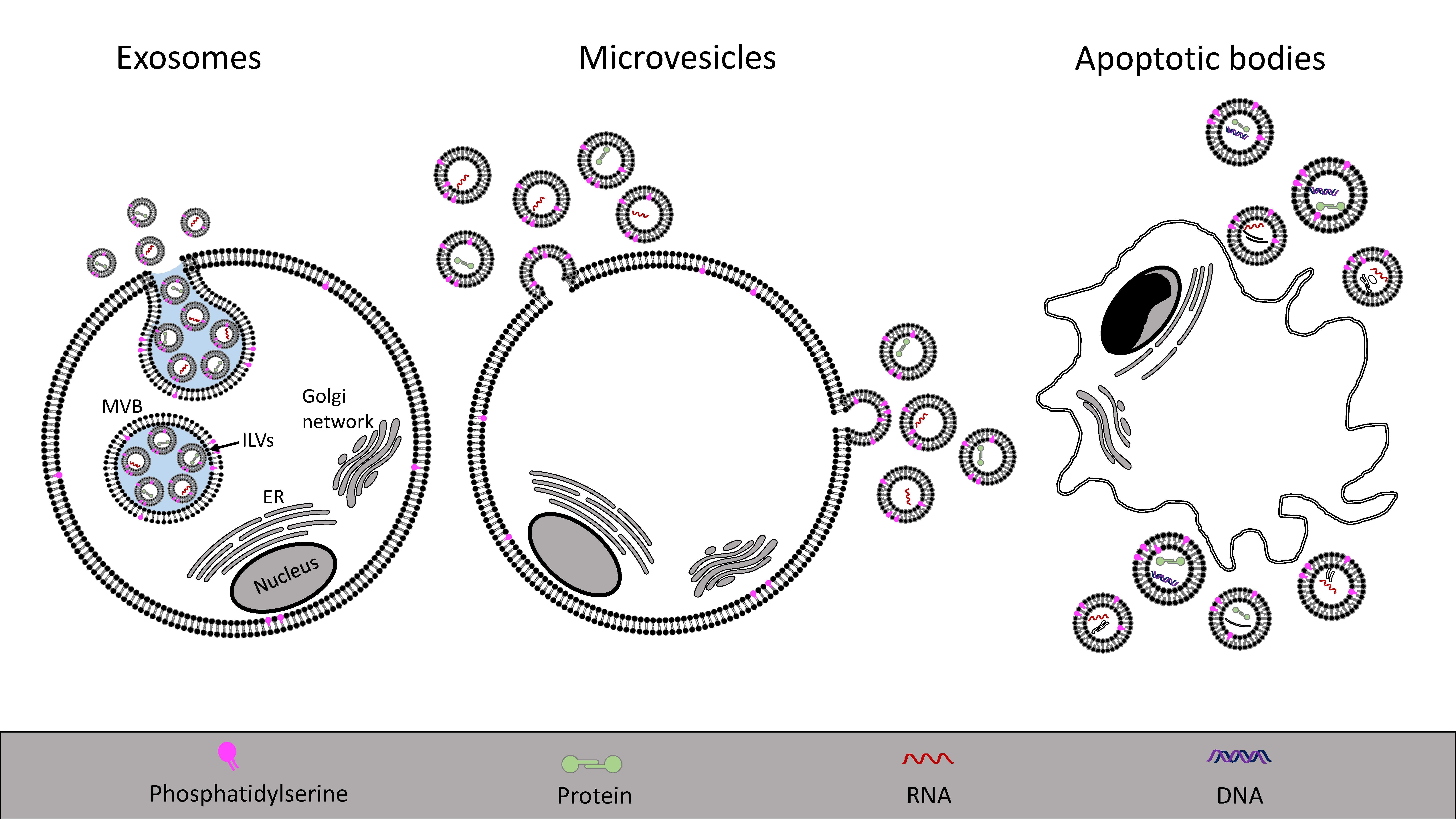{kind=link}
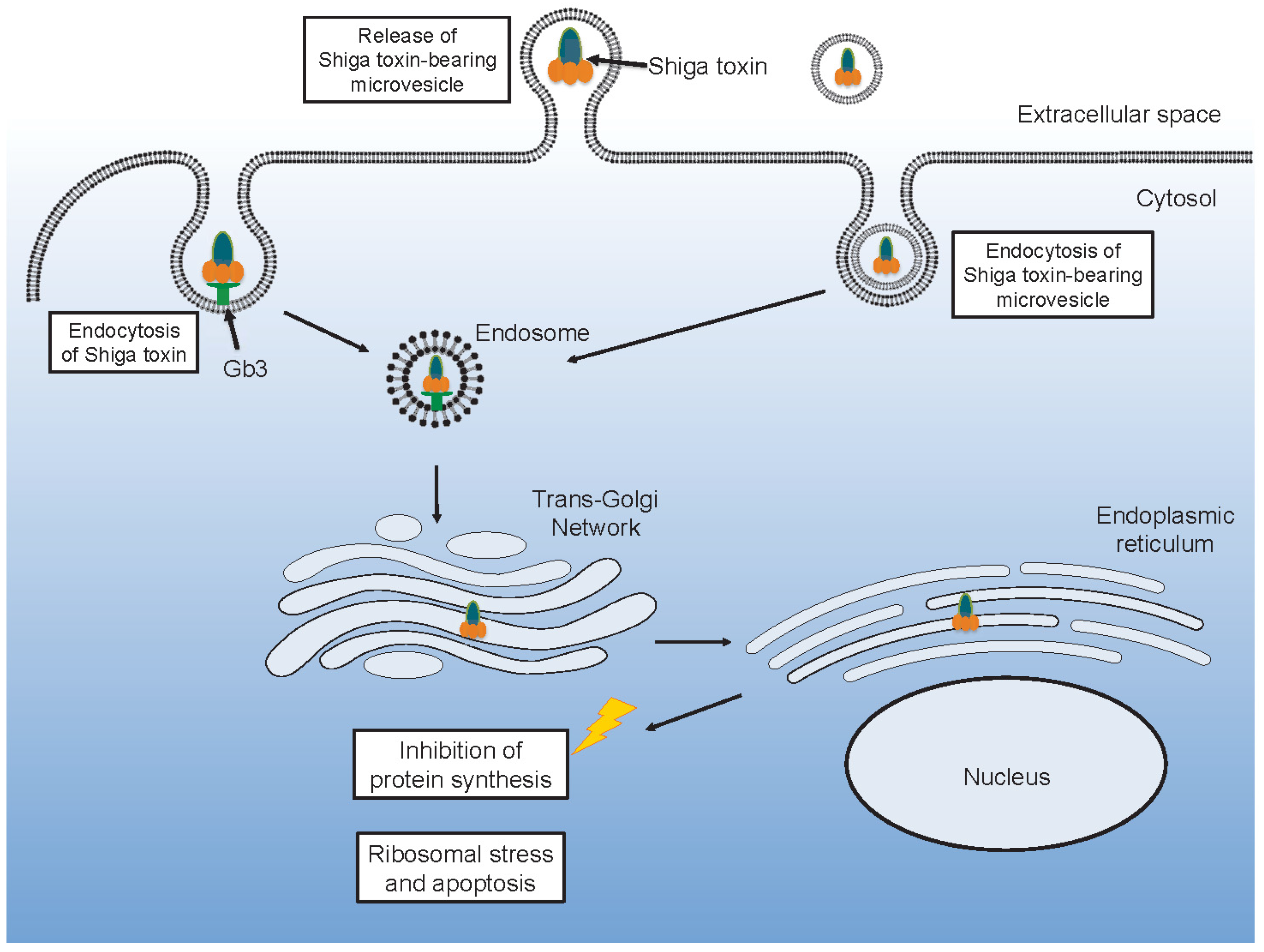{kind=link}
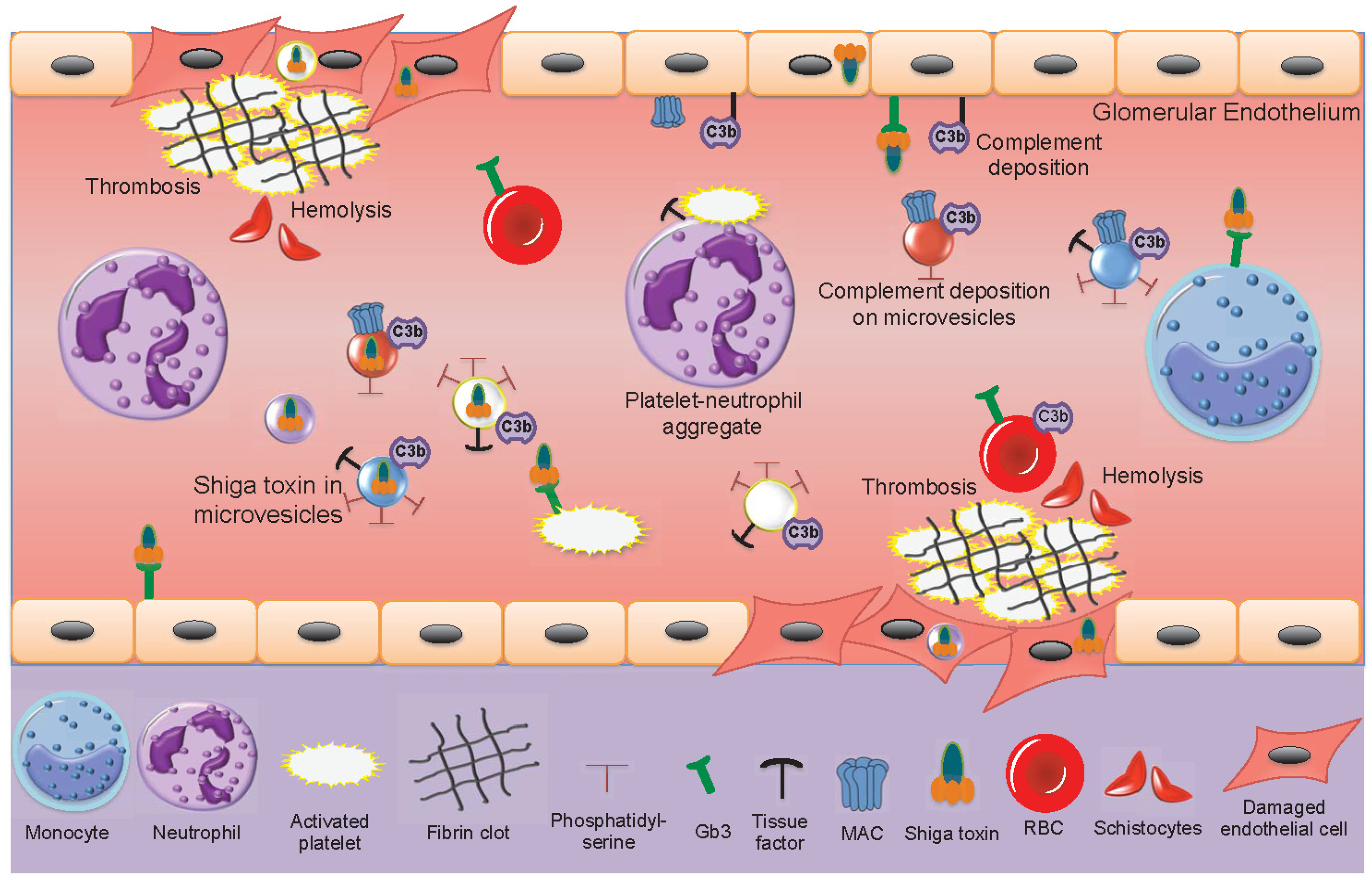{kind=link}
Table 1.
Classification of extracellular vesicles.
| Extracellular Vesicle | Origin | Size | Content | References |
|---|---|---|---|---|
| Exosome | Intraluminal vesicle within multivesicular bodies | 30–100 nm | mRNA, miRNA, proteins, lipids | [97] |
| Microvesicle | Shedding from the plasma membrane with cellular content | 100–1000 nm | mRNA, miRNA, proteins, lipids | [29] |
| Apoptotic body | Cellular breakdown and shrinkage during apoptosis | 1–5 μm | Organelles, proteins, DNA, RNAs, lipids | [97] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Villysson, A.; Tontanahal, A.; Karpman, D. Microvesicle Involvement in Shiga Toxin-Associated Infection. Toxins 2017, 9, 376. https://doi.org/10.3390/toxins9110376
AMA Style
Villysson A, Tontanahal A, Karpman D. Microvesicle Involvement in Shiga Toxin-Associated Infection. Toxins. 2017; 9(11):376. https://doi.org/10.3390/toxins9110376
Chicago/Turabian StyleVillysson, Annie, Ashmita Tontanahal, and Diana Karpman. 2017. "Microvesicle Involvement in Shiga Toxin-Associated Infection" Toxins 9, no. 11: 376. https://doi.org/10.3390/toxins9110376
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.