
Therapeutic Potential of Cholera Toxin B Subunit for the Treatment of Inflammatory Diseases of the Mucosa


1
Department of Pharmacology and Toxicology, University of Louisville School of Medicine, Louisville, KY 40202, USA
2
Center for Predictive Medicine, University of Louisville, Louisville, KY 40202, USA
3
James Graham Brown Cancer Center, University of Louisville, Louisville, KY 40202, USA
*
Author to whom correspondence should be addressed.
Toxins 2017, 9(12), 379; https://doi.org/10.3390/toxins9120379
Submission received: 18 October 2017
/
Revised: 14 November 2017
/
Accepted: 21 November 2017
/
Published: 23 November 2017
(This article belongs to the Special Issue Toxins in Drug Discovery and Pharmacology)
{kind=link}
Abstract
:Cholera toxin B subunit (CTB) is a mucosal immunomodulatory protein that induces robust mucosal and systemic antibody responses. This well-known biological activity has been exploited in cholera prevention (as a component of Dukoral® vaccine) and vaccine development for decades. On the other hand, several studies have investigated CTB’s immunotherapeutic potential in the treatment of inflammatory diseases such as Crohn’s disease and asthma. Furthermore, we recently found that a variant of CTB could induce colon epithelial wound healing in mouse colitis models. This review summarizes the possible mechanisms behind CTB’s anti-inflammatory activity and discuss how the protein could impact mucosal inflammatory disease treatment.
1. Introduction
Vibrio cholerae is a gram-negative bacterium that can colonize the gastrointestinal tract and cause life-threatening watery diarrhea. The principal virulence factor of V. cholerae is cholera toxin (CT), which consists of a catalytic A-subunit and a non-toxic homopentameric B-subunit (CTB) [1,2,3]. CTB binds cells through GM1 ganglioside receptors, which then mediates toxin entry into the cell. It has been previously shown that CTB can induce strong biological activities that can enhance or suppress immune effects under normal and various immunopathological conditions without the toxicity associated with the CTA subunit [4]. Consequently, CTB has been widely studied as a mucosal immunomodulatory agent.
In its most well-known immunostimulatory effects, CTB is used in the vaccine Dukoral®. Dukoral® is a WHO pre-qualified oral cholera vaccine which contains heat-killed whole cell V. cholerae and recombinant CTB (rCTB). Dukoral® stimulates the production of both antibacterial and antitoxin antibodies, including secretory immunoglobulin A (S-IgA) produced locally in the intestines [5]. CTB itself can induce potent mucosal and systemic antibody response upon mucosal administration in humans [6,7,8], which is largely due to the broad distribution of GM1 ganglioside on various cell types such as epithelial cells, macrophages, dendritic cells (DCs), B cells, T cells, and neurons [9,10,11,12]. Furthermore, the presence of GM1 ganglioside on the luminal surface of intestinal epithelial cells and antigen presenting cells (APCs) in the gut seems to be essential for CTB’s strong mucosal immunostimulatory effects associated with MHC class II expression and local antigen enrichment [13]. In addition, CTB stimulates specific immunosuppressive effects against autoimmune disorders, excess inflammation, and allergic reactions [4,14,15,16,17,18]. We have recently shown that oral administration of a variant of CTB mitigates colitis in chemically-induced acute and chronic colitis mouse models [19]. Although the underlying mechanisms are not well understood, recent studies have shed some light on these immunosuppressive effects induced by CTB. Thus, this review will summarize published studies on CTB’s impacts in mucosal inflammatory disease models, as well as the mechanisms associated with its therapeutic effect and the challenges that CTB faces as an immunomodulatory drug.
2. Cholera Toxin Structure and Mechanism in Gut Epithelial Cells
To reveal the mechanism of CTB-induced biological activity, we must first understand the molecule. CT is classified as an AB5 toxin family, which includes the toxins of Shigella dysenteriae and enterohaemorrhagic Escherichia coli. The toxins are usually composed of one A subunit and five B subunits (CTA and CTB, respectively, for CT). CTA consists of an enzymatically active 11-kDa N-terminal chain (CTA1) and a C-terminal chain (CTA2) that connects CTA to the central pore of CTB. CTB has the capacity to translocate the CTA across the plasma membrane, mediated by the binding of GM1 ganglioside, and then escort CTA from the plasma membrane into the endoplasmic reticulum (ER) [20,21]. The following summarizes CT’s retrograde trafficking mechanism.
The five B-subunits form a central cylindrical pore lined by five amphipathic α-helices that help form a highly stable homopentamer. The pentamer contains five GM1 binding sites that lie on the outer edge of each B subunit [1,22]. Due to an avidity effect from the pentavalent binding capacity, CTB has a very strong affinity (KD reported to be 5 pM to 1 nM) to GM1, which is mainly localized in lipid rafts on the plasma membranes of many cell types [9,10,11,12]. Once CT is bound to GM1 (up to five gangliosides at once), it is endocytosed by clathrin-dependent and independent mechanisms and trafficked via retrograde transport from the Golgi to the ER [21]. It is also known that CT can undergo transcytosis across epithelial cells from the apical to the basolateral surface. However, regardless of how the toxin enters the cell, CT travels to the trans-Golgi network via early endosomal vesicles, independent of the late endosome pathway. The C-terminus of CTA2 possesses a KDEL ER-retention signal for retrieval of CT from the cis-Golgi apparatus to the ER. Interestingly, the KDEL sequence is not vital for retrograde transport of CT to the ER. Mutations that alter the KDEL sequence on CT inhibit KDEL-dependent ER retrieval and decreased (albeit not completely) CT’s toxification [23]. Thus, it is thought that CT’s KDEL sequence—although not absolutely essential—improves the ER’s retrieval of the dissociated CT from the Golgi apparatus and prolongs the time of retention within the ER [20,23,24]. Once in the ER, the CTA1-chain is dissociated from CTA2/CTB complex by protein disulfide isomerase (PDI). Subsequently, CTA1 enters the cytosol via the ER-associated degradation pathway and escapes proteasomal degradation [1,20]. On the other hand, the fate (and remaining function, if any) of CTA2/CTB after releasing CTA1 in the ER is not well documented. Meanwhile, CTA1 reaches the inner surface of the plasma membrane and catalyzes the ADP ribosylation of Gαs, thereby continuously activating adenylate cyclase to produce cAMP. Increased intracellular cAMP impairs sodium uptake and increases chloride outflow, leading to water secretion and diarrhea [20,25].
3. At the Cellular Level—What Is Known So Far
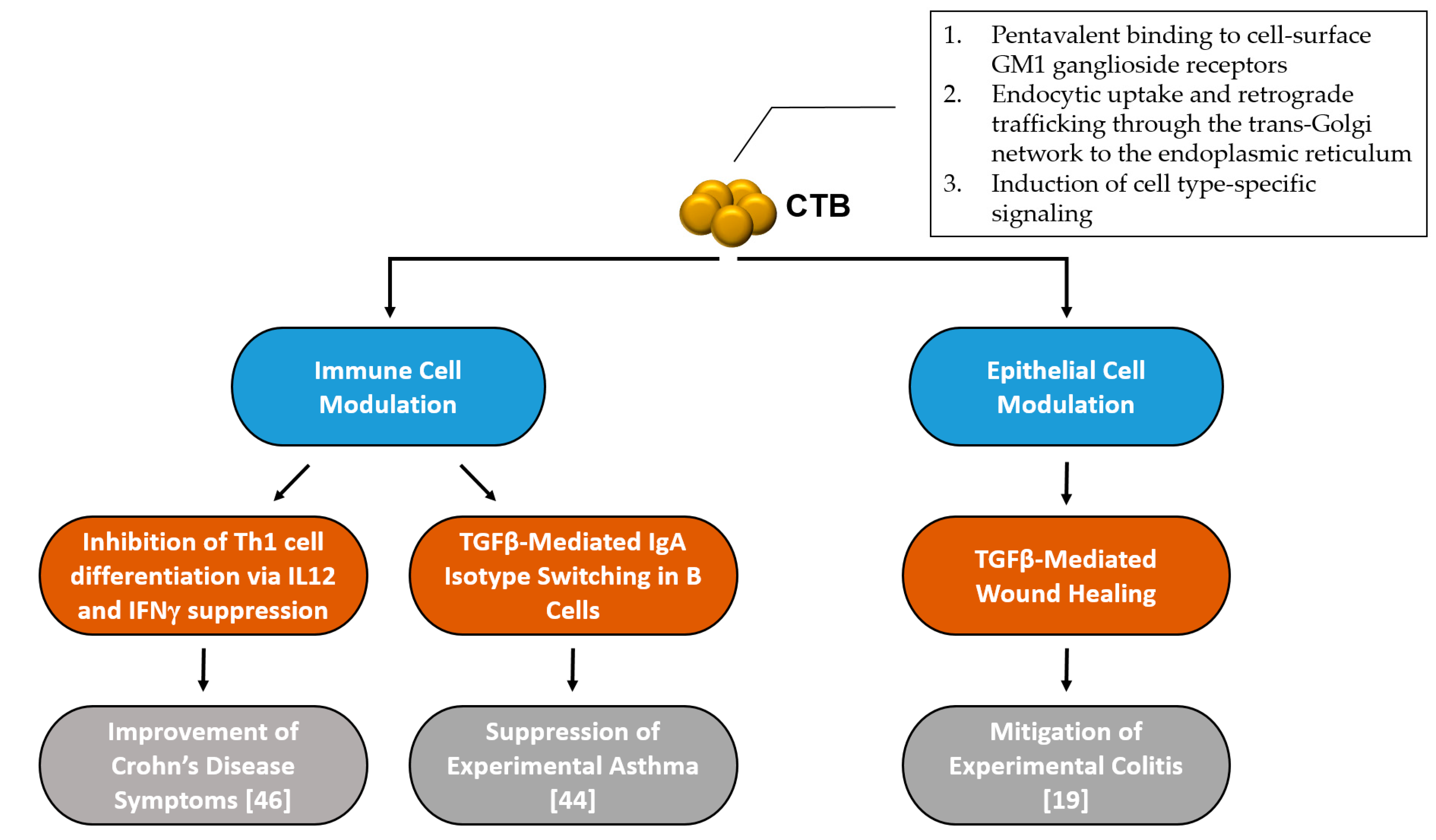
Although the virulence mechanism and intracellular trafficking of CT has been well studied, the anti-inflammatory mechanisms of CTB are much less studied and understood. After a comprehensive literature review, it seems that there are at least two separate modes of action induced by CTB to modulate inflammatory responses: one that is based on immune cell regulation, and another that is epithelial cell-mediated (Figure 1).
In 1994, the immune suppressive effects of CTB were first reported by Sun et al. [26]. This report demonstrated that oral administration of mice with CTB conjugated with antigens (sheep red blood cells, horse red blood cells, and human γ-globulin) enhanced oral tolerance to the antigens, presumably through efficient presentation of antigens to immune cells in the gut-associated lymphoid tissue and the generation of regulatory cells. In a Commentary to this article, Weiner suggested that CTB could have enhanced tolerance by serving as a “selective mucosal adjuvant” and that this unique activity could be exploited to treat autoimmunity [27]. Subsequently, this seminal finding led to a new field of studies in which CTB-antigen conjugates were applied to induce tolerogenic reactions to the conjugated antigens in various immunopathological conditions (i.e., encephalomyelitis, autoimmune diabetes, autoimmune arthritis, uveitis) and IgE-mediated allergen hypersensitivity [14,16,17,18,28,29,30,31,32,33,34,35,36,37,38]. Through these studies, it became apparent there are two unique and distinct mechanisms of CTB responsible for the suppression of immunopathological reactions in allergy and autoimmune diseases: (1) to increase antigen uptake and presentation by different APCs through binding to their cell-surface GM1 ganglioside receptors and (2) to induce anti-inflammatory and immunoregulatory activities by directly or indirectly acting on specific immune cells. The latter mechanism points to the possibility that CTB by itself may act as an immunotherapeutic agent; however, only a handful of groups have actually proven that CTB alone—without co-administration or conjugation of antigens—can induce an anti-inflammatory response. Moreover, studies conducted with non-recombinant CTB (nrCTB, prepared by chemically dissociating CTA from CTB) can have significantly skewed experimental results due to trace amounts of CT and CTA [4,39,40]. For example, we have previously shown that picomolar concentrations (<10 ng/mL) of CT significantly inhibited lipopolysaccharide (LPS)-induced TNFα production in RAW264.7 cells, while recombinant (r)CTB failed to induce such an effect at a concentration as high as 10 µg/mL [4]. Thus, the use of rCTB is required to evaluate the effects unique to CTB.
3.1. Immune Cell Modulation
With regards to CTB’s immune cell regulation, Kim et al. demonstrated in murine spleen B cells that rCTB dose-dependently increased IgA secretion and inhibited B cell growth [41]. In the presence of IL-2, rCTB significantly increased IgA isotype switching in LPS-activated B cells. These effects were reversed by the addition of an anti-TGFβ or soluble TGFβ1 receptor, which markedly inhibited rCTB-stimulated IgA response. Further analysis in the same report revealed that rCTB stimulated IgA2 B cells, upregulated TGFβ1 mRNA expression, and increased bioactive TGFβ1 levels, which is known to induce IgA isotype switching [41]. Thus, rCTB stimulated a TGFβ-mediated IgA response that was dependent on IL-2 as a cofactor. These findings have contributed to our understandings of how CTB stimulates B cell IgA production, and potentially oral tolerance as well (see below).
It is known that IgA antibodies help maintain mucosal homeostasis and play a role in immune protection [42,43]. Thus, it seems possible that rCTB administration could provide therapeutic effects in mucosal autoimmune disorders via IgA induction. For example, in an experimental mouse model of asthma, nrCTB suppressed the ability of DCs to prime for Th2 responses to inhaled allergen via an IgA-dependent manner [44]. In this study, co-administration of ovalbumin (OVA) and nrCTB suppressed classical features of asthma, including airway eosinophilia, Th2 cytokine synthesis, and bronchial hyperactivity in mice that were pre-sensitized with OVA-stimulated DCs in the lung. Furthermore, nrCTB treatment enhanced DCs’ potential to induce Treg cells in vitro; however, these Treg cells did not provide protection when transferred into the airways of naïve mice that received OVA challenge. In contrast, the transfer of B cells from OVA+CTB-DCs-immunized mice to OVA-sensitized naïve mice significantly reduced eosinophilia and lymphocytosis. It was also found that nrCTB caused a TGFβ-dependent increase in antigen-specific IgA in the airway luminal secretion, and this was attributed to nrCTB’s efficacy against the experimental asthma as the therapeutic effects were abrogated in mice lacking luminal IgA transporter (polymeric Ig receptor), which is necessary for the transport of dimeric IgA across the epithelium into the luminal mucosa [45].
Meanwhile, IgA may not be the sole factor contributing to CTB’s ability to mitigate inflammatory diseases in the mucosa. For example, in the 2,4,6-trinitrobenzene sulfonic acid (TNBS)-induced mouse model of Crohn’s disease, daily oral administration over a four-day period of 100 µg rCTB after the onset of TNBS-colitis immediately resolved weight loss and reduced inflammation [39]. In this case, the timing of mucosal restitution in regard to rCTB administration did not likely result in IgA production. In a similar TNBS-colitis study, rCTB administration reduced IL-12 and IFNγ secretion, inhibited STAT-4 and STAT-1 activation, and downregulated T-bet expression, indicating that rCTB inhibited mucosal Th1 cell signaling [46]. Moreover, these results were confirmed in a small multicenter, open-label, and nonrandomized clinical trial in which 15 patients with active CD received three oral doses of 5 mg rCTB per-week over 2 weeks (six doses total) and were examined 2, 4, 6, and 10 weeks after the start of the study. Of the 12 patients who finished the study per protocol, seven responded to treatment and five were in remission by week six and maintained remission through week 10 as defined by a CD activity index score ≤150 [47]. Of note, side effects seen in 33% of patients administered with CTB were mild (arthralgia, headache, and pruritus), and no safety concerns were raised throughout the trial [47].
Interestingly, rCTB did not reduce disease severity in an oxazolone-induced colitis model performed by the same group [39]. Oxazolone-induced colitis is mediated by IL-4 driven Th2 cells rather than IL-12/IFNγ-driven Th1 cells [39]. Thus, it appears that rCTB administration had a specific effect on specific T cell functions involved in TNBS-colitis [39]. Although the detailed mechanism by which rCTB inhibited Th1 cell was not elucidated, it is possible that the binding of CTB to GM1 ganglioside on immune cells resulted in a signaling cascade of events that led to Th1 inhibition, because non-GM1 binding CTB mutants do not modulate lymphocyte function [48]. In agreement with these findings, rCTB decreased monocyte-derived DC maturation and IL-12 production upon LPS stimulation in vitro [49]. Moreover, rCTB-pretreated, LPS-stimulated DCs induced low proliferating T cells that had enhanced production of IL-10 and reduced production of IFNγ. Rouquete-Jazdanian et al. additionally showed that the binding of rCTB to GM1 ganglioside directly prevented the activation and proliferation of CD4+ T cells [50]. This effect was induced by rCTB-mediated sphingomyelinase activation that subsequently increased the production of ceramides, which are known cell cycle arrest inducers [51]. rCTB also inhibited protein kinase Cα, a pro-growth cellular regulator, which was linked to rCTB-induced lipid raft modifications and ceramide-mediated inactivation [52,53].
3.2. Epithelial Cell Modulation
Besides serving as a barrier lining the mucosal surface, epithelial cells have multiple functions associated with the maintenance of gut homeostasis and mucosal healing, and crosstalk between epithelial and immune cells is an important component of those complex biological processes [54,55]. Even though CTB first encounters epithelial cells in the gut, the CTB-mediated modulation of epithelial cells and its consequence to the mucosal immune system have largely been ignored in comparison to the protein’s direct impacts on immune cells.
In one small study, CTB was shown to induce a dose-dependent increase of IL-10 mRNA levels in the colon epithelial cell-line T84 [56], hinting that CTB could induce epithelial cell-mediated immune modulation [57]. We have recently characterized CTB’s global impacts on the gut to further our understanding of its unique biological activities. Using a plant-made recombinant CTB (CTBp) [58,59], we have shown that oral administration of the CTB variant significantly altered several immune cell populations in the colon lamina propria [19]. Two-weeks after two oral 30 µg CTBp administrations, Th2 and Treg cells increased in the colon lamina propria. This is not the first report of CTB-induced increase in these cell types [15,36,38,60,61]. For instance, it has been shown that oral administration of a CTB–insulin conjugate in NOD mice induced a shift from Th1 to Th2 profile while generating Treg cells [15]. Additionally, intraperitoneal administration of nrCTB to rats increased Treg cells in the peripheral blood 24–72 h after ischemia [60]. Besides the specific T helper cell subsets, our study has also revealed that innate immune cells—including dendritic cells, natural killer cells and macrophages (both M1 and M2)—populations were increased in the colon lamina propria two weeks after CTBp oral administration [19]. Furthermore, a global gene expression analysis revealed that CTBp had more pronounced impacts on the colon than the small intestine, with significant activation of TGFβ-mediated pathways in the colon mucosa [19]. Given that there is a strong link between epithelial-derived TGFβ and innate immune cells in wound healing [62,63,64], the results provided implications for the potential utility of CTBp to promote colonic mucosal health. Subsequently, we found that CTBp induced TGFβ-mediated wound healing in Caco2 colon epithelial cells. Furthermore, oral administration of CTBp in mice protected against colon mucosal damage in acute colitis induced by dextran sodium sulfate (DSS). Two oral doses of as low as 1 µg of CTBp mitigated clinical signs of disease (body weight loss, decreased histopathological scores, and blunted escalation of inflammatory cytokine levels) and upregulated wound healing-related genes [19]. Interestingly, CTBp administration prevented fibrosis associated with acute colitis in mice; hence, the protein did not appear to overstimulate TGFβ signaling. In fact, TGFβ gene expression levels were high during the early inflammatory phase and became lower in the recovery phase of the acute colitis model in CTBp-treated mice.
In contrast to TNBS-induced colitis, the DSS-colitis model closely approximates human ulcerative colitis (UC) [65,66,67,68]. Thus, the results point to the possibility that CTBp could be used to facilitate mucosal healing in the management of UC. Since the main driver of intestinal inflammation in the DSS model is the damage to the epithelial barrier lining the colon that allows intestinal microbiota into submucosal compartments [69], and since therapeutic effects were observed immediately upon CTBp administration, we concluded that CTBp’s protective efficacy in the DSS colitis models were attained by the induction of TGFβ-mediated colonic epithelial wound healing. Given that UC poses an increased risk of developing colitis-associated colorectal cancer (CAC) [70,71], CTBp’s effects were also examined in the azoxymethane (AOM)/DSS mouse model of CAC. Biweekly oral administration of CTBp over 9 weeks significantly reduced inflammation and tumorigenesis in this model, again highlighting its therapeutic potential in UC treatment [19].
It is of importance to point out that many of the effects observed in the aforementioned studies using CTBp may be unique to the plant-made variant, as it has a mutation at amino acid position 4 and an ER retention signal sequence at the C-terminus (N4S-CTB-SEKDEL; [58]). The ER-retention sequence was added to CTBp to improve production in planta, while Asn4→Ser mutation was introduced to avoid N-glycosylation [58,59]. The addition of the KDEL sequence to N4S-CTB significantly reduced ER stress that otherwise caused poor production yield. It is thought that the KDEL sequence helped prolong CTBp’s residence time in the ER to allow for proper folding and assembly.
The protein ER retention mechanism involving the KDEL receptor is highly conserved among eukaryotic organisms [72]. Thus, there is a possibility that the artificial KDEL sequence of CTBp may prolong the protein’s residence in the epithelial cells upon binding to cell-surface GM1 ganglioside and retrograde transport into the ER, as has been demonstrated for CT [21,23]. Subsequently, this may induce a level of altered cell signaling. For example, interaction between CTBp’s C-terminal KDEL sequence and KDEL receptors may have an impact on ER homeostasis [73,74]. The binding of proteins to the KDEL receptor and the induction of mild UPR have been linked to TGFβ activation, wound healing, colon epithelial cell prosurvival signaling, and protection from DSS-induced colitis [73,75,76,77]. Of note, CT is a known inducer of the UPR in epithelial cells due to the KDEL sequence on CTA [23,78,79], while CTB has no effect on the UPR or ER signaling [78].
Regardless of whether the ER retention signal had a significant contribution to the mucosal healing activity in the mouse colitis models, the study has provided evidence that CTB can exhibit a therapeutic effect against colitis in an epithelia-dependent manner, warranting further investigation of CTB’s impacts on epithelial cells.
4. Conclusions—Challenges for the Use of CTB as an Immunomodulatory Drug
Although CTB has been administered in humans in the form of oral cholera vaccines over the past two decades, its development as an immunomodulatory drug will need to address unique issues associated with therapeutic use besides additional testing of safety and efficacy in specific disease indications. One of the principal questions is whether CTB’s strong mucosal immunogenicity that induces a robust IgG and IgA immune response [4,58] is beneficial or dispensable to its anti-inflammatory/immunosuppressive effects. From a conventional biopharmaceuticals development standpoint, anti-drug antibodies constitute a theoretical risk because they may affect drug efficacy and pharmacokinetics, and potentially cause immunotoxicity [80,81]. However, induction of an antibody response—particularly that of IgA isotype—may play an important role in mitigating mucosal inflammation, as illustrated in the asthma study described in Section 3.1 [44]. The CD clinical trial showed an efficacy up to 10 weeks after repeated CTB administrations over 2 weeks [47]. Although not reported, the treatment regimen must have elicited high levels of anti-CTB antibodies in the gut and blood circulation. Thus, further investigation is necessary to address long-term efficacy following repeated CTB dosing. TGFβ seems to be a major denominator of CTB-induced immunomodulatory activities. TGFβ is a pleiotropic cytokine playing critical roles in cell differentiation and proliferation, as well as dynamic biological processes in wound healing and immune responses [82,83,84]. The cytokine is also involved in various pathological conditions. For example, elevated TGFβ levels have been correlated to the development of fibrosis following injury to the skin [85]. TGFβ mediates epithelial-to-mesenchymal transition (EMT) [86], and reduction of TGFβ1 levels in a mouse model of pulmonary fibrosis blunted fibrosis [87]. TGFβ signaling also has important implications in cancer. Although the cytokine functions as a suppressor of tumorigenesis at an early stage of tumor development, its expression is correlated with tumor progression and poor prognosis at late stages [84,88]. Collectively, the double-edged sword nature of TGFβ points to the importance of careful investigation of possible consequences upon long-term CTB dosing for the treatment of chronic inflammatory diseases. As mentioned in Section 3.2, CTBp treatment significantly mitigated gut inflammation and reduced tumor development in a model of CAC [19], providing a basis for further investigations of long-term therapeutic use of CTB for the treatment of IBD.
Of considerable interest may be population-based studies investigating potential association between the Dukoral® vaccine and gastrointestinal disorders involving mucosal inflammation. In a very recent study of patients who were diagnosed with colorectal cancer from July 2005 through December 2012 in Sweden, it was revealed that those who had previously received Dukoral® had a significantly reduced risk of death from colorectal cancer (CRC) [89]. Although the underlying mechanism is not clear at this point, the authors speculated that CTB might be associated with a risk reduction of CRC [89]. This observation warrants a comprehensive investigation on this subject.
In conclusion, even though CTB has been studied since the early 1970s [3], its immunomodulatory mechanisms appear to involve complex interplay between epithelial and immune cells that requires a systematic approach for comprehensive understanding. The studies highlighted herein strongly suggest CTB’s potential as an effective mucosal anti-inflammatory agent with the potential to replace or supplement currently available therapies for the treatment of inflammatory disorders of the mucosa, such as anti-TNFα biologics used in IBD patients who are refractory to conventional medications. As anti-TNFα agents are administered systemically, these agents have limited efficacy for the induction of mucosal healing [90,91] and/or pose severe adverse reactions [92,93,94]. In contrast, CTB has few, if any, adverse effects (according to the CD clinical trial [47]), can directly heal lesions/ulcers, and blunt inflammation upon topical administration. Therefore, to aid in developing CTB-based therapeutic strategies against various mucosal immunopathological conditions, further research that delineates how CTB can modulate epithelial cell signaling and T cell functions simultaneously is warranted.
Acknowledgments
We thank Matthew Dent for critical reading of the manuscript. This manuscript is based on work supported by the Leona M. and Harry B. Helmsley Charitable Trust Fund.
Author Contributions
J.M.R. and N.M. conceived and designed the review idea and contents, and wrote the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sanchez, J.; Holmgren, J. Cholera toxin structure, gene regulation and pathophysiological and immunological aspects. Cell. Mol. Life Sci. 2008, 65, 1347–1360. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, R.A.; LoSpalluto, J.J. Pathogenesis of experimental cholera. Preparation and isolation of choleragen and choleragenoid. J. Exp. Med. 1969, 130, 185–202. [Google Scholar] [CrossRef] [PubMed]
- Lonnroth, I.; Holmgren, J. Subunit structure of cholera toxin. J. Gen. Microbiol. 1973, 76, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Baldauf, K.J.; Royal, J.M.; Hamorsky, K.T.; Matoba, N. Cholera toxin B: One subunit with many pharmaceutical applications. Toxins 2015, 7, 974–996. [Google Scholar] [CrossRef] [PubMed]
- Cholera vaccines: WHO position paper. Relev. Epidemiol. Hebd. 2010, 85, 117–128.
- Bergquist, C.; Johansson, E.L.; Lagergard, T.; Holmgren, J.; Rudin, A. Intranasal vaccination of humans with recombinant cholera toxin B subunit induces systemic and local antibody responses in the upper respiratory tract and the vagina. Infect. Immun. 1997, 65, 2676–2684. [Google Scholar] [PubMed]
- Jertborn, M.; Nordstrom, I.; Kilander, A.; Czerkinsky, C.; Holmgren, J. Local and systemic immune responses to rectal administration of recombinant cholera toxin B subunit in humans. Infect. Immun. 2001, 69, 4125–4128. [Google Scholar] [CrossRef] [PubMed]
- Kozlowski, P.A.; Cu-Uvin, S.; Neutra, M.R.; Flanigan, T.P. Comparison of the oral, rectal, and vaginal immunization routes for induction of antibodies in rectal and genital tract secretions of women. Infect. Immun. 1997, 65, 1387–1394. [Google Scholar] [PubMed]
- Cuatrecasas, P. Interaction of Vibrio cholerae enterotoxin with cell membranes. Biochemistry 1973, 12, 3547–3558. [Google Scholar] [CrossRef] [PubMed]
- Kuziemko, G.M.; Stroh, M.; Stevens, R.C. Cholera toxin binding affinity and specificity for gangliosides determined by surface plasmon resonance. Biochemistry 1996, 35, 6375–6384. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, C.R.; Hirama, T.; Lee, K.K.; Altman, E.; Young, N.M. Quantitative analysis of bacterial toxin affinity and specificity for glycolipid receptors by surface plasmon resonance. J. Biol. Chem. 1997, 272, 5533–5538. [Google Scholar] [CrossRef] [PubMed]
- Dawson, R.M. Characterization of the binding of cholera toxin to ganglioside GM1 immobilized onto microtitre plates. J. Appl. Toxicol. 2005, 25, 30–38. [Google Scholar] [CrossRef] [PubMed]
- George-Chandy, A.; Eriksson, K.; Lebens, M.; Nordstrom, I.; Schon, E.; Holmgren, J. Cholera toxin B subunit as a carrier molecule promotes antigen presentation and increases CD40 and CD86 expression on antigen-presenting cells. Infect. Immun. 2001, 69, 5716–5725. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.B.; Rask, C.; Olsson, T.; Holmgren, J.; Czerkinsky, C. Treatment of experimental autoimmune encephalomyelitis by feeding myelin basic protein conjugated to cholera toxin B subunit. Proc. Natl. Acad. Sci. USA 1996, 93, 7196–7201. [Google Scholar] [CrossRef] [PubMed]
- Ploix, C.; Bergerot, I.; Durand, A.; Czerkinsky, C.; Holmgren, J.; Thivolet, C. Oral administration of cholera toxin B-insulin conjugates protects NOD mice from autoimmune diabetes by inducing CD4+ regulatory T-cells. Diabetes 1999, 48, 2150–2156. [Google Scholar] [CrossRef] [PubMed]
- Tarkowski, A.; Sun, J.B.; Holmdahl, R.; Holmgren, J.; Czerkinsky, C. Treatment of experimental autoimmune arthritis by nasal administration of a type II collagen-cholera toxoid conjugate vaccine. Arthritis Rheum. 1999, 42, 1628–1634. [Google Scholar] [CrossRef]
- Rask, C.; Holmgren, J.; Fredriksson, M.; Lindblad, M.; Nordstrom, I.; Sun, J.B.; Czerkinsky, C. Prolonged oral treatment with low doses of allergen conjugated to cholera toxin B subunit suppresses immunoglobulin E antibody responses in sensitized mice. Clin. Exp. Allergy 2000, 30, 1024–1032. [Google Scholar] [CrossRef] [PubMed]
- Stanford, M.; Whittall, T.; Bergmeier, L.A.; Lindblad, M.; Lundin, S.; Shinnick, T.; Mizushima, Y.; Holmgren, J.; Lehner, T. Oral tolerization with peptide 336–351 linked to cholera toxin B subunit in preventing relapses of uveitis in Behcet’s disease. Clin. Exp. Immunol. 2004, 137, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Baldauf, K.J.; Royal, J.M.; Kouokam, J.C.; Haribabu, B.; Jala, V.R.; Yaddanapudi, K.; Hamorsky, K.T.; Dryden, G.W.; Matoba, N. Oral administration of a recombinant cholera toxin B subunit promotes mucosal healing in the colon. Mucosal Immunol. 2017, 10, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Wernick, N.L.; Chinnapen, D.J.; Cho, J.A.; Lencer, W.I. Cholera toxin: An intracellular journey into the cytosol by way of the endoplasmic reticulum. Toxins 2010, 2, 310–325. [Google Scholar] [CrossRef] [PubMed]
- Chinnapen, D.J.; Chinnapen, H.; Saslowsky, D.; Lencer, W.I. Rafting with cholera toxin: Endocytosis and trafficking from plasma membrane to ER. FEMS Microbiol. Lett. 2007, 266, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.G.; Scott, D.L.; Westbrook, M.L.; Nance, S.; Spangler, B.D.; Shipley, G.G.; Westbrook, E.M. The three-dimensional crystal structure of cholera toxin. J. Mol. Biol. 1995, 251, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Lencer, W.I.; Constable, C.; Moe, S.; Jobling, M.G.; Webb, H.M.; Ruston, S.; Madara, J.L.; Hirst, T.R.; Holmes, R.K. Targeting of cholera toxin and Escherichia coli heat labile toxin in polarized epithelia: Role of COOH-terminal KDEL. J. Cell Biol. 1995, 131, 951–962. [Google Scholar] [CrossRef] [PubMed]
- Fujinaga, Y.; Wolf, A.A.; Rodighiero, C.; Wheeler, H.; Tsai, B.; Allen, L.; Jobling, M.G.; Rapoport, T.; Holmes, R.K.; Lencer, W.I. Gangliosides that associate with lipid rafts mediate transport of cholera and related toxins from the plasma membrane to endoplasmic reticulm. Mol. Biol. Cell 2003, 14, 4783–4793. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, J.; Holmgren, J. Cholera toxin—A foe & a friend. Indian J. Med. Res. 2011, 133, 153–163. [Google Scholar] [PubMed]
- Sun, J.B.; Holmgren, J.; Czerkinsky, C. Cholera toxin B subunit: An efficient transmucosal carrier-delivery system for induction of peripheral immunological tolerance. Proc. Natl Acad. Sci. USA 1994, 91, 10795–10799. [Google Scholar] [CrossRef] [PubMed]
- Weiner, H.L. Oral tolerance. Proc. Natl. Acad. Sci. USA 1994, 91, 10762–10765. [Google Scholar] [CrossRef] [PubMed]
- Bublin, M.; Hoflehner, E.; Wagner, B.; Radauer, C.; Wagner, S.; Hufnagl, K.; Allwardt, D.; Kundi, M.; Scheiner, O.; Wiedermann, U.; et al. Use of a genetic cholera toxin B subunit/allergen fusion molecule as mucosal delivery system with immunosuppressive activity against Th2 immune responses. Vaccine 2007, 25, 8395–8404. [Google Scholar] [CrossRef] [PubMed]
- Ruhlman, T.; Ahangari, R.; Devine, A.; Samsam, M.; Daniell, H. Expression of cholera toxin B-proinsulin fusion protein in lettuce and tobacco chloroplasts–oral administration protects against development of insulitis in non-obese diabetic mice. Plant Biotechnol. J. 2007, 5, 495–510. [Google Scholar] [CrossRef] [PubMed]
- Carter, J.E., III; Yu, J.; Choi, N.W.; Hough, J.; Henderson, D.; He, D.; Langridge, W.H. Bacterial and plant enterotoxin B subunit-autoantigen fusion proteins suppress diabetes insulitis. Mol. Biotechnol. 2006, 32, 1–15. [Google Scholar] [CrossRef]
- Arakawa, T.; Yu, J.; Chong, D.K.; Hough, J.; Engen, P.C.; Langridge, W.H. A plant-based cholera toxin B subunit-insulin fusion protein protects against the development of autoimmune diabetes. Nat. Biotechnol. 1998, 16, 934–938. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.B.; Mielcarek, N.; Lakew, M.; Grzych, J.M.; Capron, A.; Holmgren, J.; Czerkinsky, C. Intranasal administration of a Schistosoma mansoni glutathione S-transferase-cholera toxoid conjugate vaccine evokes antiparasitic and antipathological immunity in mice. J. Immunol. (Baltimore, Md: 1950) 1999, 163, 1045–1052. [Google Scholar]
- McSorley, S.J.; Rask, C.; Pichot, R.; Julia, V.; Czerkinsky, C.; Glaichenhaus, N. Selective tolerization of Th1-like cells after nasal administration of a cholera toxoid-LACK conjugate. Eur. J. Immunol. 1998, 28, 424–432. [Google Scholar] [CrossRef]
- Czerkinsky, C.; Anjuere, F.; McGhee, J.R.; George-Chandy, A.; Holmgren, J.; Kieny, M.P.; Fujiyashi, K.; Mestecky, J.F.; Pierrefite-Carle, V.; Rask, C.; et al. Mucosal immunity and tolerance: Relevance to vaccine development. Immunol. Rev. 1999, 170, 197–222. [Google Scholar] [CrossRef] [PubMed]
- Phipps, P.A.; Stanford, M.R.; Sun, J.B.; Xiao, B.G.; Holmgren, J.; Shinnick, T.; Hasan, A.; Mizushima, Y.; Lehner, T. Prevention of mucosally induced uveitis with a HSP60-derived peptide linked to cholera toxin B subunit. Eur. J. Immunol. 2003, 33, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.B.; Xiao, B.G.; Lindblad, M.; Li, B.L.; Link, H.; Czerkinsky, C.; Holmgren, J. Oral administration of cholera toxin B subunit conjugated to myelin basic protein protects against experimental autoimmune encephalomyelitis by inducing transforming growth factor-beta-secreting cells and suppressing chemokine expression. Int. Immunol. 2000, 12, 1449–1457. [Google Scholar] [CrossRef] [PubMed]
- Bergerot, I.; Ploix, C.; Petersen, J.; Moulin, V.; Rask, C.; Fabien, N.; Lindblad, M.; Mayer, A.; Czerkinsky, C.; Holmgren, J.; et al. A cholera toxoid-insulin conjugate as an oral vaccine against spontaneous autoimmune diabetes. Proc. Natl. Acad. Sci. USA 1997, 94, 4610–4614. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.B.; Czerkinsky, C.; Holmgren, J. Mucosally induced immunological tolerance, regulatory T cells and the adjuvant effect by cholera toxin B subunit. Scand. J. Immunol. 2010, 71, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Boirivant, M.; Fuss, I.J.; Ferroni, L.; De Pascale, M.; Strober, W. Oral administration of recombinant cholera toxin subunit B inhibits IL-12-mediated murine experimental (trinitrobenzene sulfonic acid) colitis. J. Immunol. (Baltimore, Md: 1950) 2001, 166, 3522–3532. [Google Scholar] [CrossRef]
- Tamura, S.; Yamanaka, A.; Shimohara, M.; Tomita, T.; Komase, K.; Tsuda, Y.; Suzuki, Y.; Nagamine, T.; Kawahara, K.; Danbara, H.; et al. Synergistic action of cholera toxin B subunit (and Escherichia coli heat-labile toxin B subunit) and a trace amount of cholera whole toxin as an adjuvant for nasal influenza vaccine. Vaccine 1994, 12, 419–426. [Google Scholar] [CrossRef]
- Kim, P.H.; Eckmann, L.; Lee, W.J.; Han, W.; Kagnoff, M.F. Cholera toxin and cholera toxin B subunit induce IgA switching through the action of TGF-beta 1. J. Immunol. (Baltimore, Md: 1950) 1998, 160, 1198–1203. [Google Scholar]
- Reinholdt, J.; Husby, S. IgA and Mucosal Homeostasis. In Madame Curie Bioscience Database; Landes Bioscience: Austin, TX, USA, 2000–2013. [Google Scholar]
- Corthesy, B. Role of secretory IgA in infection and maintenance of homeostasis. Autoimmun. Rev. 2013, 12, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Smits, H.H.; Gloudemans, A.K.; van Nimwegen, M.; Willart, M.A.; Soullie, T.; Muskens, F.; de Jong, E.C.; Boon, L.; Pilette, C.; Johansen, F.E.; et al. Cholera toxin B suppresses allergic inflammation through induction of secretory IgA. Mucosal Immunol. 2009, 2, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M.F.; Finotto, S.; Glimcher, L.H. The role of Th1/Th2 polarization in mucosal immunity. Nat. Med. 2002, 8, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Coccia, E.M.; Remoli, M.E.; Di Giacinto, C.; Del Zotto, B.; Giacomini, E.; Monteleone, G.; Boirivant, M. Cholera toxin subunit B inhibits IL-12 and IFN-[61] production and signaling in experimental colitis and Crohn’s disease. Gut 2005, 54, 1558–1564. [Google Scholar] [CrossRef] [PubMed]
- Stal, P.; Befrits, R.; Ronnblom, A.; Danielsson, A.; Suhr, O.; Stahlberg, D.; Brinkberg Lapidus, A.; Lofberg, R. Clinical trial: The safety and short-term efficacy of recombinant cholera toxin B subunit in the treatment of active Crohn’s disease. Aliment. Pharmacol. Ther. 2010, 31, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Aman, A.T.; Fraser, S.; Merritt, E.A.; Rodigherio, C.; Kenny, M.; Ahn, M.; Hol, W.G.; Williams, N.A.; Lencer, W.I.; Hirst, T.R. A mutant cholera toxin B subunit that binds GM1-ganglioside but lacks immunomodulatory or toxic activity. Proc. Natl. Acad. Sci. USA 2001, 98, 8536–8541. [Google Scholar] [CrossRef] [PubMed]
- D’Ambrosio, A.; Colucci, M.; Pugliese, O.; Quintieri, F.; Boirivant, M. Cholera toxin B subunit promotes the induction of regulatory T cells by preventing human dendritic cell maturation. J. Leukoc. Biol. 2008, 84, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Rouquette-Jazdanian, A.K.; Foussat, A.; Lamy, L.; Pelassy, C.; Lagadec, P.; Breittmayer, J.P.; Aussel, C. Cholera toxin B-subunit prevents activation and proliferation of human CD4+ T cells by activation of a neutral sphingomyelinase in lipid rafts. J. Immunol. (Baltimore, Md: 1950) 2005, 175, 5637–5648. [Google Scholar] [CrossRef]
- Dbaibo, G.S.; Pushkareva, M.Y.; Jayadev, S.; Schwarz, J.K.; Horowitz, J.M.; Obeid, L.M.; Hannun, Y.A. Retinoblastoma gene product as a downstream target for a ceramide-dependent pathway of growth arrest. Proc. Natl. Acad. Sci. USA 1995, 92, 1347–1351. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Hannun, Y.A.; Obeid, L.M. Ceramide inactivates cellular protein kinase Calpha. J. Biol. Chem. 1996, 271, 13169–13174. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Hannun, Y.A.; Obeid, L.M. Functional dichotomy of protein kinase C (PKC) in tumor necrosis factor-alpha (TNF-alpha) signal transduction in L929 cells. Translocation and inactivation of PKC by TNF-alpha. J. Biol. Chem. 2000, 275, 29290–29298. [Google Scholar] [CrossRef] [PubMed]
- Leoni, G.; Neumann, P.A.; Sumagin, R.; Denning, T.L.; Nusrat, A. Wound repair: Role of immune-epithelial interactions. Mucosal Immunol. 2015, 8, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Kurashima, Y.; Kiyono, H. Mucosal Ecological Network of Epithelium and Immune Cells for Gut Homeostasis and Tissue Healing. Annu. Rev. Immunol. 2017, 35, 119–147. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Wolvers, D.; Stanisz, A.M.; Bienenstock, J. Interleukin-10 and nerve growth factor have reciprocal upregulatory effects on intestinal epithelial cells. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 284, R1323–R1329. [Google Scholar] [CrossRef] [PubMed]
- Stordeur, P.; Goldman, M. Interleukin-10 as a regulatory cytokine induced by cellular stress: Molecular aspects. Int. Rev. Immunol. 1998, 16, 501–522. [Google Scholar] [CrossRef] [PubMed]
- Hamorsky, K.T.; Kouokam, J.C.; Bennett, L.J.; Baldauf, K.J.; Kajiura, H.; Fujiyama, K.; Matoba, N. Rapid and scalable plant-based production of a cholera toxin B subunit variant to aid in mass vaccination against cholera outbreaks. PLoS Negl. Trop. Dis. 2013, 7, e2046. [Google Scholar] [CrossRef] [PubMed]
- Hamorsky, K.T.; Kouokam, J.C.; Jurkiewicz, J.M.; Nelson, B.; Moore, L.J.; Husk, A.S.; Kajiura, H.; Fujiyama, K.; Matoba, N. N-glycosylation of cholera toxin B subunit in Nicotiana benthamiana: Impacts on host stress response, production yield and vaccine potential. Sci. Rep. 2015, 5, 8003. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Huang, Y.; Lin, Y.; Shan, Y.; Tan, S.; Cai, W.; Li, H.; Zhang, B.; Men, X.; Lu, Z. Anti-inflammatory effect of cholera toxin B subunit in experimental stroke. J. Neuroinflamm. 2016, 13, 147. [Google Scholar] [CrossRef] [PubMed]
- Aspord, C.; Czerkinsky, C.; Durand, A.; Stefanutti, A.; Thivolet, C. alpha4 integrins and L-selectin differently orchestrate T-cell activity during diabetes prevention following oral administration of CTB-insulin. J. Autoimmun. 2002, 19, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Satoh, Y.; Ishiguro, Y.; Sakuraba, H.; Kawaguchi, S.; Hiraga, H.; Fukuda, S.; Nakane, A. Cyclosporine regulates intestinal epithelial apoptosis via TGF-beta-related signaling. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G514–G519. [Google Scholar] [CrossRef] [PubMed]
- Taverna, D.; Pollins, A.C.; Sindona, G.; Caprioli, R.M.; Nanney, L.B. Imaging mass spectrometry for assessing cutaneous wound healing: Analysis of pressure ulcers. J. Proteome Res. 2015, 14, 986–996. [Google Scholar] [CrossRef] [PubMed]
- Kiesler, P.; Fuss, I.J.; Strober, W. Experimental Models of Inflammatory Bowel Diseases. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 154–170. [Google Scholar] [CrossRef] [PubMed]
- Perse, M.; Cerar, A. Dextran sodium sulphate colitis mouse model: Traps and tricks. J. Biomed. Biotechnol. 2012, 2012, 718617. [Google Scholar] [CrossRef] [PubMed]
- Clapper, M.L.; Cooper, H.S.; Chang, W.C. Dextran sulfate sodium-induced colitis-associated neoplasia: A promising model for the development of chemopreventive interventions. Acta Pharmacol. Sin. 2007, 28, 1450–1459. [Google Scholar] [CrossRef] [PubMed]
- Jiminez, J.A.; Uwiera, T.C.; Douglas Inglis, G.; Uwiera, R.R. Animal models to study acute and chronic intestinal inflammation in mammals. Gut Pathog. 2015, 7, 29. [Google Scholar] [CrossRef] [PubMed]
- Chassaing, B.; Aitken, J.D.; Malleshappa, M.; Vijay-Kumar, M. Dextran sulfate sodium (DSS)-induced colitis in mice. Curr. Protoc. Immunol. 2014, 104. [Google Scholar] [CrossRef]
- Yashiro, M. Ulcerative colitis-associated colorectal cancer. World J. Gastroenterol. 2014, 20, 16389–16397. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I. Inflammation and colorectal cancer: Colitis-associated neoplasia. Semin. Immunopathol. 2013, 35, 229–244. [Google Scholar] [CrossRef] [PubMed]
- Denecke, J.; De Rycke, R.; Botterman, J. Plant and mammalian sorting signals for protein retention in the endoplasmic reticulum contain a conserved epitope. EMBO J. 1992, 11, 2345–2355. [Google Scholar] [PubMed]
- Yamamoto, K.; Hamada, H.; Shinkai, H.; Kohno, Y.; Koseki, H.; Aoe, T. The KDEL receptor modulates the endoplasmic reticulum stress response through mitogen-activated protein kinase signaling cascades. J. Biol. Chem. 2003, 278, 34525–34532. [Google Scholar] [CrossRef] [PubMed]
- Cancino, J.; Capalbo, A.; Di Campli, A.; Giannotta, M.; Rizzo, R.; Jung, J.E.; Di Martino, R.; Persico, M.; Heinklein, P.; Sallese, M.; et al. Control systems of membrane transport at the interface between the endoplasmic reticulum and the Golgi. Dev. Cell 2014, 30, 280–294. [Google Scholar] [CrossRef] [PubMed]
- Chusri, P.; Kumthip, K.; Hong, J.; Zhu, C.; Duan, X.; Jilg, N.; Fusco, D.N.; Brisac, C.; Schaefer, E.A.; Cai, D.; et al. HCV induces transforming growth factor beta1 through activation of endoplasmic reticulum stress and the unfolded protein response. Sci. Rep. 2016, 6, 22487. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, S.; Hiratsuka, T.; Taniguchi, M.; Shingaki, K.; Kubo, T.; Kiya, K.; Fujiwara, T.; Kanazawa, S.; Kanematsu, R.; Maeda, T.; et al. Physiological ER Stress Mediates the Differentiation of Fibroblasts. PLoS ONE 2015, 10, e0123578. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.S.; Song, B.; Kaufman, R.J. PKR protects colonic epithelium against colitis through the unfolded protein response and prosurvival signaling. Inflamm. Bowel Dis. 2012, 18, 1735–1742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, J.A.; Lee, A.H.; Platzer, B.; Cross, B.C.; Gardner, B.M.; De Luca, H.; Luong, P.; Harding, H.P.; Glimcher, L.H.; Walter, P.; et al. The unfolded protein response element IRE1alpha senses bacterial proteins invading the ER to activate RIG-I and innate immune signaling. Cell Host Microbe 2013, 13, 558–569. [Google Scholar] [CrossRef] [PubMed]
- Becker, B.; Blum, A.; Giesselmann, E.; Dausend, J.; Rammo, D.; Muller, N.C.; Tschacksch, E.; Steimer, M.; Spindler, J.; Becherer, U.; et al. H/KDEL receptors mediate host cell intoxication by a viral A/B toxin in yeast. Sci. Rep. 2016, 6, 31105. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Chen, X.; Vicini, P.; Rup, B.; Hickling, T.P. Therapeutic outcomes, assessments, risk factors and mitigation efforts of immunogenicity of therapeutic protein products. Cell. Immunol. 2015, 295, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Swanson, S.J.; Bussiere, J. Immunogenicity assessment in non-clinical studies. Curr. Opin. Microbiol. 2012, 15, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Biancheri, P.; Giuffrida, P.; Docena, G.H.; MacDonald, T.T.; Corazza, G.R.; Di Sabatino, A. The role of transforming growth factor (TGF)-beta in modulating the immune response and fibrogenesis in the gut. Cytokine Growth Factor Rev. 2014, 25, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Travis, M.A.; Sheppard, D. TGF-beta activation and function in immunity. Annu. Rev. Immunol. 2014, 32, 51–82. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-beta and the TGF-beta Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Penn, J.W.; Grobbelaar, A.O.; Rolfe, K.J. The role of the TGF-beta family in wound healing, burns and scarring: A review. Int. J. Burns Trauma 2012, 2, 18–28. [Google Scholar] [PubMed]
- Kim, M.K.; Maeng, Y.I.; Sung, W.J.; Oh, H.K.; Park, J.B.; Yoon, G.S.; Cho, C.H.; Park, K.K. The differential expression of TGF-beta1, ILK and wnt signaling inducing epithelial to mesenchymal transition in human renal fibrogenesis: An immunohistochemical study. Int. J. Clin. Exp. Pathol. 2013, 6, 1747–1758. [Google Scholar] [PubMed]
- Zhang, Y.Q.; Liu, Y.J.; Mao, Y.F.; Dong, W.W.; Zhu, X.Y.; Jiang, L. Resveratrol ameliorates lipopolysaccharide-induced epithelial mesenchymal transition and pulmonary fibrosis through suppression of oxidative stress and transforming growth factor-beta1 signaling. Clin. Nutr. (Edinburgh, Scotland) 2015, 34, 752–760. [Google Scholar] [CrossRef] [PubMed]
- Colak, S.; Ten Dijke, P. Targeting TGF-beta Signaling in Cancer. Trends Cancer 2017, 3, 56–71. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Sundquist, J.; Sundquist, K. Cholera Vaccine Use Is Associated with a Reduced Risk of Death in Patients with Colorectal Cancer: A Population-based Study. Gastroenterology 2017. [Google Scholar] [CrossRef] [PubMed]
- Villanacci, V.; Antonelli, E.; Geboes, K.; Casella, G.; Bassotti, G. Histological healing in inflammatory bowel disease: A still unfulfilled promise. World J. Gastroenterol. 2013, 19, 968–978. [Google Scholar] [CrossRef] [PubMed]
- Vaughn, B.P.; Shah, S.; Cheifetz, A.S. The role of mucosal healing in the treatment of patients with inflammatory bowel disease. Curr. Treat. Options Gastroenterol. 2014, 12, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Hansel, T.T.; Kropshofer, H.; Singer, T.; Mitchell, J.A.; George, A.J. The safety and side effects of monoclonal antibodies. Nat. Rev. Drug Discov. 2010, 9, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Murdaca, G.; Spano, F.; Contatore, M.; Guastalla, A.; Penza, E.; Magnani, O.; Puppo, F. Infection risk associated with anti-TNF-alpha agents: A review. Expert Opin. Drug Saf. 2015, 14, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Saleh, M.; Trinchieri, G. Innate immune mechanisms of colitis and colitis-associated colorectal cancer. Nat. Rev. Immunol. 2011, 11, 9–20. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Summary of mechanisms involved in cholera toxin homopentameric B-subunit (CTB)’s inflammatory disease intervention.
Figure 1.
Summary of mechanisms involved in cholera toxin homopentameric B-subunit (CTB)’s inflammatory disease intervention.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Royal, J.M.; Matoba, N. Therapeutic Potential of Cholera Toxin B Subunit for the Treatment of Inflammatory Diseases of the Mucosa. Toxins 2017, 9, 379. https://doi.org/10.3390/toxins9120379
AMA Style
Royal JM, Matoba N. Therapeutic Potential of Cholera Toxin B Subunit for the Treatment of Inflammatory Diseases of the Mucosa. Toxins. 2017; 9(12):379. https://doi.org/10.3390/toxins9120379
Chicago/Turabian StyleRoyal, Joshua M., and Nobuyuki Matoba. 2017. "Therapeutic Potential of Cholera Toxin B Subunit for the Treatment of Inflammatory Diseases of the Mucosa" Toxins 9, no. 12: 379. https://doi.org/10.3390/toxins9120379
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.