
The Potential Pathogenic Contributions of Endothelial Barrier and Arterial Contractile Dysfunction to Shock Due to B. anthracis Lethal and Edema Toxins

{kind=link}
{kind=link}
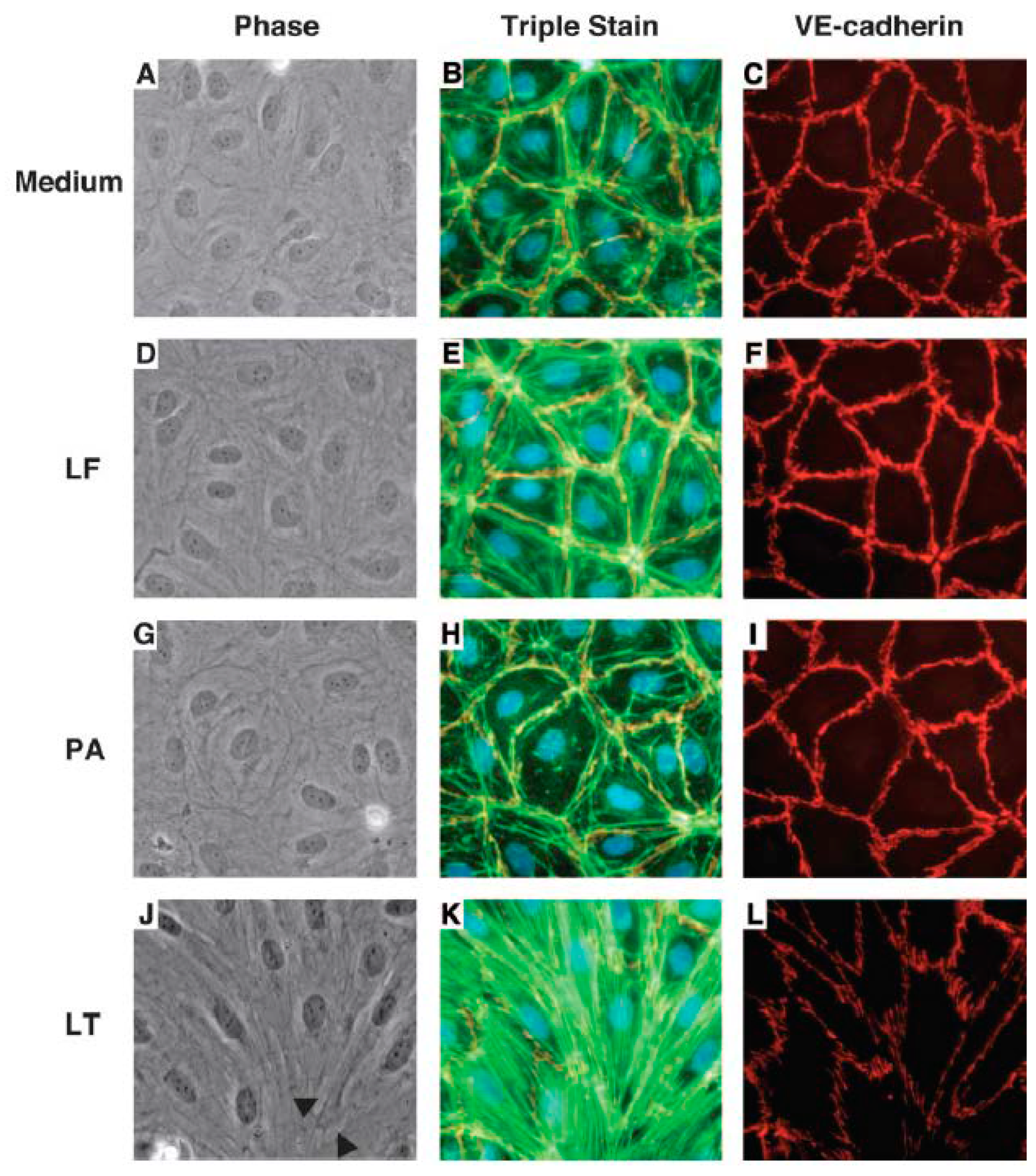{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Lethal and Edema Toxins: Structure, Receptors and Actions
3. Toxin Associated Barrier Dysfunction
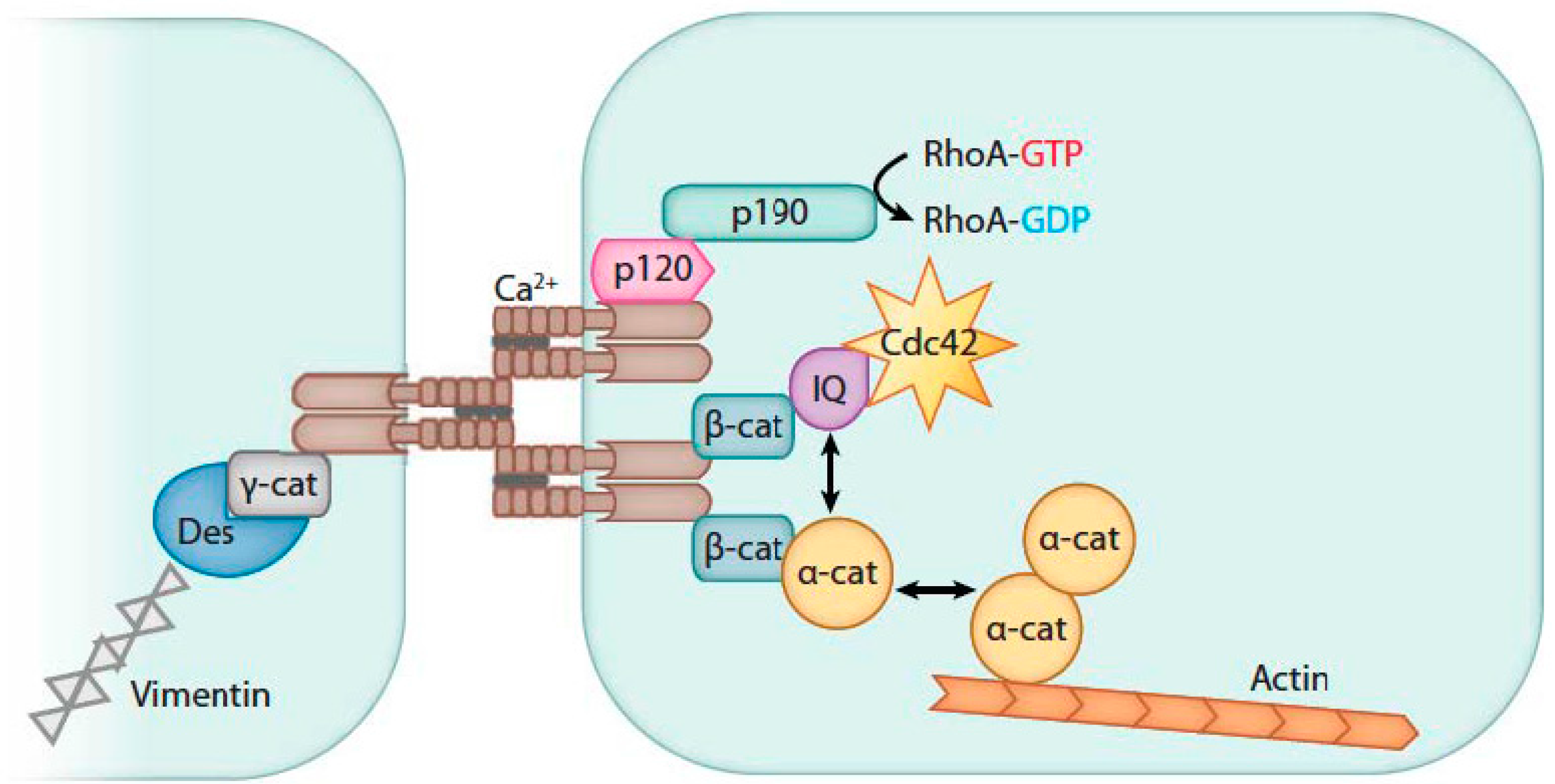
3.1. Endothelial Barrier Biology
3.2. In Vivo and In Vitro Data Implicating LT or ET in Endothelial Barrier Disruption
4. Toxin Associated Arterial Contractile Dysfunction
4.1. Arterial Contractile Biology
4.2. In Vivo and In Vitro Data Implicating LT and ET in Arterial Contractile Dysfunction
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Booth, M.G.; Hood, J.; Brooks, T.J.; Hart, A. Anthrax infection in drug users. Lancet 2010, 375, 1345–1346. [Google Scholar] [CrossRef]
- Jernigan, J.A.; Stephens, D.S.; Ashford, D.A.; Omenaca, C.; Topiel, M.S.; Galbraith, M.; Tapper, M.; Fisk, T.L.; Zaki, S.; Popovic, T.; et al. Bioterrorism-related inhalational anthrax: The first 10 cases reported in the united states. Emerg. Infect. Dis. 2001, 7, 933–944. [Google Scholar] [CrossRef] [PubMed]
- Booth, M.; Donaldson, L.; Cui, X.; Sun, J.; Cole, S.; Dailsey, S.; Hart, A.; Johns, N.; McConnell, P.; McLennan, T.; et al. Confirmed Bacillus anthracis infection among persons who inject drugs, scotland, 2009–2010. Emerg. Infect. Dis. 2014, 20, 1452–1463. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Nolen, L.D.; Sun, J.; Booth, M.; Donaldson, L.; Quinn, C.P.; Boyer, A.E.; Hendricks, K.; Shadomy, S.; Bothma, P.; et al. Analysis of anthrax immune globulin intravenous with antimicrobial treatment in injection drug users, Scotland, 2009–2010. Emerg. Infect. Dis. 2017, 23, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, E.K.; Rubenstein, A.R.; Radin, G.T.; Wiener, R.S.; Walkey, A.J. Two decades of mortality trends among patients with severe sepsis: A comparative meta-analysis*. Crit. Care Med. 2014, 42, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Angus, D.C.; van der Poll, T. Severe sepsis and septic shock. N. Engl. J. Med. 2013, 369, 840–851. [Google Scholar] [CrossRef] [PubMed]
- Epstein, L.; Dantes, R.; Magill, S.; Fiore, A. Varying estimates of sepsis mortality using death certificates and administrative codes—United States, 1999–2014. Morb. Mortal. Wkly. Rep. 2016, 65, 342–345. [Google Scholar] [CrossRef] [PubMed]
- Coggeshall, K.M.; Lupu, F.; Ballard, J.; Metcalf, J.P.; James, J.A.; Farris, D.; Kurosawa, S. The sepsis model: An emerging hypothesis for the lethality of inhalation anthrax. J. Cell. Mol. Med. 2013, 17, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Remy, K.E.; Qiu, P.; Li, Y.; Cui, X.; Eichacker, P.Q.B. Anthracis associated cardiovascular dysfunction and shock: The potential contribution of both non-toxin and toxin components. BMC Med. 2013, 11, 217. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Moayeri, M.; Purcell, R. Monoclonal antibody therapies against anthrax. Toxins 2011, 3, 1004–1019. [Google Scholar] [CrossRef] [PubMed]
- Pezard, C.; Berche, P.; Mock, M. Contribution of individual toxin components to virulence of Bacillus anthracis. Infect. Immun. 1991, 59, 3472–3477. [Google Scholar] [PubMed]
- Leysath, C.E.; Chen, K.H.; Moayeri, M.; Crown, D.; Fattah, R.; Chen, Z.; Das, S.R.; Purcell, R.H.; Leppla, S.H. Mouse monoclonal antibodies to anthrax edema factor protect against infection. Infect. Immun. 2011, 79, 4609–4616. [Google Scholar] [CrossRef] [PubMed]
- Moayeri, M.; Haines, D.; Young, H.A.; Leppla, S.H. Bacillus anthracis lethal toxin induces tnf-alpha-independent hypoxia-mediated toxicity in mice. J. Clin. Investig. 2003, 112, 670–682. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, D.A.; Cui, X.; Solomon, S.B.; Vitberg, D.A.; Migone, T.S.; Scher, D.; Danner, R.L.; Natanson, C.; Subramanian, G.M.; Eichacker, P.Q. Anthrax lethal and edema toxins produce different patterns of cardiovascular and renal dysfunction and synergistically decrease survival in canines. J. Infect. Dis. 2010, 202, 1885–1896. [Google Scholar] [CrossRef] [PubMed]
- Firoved, A.M.; Miller, G.F.; Moayeri, M.; Kakkar, R.; Shen, Y.; Wiggins, J.F.; McNally, E.M.; Tang, W.J.; Leppla, S.H. Bacillus anthracis edema toxin causes extensive tissue lesions and rapid lethality in mice. Am. J. Pathol. 2005, 167, 1309–1320. [Google Scholar] [CrossRef]
- Frankel, A.E.; Kuo, S.R.; Dostal, D.; Watson, L.; Duesbery, N.S.; Cheng, C.P.; Cheng, H.J.; Leppla, S.H. Pathophysiology of anthrax. Front. Biosci. (Landmark Ed.) 2009, 14, 4516–4524. [Google Scholar] [CrossRef] [PubMed]
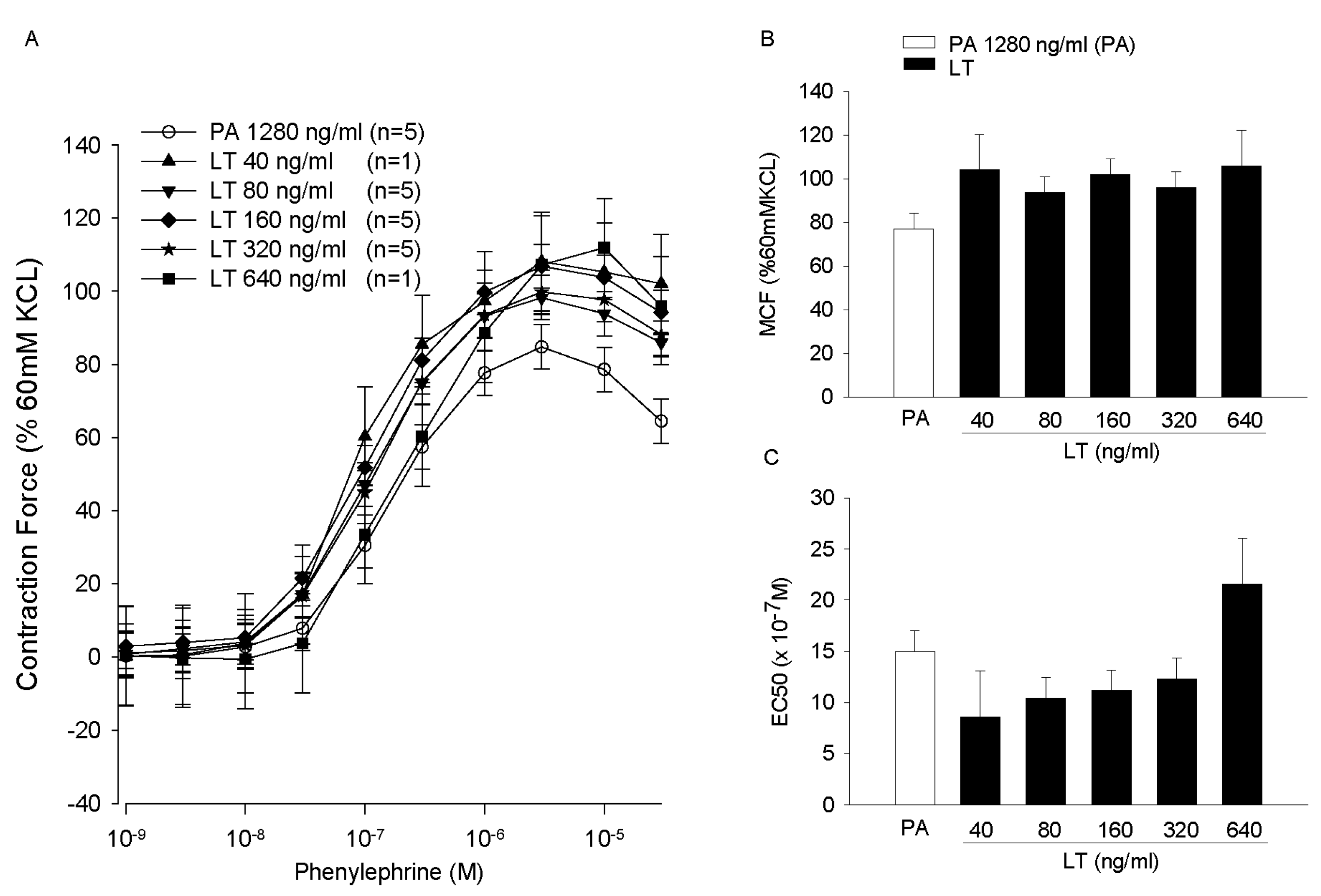
- Cui, X.; Moayeri, M.; Li, Y.; Li, X.; Haley, M.; Fitz, Y.; Correa-Araujo, R.; Banks, S.M.; Leppla, S.H.; Eichacker, P.Q. Lethality during continuous anthrax lethal toxin infusion is associated with circulatory shock but not inflammatory cytokine or nitric oxide release in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 286, R699–R709. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Li, Y.; Li, X.; Laird, M.W.; Subramanian, M.; Moayeri, M.; Leppla, S.H.; Fitz, Y.; Su, J.; Sherer, K.; et al. Bacillus anthracis edema and lethal toxin have different hemodynamic effects but function together to worsen shock and outcome in a rat model. J. Infect. Dis. 2007, 195, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Suffredini, D.A.; Sampath-Kumar, H.; Li, Y.; Ohanjanian, L.; Remy, K.E.; Cui, X.; Eichacker, P.Q. Does Bacillus anthracis lethal toxin directly depress myocardial function? A review of clinical cases and preclinical studies. Toxins 2015, 7, 5417–5434. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Moayeri, M.; Leppla, S.H. Anthrax lethal and edema toxins in anthrax pathogenesis. Trends Microbiol. 2014, 22, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Young, J.A.; Collier, R.J. Anthrax toxin: Receptor binding, internalization, pore formation, and translocation. Annu. Rev. Biochem. 2007, 76, 243–265. [Google Scholar] [CrossRef] [PubMed]
- Bradley, K.A.; Mogridge, J.; Mourez, M.; Collier, R.J.; Young, J.A. Identification of the cellular receptor for anthrax toxin. Nature 2001, 414, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Scobie, H.M.; Rainey, G.J.; Bradley, K.A.; Young, J.A. Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc. Natl. Acad. Sci. USA 2003, 100, 5170–5174. [Google Scholar] [CrossRef] [PubMed]
- Bonuccelli, G.; Sotgia, F.; Frank, P.G.; Williams, T.M.; de Almeida, C.J.; Tanowitz, H.B.; Scherer, P.E.; Hotchkiss, K.A.; Terman, B.I.; Rollman, B.; et al. ATR/TEM8 is highly expressed in epithelial cells lining bacillus anthracis’ three sites of entry: Implications for the pathogenesis of anthrax infection. Am. J. Physiol. Cell Physiol. 2005, 288, C1402–C1410. [Google Scholar] [CrossRef] [PubMed]
- Leppla, S.H. Anthrax toxin edema factor: A bacterial adenylate cyclase that increases cAMP concentrations of eukaryotic cells. Proc. Natl. Acad. Sci. USA 1982, 79, 3162–3166. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Zhukovskaya, N.L.; Guo, Q.; Florian, J.; Tang, W.J. Calcium-independent calmodulin binding and two-metal-ion catalytic mechanism of anthrax edema factor. EMBO J. 2005, 24, 929–941. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Doebele, R.C.; Lingen, M.W.; Quilliam, L.A.; Tang, W.J.; Rosner, M.R. Anthrax edema toxin inhibits endothelial cell chemotaxis via Epac and Rap1. J. Biol. Chem. 2007, 282, 19781–19787. [Google Scholar] [CrossRef] [PubMed]
- Lowe, D.E.; Glomski, I.J. Cellular and physiological effects of anthrax exotoxin and its relevance to disease. Front. Cell. Infect. Microbiol. 2012, 2, 76. [Google Scholar] [CrossRef] [PubMed]
- Pannifer, A.D.; Wong, T.Y.; Schwarzenbacher, R.; Renatus, M.; Petosa, C.; Bienkowska, J.; Lacy, D.B.; Collier, R.J.; Park, S.; Leppla, S.H.; et al. Crystal structure of the anthrax lethal factor. Nature 2001, 414, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Duesbery, N.S.; Webb, C.P.; Leppla, S.H.; Gordon, V.M.; Klimpel, K.R.; Copeland, T.D.; Ahn, N.G.; Oskarsson, M.K.; Fukasawa, K.; Paull, K.D.; et al. Proteolytic inactivation of map-kinase-kinase by anthrax lethal factor. Science 1998, 280, 734–737. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, W.; Torigoe, C.; Fang, H.; Xie, T.; Frucht, D.M. Anthrax lethal toxin inhibits translation of hypoxia-inducible factor 1-alpha and causes decreased tolerance to hypoxic stress. J. Biol. Chem. 2014, 289, 4180–4190. [Google Scholar] [CrossRef] [PubMed]
- Bromberg-White, J.; Lee, C.S.; Duesbery, N. Consequences and utility of the zinc-dependent metalloprotease activity of anthrax lethal toxin. Toxins 2010, 2, 1038–1053. [Google Scholar] [CrossRef] [PubMed]
- Moayeri, M.; Sastalla, I.; Leppla, S.H. Anthrax and the inflammasome. Microbes Infect. 2012, 14, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Moayeri, M.; Leppla, S.H.; Vrentas, C.; Pomerantsev, A.P.; Liu, S. Anthrax pathogenesis. Annu. Rev. Microbiol. 2015, 69, 185–208. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Li, Y.; Li, X.; Haley, M.; Moayeri, M.; Fitz, Y.; Leppla, S.H.; Eichacker, P.Q. Sublethal doses of Bacillus anthracis lethal toxin inhibit inflammation with lipopolysaccharide and Escherichia coli challenge but have opposite effects on survival. J. Infect. Dis. 2006, 193, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Baldari, C.T.; Tonello, F.; Paccani, S.R.; Montecucco, C. Anthrax toxins: A paradigm of bacterial immune suppression. Trends Immunol. 2006, 27, 434–440. [Google Scholar] [CrossRef] [PubMed]
- Paccani, S.R.; Baldari, C.T. T cell targeting by anthrax toxins: Two faces of the same coin. Toxins 2011, 3, 660–671. [Google Scholar] [CrossRef] [PubMed]
- Komarova, Y.; Malik, A.B. Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu. Rev. Physiol. 2010, 72, 463–493. [Google Scholar] [CrossRef] [PubMed]
- Radeva, M.Y.; Waschke, J. Mind the gap: Mechanisms regulating the endothelial barrier. Acta Physiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Sayner, S.L.; Balczon, R.; Frank, D.W.; Cooper, D.M.; Stevens, T. Filamin a is a phosphorylation target of membrane but not cytosolic adenylyl cyclase activity. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 301, L117–L124. [Google Scholar] [CrossRef] [PubMed]
- Sayner, S.L. Emerging themes of cAMP regulation of the pulmonary endothelial barrier. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 300, L667–L678. [Google Scholar] [CrossRef] [PubMed]
- Sherer, K.; Li, Y.; Cui, X.; Eichacker, P.Q. Lethal and edema toxins in the pathogenesis of Bacillus anthracis septic shock: Implications for therapy. Am. J. Respir. Crit. Care Med. 2007, 175, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Kirby, J.E. Anthrax lethal toxin induces human endothelial cell apoptosis. Infect. Immun. 2004, 72, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Warfel, J.M.; Steele, A.D.; D’Agnillo, F. Anthrax lethal toxin induces endothelial barrier dysfunction. Am. J. Pathol. 2005, 166, 1871–1881. [Google Scholar] [CrossRef]
- Warfel, J.M.; D’Agnillo, F. Anthrax lethal toxin-mediated disruption of endothelial ve-cadherin is attenuated by inhibition of the rho-associated kinase pathway. Toxins 2011, 3, 1278–1293. [Google Scholar] [CrossRef] [PubMed]
- D’Agnillo, F.; Williams, M.C.; Moayeri, M.; Warfel, J.M. Anthrax lethal toxin downregulates claudin-5 expression in human endothelial tight junctions. PLoS ONE 2013, 8, e62576. [Google Scholar] [CrossRef] [PubMed]
- Bolcome, R.E., 3rd; Sullivan, S.E.; Zeller, R.; Barker, A.P.; Collier, R.J.; Chan, J. Anthrax lethal toxin induces cell death-independent permeability in zebrafish vasculature. Proc. Natl. Acad. Sci. USA 2008, 105, 2439–2444. [Google Scholar] [CrossRef] [PubMed]
- Bolcome, R.E., 3rd; Chan, J. Constitutive Mek1 activation rescues anthrax lethal toxin-induced vascular effects in vivo. Infect. Immun. 2010, 78, 5043–5053. [Google Scholar] [CrossRef] [PubMed]
- Rolando, M.; Munro, P.; Stefani, C.; Auberger, P.; Flatau, G.; Lemichez, E. Injection of Staphylococcus aureus EDIN by the Bacillus anthracis protective antigen machinery induces vascular permeability. Infect. Immun. 2009, 77, 3596–3601. [Google Scholar] [CrossRef] [PubMed]
- Rolando, M.; Stefani, C.; Flatau, G.; Auberger, P.; Mettouchi, A.; Mhlanga, M.; Rapp, U.; Galmiche, A.; Lemichez, E. Transcriptome dysregulation by anthrax lethal toxin plays a key role in induction of human endothelial cell cytotoxicity. Cell. Microbiol. 2010, 12, 891–905. [Google Scholar] [CrossRef] [PubMed]
- Rolando, M.; Stefani, C.; Doye, A.; Acosta, M.I.; Visvikis, O.; Yevick, H.G.; Buchrieser, C.; Mettouchi, A.; Bassereau, P.; Lemichez, E. Contractile actin cables induced by Bacillus anthracis lethal toxin depend on the histone acetylation machinery. Cytoskeleton 2015, 72, 542–556. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Milia, E.; Warburton, R.R.; Hill, N.S.; Gaestel, M.; Kayyali, U.S. Anthrax lethal toxin disrupts the endothelial permeability barrier through blocking p38 signaling. J. Cell. Physiol. 2012, 227, 1438–1445. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Warburton, R.R.; Hill, N.S.; Kayyali, U.S. Anthrax lethal toxin-induced lung injury and treatment by activating Mk2. J. Appl. Physiol. 2015, 119, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Guichard, A.; Park, J.M.; Cruz-Moreno, B.; Karin, M.; Bier, E. Anthrax lethal factor and edema factor act on conserved targets in drosophila. Proc. Natl. Acad. Sci. USA 2006, 103, 3244–3249. [Google Scholar] [CrossRef] [PubMed]
- Guichard, A.; McGillivray, S.M.; Cruz-Moreno, B.; van Sorge, N.M.; Nizet, V.; Bier, E. Anthrax toxins cooperatively inhibit endocytic recycling by the Rab11/Sec15 exocyst. Nature 2010, 467, 854–858. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, C.C.; Mukherjee, A.; David, S.; Knaus, U.G.; Stearns-Kurosawa, D.J.; Kurosawa, S.; Parikh, S.M. Impaired function of the Tie-2 receptor contributes to vascular leakage and lethality in anthrax. Proc. Natl. Acad. Sci. USA 2012, 109, 10024–10029. [Google Scholar] [CrossRef] [PubMed]
- Gozes, Y.; Moayeri, M.; Wiggins, J.F.; Leppla, S.H. Anthrax lethal toxin induces ketotifen-sensitive intradermal vascular leakage in certain inbred mice. Infect. Immun. 2006, 74, 1266–1272. [Google Scholar] [CrossRef] [PubMed]
- Tessier, J.; Green, C.; Padgett, D.; Zhao, W.; Schwartz, L.; Hughes, M.; Hewlett, E. Contributions of histamine, prostanoids, and neurokinins to edema elicited by edema toxin from Bacillus anthracis. Infect. Immun. 2007, 75, 1895–1903. [Google Scholar] [CrossRef] [PubMed]
- Kamm, K.E.; Stull, J.T. Regulation of smooth muscle contractile elements by second messengers. Annu. Rev. Physiol. 1989, 51, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Sonkusare, S.K.; Bonev, A.D.; Ledoux, J.; Liedtke, W.; Kotlikoff, M.I.; Heppner, T.J.; Hill-Eubanks, D.C.; Nelson, M.T. Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science 2012, 336, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Navedo, M.F.; Amberg, G.C. Local regulation of l-type Ca2+ channel sparklets in arterial smooth muscle. Microcirculation 2013, 20, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, A.; Menice, C.B.; Laporte, R.; Morgan, K.G. Mechanisms of smooth muscle contraction. Physiol. Rev. 1996, 76, 967–1003. [Google Scholar] [PubMed]
- Somlyo, A.P.; Wu, X.; Walker, L.A.; Somlyo, A.V. Pharmacomechanical coupling: The role of calcium, g-proteins, kinases and phosphatases. Rev. Physiol. Biochem. Pharmacol. 1999, 134, 201–234. [Google Scholar] [PubMed]
- Hedges, J.C.; Oxhorn, B.C.; Carty, M.; Adam, L.P.; Yamboliev, I.A.; Gerthoffer, W.T. Phosphorylation of caldesmon by ERK MAP kinases in smooth muscle. Am. J. Physiol. Cell Physiol. 2000, 278, C718–C726. [Google Scholar] [PubMed]
- Trappanese, D.M.; Sivilich, S.; Ets, H.K.; Kako, F.; Autieri, M.V.; Moreland, R.S. Regulation of mitogen-activated protein kinase by protein kinase C and mitogen-activated protein kinase phosphatase-1 in vascular smooth muscle. Am. J. Physiol. Cell Physiol. 2016, 310, C921–C930. [Google Scholar] [CrossRef] [PubMed]
- Brozovich, F.V.; Nicholson, C.J.; Degen, C.V.; Gao, Y.Z.; Aggarwal, M.; Morgan, K.G. Mechanisms of vascular smooth muscle contraction and the basis for pharmacologic treatment of smooth muscle disorders. Pharmacol. Rev. 2016, 68, 476–532. [Google Scholar] [CrossRef] [PubMed]
- Morgado, M.; Cairrao, E.; Santos-Silva, A.J.; Verde, I. Cyclic nucleotide-dependent relaxation pathways in vascular smooth muscle. Cell. Mol. Life Sci. 2012, 69, 247–266. [Google Scholar] [CrossRef] [PubMed]
- Willoughby, D.; Cooper, D.M. Organization and Ca2+ regulation of adenylyl cyclases in cAMP microdomains. Physiol. Rev. 2007, 87, 965–1010. [Google Scholar] [CrossRef] [PubMed]
- Potter, L.R.; Abbey-Hosch, S.; Dickey, D.M. Natriuretic peptides, their receptors, and cyclic guanosine monophosphate-dependent signaling functions. Endocr. Rev. 2006, 27, 47–72. [Google Scholar] [CrossRef] [PubMed]
- Cary, S.P.; Winger, J.A.; Derbyshire, E.R.; Marletta, M.A. Nitric oxide signaling: No longer simply on or off. Trends Biochem. Sci. 2006, 31, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Akata, T. Cellular and molecular mechanisms regulating vascular tone. Part 2: Regulatory mechanisms modulating Ca2+ mobilization and/or myofilament Ca2+ sensitivity in vascular smooth muscle cells. J. Anesth. 2007, 21, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, H.; Springett, G.M.; Mochizuki, N.; Toki, S.; Nakaya, M.; Matsuda, M.; Housman, D.E.; Graybiel, A.M. A family of camp-binding proteins that directly activate Rap1. Science 1998, 282, 2275–2279. [Google Scholar] [CrossRef] [PubMed]
- Roberts, O.L.; Kamishima, T.; Barrett-Jolley, R.; Quayle, J.M.; Dart, C. Exchange protein activated by camp (Epac) induces vascular relaxation by activating Ca2+-sensitive K+ channels in rat mesenteric artery. J. Physiol. 2013, 591, 5107–5123. [Google Scholar] [CrossRef] [PubMed]
- Roberts, O.L.; Dart, C. Camp signalling in the vasculature: The role of Epac (exchange protein directly activated by camp). Biochem. Soc. Trans. 2014, 42, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhang, Y.; Moayeri, M.; Liu, J.; Crown, D.; Fattah, R.J.; Wein, A.N.; Yu, Z.X.; Finkel, T.; Leppla, S.H. Key tissue targets responsible for anthrax-toxin-induced lethality. Nature 2013, 501, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.A.; Fels, R.J.; Mosher, L.J.; Kenney, M.J. Bacillus anthracis lethal toxin alters regulation of visceral sympathetic nerve discharge. J. Appl. Physiol. 2012, 112, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
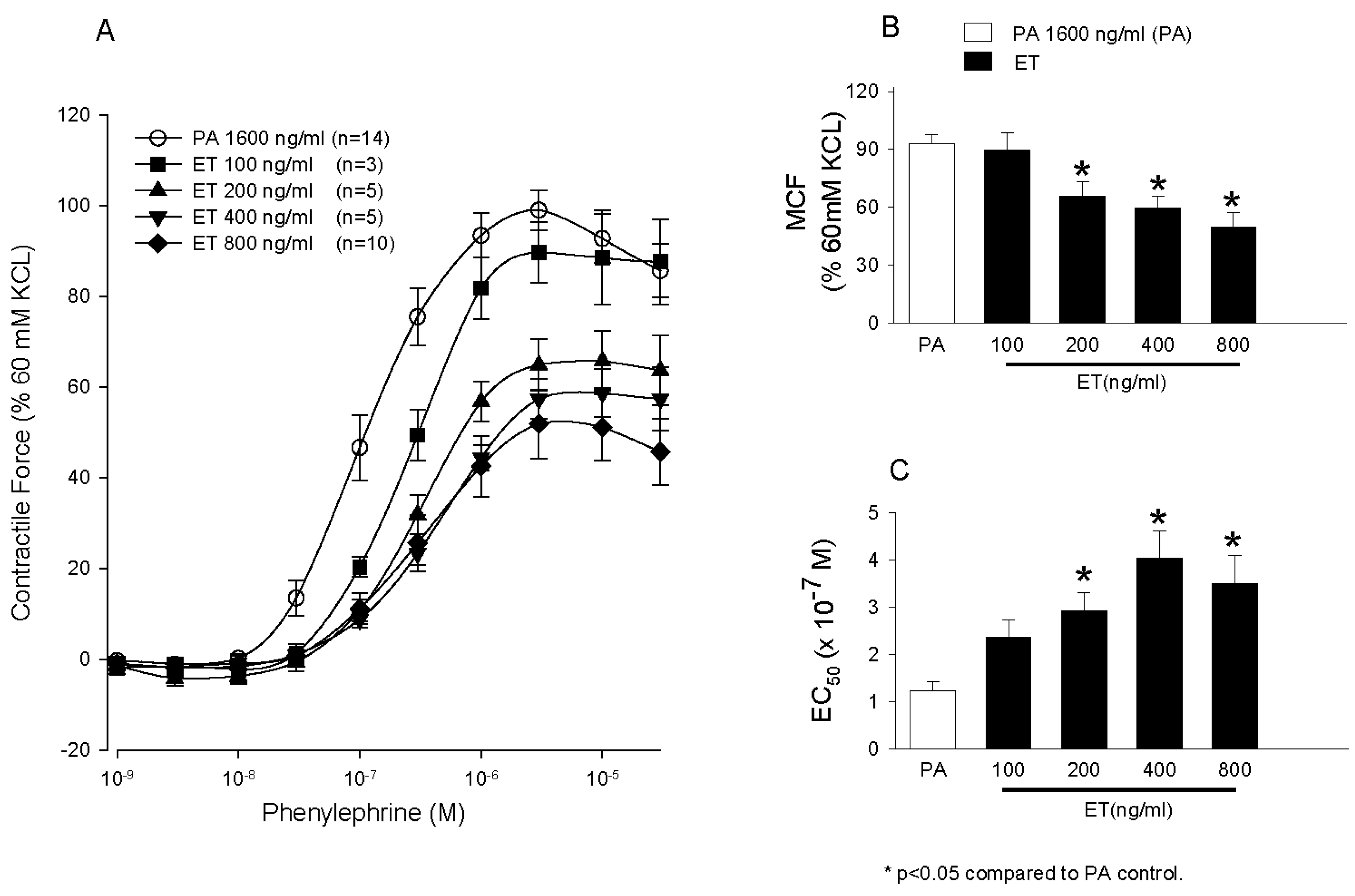
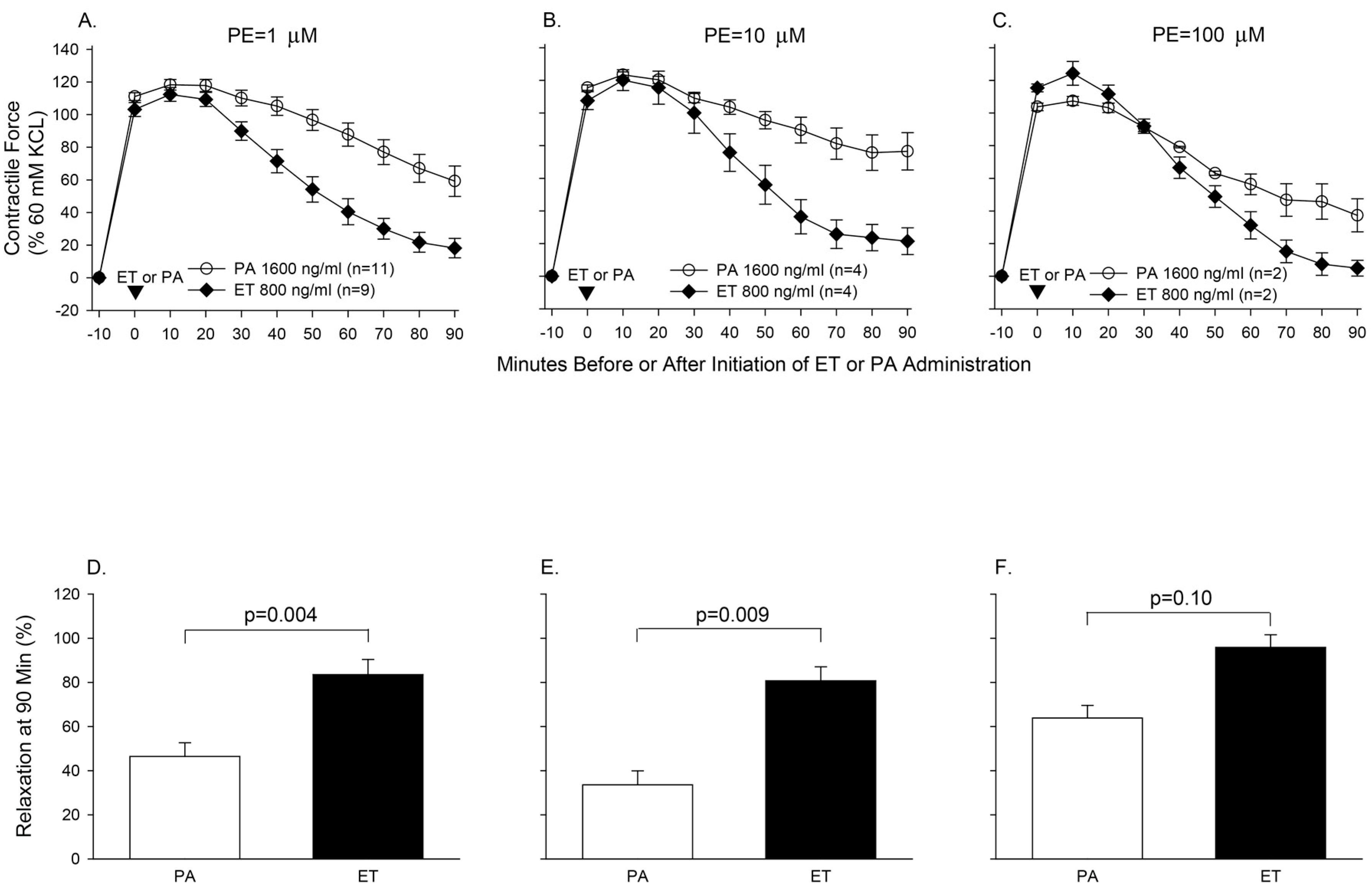
- Li, Y.; Cui, X.; Solomon, S.B.; Remy, K.; Fitz, Y.; Eichacker, P.Q.B. Bacillus anthracis edema toxin increases cAMP levels and inhibits phenylephrine-stimulated contraction in a rat aortic ring model. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H238–H250. [Google Scholar] [CrossRef] [PubMed]
- Suffredini, D.A.; Li, Y.; Xu, W.; Moayeri, M.; Leppla, S.H.; Fitz, Y.; Cui, X.; Eichacker, P.Q. Shock and lethality with anthrax edema toxin in rats are associated with reduced arterial responsiveness to phenylephrine and are reversed with adefovir. Am. J. Physiol. Heart Circ. Physiol. 2017. [Google Scholar] [CrossRef] [PubMed]
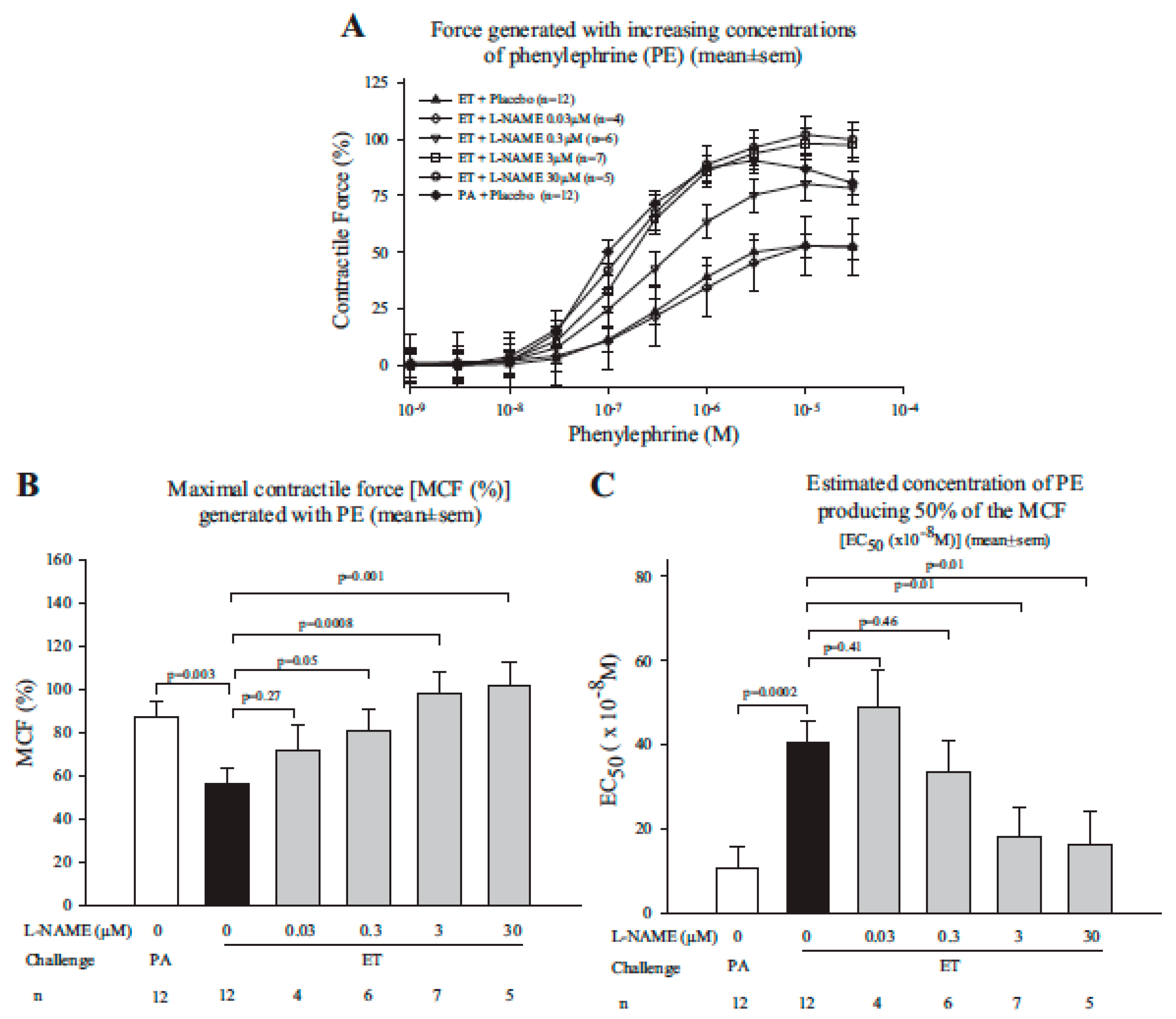
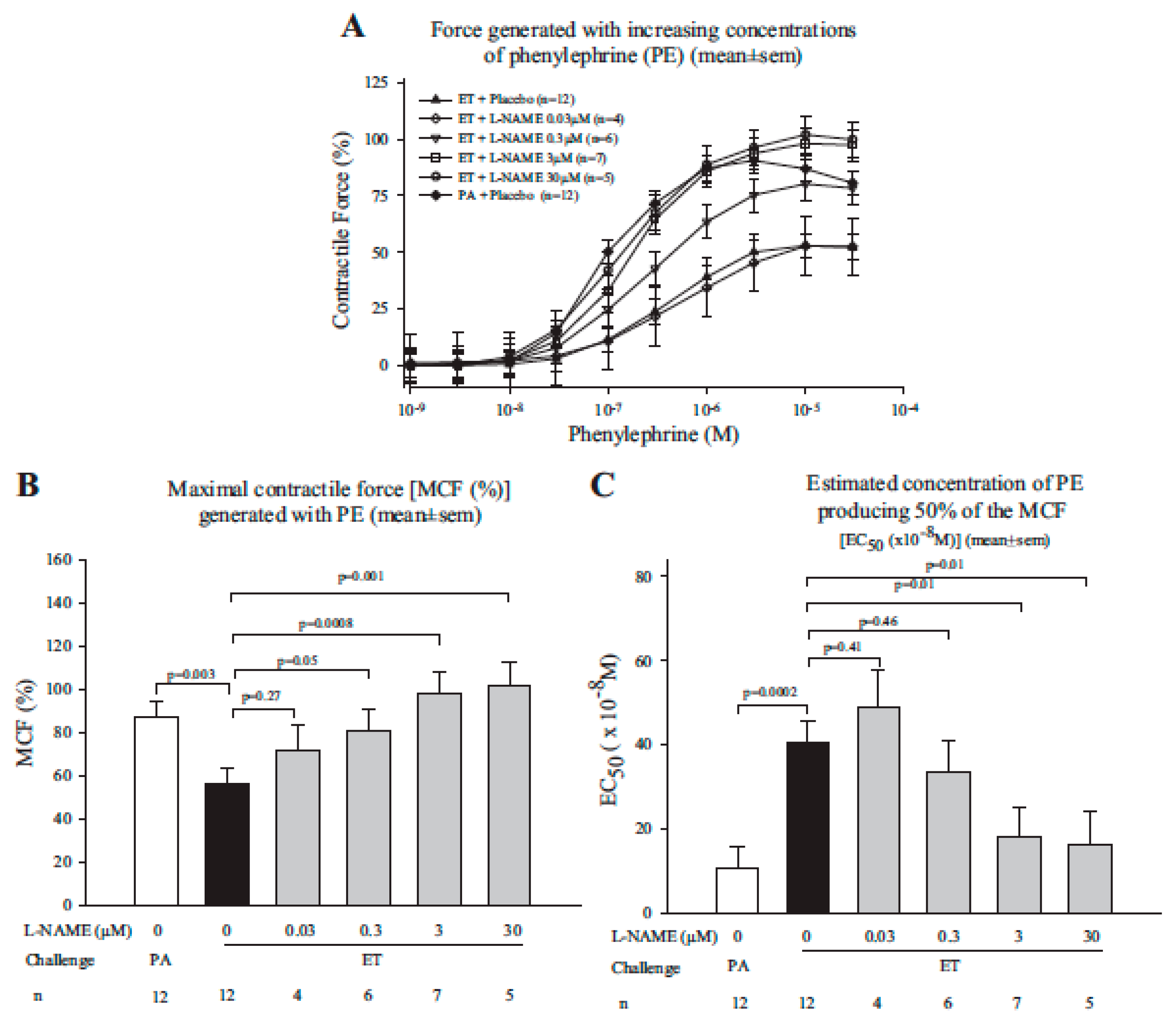
- Li, Y.; Cui, X.; Xu, W.; Ohanjanian, L.; Sampath-Kumar, H.; Suffredini, D.; Moayeri, M.; Leppla, S.; Fitz, Y.; Eichacker, P.Q. Nitric oxide production contributes to Bacillus anthracis edema toxin-associated arterial hypotension and lethality: Ex vivo and in vivo studies in the rat. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H781–H793. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suffredini, D.A.; Cui, X.; Xu, W.; Li, Y.; Eichacker, P.Q. The Potential Pathogenic Contributions of Endothelial Barrier and Arterial Contractile Dysfunction to Shock Due to B. anthracis Lethal and Edema Toxins. Toxins 2017, 9, 394. https://doi.org/10.3390/toxins9120394
Suffredini DA, Cui X, Xu W, Li Y, Eichacker PQ. The Potential Pathogenic Contributions of Endothelial Barrier and Arterial Contractile Dysfunction to Shock Due to B. anthracis Lethal and Edema Toxins. Toxins. 2017; 9(12):394. https://doi.org/10.3390/toxins9120394
Chicago/Turabian StyleSuffredini, Dante A., Xizhong Cui, Wanying Xu, Yan Li, and Peter Q. Eichacker. 2017. "The Potential Pathogenic Contributions of Endothelial Barrier and Arterial Contractile Dysfunction to Shock Due to B. anthracis Lethal and Edema Toxins" Toxins 9, no. 12: 394. https://doi.org/10.3390/toxins9120394