High Conservation of Tetanus and Botulinum Neurotoxins Cleavage Sites on Human SNARE Proteins Suggests That These Pathogens Exerted Little or No Evolutionary Pressure on Humans

 , ,
, , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
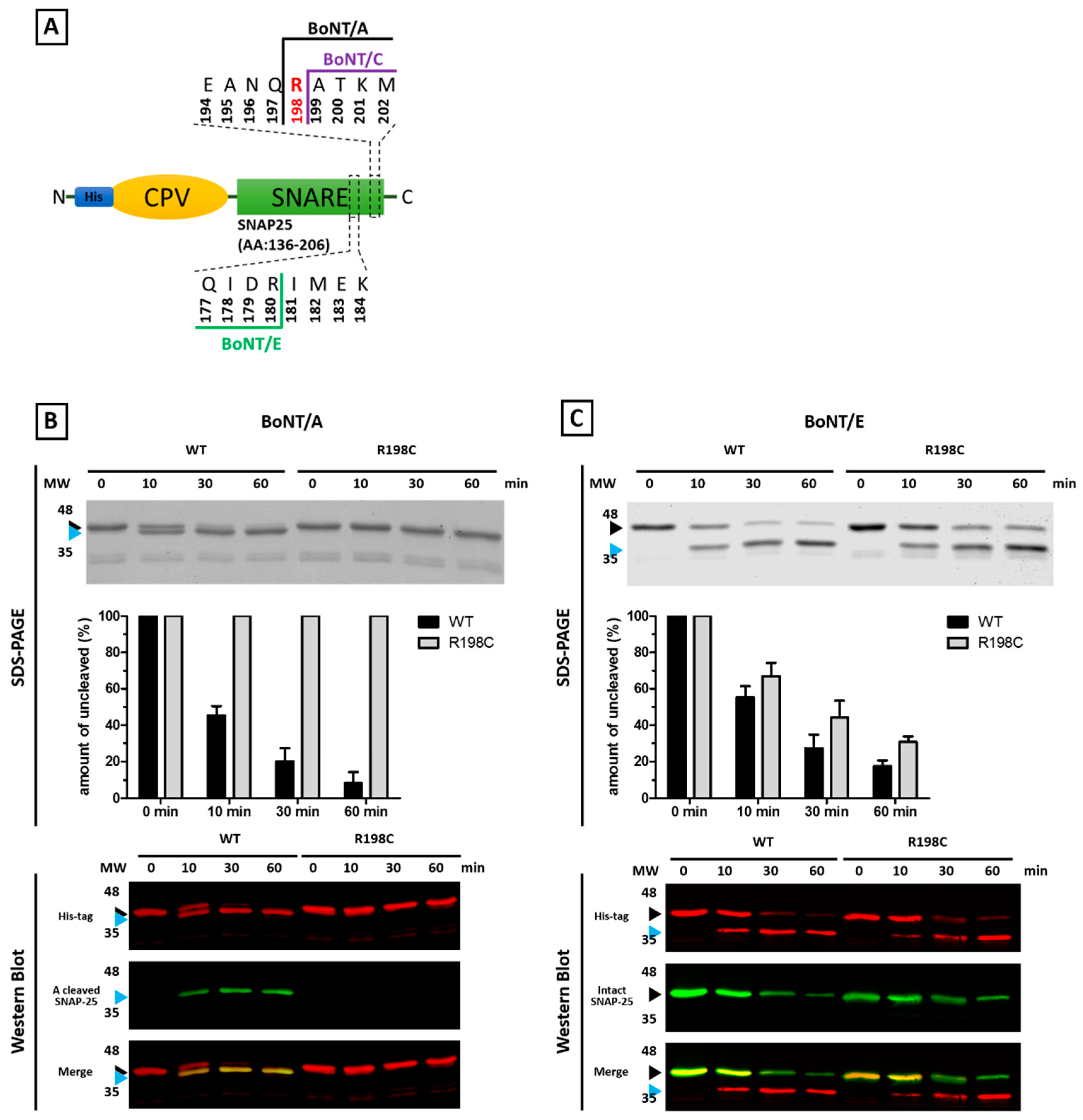
Abstract
:1. Introduction
2. Results
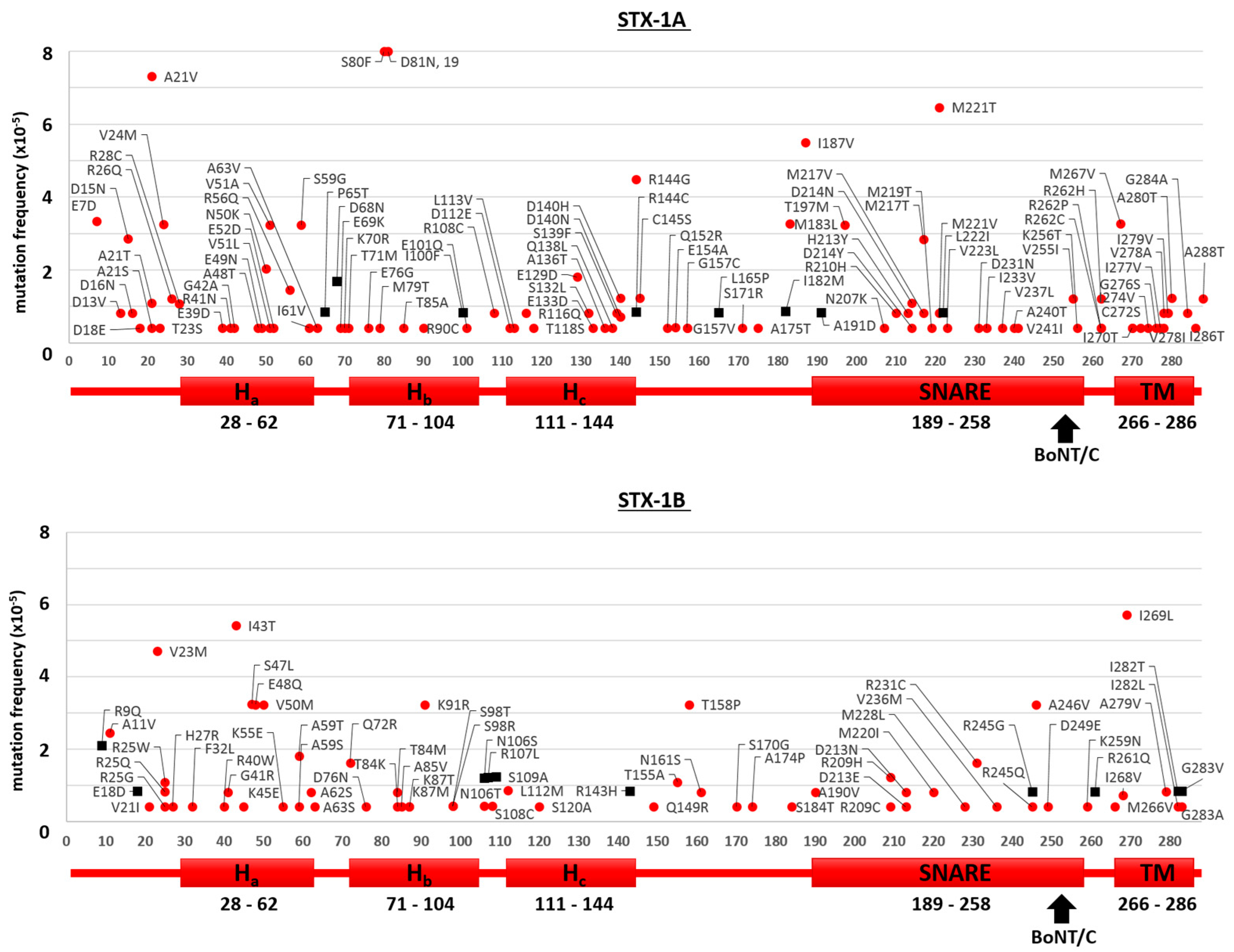
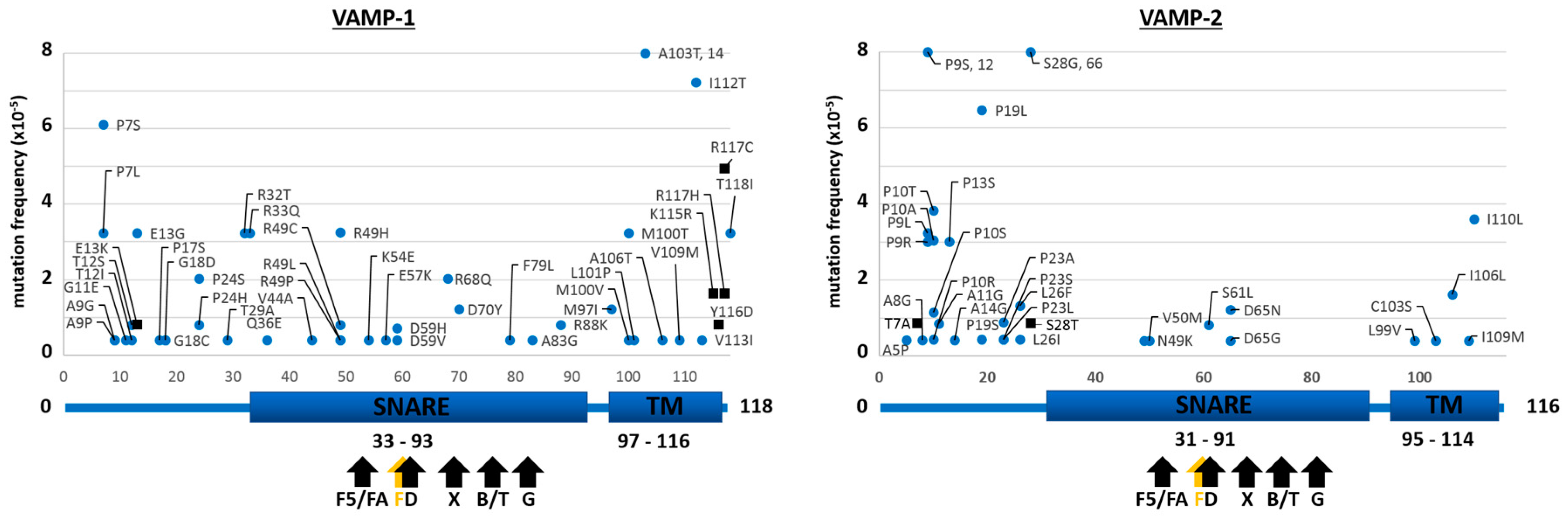
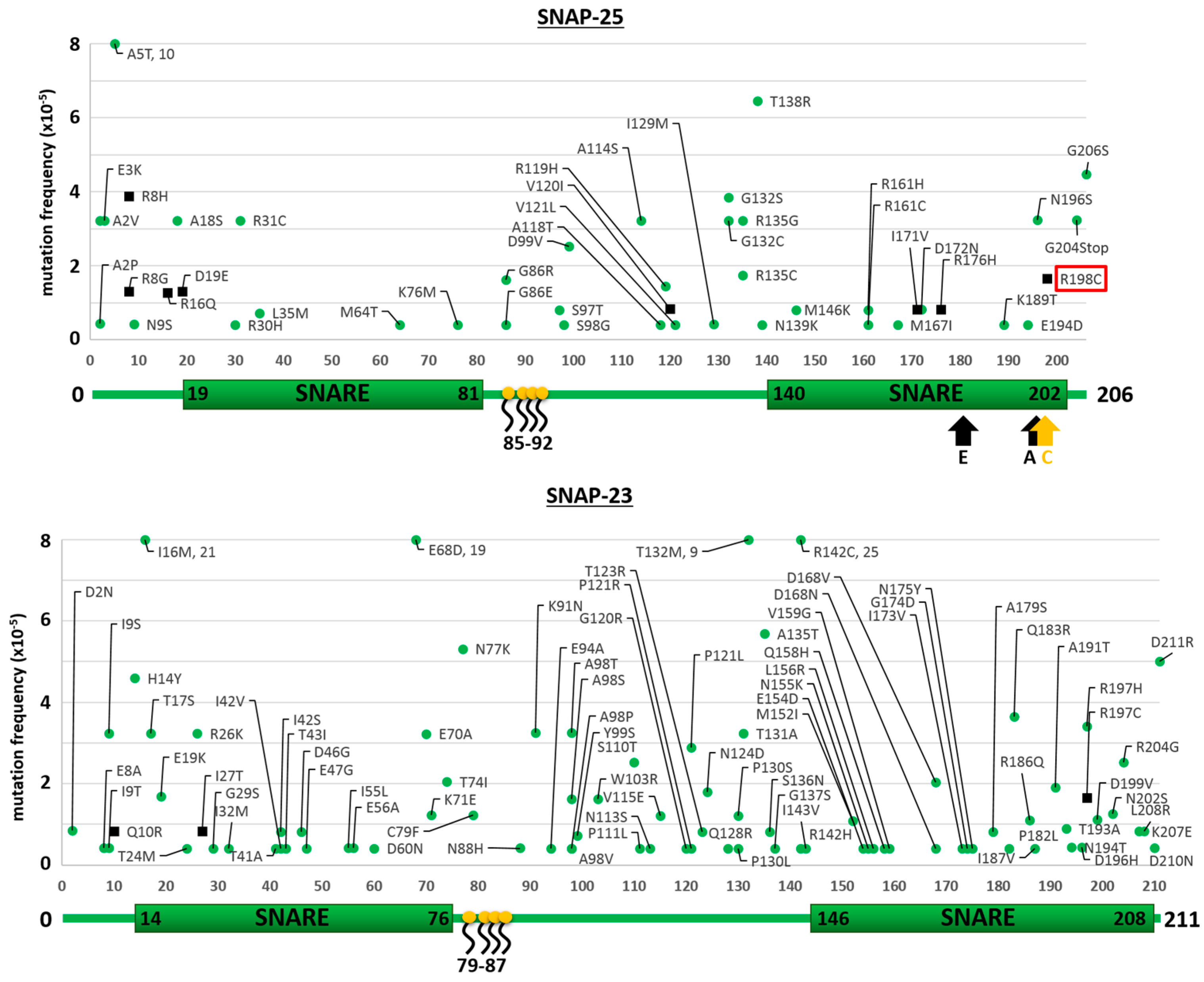
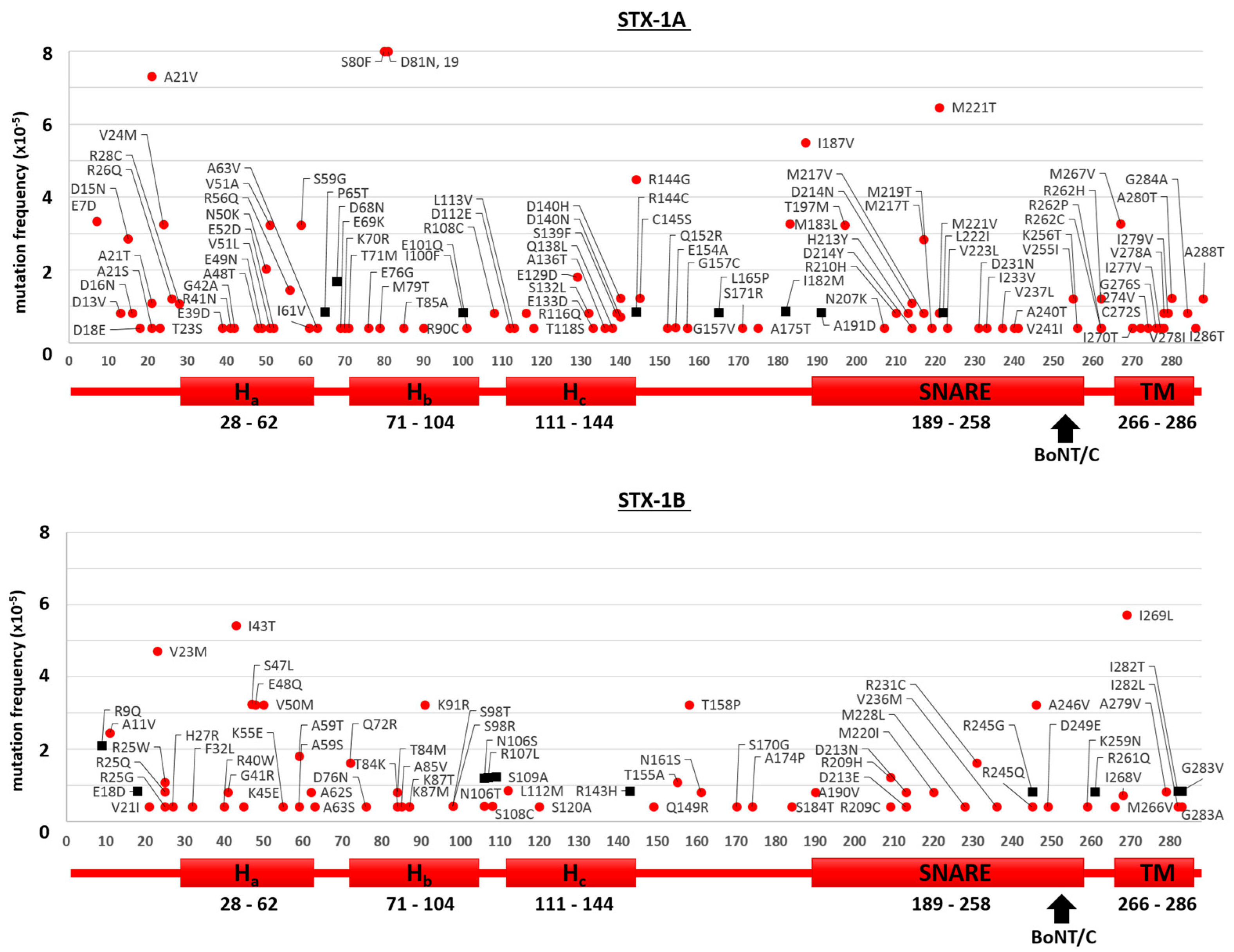
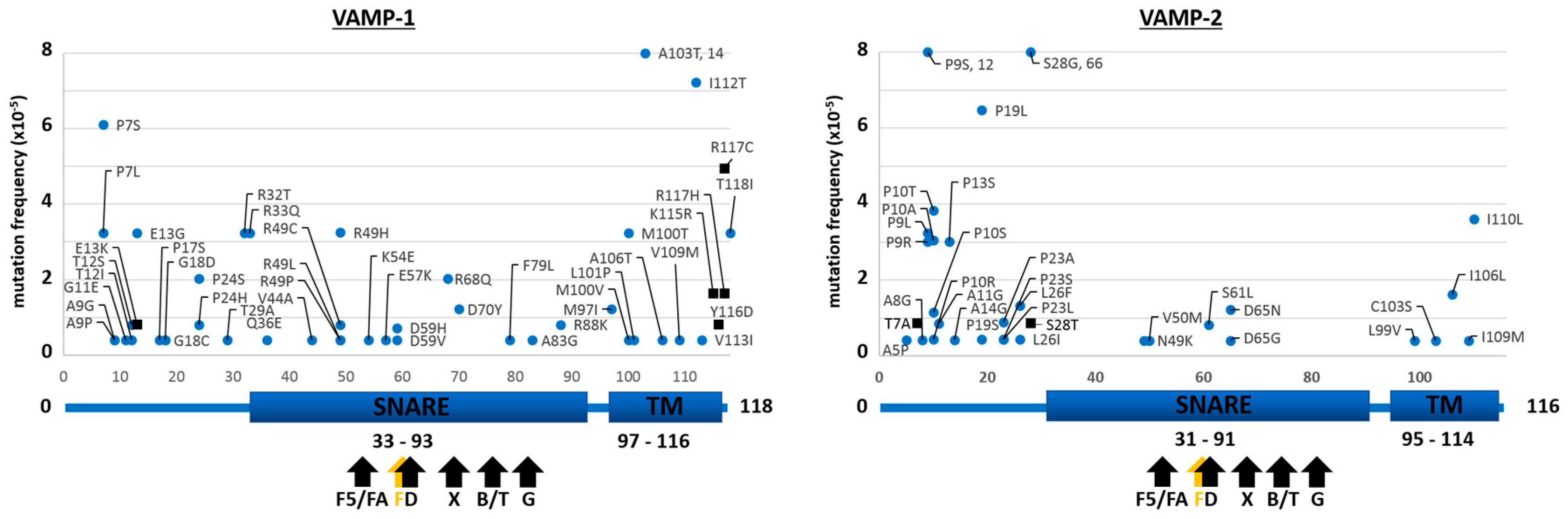
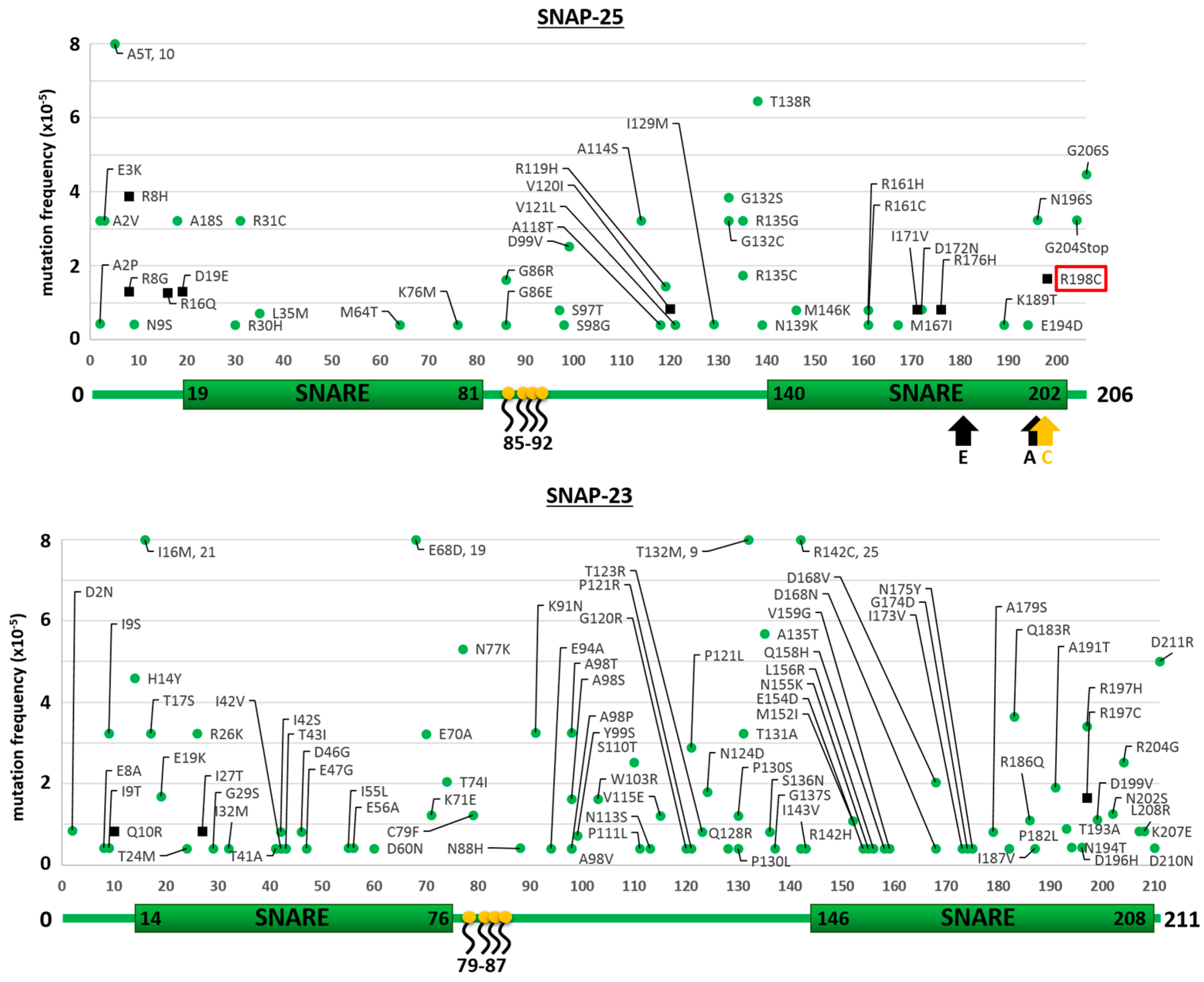
2.1. Mutants of the Neuronal SNARE Proteins in Humans
2.2. Mutations of Human SNARE Proteins at the Cleavage Sites of Tetanus and Botulinum Neurotoxins
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Generation of SNARE Protein Mutation Maps
5.2. Molecular Cloning
5.3. Expression and Affinity Purification of Recombinant His_CPV_SNAP25(136–206) Wild Type and R198C
5.4. BoNT Proteolysis Assay
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; Abecasis, G.R.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, W.; O’Connor, T.D.; Jun, G.; Kang, H.M.; Abecasis, G.; Leal, S.M.; Gabriel, S.; Altshuler, D.; Shendure, J.; Nickerson, D.A.; et al. Analysis of 6515 exomes reveals the recent origin of most human protein-coding variants. Nature 2013, 493, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Vassy, J.L.; Christensen, K.D.; Schonman, E.F.; Blout, C.L.; Robinson, J.O.; Krier, J.B.; Diamond, P.M.; Lebo, M.; Machini, K.; Azzariti, D.R.; et al. The impact of whole-genome sequencing on the primary care and outcomes of healthy adult patients: A pilot randomized trial. Ann. Intern. Med. 2017, 167, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Chen, Y.; Ayub, Q.; Huang, N.; Ball, E.V.; Mort, M.; Phillips, A.D.; Shaw, K.; Stenson, P.D.; Cooper, D.N.; et al. Deleterious- and disease-allele prevalence in healthy individuals: Insights from current predictions, mutation databases, and population-scale resequencing. Am. J. Hum. Genet. 2012, 91, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Piton, A.; Redin, C.; Mandel, J.L. Xlid-causing mutations and associated genes challenged in light of data from large-scale human exome sequencing. Am. J. Hum. Genet. 2013, 93, 368–383. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, O.; Pirazzini, M.; Montecucco, C. Botulinum neurotoxins: Genetic, structural and mechanistic insights. Nat. Rev. Microbiol. 2014, 12, 535–549. [Google Scholar] [CrossRef] [PubMed]
- Schiavo, G.; Matteoli, M.; Montecucco, C. Neurotoxins affecting neuroexocytosis. Physiol. Rev. 2000, 80, 717–766. [Google Scholar] [PubMed]
- Südhof, T.C.; Rizo, J. Synaptic vesicle exocytosis. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Pirazzini, M.; Rossetto, O.; Eleopra, R.; Montecucco, C. Botulinum neurotoxins: Biology, pharmacology, and toxicology. Pharmacol. Rev. 2017, 69, 200–235. [Google Scholar] [CrossRef] [PubMed]
- Pantano, S.; Montecucco, C. The blockade of the neurotransmitter release apparatus by botulinum neurotoxins. Cell. Mol. Life Sci. 2014, 71, 793–811. [Google Scholar] [CrossRef] [PubMed]
- Binz, T. Clostridial neurotoxin light chains: Devices for SNARE cleavage mediated blockade of neurotransmission. Curr. Top. Microbiol. Immunol. 2013, 364, 139–157. [Google Scholar] [PubMed]
- Schiavo, G.; Benfenati, F.; Poulain, B.; Rossetto, O.; Polverino de Laureto, P.; DasGupta, B.R.; Montecucco, C. Tetanus and botulinum-B neurotoxins block neurotransmitter release by proteolytic cleavage of synaptobrevin. Nature 1992, 359, 832–835. [Google Scholar] [CrossRef] [PubMed]
- Schiavo, G.; Rossetto, O.; Catsicas, S.; Polverino de Laureto, P.; DasGupta, B.R.; Benfenati, F.; Montecucco, C. Identification of the nerve terminal targets of botulinum neurotoxin serotypes A, D, and E. J. Biol. Chem. 1993, 268, 23784–23787. [Google Scholar] [PubMed]
- Yamasaki, S.; Hu, Y.; Binz, T.; Kalkuhl, A.; Kurazono, H.; Tamura, T.; Jahn, R.; Kandel, E.; Niemann, H. Synaptobrevin/vesicle-associated membrane protein (VAMP) of Aplysia californica: Structure and proteolysis by tetanus toxin and botulinal neurotoxins type D and F. Proc. Natl. Acad. Sci. USA 1994, 91, 4688–4692. [Google Scholar] [CrossRef] [PubMed]
- Schiavo, G.; Shone, C.C.; Rossetto, O.; Alexander, F.C.; Montecucco, C. Botulinum neurotoxin serotype f is a zinc endopeptidase specific for VAMP/synaptobrevin. J. Biol. Chem. 1993, 268, 11516–11519. [Google Scholar] [PubMed]
- Schiavo, G.; Malizio, C.; Trimble, W.S.; Polverino de Laureto, P.; Milan, G.; Sugiyama, H.; Johnson, E.A.; Montecucco, C. Botulinum g neurotoxin cleaves VAMP/synaptobrevin at a single Ala-Ala peptide bond. J. Biol. Chem. 1994, 269, 20213–20216. [Google Scholar] [PubMed]
- Kalb, S.R.; Baudys, J.; Webb, R.P.; Wright, P.; Smith, T.J.; Smith, L.A.; Fernández, R.; Raphael, B.H.; Maslanka, S.E.; Pirkle, J.L.; et al. Discovery of a novel enzymatic cleavage site for botulinum neurotoxin f5. FEBS Lett. 2012, 586, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Kalb, S.R.; Baudys, J.; Raphael, B.H.; Dykes, J.K.; Luquez, C.; Maslanka, S.E.; Barr, J.R. Functional characterization of botulinum neurotoxin serotype H as a hybrid of known serotypes F and A (BoNT F/A). Anal. Chem. 2015, 87, 3911–3917. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Masuyer, G.; Zhang, J.; Shen, Y.; Lundin, D.; Henriksson, L.; Miyashita, S.-I.; Martínez-Carranza, M.; Dong, M.; Stenmark, P. Identification and characterization of a novel botulinum neurotoxin. Nat. Commun. 2017, 8, 14130. [Google Scholar] [CrossRef] [PubMed]
- Zornetta, I.; Azarnia Tehran, D.; Arrigoni, G.; Anniballi, F.; Bano, L.; Leka, O.; Zanotti, G.; Binz, T.; Montecucco, C. The first non clostridial botulinum-like toxin cleaves VAMP within the juxtamembrane domain. Sci. Rep. 2016, 6, 30257. [Google Scholar] [CrossRef] [PubMed]
- Schiavo, G.; Santucci, A.; Dasgupta, B.R.; Mehta, P.P.; Jontes, J.; Benfenati, F.; Wilson, M.C.; Montecucco, C. Botulinum neurotoxins serotypes A and E cleave SNAP-25 at distinct cooh-terminal peptide bonds. FEBS Lett. 1993, 335, 99–103. [Google Scholar] [CrossRef]
- Binz, T.; Blasi, J.; Yamasaki, S.; Baumeister, A.; Link, E.; Südhof, T.C.; Jahn, R.; Niemann, H. Proteolysis of SNAP-25 by types E and A botulinal neurotoxins. J. Biol. Chem. 1994, 269, 1617–1620. [Google Scholar] [PubMed]
- Blasi, J.; Chapman, E.R.; Link, E.; Binz, T.; Yamasaki, S.; De Camilli, P.; Sudhof, T.C.; Niemann, H.; Jahn, R. Botulinum neurotoxin A selectively cleaves the synaptic protein SNAP-25. Nature 1993, 365, 160–163. [Google Scholar] [CrossRef] [PubMed]
- Blasi, J.; Chapman, E.R.; Yamasaki, S.; Binz, T.; Niemann, H.; Jahn, R. Botulinum neurotoxin c1 blocks neurotransmitter release by means of cleaving HPC-1/syntaxin. EMBO J. 1993, 12, 4821–4828. [Google Scholar] [PubMed]
- Johnson, E.A.; Montecucco, C. Botulism. Handb. Clin. Neurol. 2008, 91, 333–368. [Google Scholar] [PubMed]
- Patarnello, T.; Bargelloni, L.; Rossetto, O.; Schiavo, G.; Montecucco, C. Neurotransmission and secretion. Nature 1993, 364, 581–582. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Adler, M.; Demogines, A.; Borrell, A.; Liu, H.; Tao, L.; Tepp, W.H.; Zhang, S.-C.; Johnson, E.A.; Sawyer, S.L.; et al. Widespread sequence variations in VAMP1 across vertebrates suggest a potential selective pressure from botulinum neurotoxins. PLoS Pathog. 2014, 10, e1004177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eleopra, R.; Montecucco, C.; Devigili, G.; Lettieri, C.; Rinaldo, S.; Verriello, L.; Pirazzini, M.; Caccin, P.; Rossetto, O. Botulinum neurotoxin serotype D is poorly effective in humans: An in vivo electrophysiological study. Clin. Neurophysiol. 2013, 124, 999–1004. [Google Scholar] [CrossRef] [PubMed]
- Petrovski, S.; Wang, Q.; Heinzen, E.L.; Allen, A.S.; Goldstein, D.B. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 2013, 9, e1003709. [Google Scholar] [CrossRef]
- Sutton, R.B.; Fasshauer, D.; Jahn, R.; Brunger, A.T. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 a resolution. Nature 1998, 395, 347–353. [Google Scholar] [PubMed]
- Jahn, R.; Südhof, T.C. Membrane fusion and exocytosis. Annu. Rev. Biochem. 1999, 68, 863–911. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.J.; Tejero, R.; Arancillo, M.; Vardar, G.; Korotkova, T.; Kintscher, M.; Schmitz, D.; Ponomarenko, A.; Tabares, L.; Rosenmund, C. Syntaxin 1B is important for mouse postnatal survival and proper synaptic function at the mouse neuromuscular junctions. J. Neurophysiol. 2015, 114, 2404–2417. [Google Scholar] [CrossRef] [PubMed]
- Kofuji, T.; Fujiwara, T.; Sanada, M.; Mishima, T.; Akagawa, K. HPC-1/syntaxin 1A and syntaxin 1b play distinct roles in neuronal survival. J. Neurochem. 2014, 130, 514–525. [Google Scholar] [CrossRef] [PubMed]
- Washbourne, P.; Thompson, P.M.; Carta, M.; Costa, E.T.; Mathews, J.R.; Lopez-Bendito, G.; Molnar, Z.; Becher, M.W.; Valenzuela, C.F.; Partridge, L.D.; et al. Genetic ablation of the t-SNARE SNAP-25 distinguishes mechanisms of neuroexocytosis. Nat. Neurosci. 2002, 5, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Suh, Y.H.; Yoshimoto-Furusawa, A.; Weih, K.A.; Tessarollo, L.; Roche, K.W.; Mackem, S.; Roche, P.A. Deletion of SNAP-23 results in pre-implantation embryonic lethality in mice. PLoS ONE 2011, 6, e18444. [Google Scholar] [CrossRef] [PubMed]
- Schoch, S.; Deak, F.; Konigstorfer, A.; Mozhayeva, M.; Sara, Y.; Südhof, T.C.; Kavalali, E.T. SNARE function analyzed in synaptobrevin/VAMP knockout mice. Science 2001, 294, 1117–1122. [Google Scholar] [CrossRef] [PubMed]
- Karki, R.; Pandya, D.; Elston, R.C.; Ferlini, C. Defining “mutation” and “polymorphism” in the era of personal genomics. BMC Med. Genom. 2015, 8, 37. [Google Scholar] [CrossRef] [PubMed]
- Corradini, I.; Verderio, C.; Sala, M.; Wilson, M.C.; Matteoli, M. SNAP-25 in neuropsychiatric disorders. Ann. N. Y. Acad. Sci. 2009, 1152, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.D.; Oliver, P.L.; Davies, K.E. SNARE proteins and schizophrenia: Linking synaptic and neurodevelopmental hypotheses. Acta Biochim. Pol. 2008, 55, 619–628. [Google Scholar] [PubMed]
- Nakamura, K.; Anitha, A.; Yamada, K.; Tsujii, M.; Iwayama, Y.; Hattori, E.; Toyota, T.; Suda, S.; Takei, N.; Iwata, Y.; et al. Genetic and expression analyses reveal elevated expression of syntaxin 1a (Stx1A) in high functioning autism. Int. J. Neuropsychopharmacol. 2008, 11, 1073–1084. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, M.; Yamamori, S.; Suzuki, E.; Watanabe, S.; Sato, T.; Miyaoka, H.; Azuma, S.; Ikegami, S.; Kuwahara, R.; Suzuki-Migishima, R.; et al. A single amino acid mutation in SNAP-25 induces anxiety-related behavior in mouse. PLoS ONE 2011, 6, e25158. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.-M.; Selcen, D.; Brengman, J.; Engel, A.G. Mutant SNAP25b causes myasthenia, cortical hyperexcitability, ataxia, and intellectual disability. Neurology 2014, 83, 2247–2255. [Google Scholar] [CrossRef] [PubMed]
- Rebane, A.A.; Wang, B.; Ma, L.; Qu, H.; Coleman, J.; Krishnakumar, S.; Rothman, J.E.; Zhang, Y. Two disease-causing SNAP-25b mutations selectively impair SNARE c-terminal assembly. J. Mol. Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Catsicas, S.; Catsicas, M.; Keyser, K.T.; Karten, H.J.; Wilson, M.C.; Milner, R.J. Differential expression of the presynaptic protein SNAP-25 in mammalian retina. J. Neurosci. Res. 1992, 33, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Duc, C.; Catsicas, S. Ultrastructural localization of SNAP-25 within the rat spinal cord and peripheral nervous system. J. Comp. Neurol. 1995, 356, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Zanetti, G.; Sikorra, S.; Rummel, A.; Krez, N.; Duregotti, E.; Negro, S.; Henke, T.; Rossetto, O.; Binz, T.; Pirazzini, M. Botulinum neurotoxin C mutants reveal different effects of syntaxin or SNAP-25 proteolysis on neuromuscular transmission. PLOS Pathog. 2017, 13, e1006567. [Google Scholar] [CrossRef] [PubMed]
- Vaidyanathan, V.V.; Yoshino, K.; Jahnz, M.; Dorries, C.; Bade, S.; Nauenburg, S.; Niemann, H.; Binz, T. Proteolysis of SNAP-25 isoforms by botulinum neurotoxin types A, C, and E: Domains and amino acid residues controlling the formation of enzyme-substrate complexes and cleavage. J. Neurochem. 1999, 72, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.S.L.; Sugiyama, H. Botulism: The Organism, Its Toxins, the Disease; Charles C. Thomas Publisher, Limited: Springfield, IL, USA, 1988. [Google Scholar]
- Montecucco, C.; Rasotto, M.B. On botulinum neurotoxin variability. MBio 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Pappas, G.; Kiriaze, I.J.; Falagas, M.E. Insights into infectious disease in the era of Hippocrates. Int. J. Infect. Dis. 2008, 12, 347–350. [Google Scholar] [CrossRef] [PubMed]
- Tekiner, H. Aretaeus of Cappadocia and his treatises on diseases. Turk. Neurosurg. 2015, 25, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Kyu, H.H.; Mumford, J.E.; Stanaway, J.D.; Barber, R.M.; Hancock, J.R.; Vos, T.; Murray, C.J.L.; Naghavi, M. Mortality from tetanus between 1990 and 2015: Findings from the global burden of disease study 2015. BMC Public Health 2017, 17, 179. [Google Scholar] [CrossRef] [PubMed]
- Dietz, V.; Milstien, J.B.; van Loon, F.; Cochi, S.; Bennett, J. Performance and potency of tetanus toxoid: Implications for eliminating neonatal tetanus. Bull. World Health Organ. 1996, 74, 619–628. [Google Scholar] [PubMed]
- Peck, M.W.; Smith, T.J.; Anniballi, F.; Austin, J.W.; Bano, L.; Bradshaw, M.; Cuervo, P.; Cheng, L.W.; Derman, Y.; Dorner, B.G.; et al. Historical perspectives and guidelines for botulinum neurotoxin subtype nomenclature. Toxins 2017, 9, 38. [Google Scholar] [CrossRef] [PubMed]
- Gonzalo, S.; Linder, M.E. SNAP-25 palmitoylation and plasma membrane targeting require a functional secretory pathway. Mol. Biol. Cell 1998, 9, 585–597. [Google Scholar] [CrossRef] [PubMed]
- Salaun, C.; Greaves, J.; Chamberlain, L.H. The intracellular dynamic of protein palmitoylation. J. Cell Biol. 2010, 191, 1229–1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breidenbach, M.A.; Brunger, A.T. Substrate recognition strategy for botulinum neurotoxin serotype A. Nature 2004, 432, 925–929. [Google Scholar] [CrossRef] [PubMed]
- Schiavo, G.; Montecucco, C. Tetanus and botulism neurotoxins: Isolation and assay. Methods Enzymol. 1995, 248, 643–652. [Google Scholar] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carle, S.; Pirazzini, M.; Rossetto, O.; Barth, H.; Montecucco, C. High Conservation of Tetanus and Botulinum Neurotoxins Cleavage Sites on Human SNARE Proteins Suggests That These Pathogens Exerted Little or No Evolutionary Pressure on Humans. Toxins 2017, 9, 404. https://doi.org/10.3390/toxins9120404
Carle S, Pirazzini M, Rossetto O, Barth H, Montecucco C. High Conservation of Tetanus and Botulinum Neurotoxins Cleavage Sites on Human SNARE Proteins Suggests That These Pathogens Exerted Little or No Evolutionary Pressure on Humans. Toxins. 2017; 9(12):404. https://doi.org/10.3390/toxins9120404
Chicago/Turabian StyleCarle, Stefan, Marco Pirazzini, Ornella Rossetto, Holger Barth, and Cesare Montecucco. 2017. "High Conservation of Tetanus and Botulinum Neurotoxins Cleavage Sites on Human SNARE Proteins Suggests That These Pathogens Exerted Little or No Evolutionary Pressure on Humans" Toxins 9, no. 12: 404. https://doi.org/10.3390/toxins9120404