1. Introduction
Single-cell genomics is an emerging field that holds great promise for understanding the nature and function of genetic diversity in biological systems [
1,
2,
3]. Obviously, this fast-growing area of study relies on DNA amplification techniques that can be applied to an extremely small amount of starting material, that is, genomic DNA from one cell. Multiple displacement amplification (MDA) [
4], based on φ29 DNA polymerase and random primers, has been the method of choice for single-cell whole genome amplification (WGA) [
5,
6,
7,
8]. It generates a sufficient amount of replicated DNA of high fidelity from template DNA of unknown sequence and exhibits lower error rates and longer fragment sizes than genome-wide amplification based on polymerase chain reaction (PCR). Although a new WGA method with lower amplification bias has recently been reported [
9], MDA is still the prevailing approach because of commercially available reagents and relatively simple procedures.
Microfluidic platforms have been developed for achieving single-cell isolation and a miniaturised MDA reaction for the purpose of WGA and successfully applied to a few cell types: lab-cultured bacteria [
10], uncultured bacteria and archaea [
11,
12], and human sperm cells [
13]. However, analysis of bacterial species from environmental samples is particularly challenging because of the thick, multiple-layer cell wall structures often found in these microorganisms, which may obstruct cell lysis. Development of an effective bacterial lysis protocol is important for expanding the applicability of single-cell genomics in view of the relevance of this culture-independent approach to the hugely diverse realm of uncultured or hard-to-culture environmental prokaryotes. We chose as a model system
Synechocystis sp. PCC 6803, a unicellular cyanobacterium with a fully sequenced genome [
14]. Significant difficulty is encountered in breaking cells of this species via chemical treatment compatible with microchip MDA. It should be noted that conventional mechanical lysis methods such as French press or bead beating are not suitable for single-cell WGA.
The lysis protocol proposed in this study is based on the combined use of enzymes and detergents for disrupting the
Synechocystis cell wall structures composed of four chemically distinct layers: the external surface layers (proteins and polysaccharides), the outer lipid membrane, the crosslinked peptidoglycan layer, and the inner cytosolic membrane [
15,
16]. The order of chemical treatments, which include denaturants and proteases, was carefully designed to avoid interference with the downstream amplification activity of φ29 DNA polymerase. As a requisite for preserved single-cell identities, additional washing steps to remove extraneous genetic materials were implemented on the basis of quantitation of extracellular DNA. The efficacy of the lysis protocol for single-cell WGA was demonstrated by sequencing 15 selected genes that are nearly equally spaced across the entire chromosome using the MDA products obtained from single
Synechocystis cells.
2. Materials and Methods
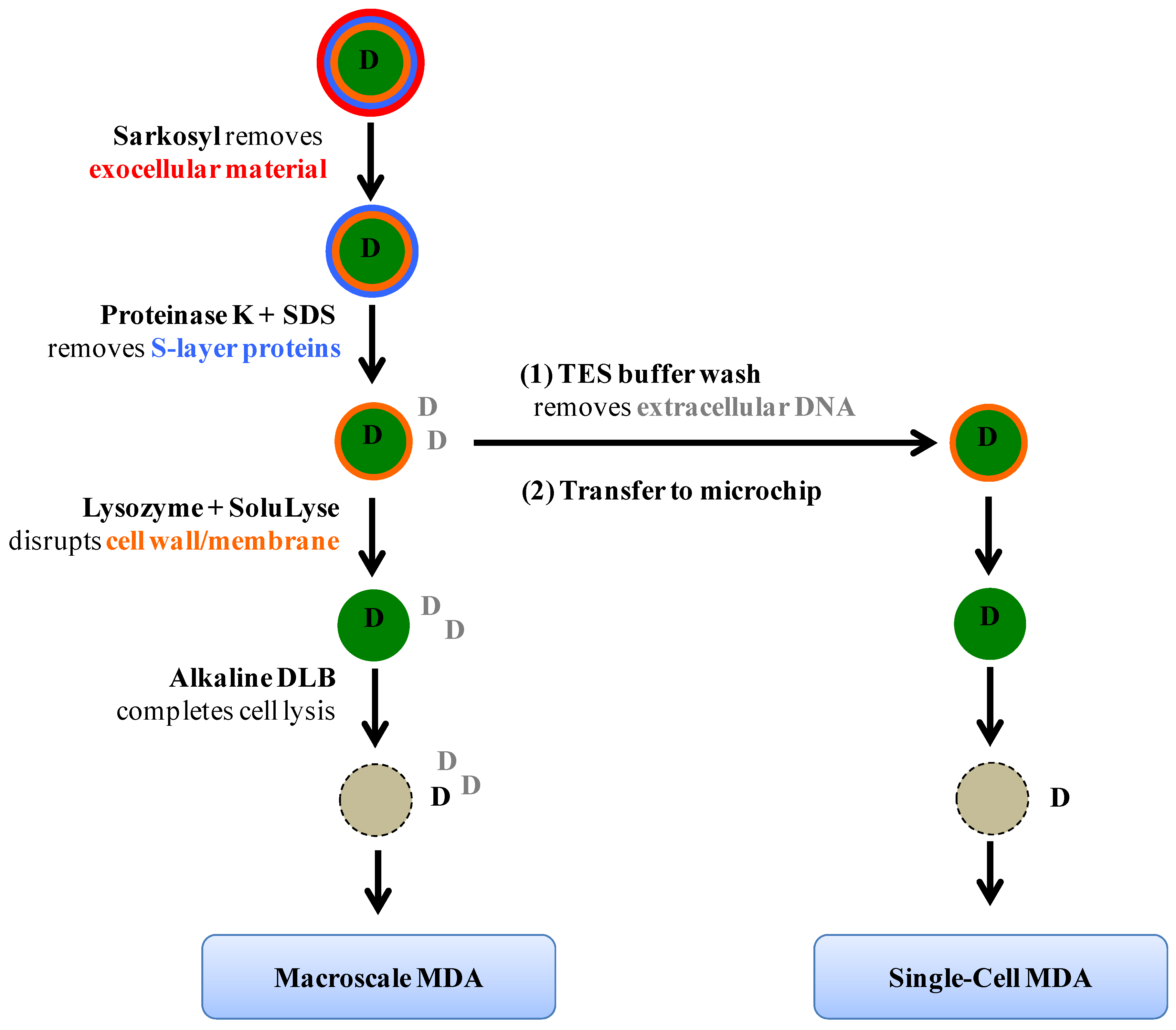
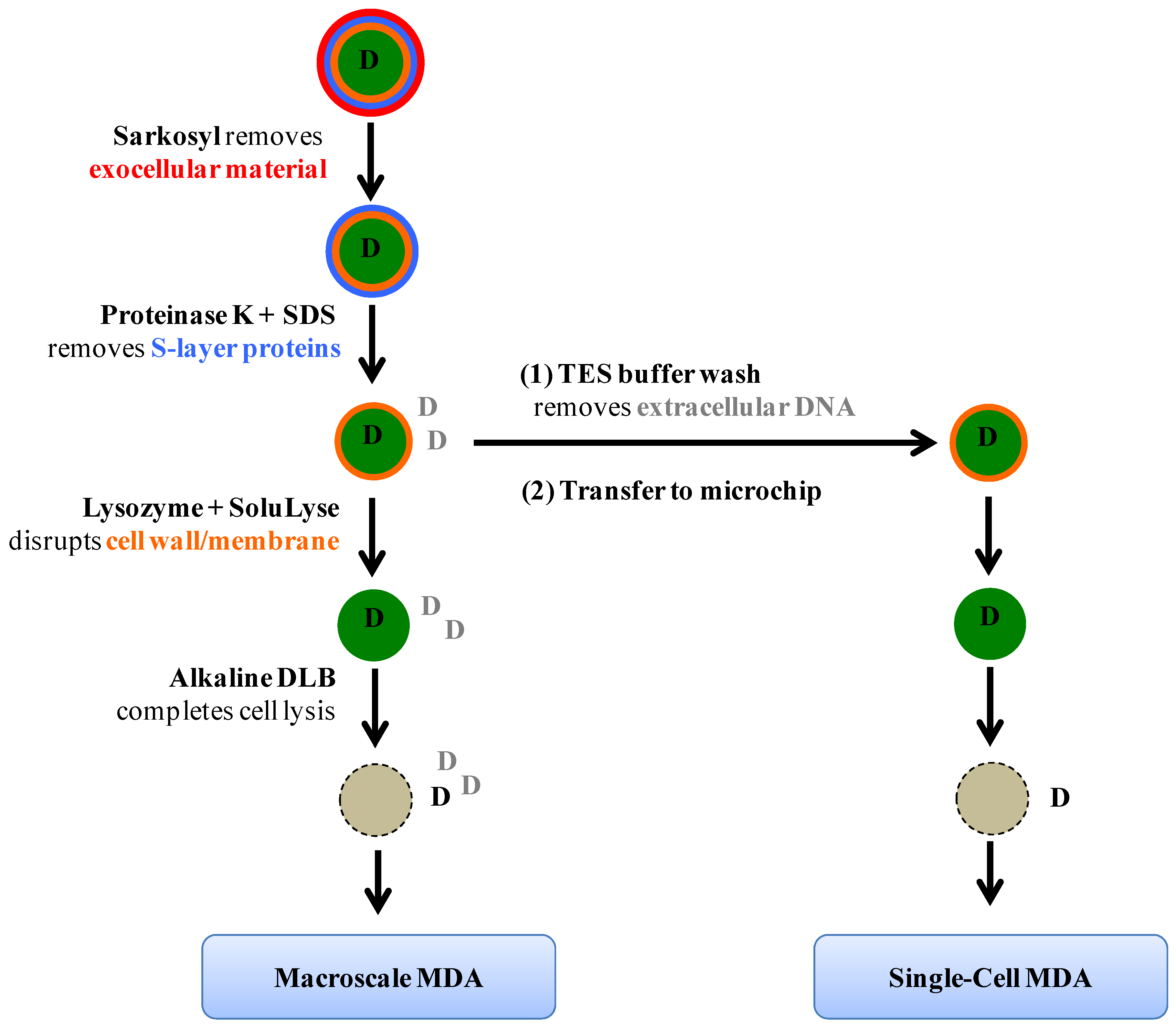
Figure 1 summarizes the lysis protocol. Briefly,
Synechocystis cells from 400 µL of liquid culture were pelleted by centrifuging at 3 krpm (RCF = 735 g) for 10 min (5145C, Eppendorf, Hamburg, Germany) and resuspended in 0.1% (w/v) Sarkosyl (Sigma, St. Louis, MO, USA) in TES Buffer (10 mM Tris (pH 8), 50 mM EDTA, and 50 mM NaCl). The cell suspension was incubated at room temperature for 10 min with gentle mixing, followed by centrifugation. Then, the pellet was resuspended in 10 µg/mL Proteinase K (RNA grade, Life Technologies, Carlsbad, CA, USA) and 0.1% (w/v) SDS in TES Buffer and the sample was incubated at 57 °C for 2 h. After centrifugation and supernatant removal, 200 U/µL lysozyme (Ready-Lyse, Epicentre, Madison, WI, USA) in SoluLyse (Genlantis, San Diego, CA, USA) was added and the suspension was incubated at 37 °C for 2 h. Finally, an equivalent volume of alkaline DLB reagent from Repli-g Midi kit (Qiagen, Venlo, The Netherlands) was added to complete cell lysis. In the case of microfluidic single-cell analysis, multiple washing steps with TES Buffer were performed immediately after the Proteinase K + SDS incubation step to remove contaminant DNA, and the cell suspension was transferred to the MDA microchip for subsequent microfluidic procedures.
Figure 1.
Schematic of the lysis protocol. Cellular layers and chemical reagents used to remove them are shown. The letter D stands for DNA molecules; D in black represents genomic materials originated from captured single cyanobacteria cells whereas D in grey indicates DNA from other cells, either cyanobacteria of interest or different species; this type of DNA is termed “contaminant DNA” in the text for describing single-cell genome amplification experiments.
Figure 1.
Schematic of the lysis protocol. Cellular layers and chemical reagents used to remove them are shown. The letter D stands for DNA molecules; D in black represents genomic materials originated from captured single cyanobacteria cells whereas D in grey indicates DNA from other cells, either cyanobacteria of interest or different species; this type of DNA is termed “contaminant DNA” in the text for describing single-cell genome amplification experiments.
The amount of DNA, either released by lysis or amplified via MDA, was quantified using a dsDNA-specific fluorescent dye (PicoGreen Kit, Life Technologies, Grand Island, NY, USA) for labeling and a 96-well plate reader (SpectraMAX Gemini EM, Molecular Devices, Sunnyvale, CA, USA) for fluorescence measurements, following the manufacturers’ protocols. The level of contaminant DNA, which was below the detection limit of the PicoGreen assay, was quantified by employing digital MDA (dMDA) as described elsewhere [
17]. The dMDA assay is based on small-scale MDA reactions performed on a commercially available microfluidic chip (765 9-nL wells as MDA microreactors; BioMark 12.765 Digital Array, Fluidigm, South San Francisco, CA, USA) and subsequent detection of fluorescent wells using the companion BioMark imaging system. The number of DNA template molecules in the original sample was calculated by counting the number of “lit” fluorescent wells and applying a Poisson correction.
Macroscale MDA (50-µL reaction) was performed using the released DNA from cell lysis as template and the Repli-g Midi kit reagents (Qiagen) as per the manufacturer’s protocol. Single-cell MDA (scMDA; 60-nL reaction) was performed on an integrated microfluidic device, which closely resembles that developed by Quake and coworkers [
10,
11]. Microchip fabrication and operation procedures were similar to those reported previously [
18] with details in Supplementary Methods (Figure S1). To obtain sufficient amounts of DNA for downstream PCR and sequencing, 1 µL of each amplification product extracted from the microchip was used as a template in a second-round 50-µL MDA reaction (33 °C incubation for 16 h) and the final amplicon was stored at 4 °C until further analysis. DNA yields from both rounds of amplification were quantified via the PicoGreen assay.
To estimate the genome coverage of the amplification product from scMDA, 15 PCR primer sets were designed for genes evenly dispersed over the main 3.57 Mbp chromosome of
Synechocystis sp. PCC 6803 (See Supplementary Table S1). Sequence specificities of the expected PCR products were checked using NCBI Primer-BLAST software [
19] (See Supplementary Table S2). Primers were synthesized at the PAN Facility of Stanford University. PCR (20 µL reactions using 25 ng of template DNA) was performed using LightCycler 480 System (Roche) with the following conditions: 95 °C for 3 min; 39 cycles of 95 °C for 30 s, 59 °C for 30 s (annealing) and 72 °C for 1 min; 72 °C for 15 min. The PCR products, the presence of which at 1-kb region was confirmed by gel electrophoresis (1% agarose, 100 V, 15 min), were submitted for Sanger sequencing at the PAN Facility.
Initial experiments on
Synechocystis sp. PCC 6803 were carried out on a cell line provided by Devaki Bhaya from the Carnegie Institution for Science, but all work reported here is based on a new culture (ATCC# 27184), which was purchased from American Type Culture Collection. The culture was maintained at 30°C in BG-11 media (C3061, Sigma, St. Louis, MO, USA). All chemicals were purchased at highest purity and care was taken to avoid introducing extraneous DNA; all of the buffers were filtered with 0.2-µm filters and exposed to UV irradiation for one hour before use, which is reported to eliminate amplification of contaminant DNA [
20].
3. Results and Discussion
The initial attempts to lyse
Synechocystis cells were made using the protocol reported by Wu
et al. [
21], which employs stepwise treatments with detergents and enzymes. Lysis effectiveness was qualitatively assessed by visual inspection of the cell pellet after each chemical treatment. The original protocol produced intact, dark green pellets, indicating that it is unsatisfactory for this type of cyanobacteria. However, improvement of cell breakage was observed when (
a) proteinase K treatment was performed in the presence of 0.1% SDS and (
b) lysozyme step was combined with SoluLyse, a proprietary detergent for bacterial lysis (See Supplementary Table S3). The amounts of DNA released into the supernatants by these modified protocols, as measured by PicoGreen assay, were comparable to the DNA level obtained from sonication-induced lysis, indicating that near-complete lysis was achieved [
22].
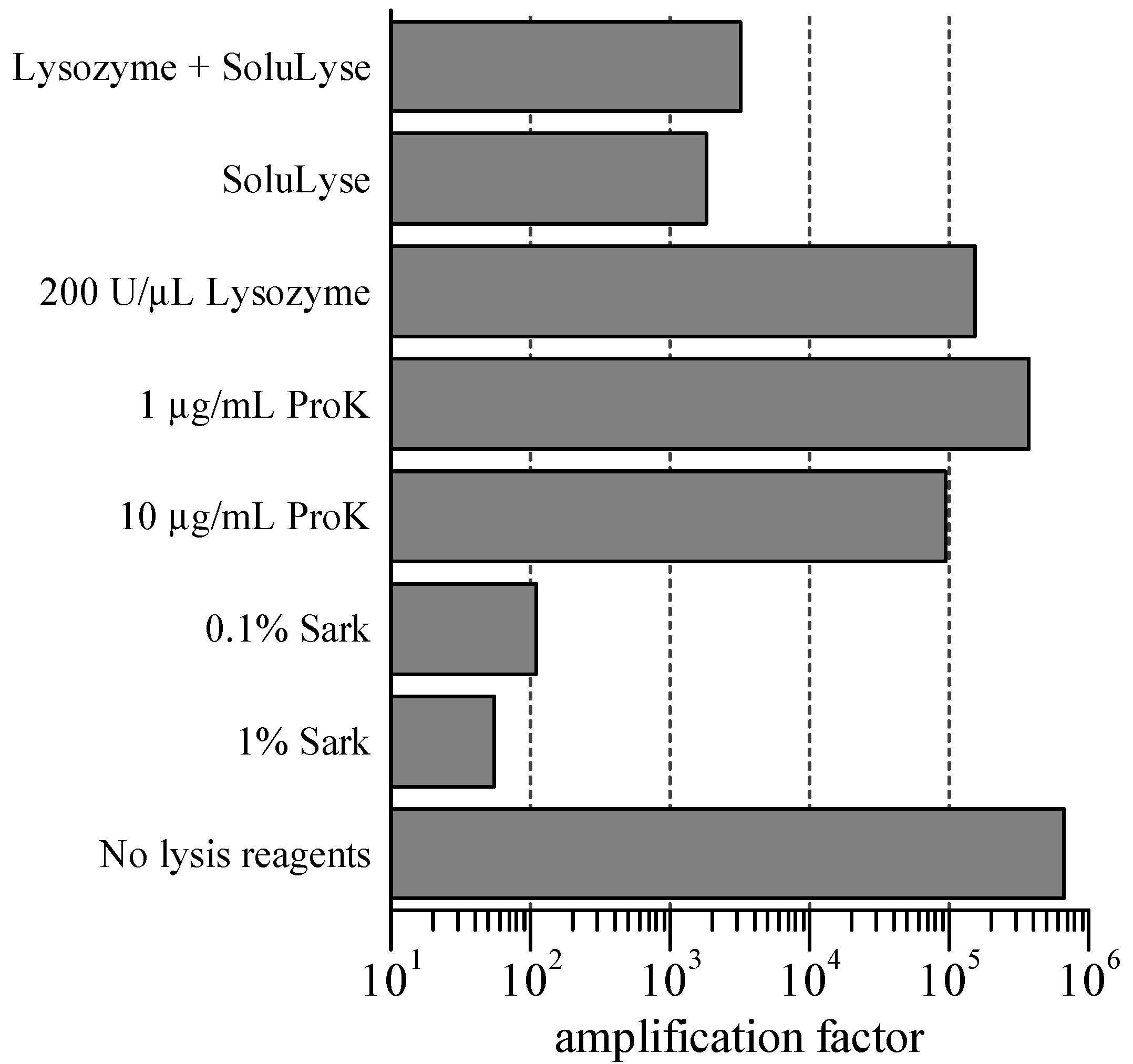
The lysis protocol was further optimized for microfluidic scMDA, our target application, by testing its compatibility with φ29 DNA polymerase activity and on-chip single-cell isolation procedure. First, we investigated the effect of the lysis reagents on the polymerase activity by performing standard macroscale MDA reactions supplemented with a series of lysis reagents and determined the resulting amplification factors (
Figure 2). The detergents were the most inhibitive against the MDA reactions, with 0.1% sarkosyl reducing the amplification by a factor of ~10
4 and 0.1% SDS suppressing the polymerase activity completely. The deleterious effects of proteinase K and lysozyme were relatively small at the tested concentrations, retaining amplification factors greater than 10
5. SoluLyse, with or without lysozyme additive, resulted in amplification factors between 10
3 and 10
4. Based on these results, we decided to carry out the first two lysis steps utilizing sarkosyl and proteinase K in SDS “off-chip” followed by supernatant removal and on-chip isolation of single cells, and finally addition of lysozyme in SoluLyse to each cell “on-chip”.
Figure 2.
Inhibition of 50-µL Multiple displacement amplification (MDA) reactions by lysis reagents. Amplification factor was calculated by dividing the amount of amplified DNA, as quantified via PicoGreen assay, with that of the starting template (50 pg). The MDA reaction with 0.1% SDS yielded an amount of product (<10 pg/µL) that could not be detected within the limits of the PicoGreen assay, meaning that its amplification factor was below 10. Sark and ProK refer to sarkosyl and proteinase K, respectively. Error bars are not shown on this figure but are approximately 10% of each value.
Figure 2.
Inhibition of 50-µL Multiple displacement amplification (MDA) reactions by lysis reagents. Amplification factor was calculated by dividing the amount of amplified DNA, as quantified via PicoGreen assay, with that of the starting template (50 pg). The MDA reaction with 0.1% SDS yielded an amount of product (<10 pg/µL) that could not be detected within the limits of the PicoGreen assay, meaning that its amplification factor was below 10. Sark and ProK refer to sarkosyl and proteinase K, respectively. Error bars are not shown on this figure but are approximately 10% of each value.
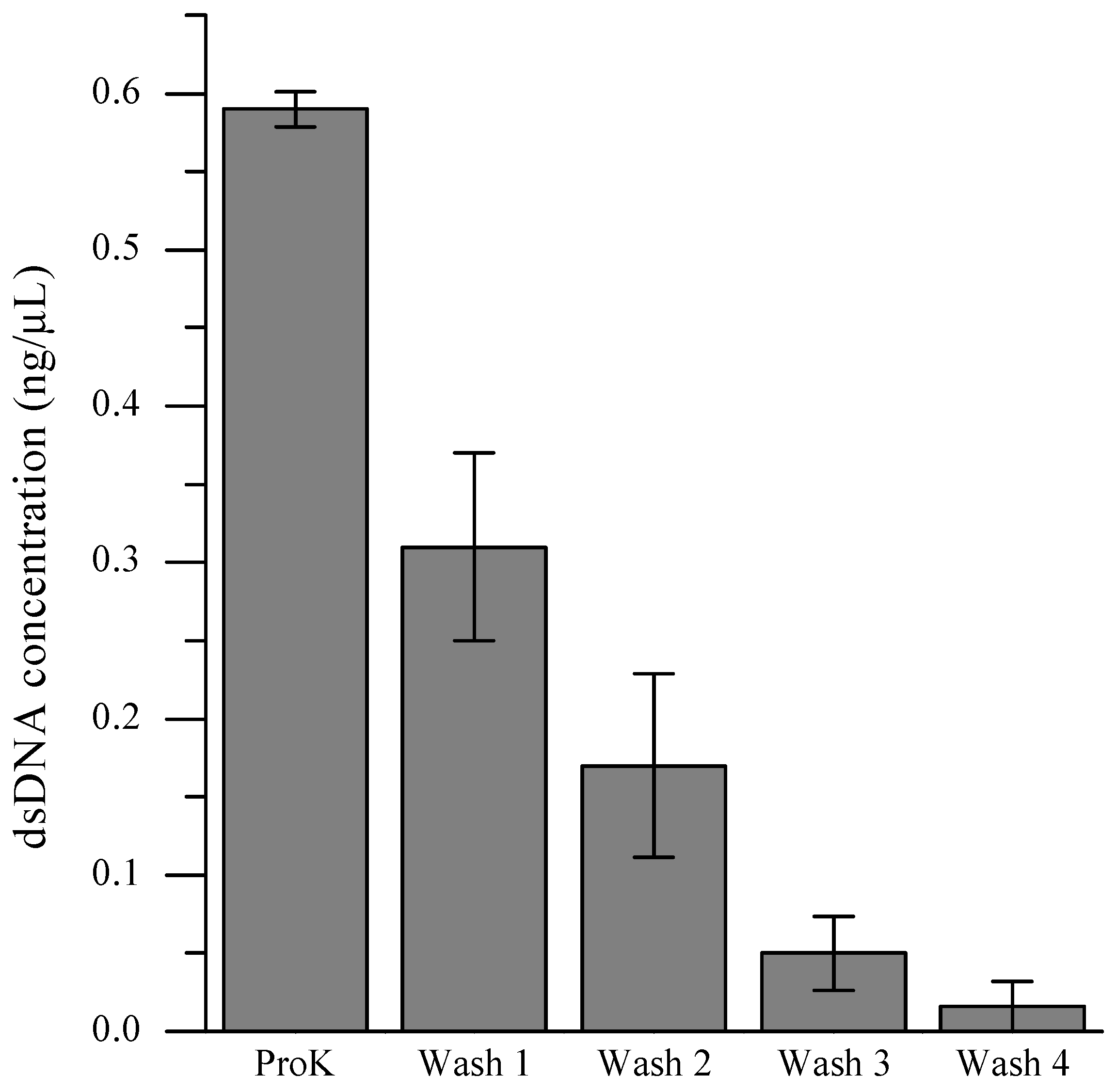
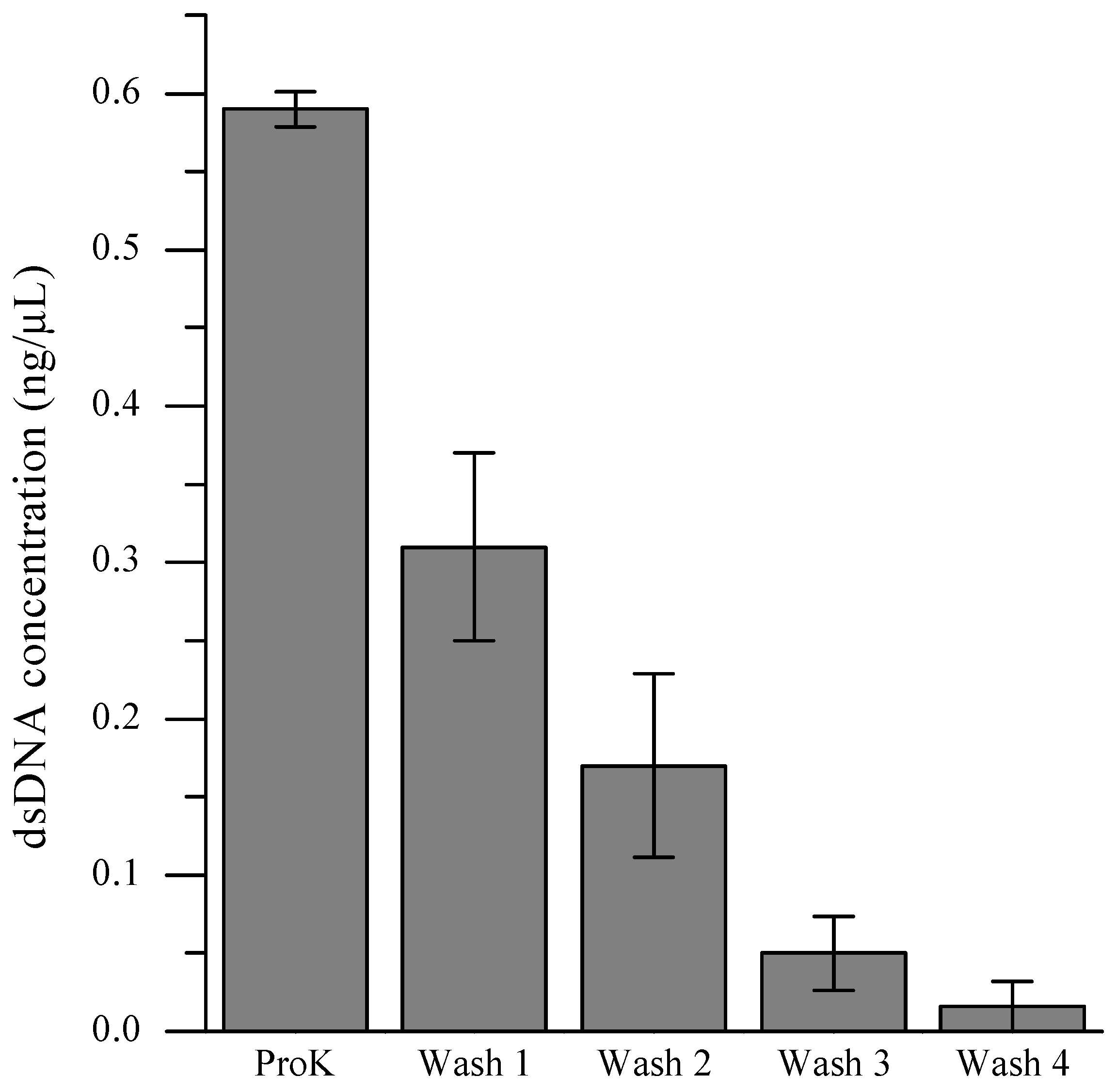
In order to retain cellular identities for each MDA reaction, it is crucial to prevent extraneous DNA from entering a microchamber together with the desired cell during the single-cell isolation process. Even a minuscule amount of DNA, either from the same organism or foreign species, can interfere with the analysis by competing with the targeted intracellular DNA during the amplification reaction. A facile strategy involving multiple washings of the cell pellet immediately prior to introduction of the cell suspension to the microfluidic device was developed in order to eliminate contaminant DNA from the environmental samples. To determine the number of washing steps necessary to eliminate extraneous DNA, we quantified the level of DNA present in the supernatant after each washing step (
Figure 3). Although the reduction of DNA amounts within the supernatant by consecutive washings was obvious, the PicoGreen assay was not sensitive enough to detect DNA present below single-cell quantities. When the dMDA assay was employed to quantify DNA present in the supernatants, the level of extracellular DNA in the cell suspension after the fourth TES wash and subsequent 200-fold dilution with Injection Buffer (PBS pH 7.4 with 0.1% Tween-20) was comparable to that of the no-template-DNA control sample (
Figure 4). Therefore, for scMDA experiments, five TES washing steps were inserted between the off-chip and on-chip lysis procedures to preclude DNA contamination.
Figure 3.
Removal of extracellular DNA via multiple washings of cell pellets before injecting into the microfluidic device. dsDNA concentration of the supernatant solutions were determined via PicoGreen fluorescence assay. ProK refers to proteinase K.
Figure 3.
Removal of extracellular DNA via multiple washings of cell pellets before injecting into the microfluidic device. dsDNA concentration of the supernatant solutions were determined via PicoGreen fluorescence assay. ProK refers to proteinase K.
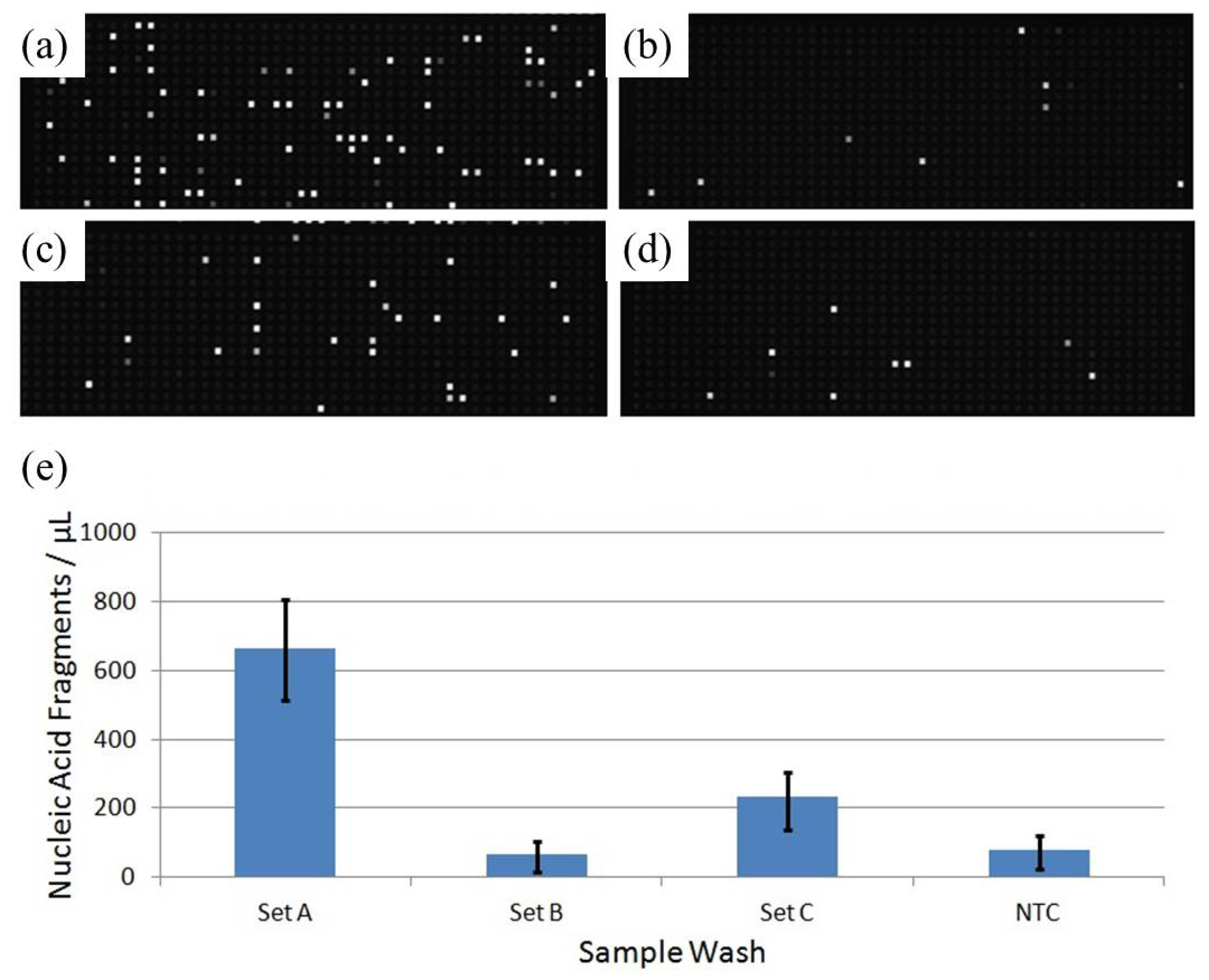
Figure 4.
dMDA confirmation of extracellular DNA removal via multiple washings of cell pellets before injection into the microfluidic device. The amounts of DNA fragments in the supernatants from the third and fourth successive TES Buffer washes were quantified with dMDA (as explained in Materials & Methods). Fluorescence images of the dMDA chips containing (a) the supernatant from the third wash and (b) its 200-fold dilution with Injection Buffer; (c) and (d) show the same set from the fourth wash, and (e) is the no-template-DNA control result.
Figure 4.
dMDA confirmation of extracellular DNA removal via multiple washings of cell pellets before injection into the microfluidic device. The amounts of DNA fragments in the supernatants from the third and fourth successive TES Buffer washes were quantified with dMDA (as explained in Materials & Methods). Fluorescence images of the dMDA chips containing (a) the supernatant from the third wash and (b) its 200-fold dilution with Injection Buffer; (c) and (d) show the same set from the fourth wash, and (e) is the no-template-DNA control result.
With the established lysis protocol, microfluidic scMDA of
Synechocystis was performed. Three independent experimental sets (Sets A–C) were prepared, each of which consisted of six single-cell-containing 60-nL microchambers and two negative control chambers (that is, containing no cell). The amplification factors for the first-round on-chip MDA reactions were estimated to be ~10
5, which are significantly greater than those of macroscale reactions under the same conditions (See Supplementary Figure S2). This increase is consistent with the previous reports [
10] that a confined reaction volume increases the yield of MDA. It is also notable that the inhibitory effects of added lysis reagents do not seem to be similarly enhanced.
The MDA amplicons from the “on-chip” experiment were amplified via macroscale 50-µL MDA reactions and then PCR-amplified for sequencing using 15
Synechocystis-specific primer pairs targeting genes across the entire chromosome (See Supplementary Table S1). When PCR products of expected fragment sizes, assessed from gel electrophoresis results, were Sanger-sequenced and their sequences were compared against the reference genome, 13 out of 17 single-cell amplicons (one sample lost) and 3 out of 6 negative controls were found to contain at least one
Synechocystis-specific target sequence. Sequences matching organisms other than
Synechocystis were not found via BLAST searches against known genome sequences.
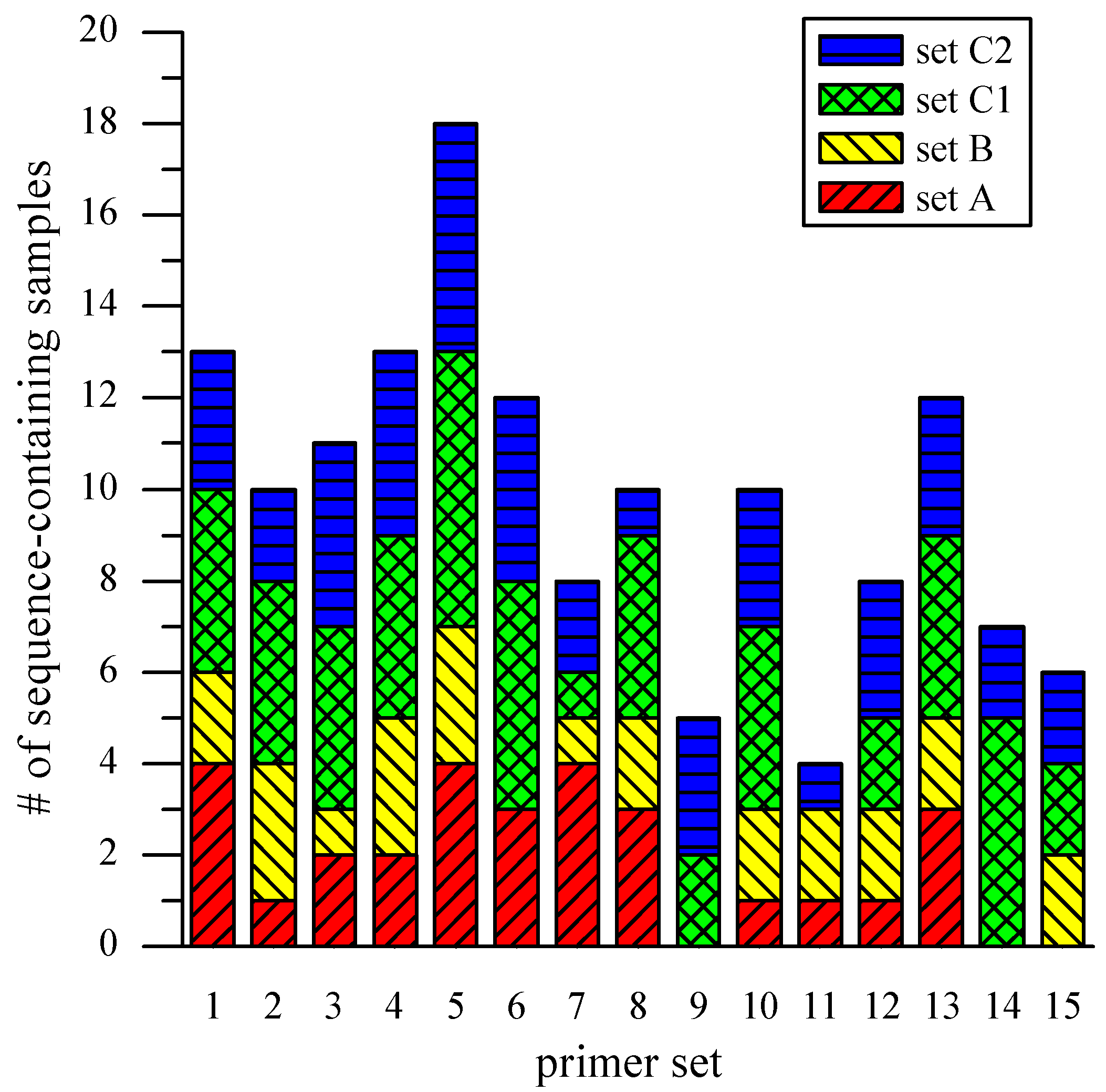
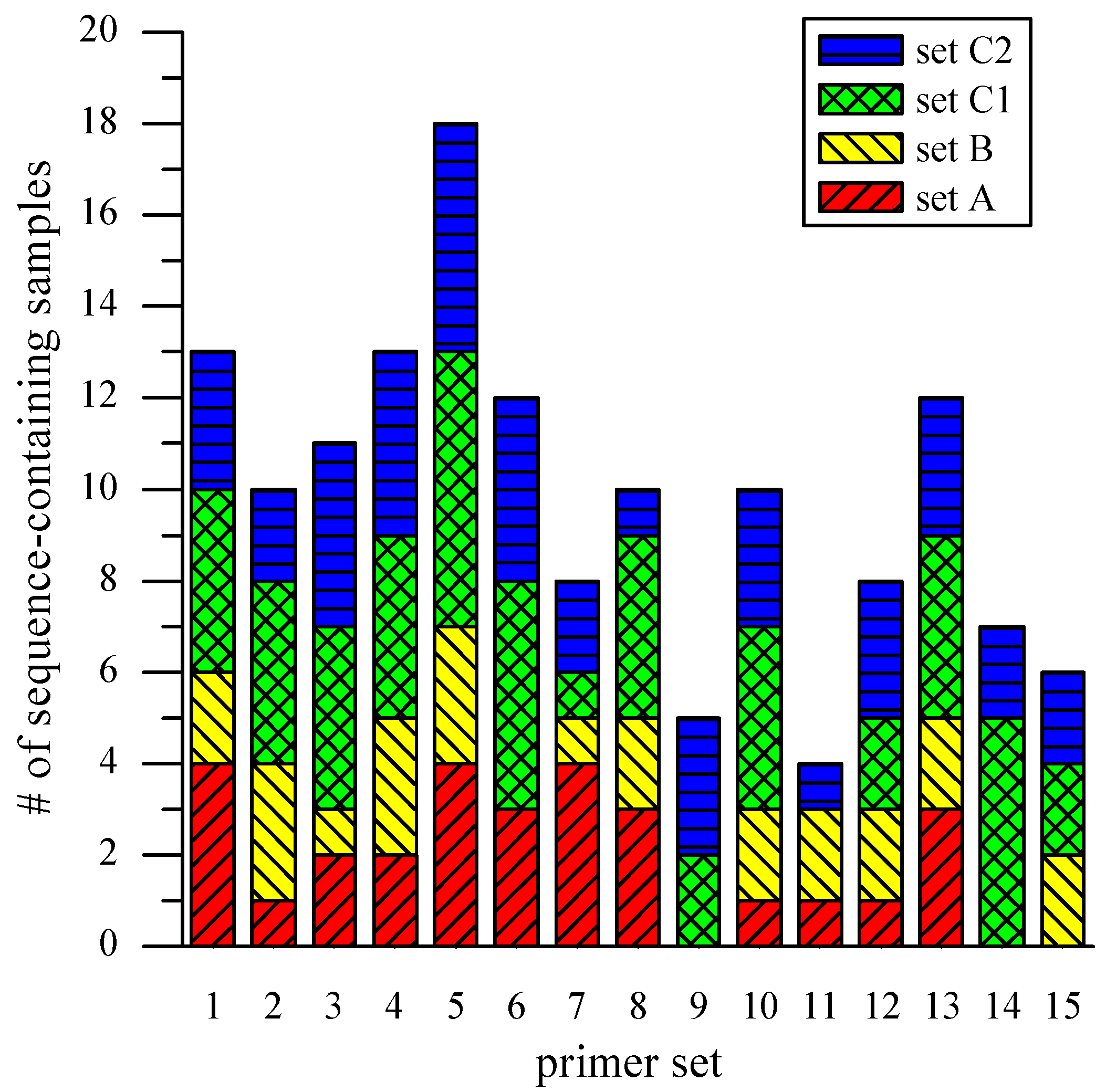
Figure 5 is a graphical representation of the results from all samples, broken down by sample sets and primer sets. The average occurrence of the specific target sequence from the three sets was 10 (out of 17 single-cell amplicons) with a standard deviation of 4. In an effort to gain preliminary insights into possible sources of variable genome coverage, the “on-chip” amplicons of Set C were divided and subjected to two parallel, off-chip 50-µL reactions (Set C1 and C2). Among the 67
Synechocystis-specific sequences produced by the two sets, 32 (48%) were present in both, 22 (33%) were produced only by Set C1, and 13 (19%) only by Set C2. These results indicate that the observed variation of genome coverage may be attributed partly to the stochastic property of the MDA reaction itself, and not those factors that might be the result of microchip complications such as incomplete lysis, obstruction of the template by cell debris, and a small starting amount of template [
10].
Figure 5.
Loci coverage across scMDA samples. The bar chart presents the occurrence of Synechocystis-specific sequences across scMDA samples, broken down by sample and primer sets.
Figure 5.
Loci coverage across scMDA samples. The bar chart presents the occurrence of Synechocystis-specific sequences across scMDA samples, broken down by sample and primer sets.
As mentioned above, half of all negative controls across the three sets produced
Synechocystis-specfic target amplicons. To characterize the amount of extracellular DNA in the injected sample sets, the supernatants from the final TES washes of each sample were analyzed via dMDA (
Figure 6). It should be noted that Set B’s free DNA content is indistinguishable from the no-template control, which is consistent with the observation that no negative controls from Set B produced
Synechocystis-specific amplicons. The no-template control is prepared according to all steps of the lysis protocol presented here, but no cells were added. We found that the no-template control gave negligible response after MDA amplification, demonstrating that our lysis reagents and sample handling do not introduce contaminating DNA. Therefore, the data from Set B represent a best-controlled single-cell experiment while other sets appear to still contain extracellular DNA, which must come from weakened cells leaking DNA into the solution,. In spite of these complications, we believe our data present a successful demonstration of the efficacy of our lysis protocol for
Synechocystis and its compatibility with microfluidic scMDA. It should be recognized that prevention of leakage of genetic material into the isolation volume will be crucial to extend our lysis protocol toward the analysis of heterogeneous samples, such as those found in environmental bacterial communities.
Figure 6.
Nucleic acid fragment content of final sample set washes. The supernatant of the final wash for each sample set was saved and diluted by a factor of 200. Nucleic acid fragments of the supernatants were quantified to obtain an idea of the level of sample exogenous contamination present in each set injection. Panels are lettered by sample ID (a) through (c) while the fourth (d) is a no-template control. Target counts are quantified per microliter of wash analyte (e), with error bars representing upper and lower 95% confidence interval estimates.
Figure 6.
Nucleic acid fragment content of final sample set washes. The supernatant of the final wash for each sample set was saved and diluted by a factor of 200. Nucleic acid fragments of the supernatants were quantified to obtain an idea of the level of sample exogenous contamination present in each set injection. Panels are lettered by sample ID (a) through (c) while the fourth (d) is a no-template control. Target counts are quantified per microliter of wash analyte (e), with error bars representing upper and lower 95% confidence interval estimates.
4. Conclusions
Whole genome analysis from single cells remains a topic of great interest. Significant progress has been made by combining microfluidic platforms with different kinds of gene amplification procedures [
2,
11,
12]. For cells that easily lyse, such as mammalian cells, much progress has been achieved [
13], but for cells having multiple cell wall layers, very few reports exist of their successful genomic analysis. This article has addressed this last issue by developing a lysis protocol that consists of off-chip partial removal and weakening of cell wall layers followed by on-chip lysis using reagents that do not interfere with the multiple displacement amplification reaction. This technique has been applied to
Synechocystis sp. PCC 6803, a fully-genome-sequenced strain of photosynthetic cyanobacteria that commonly occurs in freshwater [
14]. The challenge of single-cell genomic amplification is severe because of two types of interference, unwanted extraneous DNA arising from foreign sources and those arising from leaky cells [
17,
20]. These can dominate the amplification products because the lysing of a single cell releases so little genomic DNA. Consequently, much care has been taken to eliminate as best as we can these types of interference.
We have developed an effective lysis protocol for the model system Synechocystis and demonstrated its compatibility with microfluidic scMDA, thus extending this whole genome amplification technique to a strain of cyanobacteria. The protocol is both straightforward and flexible; it remains to be demonstrated that it is applicable with minor modifications to other prokaryotic species that are resistant to traditional lysis strategies. This chemical method, which is equally effective at a macro scale, also constitutes an alternative to conventional methods for preparing genomic DNA, which rely on mechanical cell breakage and extraction with organic solvents. We have shown that high-fidelity genome sequencing of single cells of Synechocystis can be achieved by performing microfluidic MDA reactions using this protocol, at least, as judged by performing sequencing on 15 loci that are widely separated. Whole genome sequencing was not performed in this study, but shotgun and next-generation sequencing and assembly might be done to assess how well this technique can cover the whole genome.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}