Hydrodynamic Cell Trapping for High Throughput Single-Cell Applications

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Procedure
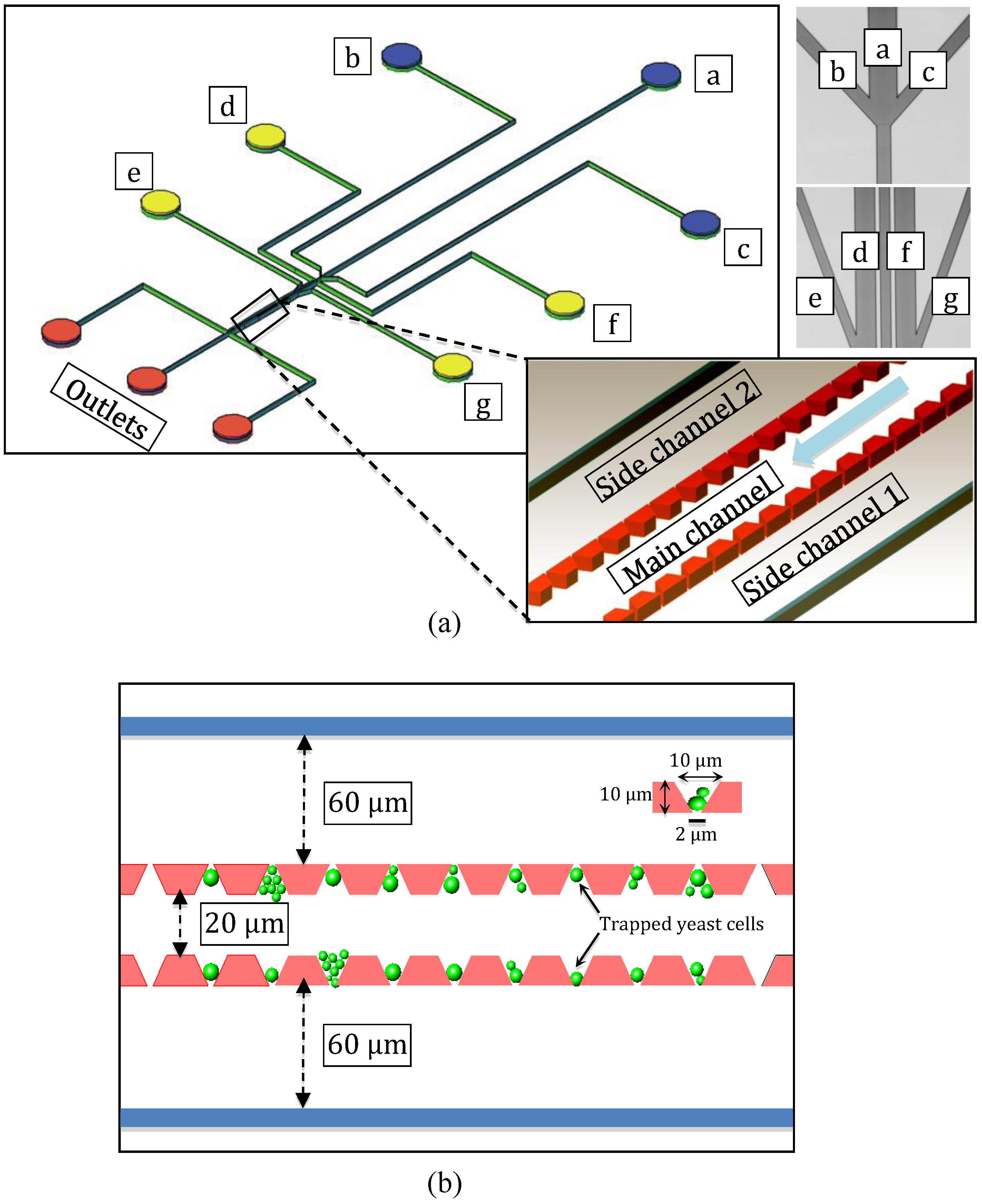
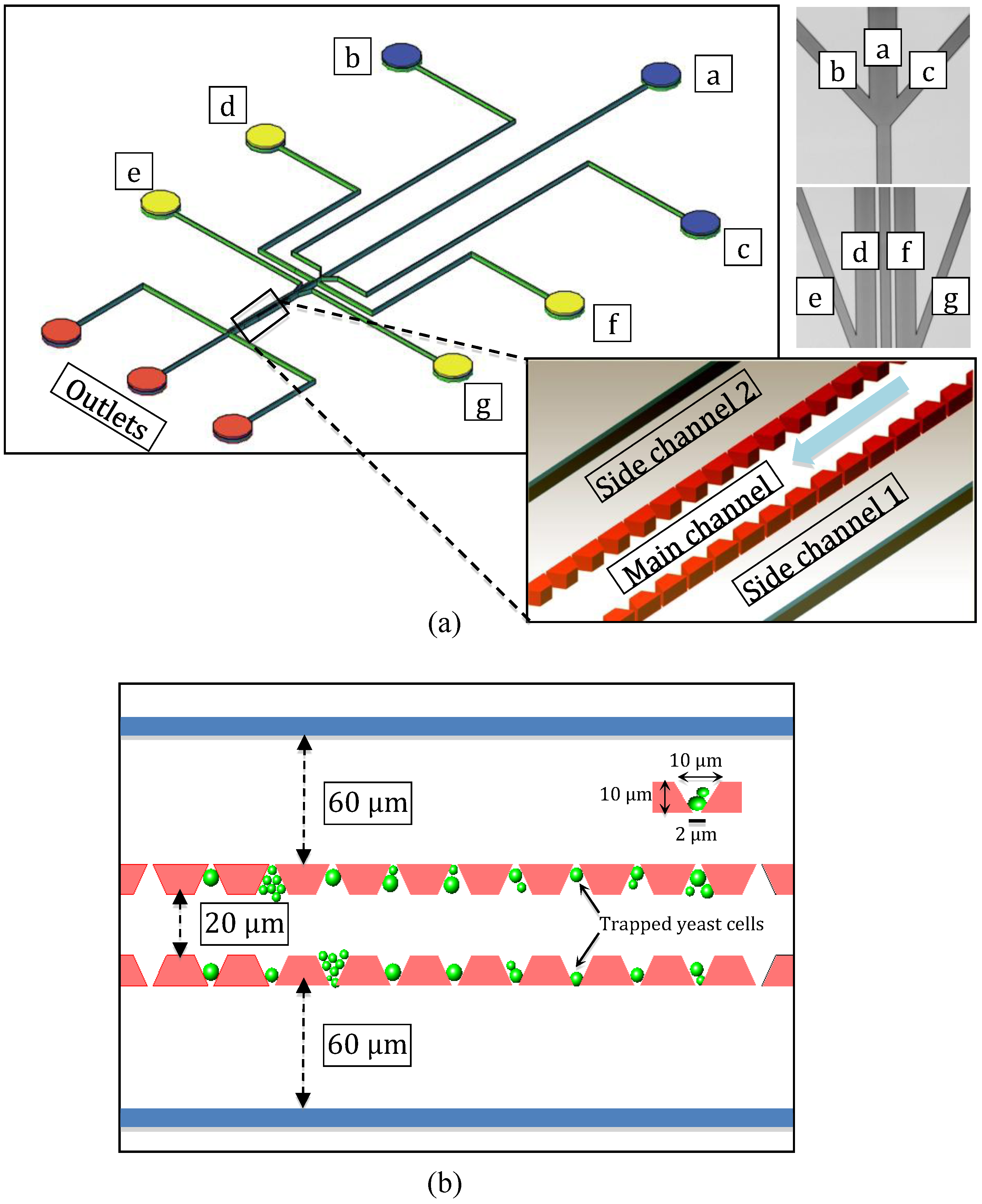
2.1. Microfluidic System Design and Fabrication
2.2. Numerical Simulations Using COMSOL Multiphysics
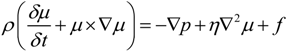
2.3. Cell Preparation
2.4. Sodium Arsenite Solution Preparation
3. Experimental Setup, Data Acquisition and Analysis
3.1. Microfluidic System Operation
3.2. Image Acquisition and Analysis
4. Results and Discussion
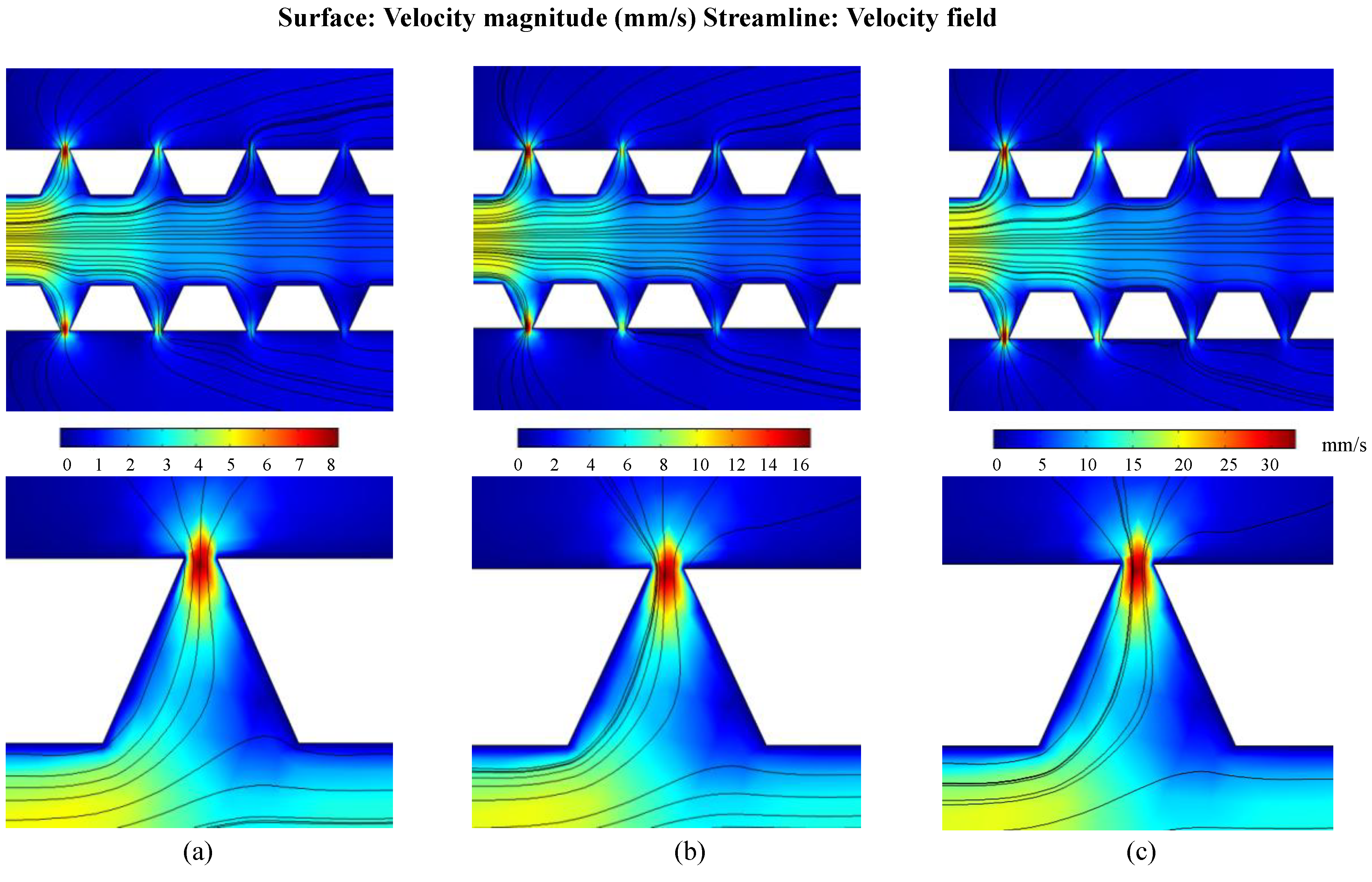
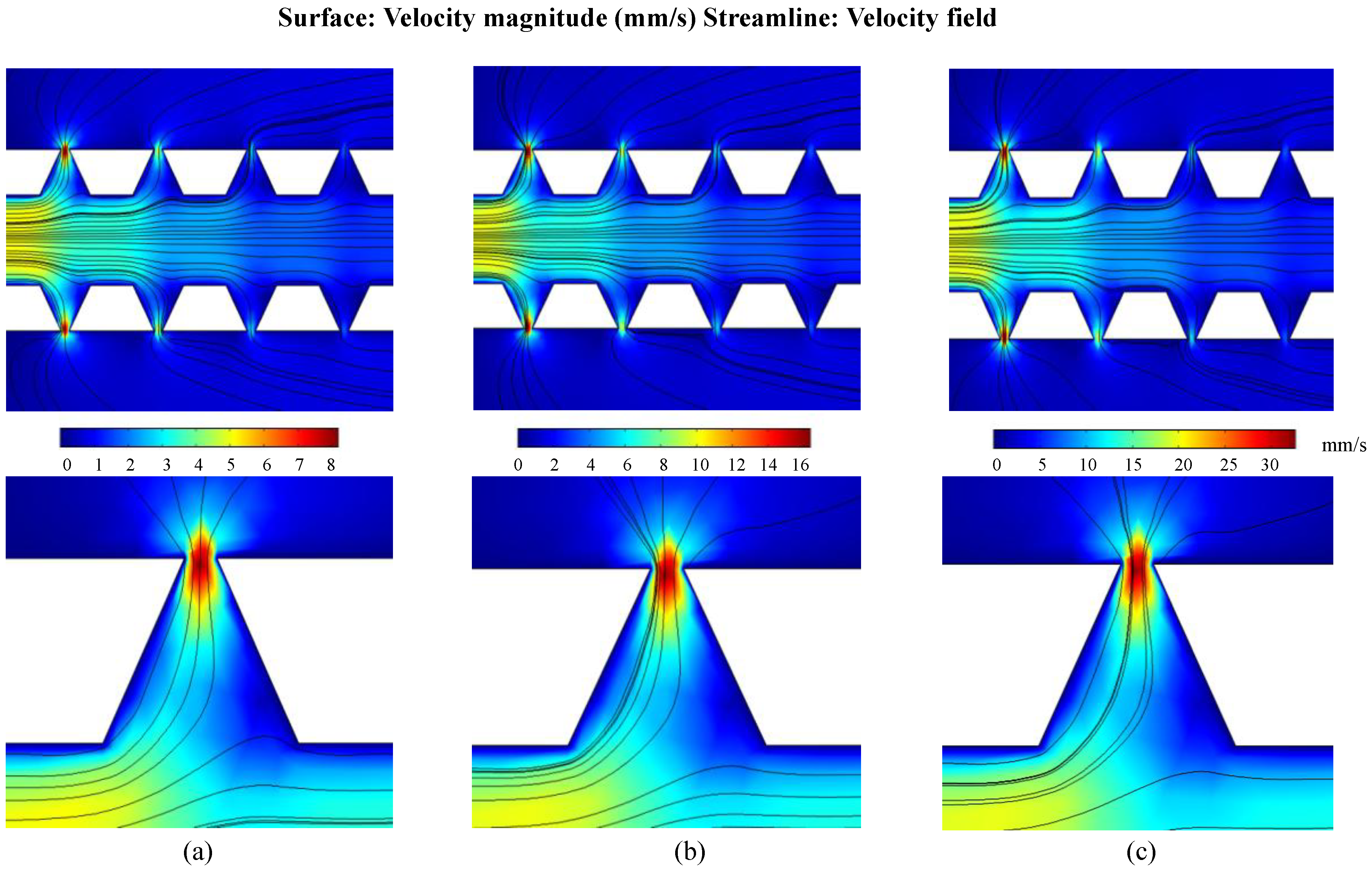
4.1. Velocity Field Simulations
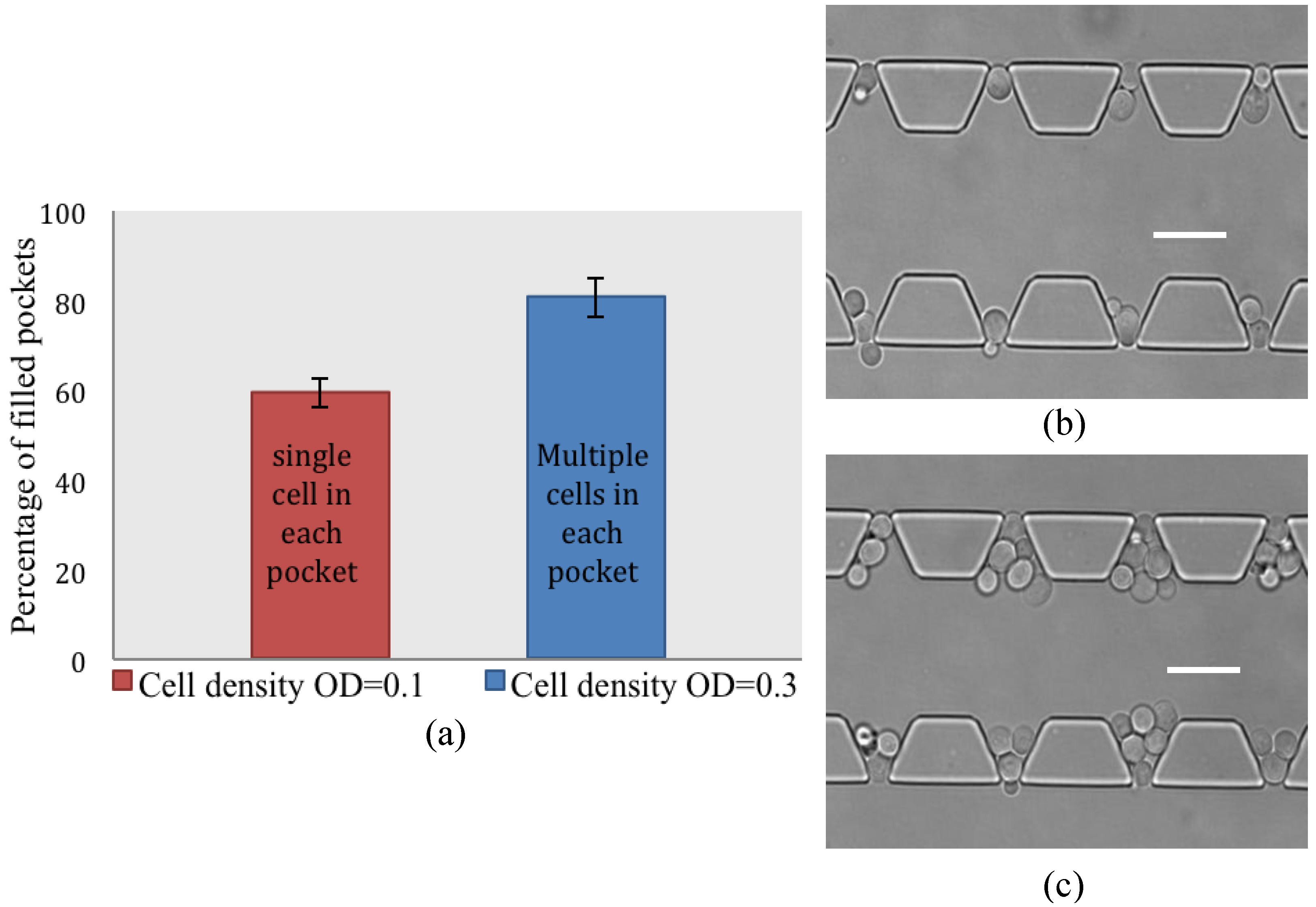
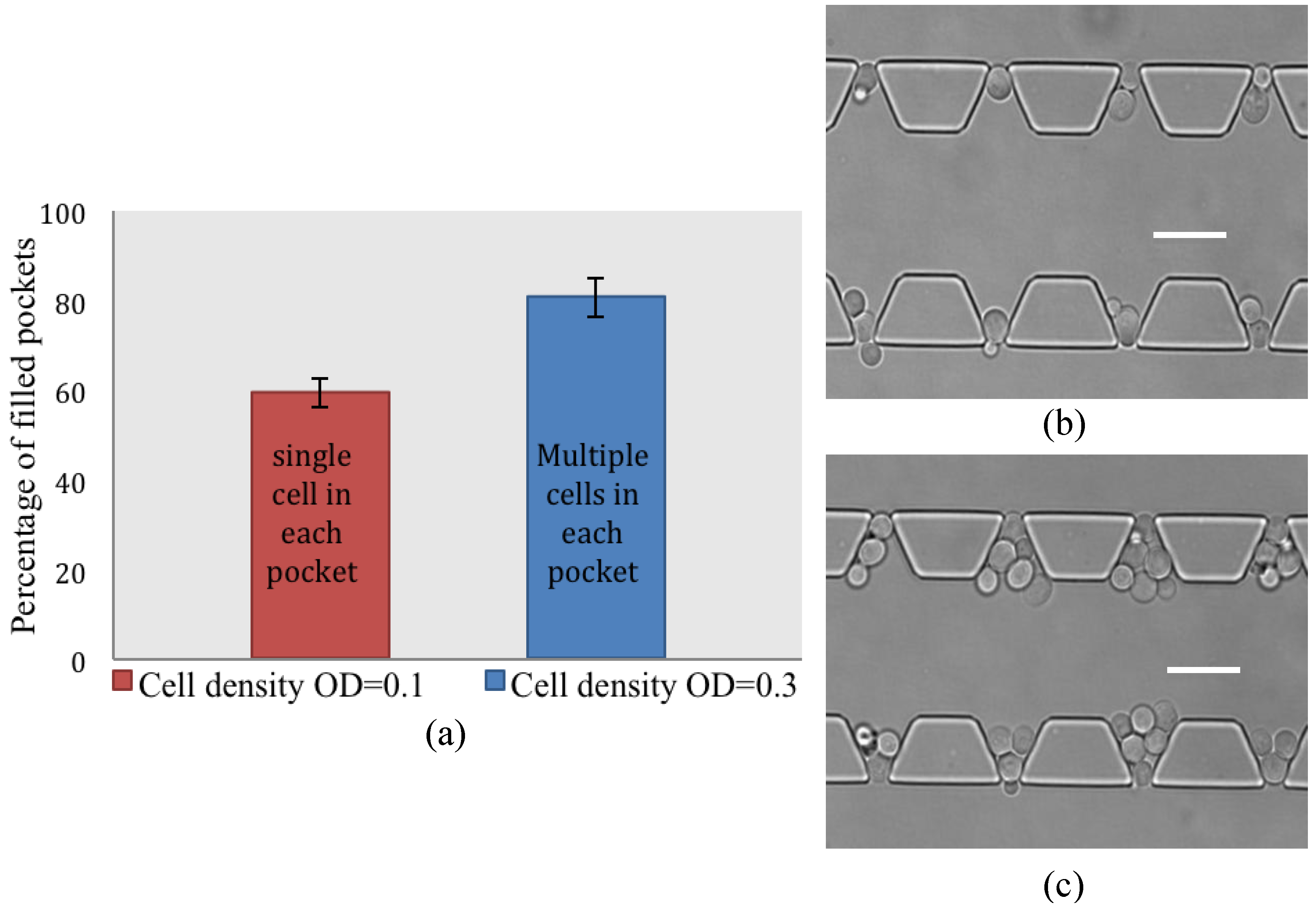
4.2. Cell Loading Efficiency
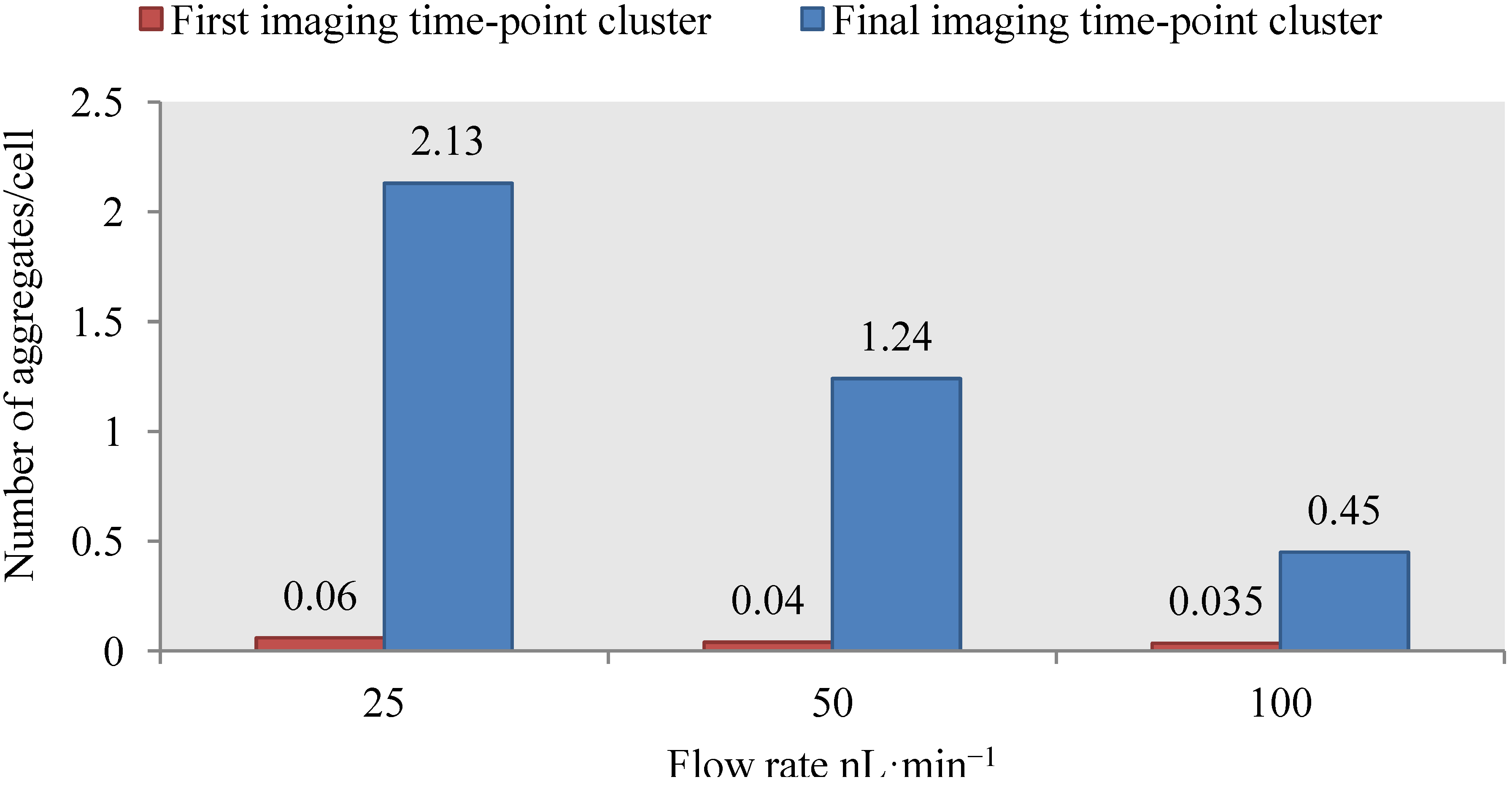
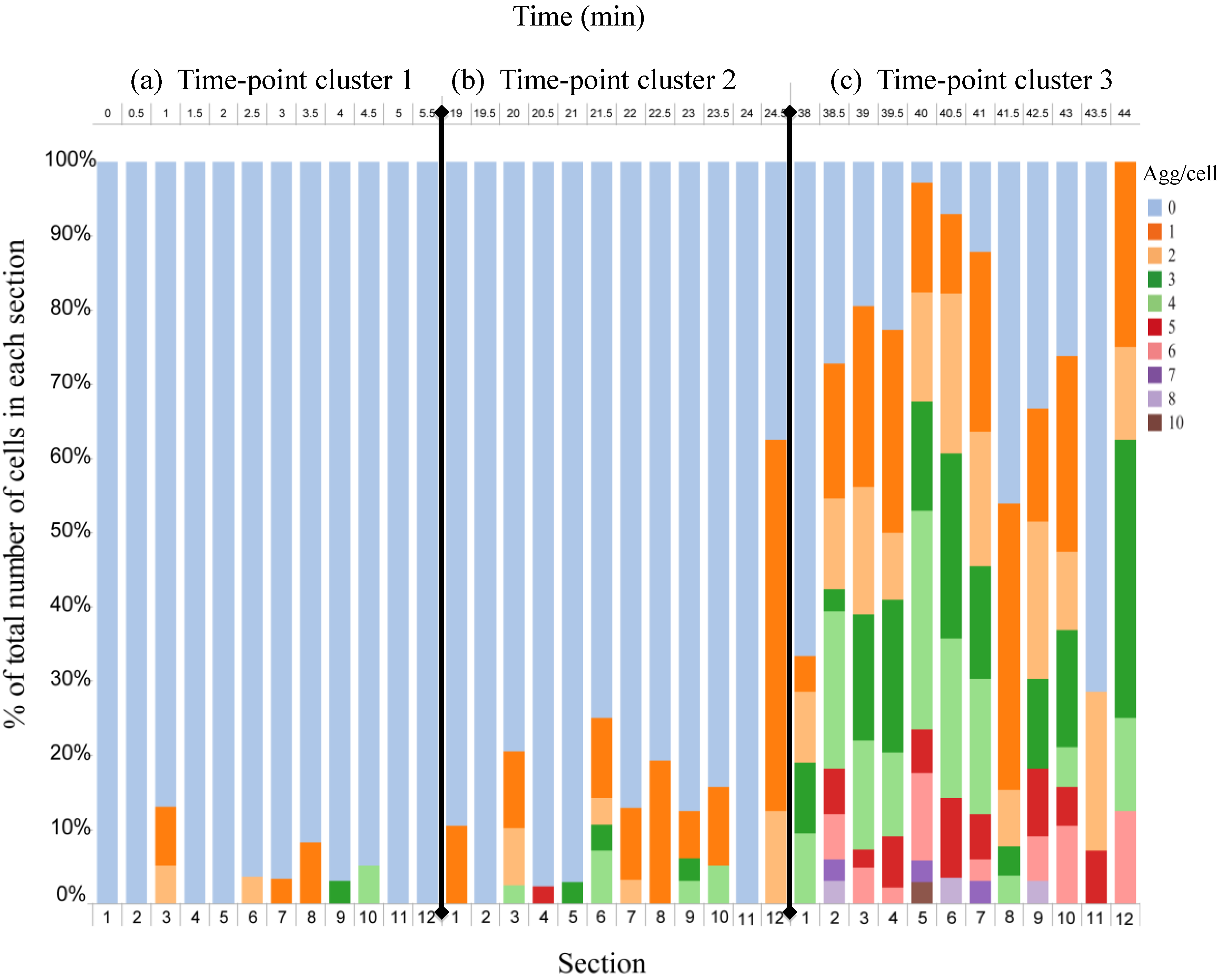
4.3. Effect of Flow Rate Modulation on Arsenite Uptake in Yeast
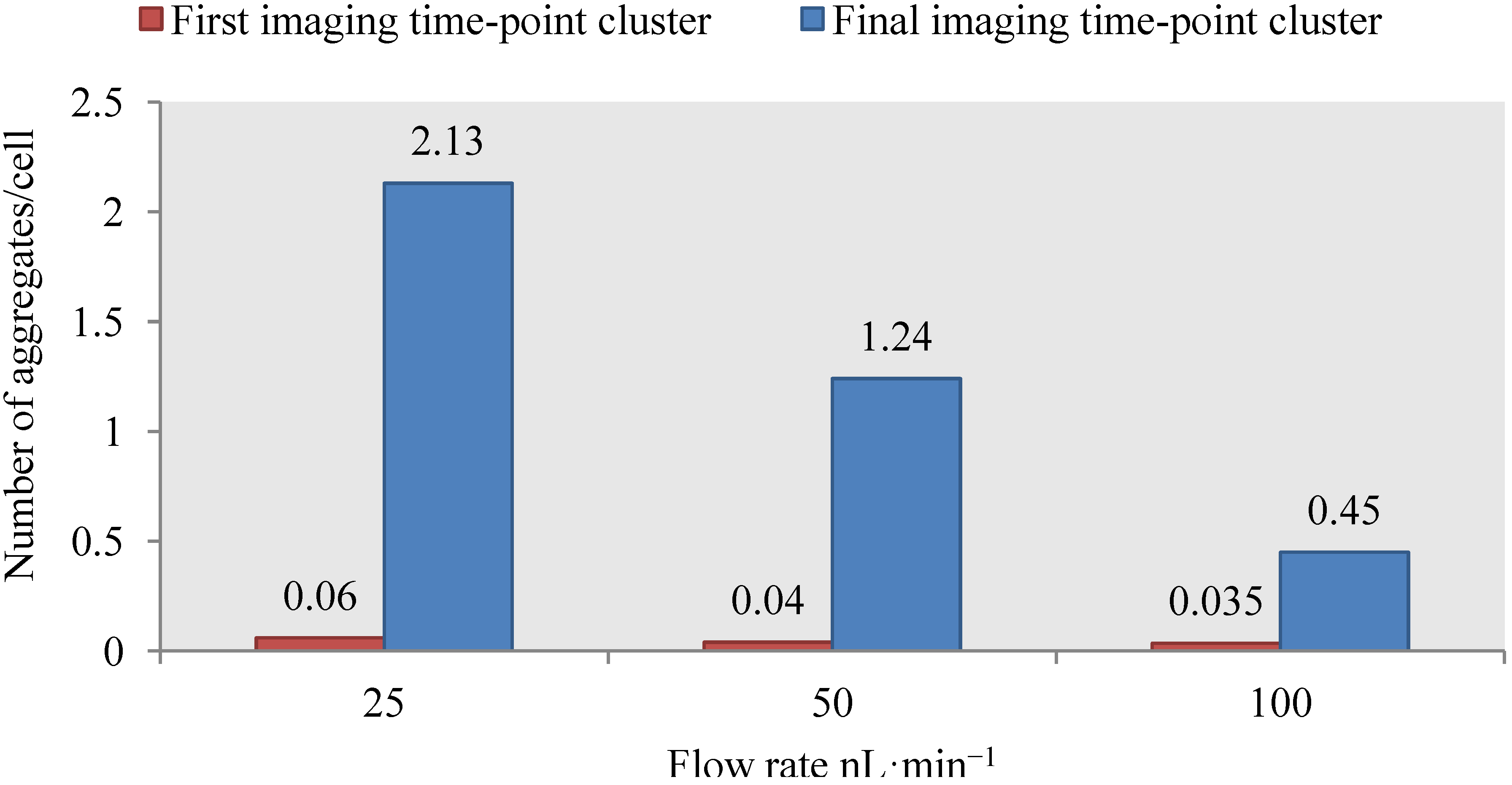

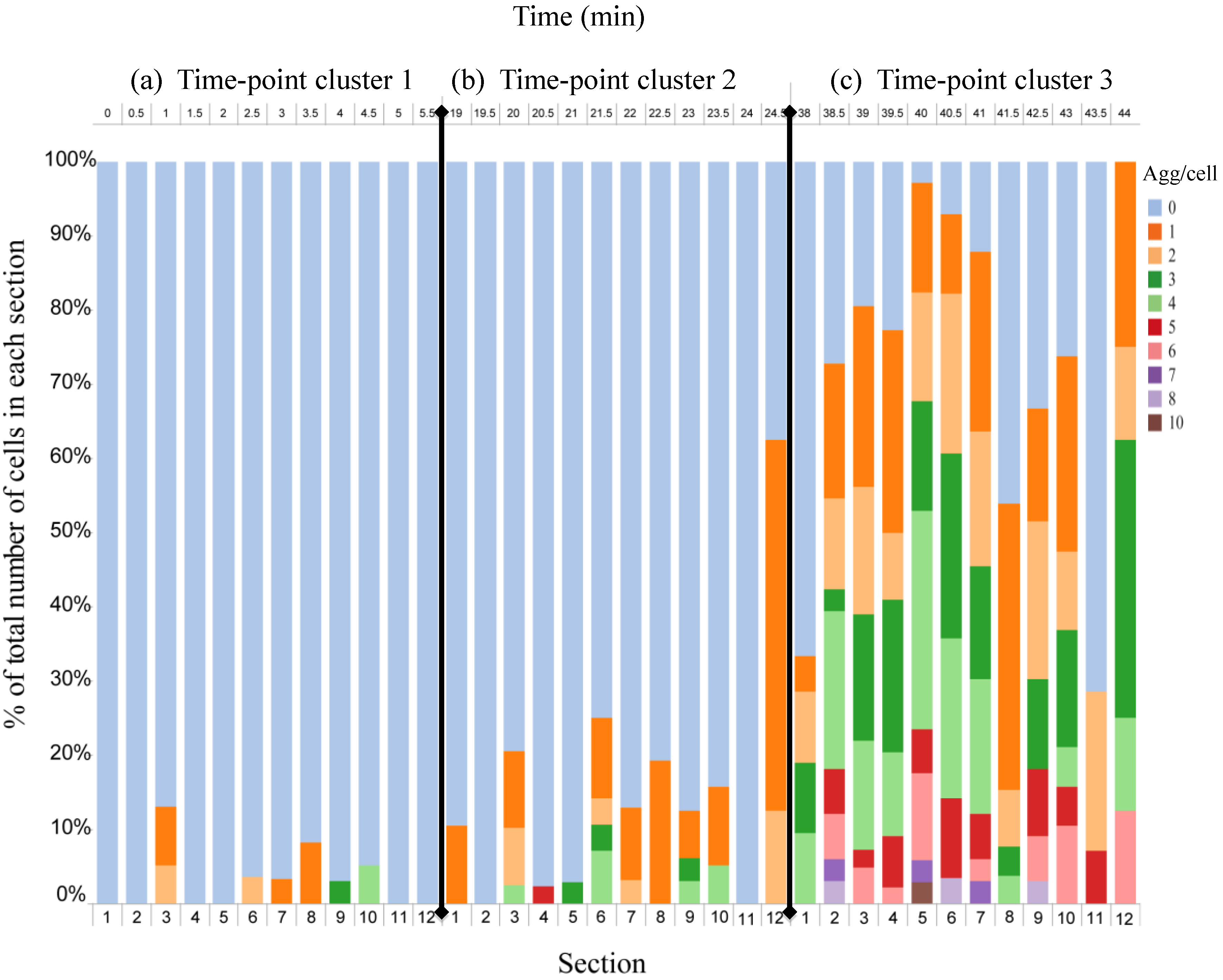
4.4. Discussion
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Figeys, D.; Pinto, D. Lab-on-a-chip: A revolution in biological and medical sciences. Anal. Chem. 2000, 72, 330A–335A. [Google Scholar]
- Haeberle, S.; Zengerle, R. Microfluidic platforms for lab-on-a-chip applications. Lab Chip 2007, 7, 1094–1110. [Google Scholar] [CrossRef]
- Squires, T.M.; Quake, S.R. Microfluidics: Fluid physics at the nanoliter scale. Rev. Mod. Phys. 2005, 77, 977–1026. [Google Scholar] [CrossRef]
- Elowitz, M.B.; Levine, A.J.; Siggia, E.D.; Swain, P.S. Stochastic gene expression in a single cell. Sci. Signal. 2002, 297, 1183–1186. [Google Scholar]
- Kærn, M.; Elston, T.C.; Blake, W.J.; Collins, J.J. Stochasticity in gene expression: From theories to phenotypes. Nat. Rev. Genet. 2005, 6, 451–464. [Google Scholar] [CrossRef]
- Burnette, W.N. “Western blotting”: Electrophoretic transfer of proteins from sodium dodecyl sulfate-polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein a. Anal. Biochem. 1981, 112, 195–203. [Google Scholar] [CrossRef]
- Dorak, M.T. Real-Time PCR; Taylor & Francis: New York, NY, USA, 2006. [Google Scholar]
- Svahn, H.A.; van den Berg, A. Single cells or large populations? Lab Chip 2007, 7, 544–546. [Google Scholar] [CrossRef]
- Breslauer, D.N.; Lee, P.J.; Lee, L.P. Microfluidics-based systems biology. Mol. Biosyst. 2006, 2, 97–112. [Google Scholar] [CrossRef]
- Muzzey, D.; van Oudenaarden, A. Quantitative time-lapse fluorescence microscopy in single cells. Ann. Rev. Cell Dev. Biol. 2009, 25, 301–327. [Google Scholar] [CrossRef]
- Jen, C.-P.; Hsiao, J.-H.; Maslov, N.A. Single-cell chemical lysis on microfluidic chips with arrays of microwells. Sensors 2011, 12, 347–358. [Google Scholar] [CrossRef]
- Rettig, J.R.; Folch, A. Large-scale single-cell trapping and imaging using microwell arrays. Anal. Chem. 2005, 77, 5628–5634. [Google Scholar] [CrossRef]
- Yamamura, S.; Kishi, H.; Tokimitsu, Y.; Kondo, S.; Honda, R.; Rao, S.R.; Omori, M.; Tamiya, E.; Muraguchi, A. Single-cell microarray for analyzing cellular response. Anal. Chem. 2005, 77, 8050–8056. [Google Scholar] [CrossRef]
- Irimia, D.; Toner, M. Cell handling using microstructured membranes. Lab Chip 2006, 6, 345–352. [Google Scholar] [CrossRef]
- Wheeler, A.R.; Throndset, W.R.; Whelan, R.J.; Leach, A.M.; Zare, R.N.; Liao, Y.H.; Farrell, K.; Manger, I.D.; Daridon, A. Microfluidic device for single-cell analysis. Anal. Chem. 2003, 75, 3581–3586. [Google Scholar] [CrossRef]
- Ashkin, A.; Dziedzic, J.; Yamane, T. Optical trapping and manipulation of single cells using infrared laser beams. Nature 1987, 330, 769–771. [Google Scholar] [CrossRef]
- Eriksson, E.; Scrimgeour, J.; Graneli, A.; Ramser, K.; Wellander, R.; Enger, J.; Hanstorp, D.; Goksör, M. Optical manipulation and microfluidics for studies of single cell dynamics. J. Opt. A Pure Appl. Opt. 2007, 9, S113. [Google Scholar] [CrossRef]
- Ramser, K.; Hanstorp, D. Optical manipulation for single-cell studies. J. Biophotonics 2010, 3, 187–206. [Google Scholar] [CrossRef]
- Taff, B.M.; Voldman, J. A scalable addressable positive-dielectrophoretic cell-sorting array. Anal. Chem. 2005, 77, 7976–7983. [Google Scholar] [CrossRef]
- Gascoyne, P.; Mahidol, C.; Ruchirawat, M.; Satayavivad, J.; Watcharasit, P.; Becker, F.F. Microsample preparation by dielectrophoresis: Isolation of malaria. Lab Chip 2002, 2, 70–75. [Google Scholar] [CrossRef]
- Gagnon, Z.R. Cellular dielectrophoresis: Applications to the characterization, manipulation, separation and patterning of cells. Electrophoresis 2011, 32, 2466–2487. [Google Scholar] [CrossRef]
- Falconnet, D.; Csucs, G.; Michelle Grandin, H.; Textor, M. Surface engineering approaches to micropattern surfaces for cell-based assays. Biomaterials 2006, 27, 3044–3063. [Google Scholar] [CrossRef]
- Lim, J.Y.; Donahue, H.J. Cell sensing and response to micro-and nanostructured surfaces produced by chemical and topographic patterning. Tissue Eng. 2007, 13, 1879–1891. [Google Scholar] [CrossRef]
- Di Carlo, D.; Wu, L.Y.; Lee, L.P. Dynamic single cell culture array. Lab Chip 2006, 6, 1445–1449. [Google Scholar] [CrossRef]
- Zhu, Z.; Frey, O.; Ottoz, D.S.; Rudolf, F.; Hierlemann, A. Microfluidic single-cell cultivation chip with controllable immobilization and selective release of yeast cells. Lab Chip 2012, 12, 906–915. [Google Scholar] [CrossRef]
- Van den Brink, F.T.; Gool, E.; Frimat, J.P.; Bomer, J.; van den Berg, A.; le Gac, S. Parallel single-cell analysis microfluidic platform. Electrophoresis 2011, 32, 3094–3100. [Google Scholar] [CrossRef]
- Tan, W.-H.; Takeuchi, S. A trap-and-release integrated microfluidic system for dynamic microarray applications. Proc. Natl. Acad. Sci. USA 2007, 104, 1146–1151. [Google Scholar] [CrossRef]
- Kobel, S.; Valero, A.; Latt, J.; Renaud, P.; Lutolf, M. Optimization of microfluidic single cell trapping for long-term on-chip culture. Lab Chip 2010, 10, 857–863. [Google Scholar] [CrossRef]
- Chung, K.; Rivet, C.A.; Kemp, M.L.; Lu, H. Imaging single-cell signaling dynamics with a deterministic high-density single-cell trap array. Anal. Chem. 2011, 83, 7044–7052. [Google Scholar] [CrossRef]
- Eriksson, E.; Enger, J.; Nordlander, B.; Erjavec, N.; Ramser, K.; Goksör, M.; Hohmann, S.; Nyström, T.; Hanstorp, D. A microfluidic system in combination with optical tweezers for analyzing rapid and reversible cytological alterations in single cells upon environmental changes. Lab Chip 2006, 7, 71–76. [Google Scholar]
- Eriksson, E.; Sott, K.; Lundqvist, F.; Sveningsson, M.; Scrimgeour, J.; Hanstorp, D.; Goksör, M.; Granéli, A. A microfluidic device for reversible environmental changes around single cells using optical tweezers for cell selection and positioning. Lab Chip 2010, 10, 617–625. [Google Scholar] [CrossRef]
- Jacobson, T.; Navarrete, C.; Sharma, S.K.; Sideri, T.C.; Ibstedt, S.; Priya, S.; Grant, C.M.; Christen, P.; Goloubinoff, P.; Tamás, M.J. Arsenite interferes with protein folding and triggers formation of protein aggregates in yeast. J. Cell Sci. 2012, 125, 5073–5083. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.H.; Nguyen, N.-T.; Chua, Y.C.; Kang, T.G. Oxygen plasma treatment for reducing hydrophobicity of a sealed polydimethylsiloxane microchannel. Biomicrofluidics 2010, 4, 32204. [Google Scholar] [CrossRef]
- Sott, K.; Eriksson, E.; Goksör, M. Acquisition of Single Cell Data in an Optical Microscope. In Lab on a Chip Technology: Biomolecular Separation and Analysis; Caister Academic Press: Norfolk, UK, 2009; pp. 151–166. [Google Scholar]
- Smedh, M.; Beck, C.; Sott, K.; Goksör, M. Cellstress-Open Source Image Analysis Program for Single-Cell Analysis. In Proceedings of SPIE 7762, Optical Trapping and Optical Micromanipulation VII, 77622N, San Diego, CA, USA, 27 August 2010; International Society for Optics and Photonics: Bellingham, WA, USA, 2010. [Google Scholar] [CrossRef]
- Wysocki, R.; Chéry, C.C.; Wawrzycka, D.; van Hulle, M.; Cornelis, R.; Thevelein, J.M.; Tamás, M.J. The glycerol channel fps1p mediates the uptake of arsenite and antimonite in Saccharomyces cerevisiae. Mol. Microbiol. 2001, 40, 1391–1401. [Google Scholar] [CrossRef]
- Tamás, M.J.; Karlgren, S.; Bill, R.M.; Hedfalk, K.; Allegri, L.; Ferreira, M.; Thevelein, J.M.; Rydström, J.; Mullins, J.G.; Hohmann, S. A short regulatory domain restricts glycerol transport through yeast fps1p. J. Biol. Chem. 2003, 278, 6337–6345. [Google Scholar] [CrossRef]
- Maciaszczyk-Dziubinska, E.; Migdal, I.; Migocka, M.; Bocer, T.; Wysocki, R. The yeast aquaglyceroporin Fps1p is a bidirectional arsenite channel. FEBS Lett. 2010, 584, 726–732. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Banaeiyan, A.A.; Ahmadpour, D.; Adiels, C.B.; Goksör, M. Hydrodynamic Cell Trapping for High Throughput Single-Cell Applications. Micromachines 2013, 4, 414-430. https://doi.org/10.3390/mi4040414
Banaeiyan AA, Ahmadpour D, Adiels CB, Goksör M. Hydrodynamic Cell Trapping for High Throughput Single-Cell Applications. Micromachines. 2013; 4(4):414-430. https://doi.org/10.3390/mi4040414
Chicago/Turabian StyleBanaeiyan, Amin Abbaszadeh, Doryaneh Ahmadpour, Caroline Beck Adiels, and Mattias Goksör. 2013. "Hydrodynamic Cell Trapping for High Throughput Single-Cell Applications" Micromachines 4, no. 4: 414-430. https://doi.org/10.3390/mi4040414