JNK, p38, ERK, and SGK1 Inhibitors in Cancer
by
 ,
,
Jonas Cicenas
1,2,3,* ,
,
Egle Zalyte
2, 
Arnas Rimkus
4,
Dalius Dapkus
5,
Remigijus Noreika
5 and
Sigitas Urbonavicius
6 1
Department for Microbiology, Immunbiology und Genetics, Max F. Perutz Laboratories, University of Vienna, Vienna AT-1030, Austria
2
Proteomics Centre, Institute of Biochemistry, Vilnius University, 01513 Vilnius, Lithuania
3
MAP Kinase Resource, Bioinformatics, Melchiorstrasse 9, CH-3027 Bern, Switzerland
4
Faculty of Medicine, Vilnius University, 01513 Vilnius, Lithuania
5
Department of Biology and Chemistry, Lithuanian University of Educational Sciences, 08106 Vilnius, Lithuania
6
Cardiovascular Research Centre, Viborg Hospital, Heibergs Alle 4, 8800 Viborg, Denmark
*
Author to whom correspondence should be addressed.
Cancers 2018, 10(1), 1; https://doi.org/10.3390/cancers10010001
Submission received: 25 October 2017
/
Revised: 14 December 2017
/
Accepted: 19 December 2017
/
Published: 21 December 2017
(This article belongs to the Special Issue Kinases and Cancer)
Abstract
:Mitogen-activated protein kinases (MAP kinases) are a family of kinases that regulates a range of biological processes implicated in the response to growth factors like latelet-derived growth factor (PDGF), epidermal growth factor (EGF), vascular endothelial growth factor (VEGF), and stress, such as ultraviolet irradiation, heat shock, and osmotic shock. The MAP kinase family consists of four major subfamilies of related proteins (extracellular regulated kinases 1/2 (ERK1/2), c-Jun N-terminal kinase (JNK), p38, and extracellular regulated kinase 5 (ERK5)) and regulates numerous cellular activities, such as apoptosis, gene expression, mitosis, differentiation, and immune responses. The deregulation of these kinases is shown to be involved in human diseases, such as cancer, immune diseases, inflammation, and neurodegenerative disorders. The awareness of the therapeutic potential of the inhibition of MAP kinases led to a thorough search for small-molecule inhibitors. Here, we discuss some of the most well-known MAP kinase inhibitors and their use in cancer research.
1. Introduction
Protein kinases are a family of enzymes that phosphorylate proteins on serine, threonine, or tyrosine. Protein phosphorylation brings about changes of their functions, such as their interaction with other proteins, localization, or enzymatic activity. Our genome contains more than 500 protein kinase genes as well as some pseudogenes. Protein phosphorylation plays a critical role in the regulation of numerous cellular properties such as proliferation, differentiation, apoptosis, migration, and adhesion. Therefore, the wrong kinase activity can result in exceptional alterations of these processes. In fact, defective kinases are often found to be oncogenic and can be important for the existence of cancer cells. In addition, the phosphorylation of some proteins, such as protein kinase B, also known as Akt (Akt) [1,2], epidermal growth factor (EGF), [3], ErbB receptor family member 2 (ERBB2) [4,5], extracellular regulated kinase (ERK) [6,7], p38 [8], and Src homology 2 domain-containing-transforming protein C-A (SchA) [9], is associated with prognosis in several human cancers. The first MAP kinase network to be discovered was the GTPase Ras-RAF proto-oncogene serine/threonine-protein kinase-extracellular regulated kinase (RAS-RAF-ERK) signal transduction cascade (Figure 1), defined by ERK1 and ERK2 [10,11]. The ERK cascade functions in cellular proliferation, differentiation, and survival and deregulation of it is common in cancer. In many cases, the activity of ERKs in cancer depends on mutations in RAS and RAF kinases [12,13]. c-Jun N-terminal kinase (JNK) is a subfamily of mitogen-activated protein (MAP) (Figure 1) kinases originally identified as kinases that bind and phosphorylate transcription factor AP-1 (JUN) on S63 and S73 within its transcriptional activation domain [14]. There are three differently spliced genes in the subfamily, namely, JNK1, JNK2, and JNK3. Aberrant activation of JNKs is found in many cancers, as well as inflammatory and neurodegenerative disorders. p38 is yet another subfamily (Figure 1), consisting of four isoforms: α, β, γ, and δ [15]. Pathogens or inflammatory stimuli initiate a cascade mediated by p38 kinases and abnormal activity of these kinases is observed in inflammatory diseases and cancers. One of the major substrates and further signal transducers is serum/glucocorticoid-regulated kinase 1 (SGK1) kinase, which is also important in cancer development. The last of four major MAP kinase pathways is the mitogen-activated protein kinase kinase 5- extracellular regulated kinase 5 (MEK5-ERK5) cascade (Figure 1). Activation of this pathway is a common event in tumor development and it is involved in anti-apoptotic signaling and chemoresistance [16]. There are many inhibitors, which are quite specific and quite a few of them are already approved for cancer therapy or at least in clinical studies of different phases. Thus far, several kinases are quite popular targets of inhibition in cancers, such as tyrosine kinases [17], cyclin-dependent kinases (CDKs) [18,19,20] and aurora kinases [21,22]. In some cases, however, clinical trials have failed, leading to the reevaluation or redesign of inhibitors. This review discusses some of interesting MAP kinase inhibitors used for cancer research.
2. MAP Kinase Inhibitors in Cancer Research
SP600125 is a selective and reversible inhibitor of JNK kinases, which has an IC50 for JNK1, JNK2 = 40 nM, and JNK3 = 90 nM. It induces cell death selectively in undifferentiated thyroid cancer cell lines [23], reduces the viability of doxorubicin-resistant stomach cancer cells [24], sensitizes the multidrug-resistant KBV20C human oral squamous carcinoma cell line [25], enhances dihydroartemisinin-induced apoptosis in human lung adenocarcinoma cells [26], enhances transforming growth factor beta (TGF-β)-induced apoptosis in human cholangiocarcinoma cell line RBE [27], selectively kills p53-deficient human colon carcinoma cells in a mouse xenograft model [28], affects the regulation of the epithelial barrier function and cell shape during the remodeling of pancreatic cancer cells [29], and suppresses glioblastoma cells [30]. Apart from anticancer properties, this inhibitor is also used in inflammation research [31,32], and neuroprotection [33,34].
AS601245 is a cell-permeable JNK inhibitor (Figure 2). The IC50 for JNK1 = 150 nM, for JNK2 = 220 nM, and for JNK3 = 70 nM. AS601245 affects the proliferation of colon cancer cell lines [35] and decreases cell adhesion and migration via a decrease in the fibrinogen release in human colon cancer cells [36]. AS601245 also has an effect on leukemia by leading T-cell acute lymphoblastic leukemia cells to cell cycle arrest and apoptosis [37] and sensitizing promonocytic leukemia cells to arsenic trioxide-induced apoptosis [38]. In addition to cancer, AS601245 is also used in inflammation [39] and as antiviral agent [40,41].
CC-401 is a specific inhibitor of JNK which has an IC50 for these kinase in a range of 25–50 nM. CC-401 in combination with oxaliplatin shows synergism in colon cancer cell lines SW620 and HT29 in vitro and in mouse xenografts [42]. It is also used for glomerulonephritis [43,44] and hepatic ischemia reperfusion injury [45,46].
Several other JNK inhibitors show promise in cancer cells: AS602801 (IC50 for JNK1 = 80 nM, for JNK2 = 90 nM, and for JNK3 = 230 nM) inhibits cancer stem cells in vitro and in vivo [47], D-JNKI-1 (IC50 for JNKs is 2.31 μM) reduces tumor growth in a mouse skin cancer model [48], and BI-78D3 (IC50 for JNKs is 280 nM) sensitizes osteosarcoma to doxorubicin [49].
SCIO-469 (Talmapimod) is a selective p38 inhibitor. The IC50 for p38α = 9 nM and for p38β = 90 nM. SCIO-469 enhances bortezomib-induced cytotoxicity against multiple myeloma cells [50], reduces multiple myeloma cell proliferation and adhesion [51], enhances the apoptosis of myeloma cells and inhibits tumor growth [52], and decreases tumor burden and angiogenesis in murine models of multiple myeloma [53,54]. In addition, it enhances the arsenic trioxide-dependent induction of apoptosis in chronic myelogenous leukemia or acute promyelocytic leukemia-derived cell lines [55]. This inhibitor is also used in phase II human clinical trials for the treatment of rheumatoid arthritis [56], myelodysplastic syndrome [57], and acute dental pain [58].
BIRB-796 (Doramapimod) is a p38 inhibitor (Figure 2) which has an IC50 for p38α = 38 nM, for p38β = 65 nM, for p38γ = 200 nM, and for p38δ = 520 nM. BIRB-796 enhances cytotoxicity and inhibits paracrine tumor growth in multiple myeloma [59], enhances the efficacy of chemotherapeutic agents in multidrug resistance protein 1 (ABCB1) overexpressing oral epidermoid carcinoma cells [60], and enhances the antitumor effects of aurora kinase inhibitor VX680 in cervical cancer [61]. It is also used in inflammation research [62,63].
LY2228820 (Ralimetinib) is a selective p38 inhibitor. The IC50 for p38α = 5.3 nM and for p38β = 3.2 nM. LY2228820 enhances bortezomib-induced cytotoxicity and inhibits osteoclastogenesis in multiple myeloma [64], produces significant tumor growth delay in multiple in vivo cancer models (melanoma, non-small cell lung cancer, ovarian, glioma, myeloma, breast) [65], and inhibits the Ras-related C3 botulinum toxin substrate 3 (Rac3)-induced cell invasion and migration of lung adenocarcinoma [66]. A Phase I clinical trial in patients with advanced cancer (colorectal, breast, sarcoma, non small cell lung, renal, pancreatic, melanoma, and ovarian) demonstrated acceptable safety, tolerability, and pharmacokinetics [67]. Although none of the patients had either full or partial remission, 19 (23.3%) patients had a stable disease within a median time of 3.7 months. That shows some promise using this inhibitor either as a single agent or in combination with chemotherapeutic agents; however, additional studies are required to find biomarkers that predict the clinical efficiency of LY2228820 for patients with advanced cancer. Another Phase I/II trial study of LY2228820 plus gemcitabine and carboplatin for platinum-sensitive ovarian cancer is still ongoing [68].
A couple other p38 inhibitors show promise in cancer cells: VX-745 (IC50 for p38α = 10 nM and for p38β = 220 nM) inhibits multiple myeloma cell growth [69] and PH-797804 (IC50 for p38α = 26 nM and for p38β = 102 nM) reduces tumor growth in colon tumor xenografts [70].
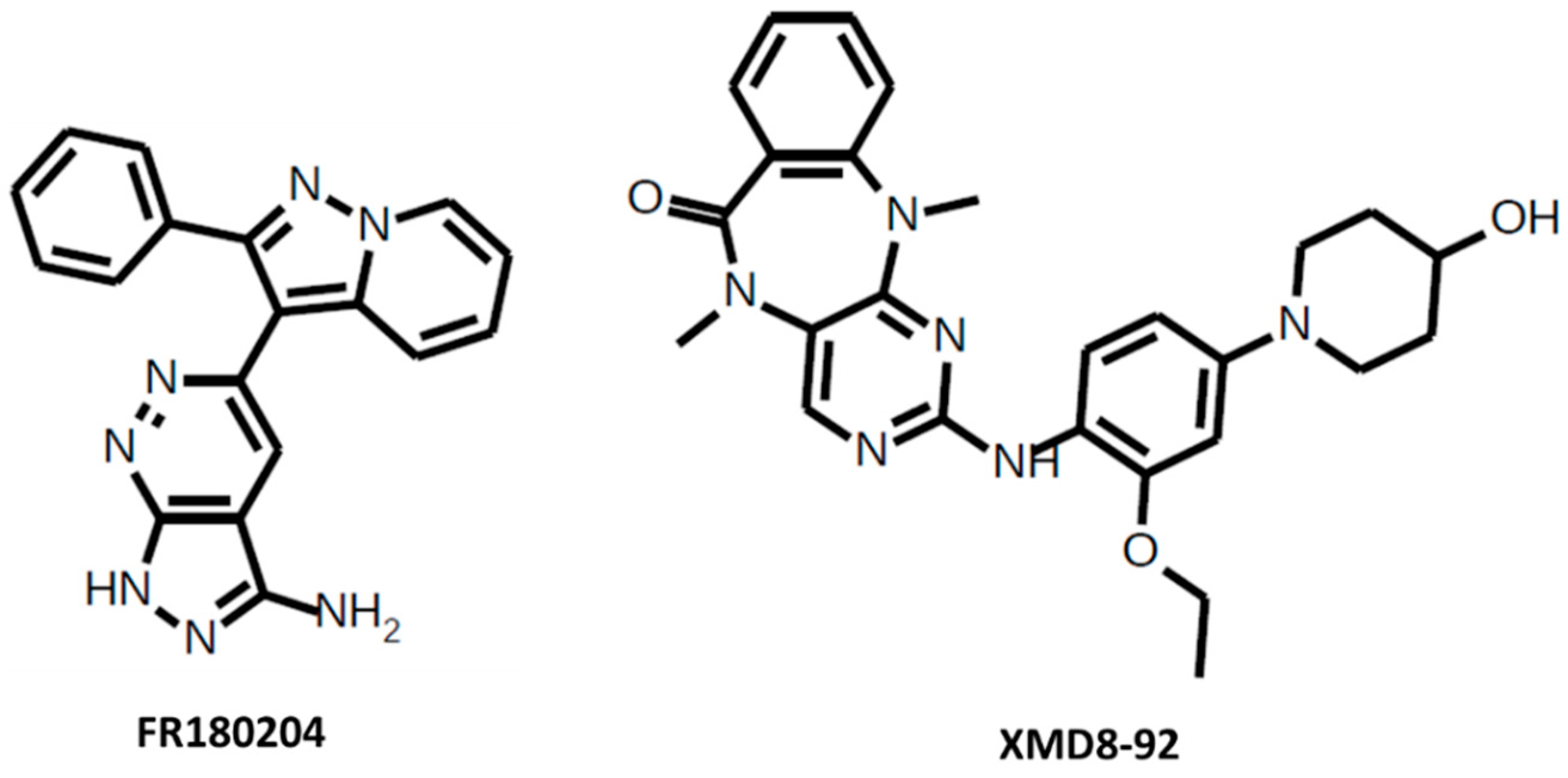
FR180204 is a selective ERK1/2 inhibitor (Figure 3). The IC50 for ERK1 = 0.14 μM and for ERK2 = 0.31 μM. FR180204 attenuates mesothelioma cell proliferation [71], decreases cell viability colorectal cancer cell lines [72], decreases cell proliferation and increases apoptosis in colorectal cancer cells in combination with AKT inhibitor API-1 [73], and inhibits intestinal myofibroblast migration induced by KRAS-mutated colorectal cancer cells [74]. This inhibitor is also used in arthritis research [75].
XMD8-92 is a selective ERK5 inhibitor (Figure 3), which has an IC50 for ERK5 = 300 nM. XMD8-92 blocks tumor cell proliferation in vitro and significantly inhibits tumor growth in a lung and cervical tumor xenograft model by 95% [76], causes cell cycle arrest in G2 phase in acute myeloid leukemia cells [77], inhibits pancreatic tumor xenograft growth [78], reduces proliferation, cell cycle progression, cell migration, and invasion in hepatocellular carcinoma cells, reduces tumor growth in xenografts [79], increases colon cancer cell sensitivity to 5-fluorouracil in a murine subcutaneous xenograft model [80], and impairs resistance to the combined inhibition of RAF proto-oncogene serine/threonine kinase B (BRAF) and MEK1/2 and the proliferation of resistant cells [81].
SI113 is a selective SGK1 inhibitor, which has an IC50 for SGK1 = 600 nM. This inhibitor induces autophagy, apoptosis, and cell viability through endoplasmic reticulum stress in endometrial cancer cells [82], induces apoptosis and cytotoxic autophagy as well as increases the effects of radiotherapy and the response to oxidative stress in glioblastoma cells [83], blocks tumor progression in vitro and in liver hepatocellular carcinoma xenograft mice models, and synergizes with radiotherapy [84] (Table 1).
3. Conclusions and Future Perspectives
In the past couple of decades, developments in the small-molecule MAP kinase inhibitor field have led to quite a number of marketed products with a diverse range of the inhibited targets. In addition, many more are still in development and/or improvement. The assortment of these inhibitors allows researchers to choose the most effective and appropriate methodologies suitable for the specific experiments. Preclinical studies both in cell lines as well as in proper animal models provide essential information for the design of clinical studies evaluating the improved efficiency of these agents. Clinical studies with MAP kinase inhibitors ought to determine which MAP kinase inhibitors are most effective for anticancer therapy. Many types of cancers can be targeted by inhibitors, as can be seen in this review. The particular use of MAP kinase inhibitors greatly depends on the genetic background and the precise signaling pathways that direct the cancerogenous properties of the cells in a given cancer type.
Although there has been significant progress lately in the development of MAP kinase inhibitors, there remains much to be improved. Firstly, for the treatment of cancers, kinase inhibitors have to have high specificity in order to avoid off-target inhibition. However, keeping in mind that there are more than 510 kinases in our cells, it is not an easy task. Sequence similarities between kinases and especially between isoforms of the same family could make it difficult to design specific inhibitors. Peptide inhibitors or aptamers [85] could be a solution. D-JNKI-1 is a good example showing that peptide inhibitors can go as far as clinical trials [86]. On the other hand, it is known that kinase inhibitors can inhibit other non-kinase targets. One good example is an NAD(P)H dehydrogenase 2 (NQO2), which can be inhibited by kinase inhibitors imatinib, nilotinib, TBB, and DMAT [87]. Therefore, more extensive research on off-target possibilities is needed. So far, after a new inhibitor is designed, it is common practice to test it on a panel of other kinases. However, kinase inhibitors are not tested against other proteins. Probably proteomics or similar high throughput techniques could help to achieve that. Then again, new approaches, such as computer-assisted, 3D structure-based approaches to generate new generations of kinase inhibitors, could be a solution for the future of MAP kinase inhibitors [87,88]. Structural insights into the distinctive inhibitor-kinase binding mechanisms have led to the discovery of several highly selective inhibitors [89,90,91]. Besides, in many cases, combination therapies using kinase inhibitors and chemotherapeutic agents or radiotherapy are even more encouraging than the use of these inhibitors as single agents. The achievements of combination therapies have already been shown both for MAP kinase (as mentioned in this review) and other kinases, such as CDKs, aurora kinase A (AURKA), tyrosine kinase, or multi-kinase inhibitors. Therefore, this field of both preclinical and clinical research should be further advanced. The other problem in kinase inhibitor therapies is the resistance of some tumors to these inhibitors. There are many pathways that lead to resistance and each requires a different solution. One of the problems could arise because of the overactivation of downstream effectors, the solution to which could be the inhibition of these factors. Activation of compensatory pathways could be another problem, possibly solved by dual inhibition. Mutations in target kinases could also render inhibitors insensitive, which again requires alternative inhibitors or at least combinatorial treatment. Some other problems, such as drug inactivation, multidrug resistance, or epigenetics, could be more problematic and less effectively solved.
Interestingly, there are several alternative compounds which show some potential as MAP kinase inhibitors in cancer research. The natural compound Sulforaphane, extracted from cruciferous vegetables, suppresses ERK and AKT phosphorylation and induces apoptosis through G2/M phase arrest in osteosarcoma cells [92]. Metformin, a known diabetes type II drug, together with arsenic trioxide, which is known for acute myeloid leukemia treatment, suppresses intrahepatic cholangiocarcinoma cell proliferation via the regulation of AMP-activated protein kinase (AMPK), p38, ERK3, and mammalian target of rapamycin complex 1 (mTORC1) pathways [93]. RH1, a compound shown to inhibit the proliferation of several cancers, was also bioinformatically shown to potentially inhibit several kinases, including ERK2 [87].
In conclusion, the future seems to be bright for MAP kinase inhibitors and they definitely should not be dismissed.
Acknowledgments
The research of Jonas Cicenas and Egle Zalyte was funded by Scientific Council of Lithuania (Project #SEN-01/2016). Ethical approval: This article does not contain any studies with human participants or animals performed by any of the authors.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cicenas, J. The potential role of Akt phosphorylation in human cancers. Int. J. Biol. Markers 2008, 23, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Cicenas, J.; Urban, P.; Vuaroqueaux, V.; Labuhn, M.; Kung, W.; Wight, E.; Mayhew, M.; Eppenberger, U.; Eppenberger-Castori, S. Increased level of phosphorylated akt measured by chemiluminescence-linked immunosorbent assay is a predictor of poor prognosis in primary breast cancer overexpressing erbb-2. Breast Cancer Res. 2005, 7, R394–R401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyndall, C.; Watt, F.; Molloy, P.L.; Vincent, P.C.; Frommer, M. Binding of proteins from embryonic and differentiated cells to a bidirectional promoter contained within a CpG island. J. Mol. Biol. 1992, 226, 289–299. [Google Scholar] [CrossRef]
- Cicenas, J.; Urban, P.; Kung, W.; Vuaroqueaux, V.; Labuhn, M.; Wight, E.; Eppenberger, U.; Eppenberger-Castori, S. Phosphorylation of tyrosine 1248-erbb2 measured by chemiluminescence-linked immunoassay is an independent predictor of poor prognosis in primary breast cancer patients. Eur. J. Cancer 2006, 42, 636–645. [Google Scholar] [CrossRef] [PubMed]
- DiGiovanna, M.P.; Stern, D.F.; Edgerton, S.M.; Whalen, S.G.; Moore, D., 2nd; Thor, A.D. Relationship of epidermal growth factor receptor expression to erbb-2 signaling activity and prognosis in breast cancer patients. J. Clin. Oncol. 2005, 23, 1152–1160. [Google Scholar] [CrossRef] [PubMed]
- Milde-Langosch, K.; Bamberger, A.M.; Rieck, G.; Grund, D.; Hemminger, G.; Muller, V.; Loning, T. Expression and prognostic relevance of activated extracellular-regulated kinases (erk1/2) in breast cancer. Br. J. Cancer 2005, 92, 2206–2215. [Google Scholar] [CrossRef] [PubMed]
- Bergqvist, J.; Elmberger, G.; Ohd, J.; Linderholm, B.; Bjohle, J.; Hellborg, H.; Nordgren, H.; Borg, A.L.; Skoog, L.; Bergh, J. Activated erk1/2 and phosphorylated oestrogen receptor alpha are associated with improved breast cancer survival in women treated with tamoxifen. Eur. J. Cancer 2006, 42, 1104–1112. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.J.; Wan, X.B.; Fu, X.H.; Wu, P.H.; Chen, D.K.; Wang, P.N.; Jiang, L.; Wang, D.H.; Chen, Z.T.; Huang, Y.; et al. Phosphorylated p38, a negative prognostic biomarker, complements TNM staging prognostication in colorectal cancer. Tumour Biol. 2014, 35, 10487–10495. [Google Scholar] [CrossRef] [PubMed]
- Cicenas, J.; Kung, W.; Eppenberger, U.; Eppenberger-Castori, S. Increased level of phosphorylated ShcA measured by chemiluminescence-linked immunoassay is a predictor of good prognosis in primary breast cancer expressing low levels of estrogen receptor. Cancers 2010, 2, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Seger, R.; Krebs, E.G. The MAPK signaling cascade. FASEB J. 1995, 9, 726–735. [Google Scholar] [PubMed]
- Campbell, J.S.; Seger, R.; Graves, J.D.; Graves, L.M.; Jensen, A.M.; Krebs, E.G. The map kinase cascade. Recent Prog. Horm. Res. 1995, 50, 131–159. [Google Scholar] [PubMed]
- Cicenas, J.; Tamosaitis, L.; Kvederaviciute, K.; Tarvydas, R.; Staniute, G.; Kalyan, K.; Meskinyte-Kausiliene, E.; Stankevicius, V.; Valius, M. KRAS, NRAS and BRAF mutations in colorectal cancer and melanoma. Med. Oncol. 2017, 34, 26. [Google Scholar] [CrossRef] [PubMed]
- Cicenas, J.; Kvederaviciute, K.; Meskinyte, I.; Meskinyte-Kausiliene, E.; Skeberdyte, A.; Cicenas, J. KRAS, TP53, CDKN2A, SMAD4, BRCA1, and BRCA2 mutations in pancreatic cancer. Cancers 2017, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Derijard, B.; Hibi, M.; Wu, I.H.; Barrett, T.; Su, B.; Deng, T.; Karin, M.; Davis, R.J. Jnk1: A protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell 1994, 76, 1025–1037. [Google Scholar] [CrossRef]
- Zarubin, T.; Han, J. Activation and signaling of the p38 map kinase pathway. Cell Res. 2005, 15, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Drew, B.A.; Burow, M.E.; Beckman, B.S. MEK5/ERK5 pathway: The first fifteen years. Biochim. Biophys. Acta 2012, 1825, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Gharwan, H.; Groninger, H. Kinase inhibitors and monoclonal antibodies in oncology: Clinical implications. Nat. Rev. Clin. Oncol. 2016, 13, 209–227. [Google Scholar] [CrossRef] [PubMed]
- Cicenas, J.; Valius, M. The cdk inhibitors in cancer research and therapy. J. Cancer Res. Clin. Oncol. 2011, 137, 1409–1418. [Google Scholar] [CrossRef] [PubMed]
- Cicenas, J.; Kalyan, K.; Sorokinas, A.; Jatulyte, A.; Valiunas, D.; Kaupinis, A.; Valius, M. Highlights of the latest advances in research on CDK inhibitors. Cancers 2014, 6, 2224–2242. [Google Scholar] [CrossRef] [PubMed]
- Cicenas, J.; Kalyan, K.; Sorokinas, A.; Stankunas, E.; Levy, J.; Meskinyte, I.; Stankevicius, V.; Kaupinis, A.; Valius, M. Roscovitine in cancer and other diseases. Ann. Transl. Med. 2015, 3, 135. [Google Scholar] [CrossRef] [PubMed]
- Cicenas, J. The aurora kinase inhibitors in cancer research and therapy. J. Cancer Res. Clin. Oncol. 2016, 142, 1995–2012. [Google Scholar] [CrossRef] [PubMed]
- Cicenas, J.; Cicenas, E. Multi-kinase inhibitors, AURKs and cancer. Med. Oncol. 2016, 33, 43. [Google Scholar] [CrossRef] [PubMed]
- Grassi, E.S.; Vezzoli, V.; Negri, I.; Labadi, A.; Fugazzola, L.; Vitale, G.; Persani, L. Sp600125 has a remarkable anticancer potential against undifferentiated thyroid cancer through selective action on rock and p53 pathways. Oncotarget 2015, 6, 36383–36399. [Google Scholar] [PubMed]
- Kim, J.H.; Kim, T.H.; Kang, H.S.; Ro, J.; Kim, H.S.; Yoon, S. Sp600125, an inhibitor of Jnk pathway, reduces viability of relatively resistant cancer cells to doxorubicin. Biochem. Biophys. Res. Commun. 2009, 387, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Chae, M.; Choi, A.R.; Sik Kim, H.; Yoon, S. Sp600125 overcomes antimitotic drug-resistance in cancer cells by increasing apoptosis with independence of P-gp inhibition. Eur. J. Pharmacol. 2014, 723, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.Y.; Chen, T.S.; Wang, X.P.; Qu, J.L.; Chen, M. The JNK inhibitor sp600125 enhances dihydroartemisinin-induced apoptosis by accelerating Bax translocation into mitochondria in human lung adenocarcinoma cells. FEBS Lett. 2010, 584, 4019–4026. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Zhang, B.; Liang, H.; Lu, Y.; Ai, X.; Zhang, B.; Chen, X. JNK inhibitor SP600125 enhances TGF-beta-induced apoptosis of RBE human cholangiocarcinoma cells in a sSmad-dependent manner. Mol. Med. Rep. 2013, 8, 1623–1629. [Google Scholar] [CrossRef] [PubMed]
- Jemaa, M.; Vitale, I.; Kepp, O.; Berardinelli, F.; Galluzzi, L.; Senovilla, L.; Marino, G.; Malik, S.A.; Rello-Varona, S.; Lissa, D.; et al. Selective killing of p53-deficient cancer cells by sp600125. EMBO Mol. Med. 2012, 4, 500–514. [Google Scholar] [CrossRef] [PubMed]
- Konno, T.; Ninomiya, T.; Kohno, T.; Kikuchi, S.; Sawada, N.; Kojima, T. c-Jun n-terminal kinase inhibitor sp600125 enhances barrier function and elongation of human pancreatic cancer cell line HPAC in a Ca-switch model. Histochem. Cell Biol. 2015, 143, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Huang, J.Y.; Xing, B.; Ren, K.W.; Li, M.; Wei, D.; Gu, P.Y.; Chen, G.; Gu, B.; Zhang, G.F.; et al. Sp600125, a JNK inhibitor, suppresses growth of JNK-inactive glioblastoma cells through cell-cycle g2/m phase arrest. Die Pharm. 2012, 67, 942–946. [Google Scholar] [PubMed]
- Wu, H.M.; Fang, L.; Shen, Q.Y.; Liu, R.Y. Sp600125 promotes resolution of allergic airway inflammation via tlr9 in an ova-induced murine acute asthma model. Mol. Immunol. 2015, 67, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Minutoli, L.; Altavilla, D.; Marini, H.; Passaniti, M.; Bitto, A.; Seminara, P.; Venuti, F.S.; Famulari, C.; Macri, A.; Versaci, A.; et al. Protective effects of sp600125 a new inhibitor of c-Jun n-terminal kinase (JNK) and extracellular-regulated kinase (erk1/2) in an experimental model of cerulein-induced pancreatitis. Life Sci. 2004, 75, 2853–2866. [Google Scholar] [CrossRef] [PubMed]
- Anand, S.S.; Maruthi, M.; Babu, P.P. The specific, reversible JNK inhibitor sp600125 improves survivability and attenuates neuronal cell death in experimental cerebral malaria (ECM). Parasitol. Res. 2013, 112, 1959–1966. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Shi, L.; Xie, Y.; Ma, C.; Li, W.; Su, X.; Huang, S.; Chen, R.; Zhu, Z.; Mao, Z.; et al. Sp600125, a new JNK inhibitor, protects dopaminergic neurons in the MPTP model of parkinson’s disease. Neurosci. Res. 2004, 48, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Cerbone, A.; Toaldo, C.; Pizzimenti, S.; Pettazzoni, P.; Dianzani, C.; Minelli, R.; Ciamporcero, E.; Roma, G.; Dianzani, M.U.; Canaparo, R.; et al. As601245, an anti-inflammatory jnk inhibitor, and clofibrate have a synergistic effect in inducing cell responses and in affecting the gene expression profile in caco-2 colon cancer cells. PPAR Res. 2012, 2012, 269751. [Google Scholar] [CrossRef] [PubMed]
- Cerbone, A.; Toaldo, C.; Minelli, R.; Ciamporcero, E.; Pizzimenti, S.; Pettazzoni, P.; Roma, G.; Dianzani, M.U.; Ullio, C.; Ferretti, C.; et al. Rosiglitazone and as601245 decrease cell adhesion and migration through modulation of specific gene expression in human colon cancer cells. PLoS ONE 2012, 7, e40149. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Wang, Q.; Wang, J.; Lv, M.; Zhu, N.; Li, Y.; Feng, J.; Shen, B.; Zhang, J. Basal c-jun nh2-terminal protein kinase activity is essential for survival and proliferation of t-cell acute lymphoblastic leukemia cells. Mol. Cancer Ther. 2009, 8, 3214–3222. [Google Scholar] [CrossRef] [PubMed]
- Ramos, A.M.; Fernandez, C.; Amran, D.; Esteban, D.; de Blas, E.; Palacios, M.A.; Aller, P. Pharmacologic inhibitors of extracellular signal-regulated kinase (ERKs) and c-Jun NH(2)-terminal kinase (JNK) decrease glutathione content and sensitize human promonocytic leukemia cells to arsenic trioxide-induced apoptosis. J. Cell. Physiol. 2006, 209, 1006–1015. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, P.; Jeanclaude-Etter, I.; Ardissone, V.; Arkinstall, S.; Cambet, Y.; Camps, M.; Chabert, C.; Church, D.; Cirillo, R.; Gretener, D.; et al. Design and synthesis of the first generation of novel potent, selective, and in vivo active (benzothiazol-2-yl)acetonitrile inhibitors of the c-Jun n-terminal kinase. J. Med. Chem. 2005, 48, 4596–4607. [Google Scholar] [CrossRef] [PubMed]
- Wolschendorf, F.; Bosque, A.; Shishido, T.; Duverger, A.; Jones, J.; Planelles, V.; Kutsch, O. Kinase control prevents hiv-1 reactivation in spite of high levels of induced NF-κB activity. J. Virol. 2012, 86, 4548–4558. [Google Scholar] [CrossRef] [PubMed]
- Nacken, W.; Ehrhardt, C.; Ludwig, S. Small molecule inhibitors of the c-Jun n-terminal kinase (JNK) possess antiviral activity against highly pathogenic avian and human pandemic influenza a viruses. Biol. Chem. 2012, 393, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Vasilevskaya, I.A.; Selvakumaran, M.; Hierro, L.C.; Goldstein, S.R.; Winkler, J.D.; O’Dwyer, P.J. Inhibition of JNK sensitizes hypoxic colon cancer cells to DNA-damaging agents. Clin. Cancer Res. 2015, 21, 4143–4152. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.Y.; Flanc, R.S.; Tesch, G.H.; Bennett, B.L.; Friedman, G.C.; Nikolic-Paterson, D.J. Blockade of the c-Jun amino terminal kinase prevents crescent formation and halts established anti-GBM glomerulonephritis in the rat. Lab. Investig. 2009, 89, 470–484. [Google Scholar] [CrossRef] [PubMed]
- Flanc, R.S.; Ma, F.Y.; Tesch, G.H.; Han, Y.; Atkins, R.C.; Bennett, B.L.; Friedman, G.C.; Fan, J.H.; Nikolic-Paterson, D.J. A pathogenic role for JNK signaling in experimental anti-GBM glomerulonephritis. Kidney Int. 2007, 72, 698–708. [Google Scholar] [CrossRef] [PubMed]
- Uehara, T.; Bennett, B.; Sakata, S.T.; Satoh, Y.; Bilter, G.K.; Westwick, J.K.; Brenner, D.A. Jnk mediates hepatic ischemia reperfusion injury. J. Hepatol. 2005, 42, 850–859. [Google Scholar] [CrossRef] [PubMed]
- Kanellis, J.; Ma, F.Y.; Kandane-Rathnayake, R.; Dowling, J.P.; Polkinghorne, K.R.; Bennett, B.L.; Friedman, G.C.; Nikolic-Paterson, D.J. Jnk signalling in human and experimental renal ischaemia/reperfusion injury. Nephrol. Dial. Transplant. 2010, 25, 2898–2908. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Kuramoto, K.; Takeda, H.; Watarai, H.; Sakaki, H.; Seino, S.; Seino, M.; Suzuki, S.; Kitanaka, C. The novel jnk inhibitor as602801 inhibits cancer stem cells in vitro and in vivo. Oncotarget 2016, 7, 27021–27032. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.J.; Cheng, J.K.; Zeng, Q.; Xu, Z.Z.; Decosterd, I.; Xu, X.; Ji, R.R. Selective inhibition of jnk with a peptide inhibitor attenuates pain hypersensitivity and tumor growth in a mouse skin cancer pain model. Exp. Neurol. 2009, 219, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Posthumadeboer, J.; van Egmond, P.W.; Helder, M.N.; de Menezes, R.X.; Cleton-Jansen, A.M.; Belien, J.A.; Verheul, H.M.; van Royen, B.J.; Kaspers, G.J.; van Beusechem, V.W. Targeting jnk-interacting-protein-1 (jip1) sensitises osteosarcoma to doxorubicin. Oncotarget 2012, 3, 1169–1181. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Podar, K.; Chauhan, D.; Ishitsuka, K.; Mitsiades, C.; Tai, Y.T.; Hamasaki, M.; Raje, N.; Hideshima, H.; Schreiner, G.; et al. P38 MAPK inhibition enhances PS-341 (bortezomib)-induced cytotoxicity against multiple myeloma cells. Oncogene 2004, 23, 8766–8776. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.N.; Stebbins, E.G.; Henson, M.; O’Young, G.; Choi, S.J.; Quon, D.; Damm, D.; Reddy, M.; Ma, J.Y.; Haghnazari, E.; et al. Normalizing the bone marrow microenvironment with p38 inhibitor reduces multiple myeloma cell proliferation and adhesion and suppresses osteoclast formation. Exp. Cell Res. 2006, 312, 1909–1923. [Google Scholar] [CrossRef] [PubMed]
- Navas, T.A.; Nguyen, A.N.; Hideshima, T.; Reddy, M.; Ma, J.Y.; Haghnazari, E.; Henson, M.; Stebbins, E.G.; Kerr, I.; O’Young, G.; et al. Inhibition of p38alpha MAPK enhances proteasome inhibitor-induced apoptosis of myeloma cells by modulating hsp27, Bcl-X(L), Mcl-1 and p53 levels in vitro and inhibits tumor growth in vivo. Leukemia 2006, 20, 1017–1027. [Google Scholar] [CrossRef] [PubMed]
- Vanderkerken, K.; Medicherla, S.; Coulton, L.; De Raeve, H.; Willems, A.; Lawson, M.; Van Camp, B.; Protter, A.A.; Higgins, L.S.; Menu, E.; et al. Inhibition of p38alpha mitogen-activated protein kinase prevents the development of osteolytic bone disease, reduces tumor burden, and increases survival in murine models of multiple myeloma. Cancer Res. 2007, 67, 4572–4577. [Google Scholar] [CrossRef] [PubMed]
- Medicherla, S.; Reddy, M.; Ying, J.; Navas, T.A.; Li, L.; Nguyen, A.N.; Kerr, I.; Hanjarappa, N.; Protter, A.A.; Higgins, L.S. P38alpha-selective map kinase inhibitor reduces tumor growth in mouse xenograft models of multiple myeloma. Anticancer Res. 2008, 28, 3827–3833. [Google Scholar] [PubMed]
- Giafis, N.; Katsoulidis, E.; Sassano, A.; Tallman, M.S.; Higgins, L.S.; Nebreda, A.R.; Davis, R.J.; Platanias, L.C. Role of the p38 mitogen-activated protein kinase pathway in the generation of arsenic trioxide-dependent cellular responses. Cancer Res. 2006, 66, 6763–6771. [Google Scholar] [CrossRef] [PubMed]
- Genovese, M.C.; Cohen, S.B.; Wofsy, D.; Weinblatt, M.E.; Firestein, G.S.; Brahn, E.; Strand, V.; Baker, D.G.; Tong, S.E. A 24-week, randomized, double-blind, placebo-controlled, parallel group study of the efficacy of oral scio-469, a p38 mitogen-activated protein kinase inhibitor, in patients with active rheumatoid arthritis. J. Rheumatol. 2011, 38, 846–854. [Google Scholar] [CrossRef] [PubMed]
- Sokol, L.; Cripe, L.; Kantarjian, H.; Sekeres, M.A.; Parmar, S.; Greenberg, P.; Goldberg, S.L.; Bhushan, V.; Shammo, J.; Hohl, R.; et al. Randomized, dose-escalation study of the p38alpha mapk inhibitor scio-469 in patients with myelodysplastic syndrome. Leukemia 2013, 27, 977–980. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.E.; Daniels, S.E.; Black, P.; Chang, S.; Protter, A.; Desjardins, P.J. Novel p38alpha mitogen-activated protein kinase inhibitor shows analgesic efficacy in acute postsurgical dental pain. J. Clin. Pharmacol. 2012, 52, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Yasui, H.; Hideshima, T.; Ikeda, H.; Jin, J.; Ocio, E.M.; Kiziltepe, T.; Okawa, Y.; Vallet, S.; Podar, K.; Ishitsuka, K.; et al. Birb 796 enhances cytotoxicity triggered by bortezomib, heat shock protein (Hsp) 90 inhibitor, and dexamethasone via inhibition of p38 mitogen-activated protein kinase/hsp27 pathway in multiple myeloma cell lines and inhibits paracrine tumour growth. Br. J. Haematol. 2007, 136, 414–423. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Zhao, X.Q.; Chen, X.G.; Fang, Y.; Singh, S.; Talele, T.T.; Qiu, H.J.; Liang, Y.J.; Wang, X.K.; Zhang, G.Q.; et al. Birb796, the inhibitor of p38 mitogen-activated protein kinase, enhances the efficacy of chemotherapeutic agents in abcb1 overexpression cells. PLoS ONE 2013, 8, e54181. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Mo, Q.; Zhang, Y.; Gao, Y.; Wu, Y.; Li, J.; Hao, X.; Ma, D.; Gao, Q.; Chen, P. The p38 MAPK inhibitor birb796 enhances the antitumor effects of vx680 in cervical cancer. Cancer Biol. Ther. 2016, 17, 566–576. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, S.; Choi, J.; Kim, J.; Bae, S.; Hong, J.; Jo, S.; Kim, S.; Lee, Y. Birb 796 has distinctive anti-inflammatory effects on different cell types. Immune Netw. 2013, 13, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, S.; Feagan, B.; D’Haens, G.; Colombel, J.F.; Geboes, K.; Yurcov, M.; Isakov, V.; Golovenko, O.; Bernstein, C.N.; Ludwig, D.; et al. Oral p38 mitogen-activated protein kinase inhibition with BIRB 796 for active crohn’s disease: A randomized, double-blind, placebo-controlled trial. Clin. Gastroenterol. Hepatol. 2006, 4, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Ishitsuka, K.; Hideshima, T.; Neri, P.; Vallet, S.; Shiraishi, N.; Okawa, Y.; Shen, Z.; Raje, N.; Kiziltepe, T.; Ocio, E.M.; et al. P38 mitogen-activated protein kinase inhibitor ly2228820 enhances bortezomib-induced cytotoxicity and inhibits osteoclastogenesis in multiple myeloma; therapeutic implications. Br. J. Haematol. 2008, 141, 598–606. [Google Scholar] [CrossRef] [PubMed]
- Campbell, R.M.; Anderson, B.D.; Brooks, N.A.; Brooks, H.B.; Chan, E.M.; De Dios, A.; Gilmour, R.; Graff, J.R.; Jambrina, E.; Mader, M.; et al. Characterization of ly2228820 dimesylate, a potent and selective inhibitor of p38 MAPK with antitumor activity. Mol. Cancer Ther. 2014, 13, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, T.; Wang, G.; Wang, H.; Che, X.; Gao, X.; Liu, H. Rac3 regulates cell invasion, migration and EMT in lung adenocarcinoma through p38 MAPK pathway. J. Cancer 2017, 8, 2511–2522. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, A.; Haluska, P.; Tolcher, A.W.; Erlichman, C.; Papadopoulos, K.P.; Lensing, J.L.; Beeram, M.; Molina, J.R.; Rasco, D.W.; Arcos, R.R.; et al. A first-in-human phase i study of the oral p38 mapk inhibitor, ralimetinib (ly2228820 dimesylate), in patients with advanced cancer. Clin. Cancer Res. 2016, 22, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- A Study of LY2228820 for Recurrent Ovarian Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT01663857 (accessed on 8 September 2017).
- Hideshima, T.; Akiyama, M.; Hayashi, T.; Richardson, P.; Schlossman, R.; Chauhan, D.; Anderson, K.C. Targeting p38 MAPK inhibits multiple myeloma cell growth in the bone marrow milieu. Blood 2003, 101, 703–705. [Google Scholar] [CrossRef] [PubMed]
- Gupta, J.; Igea, A.; Papaioannou, M.; Lopez-Casas, P.P.; Llonch, E.; Hidalgo, M.; Gorgoulis, V.G.; Nebreda, A.R. Pharmacological inhibition of p38 MAPK reduces tumor growth in patient-derived xenografts from colon tumors. Oncotarget 2015, 6, 8539–8551. [Google Scholar] [CrossRef] [PubMed]
- Honda, M.; Kanno, T.; Fujita, Y.; Gotoh, A.; Nakano, T.; Nishizaki, T. Mesothelioma cell proliferation through autocrine activation of pdgf-betabeta receptor. Cell. Physiol. Biochem. 2012, 29, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Ragusa, M.; Statello, L.; Maugeri, M.; Majorana, A.; Barbagallo, D.; Salito, L.; Sammito, M.; Santonocito, M.; Angelica, R.; Cavallaro, A.; et al. Specific alterations of the microRNA transcriptome and global network structure in colorectal cancer after treatment with MAPK/ERK inhibitors. J. Mol. Med. 2012, 90, 1421–1438. [Google Scholar] [CrossRef] [PubMed]
- Saglam, A.S.; Alp, E.; Elmazoglu, Z.; Menevse, E.S. Effect of api-1 and fr180204 on cell proliferation and apoptosis in human dld-1 and lovo colorectal cancer cells. Oncol. Lett. 2016, 12, 2463–2474. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, H.; Saotome, T.; Usui, T.; Ohama, T.; Sato, K. Regulation of intestinal myofibroblasts by KRas-mutated colorectal cancer cells through heparin-binding epidermal growth factor-like growth factor. Oncol. Rep. 2017, 37, 3128–3136. [Google Scholar] [CrossRef] [PubMed]
- Ohori, M.; Takeuchi, M.; Maruki, R.; Nakajima, H.; Miyake, H. Fr180204, a novel and selective inhibitor of extracellular signal-regulated kinase, ameliorates collagen-induced arthritis in mice. Naunyn Schmiedebergs Arch. Pharmacol. 2007, 374, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Deng, X.; Lu, B.; Cameron, M.; Fearns, C.; Patricelli, M.P.; Yates, J.R., 3rd; Gray, N.S.; Lee, J.D. Pharmacological inhibition of bmk1 suppresses tumor growth through promyelocytic leukemia protein. Cancer Cell 2010, 18, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Pesakhov, S.; Weng, A.; Kafka, M.; Gocek, E.; Nguyen, M.; Harrison, J.S.; Danilenko, M.; Studzinski, G.P. ERK 5/mapk pathway has a major role in 1alpha,25-(oh)2 vitamin d3-induced terminal differentiation of myeloid leukemia cells. J. Steroid Biochem. Mol. Biol. 2014, 144, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Sureban, S.M.; May, R.; Weygant, N.; Qu, D.; Chandrakesan, P.; Bannerman-Menson, E.; Ali, N.; Pantazis, P.; Westphalen, C.B.; Wang, T.C.; et al. Xmd8-92 inhibits pancreatic tumor xenograft growth via a dclk1-dependent mechanism. Cancer Lett. 2014, 351, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Rovida, E.; Di Maira, G.; Tusa, I.; Cannito, S.; Paternostro, C.; Navari, N.; Vivoli, E.; Deng, X.; Gray, N.S.; Esparis-Ogando, A.; et al. The mitogen-activated protein kinase erk5 regulates the development and growth of hepatocellular carcinoma. Gut 2015, 64, 1454–1465. [Google Scholar] [CrossRef] [PubMed]
- Pereira, D.M.; Simoes, A.E.; Gomes, S.E.; Castro, R.E.; Carvalho, T.; Rodrigues, C.M.; Borralho, P.M. Mek5/erk5 signaling inhibition increases colon cancer cell sensitivity to 5-fluorouracil through a p53-dependent mechanism. Oncotarget 2016, 7, 34322–34340. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Wang, L.; Xu, Q.; Wang, K.; Xie, D.; Yu, Z.; Jiang, K.; Liao, L.; Yates, J.R.; Lee, J.D.; et al. Targeting bmk1 impairs the drug resistance to combined inhibition of BRAF and MEK1/2 in melanoma. Sci. Rep. 2017, 7, 46244. [Google Scholar] [CrossRef] [PubMed]
- Conza, D.; Mirra, P.; Cali, G.; Tortora, T.; Insabato, L.; Fiory, F.; Schenone, S.; Amato, R.; Beguinot, F.; Perrotti, N.; et al. The sgk1 inhibitor si113 induces autophagy, apoptosis, and endoplasmic reticulum stress in endometrial cancer cells. J. Cell. Physiol. 2017, 232, 3735–3743. [Google Scholar] [CrossRef] [PubMed]
- Talarico, C.; Dattilo, V.; D’Antona, L.; Barone, A.; Amodio, N.; Belviso, S.; Musumeci, F.; Abbruzzese, C.; Bianco, C.; Trapasso, F.; et al. Si113, a sgk1 inhibitor, potentiates the effects of radiotherapy, modulates the response to oxidative stress and induces cytotoxic autophagy in human glioblastoma multiforme cells. Oncotarget 2016, 7, 15868–15884. [Google Scholar] [CrossRef] [PubMed]
- Talarico, C.; D’Antona, L.; Scumaci, D.; Barone, A.; Gigliotti, F.; Fiumara, C.V.; Dattilo, V.; Gallo, E.; Visca, P.; Ortuso, F.; et al. Preclinical model in HCC: The sgk1 kinase inhibitor si113 blocks tumor progression in vitro and in vivo and synergizes with radiotherapy. Oncotarget 2015, 6, 37511–37525. [Google Scholar] [CrossRef] [PubMed]
- Seiwert, S.D.; Stines Nahreini, T.; Aigner, S.; Ahn, N.G.; Uhlenbeck, O.C. Rna aptamers as pathway-specific map kinase inhibitors. Chem. Biol. 2000, 7, 833–843. [Google Scholar] [CrossRef]
- Suckfuell, M.; Lisowska, G.; Domka, W.; Kabacinska, A.; Morawski, K.; Bodlaj, R.; Klimak, P.; Kostrica, R.; Meyer, T. Efficacy and safety of am-111 in the treatment of acute sensorineural hearing loss: A double-blind, randomized, placebo-controlled phase ii study. Otol. Neurotol. 2014, 35, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.P.; Bastikar, V.A.; Kuciauskas, D.; Kothari, S.L.; Cicenas, J.; Valius, M. Molecular modeling and structure-based drug discovery approach reveals protein kinases as off-targets for novel anticancer drug rh1. Med. Oncol. 2017, 34, 176. [Google Scholar] [CrossRef] [PubMed]
- Duchowicz, P.R.; Castro, E.A. Qsar studies for the pharmacological inhibition of glycogen synthase kinase-3. Med. Chem. 2007, 3, 393–417. [Google Scholar] [CrossRef] [PubMed]
- Selness, S.R.; Devraj, R.V.; Devadas, B.; Walker, J.K.; Boehm, T.L.; Durley, R.C.; Shieh, H.; Xing, L.; Rucker, P.V.; Jerome, K.D.; et al. Discovery of ph-797804, a highly selective and potent inhibitor of p38 map kinase. Bioorg. Med. Chem. Lett. 2011, 21, 4066–4071. [Google Scholar] [CrossRef] [PubMed]
- Zheng, K.; Iqbal, S.; Hernandez, P.; Park, H.; LoGrasso, P.V.; Feng, Y. Design and synthesis of highly potent and isoform selective jnk3 inhibitors: SAR studies on aminopyrazole derivatives. J. Med. Chem. 2014, 57, 10013–10030. [Google Scholar] [CrossRef] [PubMed]
- Ortuso, F.; Amato, R.; Artese, A.; D’Antona, L.; Costa, G.; Talarico, C.; Gigliotti, F.; Bianco, C.; Trapasso, F.; Schenone, S.; et al. In silico identification and biological evaluation of novel selective serum/glucocorticoid-inducible kinase 1 inhibitors based on the pyrazolo-pyrimidine scaffold. J. Chem. Inf. Model. 2014, 54, 1828–1832. [Google Scholar] [CrossRef] [PubMed]
- Angulo, P.; Kaushik, G.; Subramaniam, D.; Dandawate, P.; Neville, K.; Chastain, K.; Anant, S. Natural compounds targeting major cell signaling pathways: A novel paradigm for osteosarcoma therapy. J. Hematol. Oncol. 2017, 10, 10. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.; Xie, H.; Yang, F.; Shan, Q.; Dai, H.; Zhuo, J.; Wei, X.; Song, P.; Zhou, L.; Xu, X.; et al. Metformin potentiates the effect of arsenic trioxide suppressing intrahepatic cholangiocarcinoma: Roles of p38 MAPK, erk3, and mtorc1. J. Hematol. Oncol. 2017, 10, 59. [Google Scholar] [CrossRef] [PubMed]
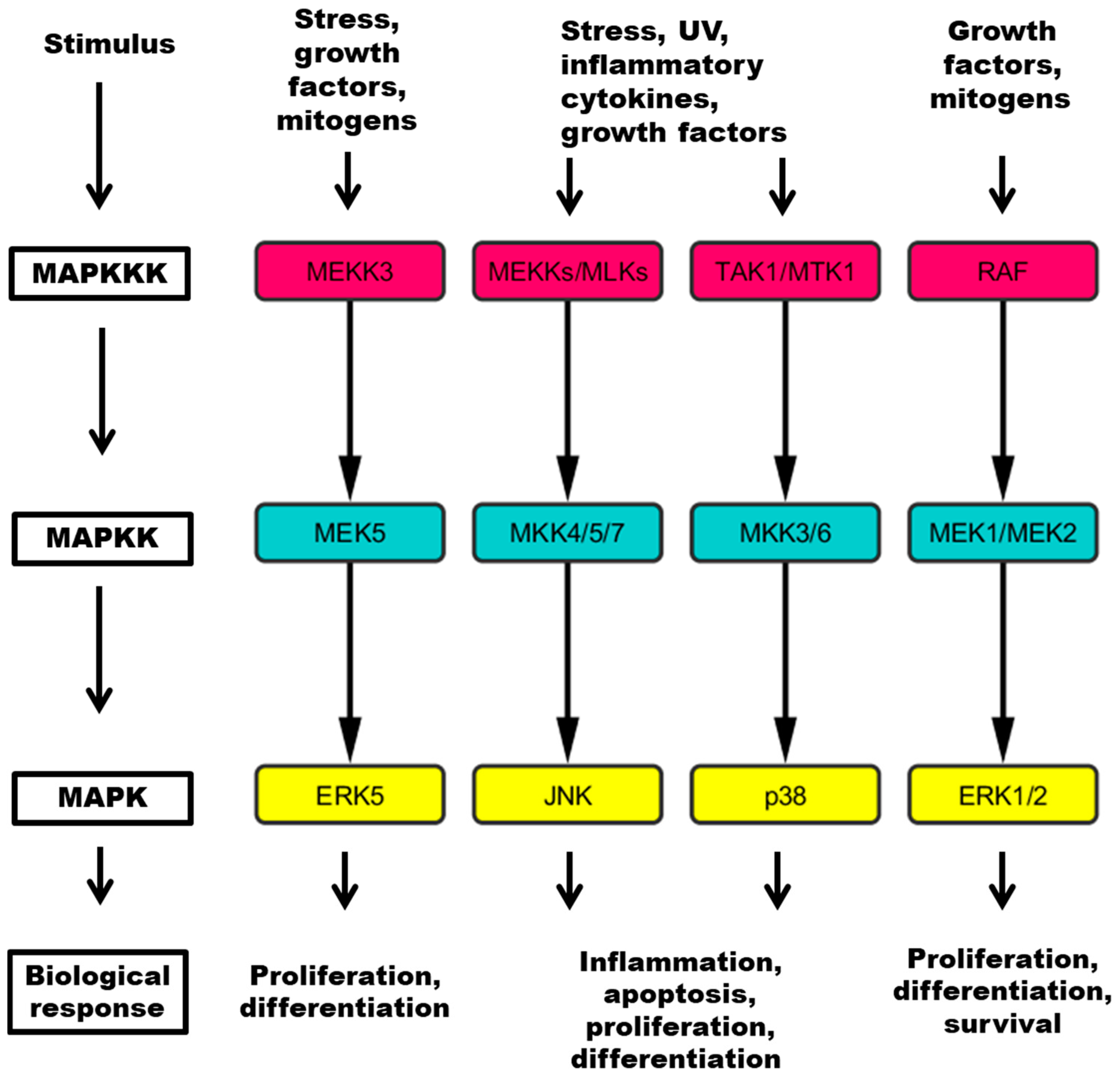
Figure 1.
Four major pathways contain a three tiered kinase cascade comprising a MAP kinase kinase kinase (MAPKKK), a MAP kinase kinase (MAPKK) and the MAPK, which mediates responses to specific stimuli. MEKK: mitogen-activated kinase kinase kinase; MLK: mixed lineage kinase; TAK: Tat-associated kinase; MTK: mitogen-activated protein kinase kinase kinase 4; RAF: RAF proto-oncogene serine/threonine kinase; MEK: mitogen-activated protein kinase kinase; MKK: mitogen-activated protein kinase kinase; ERK: extracellular regulated kinase; JNK: c-Jun N-terminal kinase; UV: ultraviolet light.
Figure 1.
Four major pathways contain a three tiered kinase cascade comprising a MAP kinase kinase kinase (MAPKKK), a MAP kinase kinase (MAPKK) and the MAPK, which mediates responses to specific stimuli. MEKK: mitogen-activated kinase kinase kinase; MLK: mixed lineage kinase; TAK: Tat-associated kinase; MTK: mitogen-activated protein kinase kinase kinase 4; RAF: RAF proto-oncogene serine/threonine kinase; MEK: mitogen-activated protein kinase kinase; MKK: mitogen-activated protein kinase kinase; ERK: extracellular regulated kinase; JNK: c-Jun N-terminal kinase; UV: ultraviolet light.
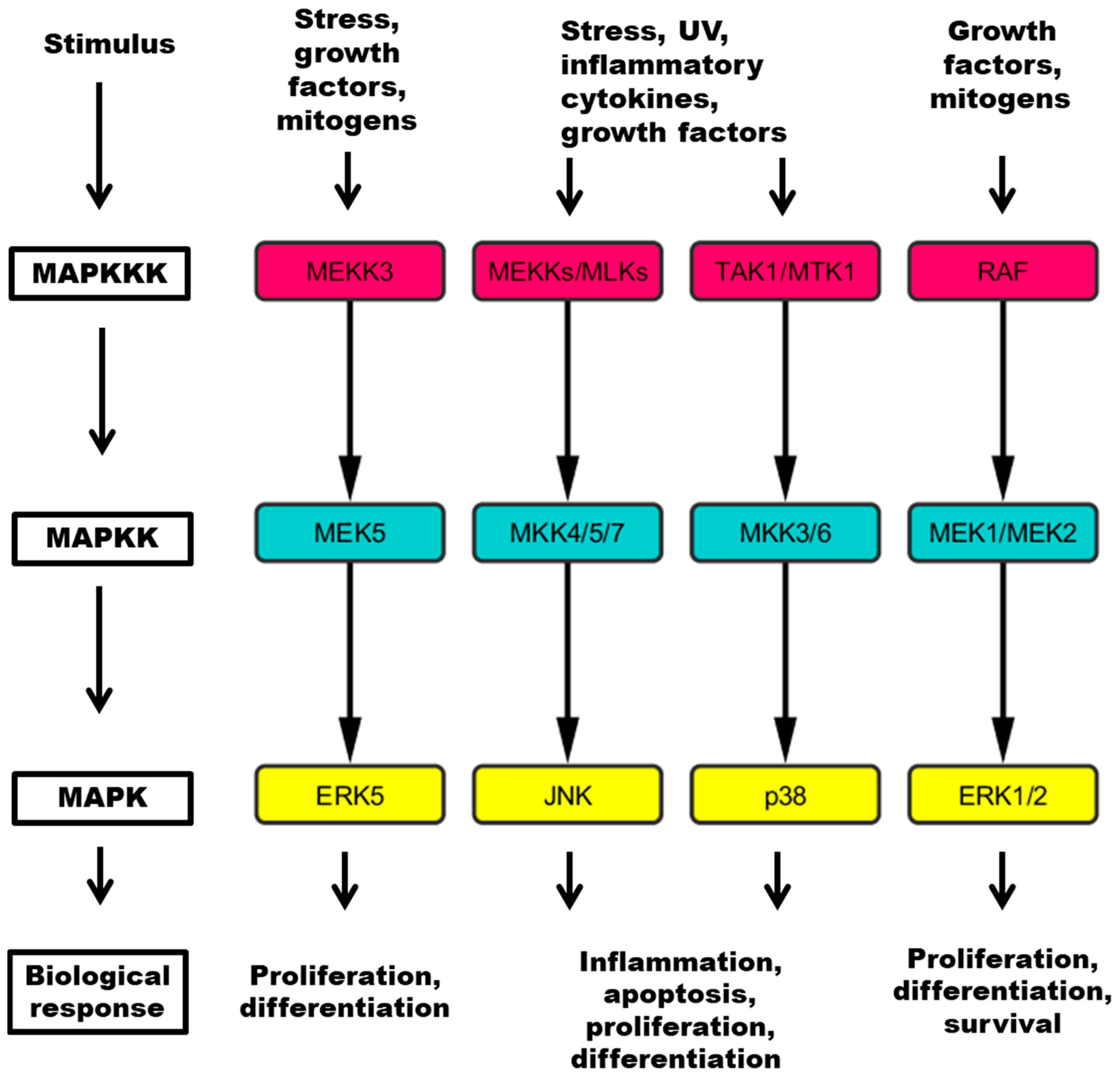
Figure 2.
AS601245 and BIRB796.
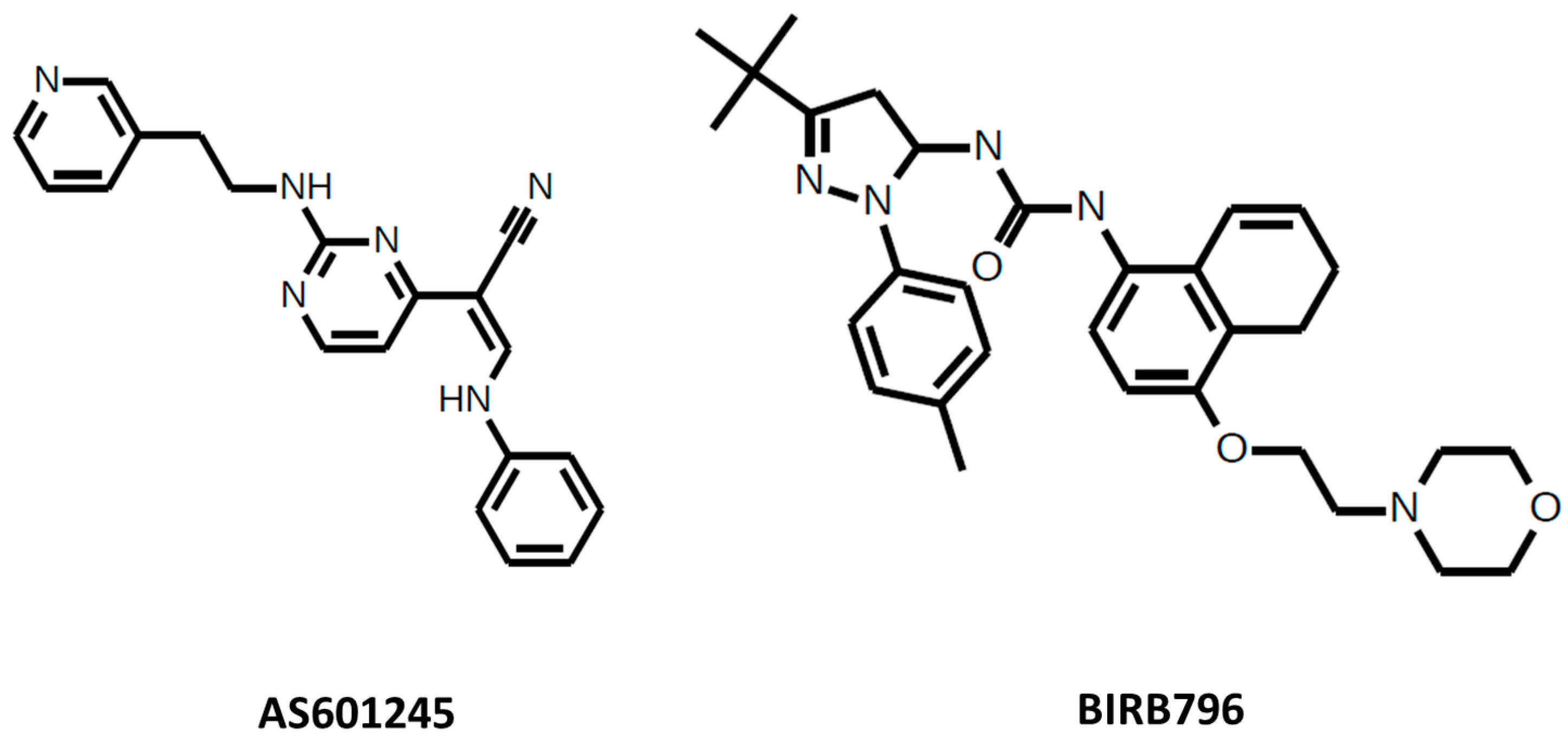
Figure 3.
FR180204 and XMD8-92.

{kind=link}
{kind=link}
{kind=link}
Table 1.
The summary of small molecule inhibitors with their potential usages.
| Ihibitor | Target | Potential Usages in Cancer |
|---|---|---|
| SP600125 | JNK | stomach cancer [24], oral squamous carcinoma [25], lung adenocarcinoma [26], cholangiocarcinoma [27], colon carcinoma [28], pancreatic cancer [29], glioblastoma [30] |
| AS601245 | JNK | colon cancer [35], leukemia [37,38] |
| CC-401 | JNK | colon cancer [42] |
| SCIO-469 | p38 | multiple myeloma [50], leukemia [55] |
| BIRB-796 | p38 | multiple myeloma [59], oral epidermoid carcinoma [60], cervical cancer [61] |
| LY2228820 | p38 | melanoma, non-small cell lung cancer, ovarian cancer, glioma, myeloma, breast cancer [65], lung adenocarcinoma [66], phase I clinical trial in colorectal, breast, sarcoma, NSCLC, renal, pancreatic, melanoma and ovarian [67], phase I/II trial [68] |
| FR180204 | ERK1/2 | colorectal cancer [72] |
| XMD8-92 | ERK5 | lung cancer, cervical cancer [76], acute myeloid leukemia [77], pancreatic cancer [78], hepatocellular carcinoma [79], colon cancer [80] |
| SI113 | SGK1 | endometrial cancer [82], glioblastoma [83], hepatocellular carcinoma [84] |
JNK: c-Jun N-terminal kinase; ERK: extracellular regulated kinase; SGK1: serum/glucocorticoid-regulated kinase 1; NSCLC: non small cell lung cancer.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Cicenas, J.; Zalyte, E.; Rimkus, A.; Dapkus, D.; Noreika, R.; Urbonavicius, S. JNK, p38, ERK, and SGK1 Inhibitors in Cancer. Cancers 2018, 10, 1. https://doi.org/10.3390/cancers10010001
AMA Style
Cicenas J, Zalyte E, Rimkus A, Dapkus D, Noreika R, Urbonavicius S. JNK, p38, ERK, and SGK1 Inhibitors in Cancer. Cancers. 2018; 10(1):1. https://doi.org/10.3390/cancers10010001
Chicago/Turabian StyleCicenas, Jonas, Egle Zalyte, Arnas Rimkus, Dalius Dapkus, Remigijus Noreika, and Sigitas Urbonavicius. 2018. "JNK, p38, ERK, and SGK1 Inhibitors in Cancer" Cancers 10, no. 1: 1. https://doi.org/10.3390/cancers10010001
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.