The Role of Oncogenic Tyrosine Kinase NPM-ALK in Genomic Instability


1
Central European Institute of Technology (CEITEC), Masaryk University, Kamenice 5, 62500 Brno, Czech Republic
2
Department of Internal Medicine—Hematology and Oncology, University Hospital Brno, 62500 Brno, Czech Republic
*
Author to whom correspondence should be addressed.
Cancers 2018, 10(3), 64; https://doi.org/10.3390/cancers10030064
Submission received: 26 January 2018
/
Revised: 20 February 2018
/
Accepted: 27 February 2018
/
Published: 5 March 2018
(This article belongs to the Special Issue Advances in Our Understanding of ALK-Related Cancers: A Selection of Papers from the Joint Annual Meeting of the European Research Initiative for ALK-Related Malignancies (ERIA) and the European Union Marie Curie European Training Network ALKATRAS)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Genomic stability is crucial for cell life and transmitting genetic material is one of the primary tasks of the cell. The cell needs to be able to recognize any possible error and quickly repair it, and thus, cells have developed several mechanisms to detect DNA damage and promote repair during evolution. The DNA damage response (DDR) and DNA repair pathways ensure the control of possible errors that could impair the duplication of genetic information and introduce variants in the DNA. Endogenous and exogenous factors compromise genomic stability and cause dysregulation in the DDR and DNA repair pathways. Cancer cells often impair these mechanisms to overcome cellular barriers (cellular senescence and/or apoptosis), leading to malignancy. NPM (nucleophosmin)-ALK (anaplastic lymphoma kinase) is an oncogenic tyrosine kinase that is involved in the development of anaplastic large cell lymphoma (ALCL). NPM-ALK is known to be involved in the activation of proliferative and anti-apoptotic signaling pathways. New evidence reveals that NPM-ALK translocation also impairs the ability of cells to maintain the genomic stability through both DDR and DNA repair pathways. This review aims to highlight the role of the oncogenic tyrosine kinase NPM-ALK in the cell, and pointing to new possible therapeutic strategies.
1. Introduction
Cells have to robustly counteract against events that can cause DNA damage, and, be ready to recognize and repair any possible error in genetic material. They are subjected to a high degree of external stress daily, from sources such as irradiation, UV light, reactive oxygen species (ROS), tobacco smoke, or from the cellular environment, such as errors that are formed during DNA replication by DNA polymerases. Since there are a plethora of possible factors that can induce different types of damage in the cell’s genomic integrity, each error has a specific way to be recognized and treated. The genome-maintenance machinery is well organized: cells need to recognize the error by sensor proteins, communicate the problem using signal transducers, and then fix it through effectors [1]. There are two main mechanisms responsible for these essential tasks: DNA damage response and DNA repair pathways. Damage or mutations in these two pathways are often related to genomic instability and cancer development. A case in point, as has been shown, is that oncogenic tyrosine kinases, such as NPM (nucleophosmin)-ALK (anaplastic lymphoma kinase) (and also BCR (breakpoint cluster region)-ABL (abelson murine leukemia viral oncogene homolog 1), TEL (translocation ETS leukemia)-ABL, TEL-JAK2 (janus kinase 2)) induce DNA damage, and directly impair the normal function of proteins that are involved in these pathways [2]. For instance, BCR-ABL was found to be directly involved in the regulation of DNA repair decreasing the expression of DNA-dependent protein kinase, catalytic subunit (DNA-PKcs) in patients with chronic myeloid leukemia (CML) [3]. DNA-PKcs is a kinase protein that is involved in the non-homologous end joining (NHEJ) pathway of DNA repair. Down-regulation of DNA-PKcs by BCR-ABL is related to DNA repair deficiency and it plays a role in CML progression [4].
The Oncogenic Tyrosine Kinase NPM-ALK
The translocation t(2;5) (p23;q35) was discovered for the first time in the late 1980s in anaplastic large cell lymphoma (ALCL) [5]. Subsequently, in 1994, the product of this translocation was identified as the fusion between the receptor anaplastic lymphoma kinase (ALK) and nucleophosmin (NPM1) loci, on chromosomes 2 and 5, respectively [6]. Since that time, many studies were focused on understanding of the role of NPM-ALK translocation. ALK is a receptor protein-tyrosine kinase and its aberration is observed in several human malignancies, such as ALCL, neuroblastoma, inflammatory myofibroblastic tumor, or non-small cell lung carcinoma (NSCLC) [7]. The translocation with NPM is the most common in ALCL, as it is present in about 80% of ALK-positive ALCL [8], although several other ALK partner genes are described, such as TFG, TPM3, ATIC [9,10,11]. NPM-ALK fusion protein is constitutively activated in ALCL, and after its dimerization it activates various signaling pathways, including phosphatidylinositol-3-kinase(PI3K)/AKT/mTOR, JAK/signal transducer and activator of transcription 3 (STAT3), RAS/ERK, and phospholipase C (PLCγ) [12,13,14]. Through the deregulated activation of these pathways, NPM-ALK induces cell-cycle progression, proliferation, cell survival and anti-apoptotic functions [15]. In this review, we focus on the involvement of NPM-ALK translocation in the tumor pathogenesis, by influencing pathways that are dedicated to the maintenance of genomic stability.
2. DNA Damage Response and Role of Tumor Suppressor p53
The cell evolved extremely powerful machinery to protect itself from damage—DNA damage response (DDR) pathways. Various types of damage can affect the cell: single point mutations, single-strand DNA breaks (SSBs) or double-strand DNA breaks (DSBs). The main actors in DNA damage response are two kinases: ATM (ataxia telangiectasia mutated) and ATR (ATM-Rad3-related) [16]. ATM and ATR are upstream molecules of the main DDR pathways dedicated to recognizing DSBs and SSBs, respectively [17]. Once the sensor proteins detect the DNA damage, these two kinases are recruited and trigger DNA damage response cascades. These cascades include (i) cellular checkpoints, able to stop cell cycle progression, (ii) the DNA repair pathway, to rectify possible mistake(s), and (iii) the apoptotic pathway, to induce cell death when the damage resolution is not possible. In recent decades, several studies clarified how these kinases are initially triggered, and identified a pool of proteins that directly bind the DNA in the broken sites. The Mre11-Rad50-Nbs1 (MRN) mediator complex identifies the DSB and recruits ATM to the broken DNA site [18,19]. Another complex mechanism is required for ATR recruitment; when the replication protein A (RPA) binds the ssDNA [20], it triggers the enrollment of ATR through ATR interacting protein (ATRIP) [21] and the activation of Rad9-Rad1-HUS1 and Rad17-RFC complexes [16,17], allowing for ATR to interact with the damage. ATM and ATR share downstream targets, including checkpoint kinase 1 and 2 (CHK1and CHK2) [22] and components of p53 pathway [23]. Indeed, ATM and ATR mediate the binding degradation of p53 with its negative regulators, murine double minute 2 (MDM2), or murine double minute 4 (MDM4) [24,25]. Though it is still not entirely clear how p53 is recruited, evidence suggests that DNA damage and genomic instability might induce the activation of p53 to accomplish tumor suppression [26]. In this scenario, the tumor suppressor p53 plays a central role in the cell’s fate. p53 is a transcription factor and a master regulator of the cell cycle. After a stressful event, such as DNA damage, the cell can go through either cell-cycle arrest or apoptosis, and p53 is a crucial protein for this decision [27]. In stress conditions, it receives the signal and induces the activation of a spectrum of cellular response involved in cell cycle arrest, DNA repair, autophagy, apoptosis and other activities according to the level of damage suffered by the cell. The transcription factor p53 has different domains: a N-terminal transactivation domain (TAD), a proline-rich domain (PRD), a core DNA binding domain (DBD), a tetramerization domain (4D), and a C-terminal regulatory domain (CTD) [28]. Somatic mutation or deletion of the TP53 gene are the most common and well-documented mechanisms by which p53 activity is deregulated. Moreover, damage in the p53-regulator pathways, such as the overexpression of its negative regulators MDM2 or MDM4, but also epigenetic modification, miRNAs alteration or TP53 splicing deregulation, can impair p53 activity [29]. The level of p53 is essential and is strictly controlled by the cell. Under normal conditions, p53 is negatively regulated by MDM2 or MDM4, which bind the TAD domain of p53, inducing the degradation of the protein by ubiquitination [30,31]. The balance between p53 and MDM2 is crucial for p53 activation. In fact, p53 activates MDM2 transcription, inducing negative feedback on its expression. This balance is altered by DNA damage that increases p53 levels and induces post-translational modification of MDM2. In this situation, MDM2 is not able to negatively regulate p53, allowing for the activation of p53 gene targets [32,33]. Activated p53 regulates the expression of a plethora of genes that are involved in multiple cellular functions, such as (i) cyclin dependent kinase inhibitor 1A (CDKN1A), by the transcription regulation of which it is able to halt the cell at the G1 phase, allowing to the cell to have sufficient time to repair the DNA damage and restore genomic stability, (ii) Bcl-2-binding component 3 (BBC3) and Bcl-2-associated X (BAX) in apoptosis or (iii) promyelocytic leukemia protein (PML) in cellular senescence [34]. Defects in ATM, ATR, and p53 have been described in B and T-cell lymphoma [35,36]. For instance, alterations in TP53 and ATM, due to deletions or mutations, have been associated with poor prognosis and chemoresistance in chronic lymphocytic leukemia (CLL) and their detection has become clinically necessary [37,38,39]
p53 in ALK-Positive Anaplastic Large Cell Lymphoma
The emerging evidence of the role of p53 role in cancer development has prompted researchers to investigate this transcription factor in various tumors, including anaplastic large cell lymphoma. Early genetic studies showed that p53 is uncommonly mutated in ALCL (<10%) and that it is frequently expressed [40,41]. A more recent study, using high throughput technologies, showed that the loss at 17p13 encompassing TP53 gene, together with the loss at 6q21, are the most frequent lesions in ALCL [42]. The most common ways that are used by cancer cells to inactivate p53 are by mutating TP53 gene or over-expressing its negative regulator (MDM2). Usually, ALK-positive ALCL carries wild-type p53 and does not over-express MDM2, suggesting that, in this tumor, p53 activity is controlled in an alternative way. It has been shown that NPM-ALK induces phosphoinositide 3-kinase (PI3K) [13] and Jun N-terminal kinase (JNK) [43] and by interaction with these molecules is capable of regulating p53 activity. The transcription factor p53 needs to be localized in the nucleus to carry out its tumor suppressor function. Recent studies suggest that NPM-ALK translocation disrupts p53 function by sequestering p53 in the cytoplasm and by inducing its degradation through JNK and MDM2 activities [44,45]. In particular, Cui and colleagues [44] demonstrated that PI3K phosphorylates MDM2 on serine 166, increasing its stabilization and this leads to an increment of p53-MDM2 binding. As is known, this binding leads to p53 localization in the cytoplasm, and thus to its inhibition. Moreover, the phosphorylation of JNK by NPM-ALK translocation influences also p53 activity. Indeed, p-JNK sequesters the tumor suppressor p53 and induces its degradation (Figure 1B). Further proof of the importance of p53 in NPM-ALK malignancies comes from the murine embryonic fibroblast (MEFs) cell line deficient for p53 and transfected with NPM-ALK. Indeed, p53 seems to play a role in blocking the proliferation-inducing senescence. Loss of p53 allows for the NPM-ALK cells to bypass the senescence and manifest a tumor phenotype [45].
Since the transcription factor p53 prevents tumor progression [46] restoring its expression can be used to promote tumor regression [47]. The wild-type status of p53 in ALK-expressing ALCL may represent an important ally in the struggle against cancer. Indeed, the re-activation of p53 suggests a potential strategy for ALK-positive ALCL treatment. Nutlin-3a is a small molecule able to target MDM2 and disrupt p53-MDM2 bound [48]. Drakos and colleagues showed that nutlin-3a inhibits the interaction p53-MDM2 leading to cell cycle arrest and apoptosis induced by p53 re-activation in one ALK-positive ALCL in vitro system [49]. Their data suggest that the p53-MDM2 interaction could become a novel potential therapeutic target in ALK-positive ALCL.
3. NPM-ALK Induces Cellular Senescence in Anaplastic Large Cell Lymphoma
Cellular senescence is a stress response resulting from several mechanisms (e.g., DNA damage, oncogene activation, or telomere shortening) that is aimed at protecting the cell against cancer development or organismal aging [50]. A particular type of cellular senescence mechanism is the oncogene-induced senescence (OIS). In response to DNA damage or various forms of stress induced by expression of activated oncogenes, particularly in the early stage of tumorigenesis, the cell activates cellular senescence pathways inducing irreversible growth arrest [51]. Two of the primary pathways that are involved in OIS mechanisms are p16INK4a/retinoblastoma (RB) and alternative reading frame (ARF)/p53. These proteins trigger the senescence cascade and their high expression can be used as senescence markers, although there are cases in which the overexpression of these markers is not correlated with senescence status, because downstream mutations allow the malignant tumor to overcome the senescence [52]. Oncogenes induce accumulation of DNA damage with consequent activation of ARF and p16INK4a, which respectively stabilize p53 and maintain RB in its active form (hypophosphorylated RB). These two pathways, ARF/p53 and p16INK4a/RB, are frequently mutated in human tumors, and inactivation of these pathways allows the tumor cell to overcome the senescence barrier and induce tumor transformation [53]. Attenuation of ARF and p53 is a typical behavior of several oncogenes in hematological malignancies, such as BCR-ABL in CLL [54], PML-RARα in promyelocytic leukemia [55], and specifically NPM-ALK in ALCL [44]. Transfection of NPM-ALK in primary MEFs shows that cells try to counteract this transformation using cellular senescence mechanisms, in particular p53 and RB pathways [45,53]. As previously described, NPM-ALK inhibits p53 through MDM2 and JNK [44] and ectopic expression of ALK translocation in MEFs shows the deregulation of not only p53, but also the p16INK4a/RB pathway. Indeed, Martinelli et al. [53], showed, in vivo and in vitro, that NPM-ALK induces DNA damage and senescence by activation of p16INK4a, and that consequently, its loss is necessary for ALCL development. Supporting this hypothesis, are the expression data of p16INK4a and RB in ALCL. Low levels or no expression of p16INK4a and expression of inactive RB (phosphorylated RB) were found in NPM-ALK human samples and ALCL cell lines. This study also investigated the mechanisms through NPM-ALK induces the OIS in ALCL, showing that p16INK4a is repressed by methylation in its promoter and oncogenic tyrosine kinase induces de-methylation by Jmjd3, a histone lysine demethylase, and then p16INK4a transcription. Further data shows that the activation of p16INK4a is due to multiple signals, including Jmjd3 and STAT3, the second one known to be expressed by NPM-ALK [12] (Figure 2). Taken together, these data reveal the ambivalent behavior of NPM-ALK, acting on pathways that are predisposed to arrest the proliferation and cancer development. These cellular senescence pathways are frequently silenced in ALCL and reactivation of their activity, for instance, with specific de-methylating agents, might be a target for a novel therapeutic strategy.
4. NPM-ALK Overexpression Lead to DNA Damage Response activation
As we have described so far, NPM-ALK interferes with the DNA damage response pathway, aiming to circumvent cellular senescence and inducing cancer progression. Surprisingly, a recent study reported an excess of NPM-ALK as leading to the activation of ATM/Chk2 pathway, involved in DDR signaling, and promoting apoptosis [56]. NPM-ALK is equally distributed between nucleus and cytoplasm, but it has been shown that only the cytoplasmic portion is active and that the nuclear portion is sequestered in the nucleus together with the wild-type NPM1 [56]. Indeed, the NPM portion in the translocation is lacking in nuclear localization sequence (NLS) and nucleolar localization signal (NuLS), domains that allow wild-type NPM1 to translocate in the nucleus. Overexpression of NPM-ALK, in vitro and in vivo, increases the cytoplasmic portion of the translocation leading to activation of caspase 3 and 7 and apoptosis. As in ALCL, these results showed that the balance of cytoplasmic and nucleus portion of NPM-ALK is crucial. Moreover, the stress that was caused by NPM-ALK overexpression led to the DNA damage response activation, mainly through ATM/Chk2 pathway and H2A histone family member X (H2AX) phosphorylation caused by the increase in MAPK/ERK1/2 activation. Notably, genomic amplification of the ALK locus has been described in ALK tyrosine kinase inhibitors (TKI) resistance [57,58]. Indeed, in NPM-ALK cell lines harboring ALK amplification showed a higher localization of ALK-translocation in the cytoplasm and TKIs resistance. Interestingly, and necessary for further investigation, is the behavior of NPM-ALK amplified cells treated with TKIs upon drug withdrawal. In this condition, the cells activate the pathways that are previously described in the presence of NPM-ALK overexpression, with high activation of MAPK/ERK1/2, phosphorylation of H2AX and increased cell death [56]. This remarkable behavior of NPM-ALK overexpression after TKIs suspension warrants further investigation and it could represent another valid therapeutic option in TKIs resistance ALK-amplified ALCL using the DNA damage response pathway as an ally.
5. DNA Repair
DNA damage can be manifested in a variety of forms; hence, the cell developed a complex DNA repair system specific to different types of DNA lesions. Concerning mammalian cells, one can identify five distinct mechanisms, by which the DNA repair machinery works; base excision repair (BER), DNA mismatch repair (MMR), nucleotide excision repair (NER), DNA strand break repair, including homologous recombination (HR), and nonhomologous end joining (NHEJ) [59,60]. For the aim of this review, we will focus mainly on DNA mismatch repair in which NPM-ALK appears to play a part.
5.1. DNA Mismatch Repair
During DNA synthesis, the fidelity with which each copy of DNA is duplicated is critical. Base-base mismatches or insertion/deletion loops (IDLs) can possibly occur, particularly in DNA regions with repeated-sequence motifs. If these errors remain unrepaired, they can evolve in single nucleotide variations (SNVs), modify the phenotype and lead to disease. To prevent these kinds of events during DNA synthesis, and to maintain genomic stability, the cells developed a specific mechanism—DNA mismatch repair (MMR) [61]. The MMR pathway is highly conserved across species and its homologous MMR components that are present in Escherichia coli have also been found in the mammalian cell [62,63]. The MMR pathway needs first to recognize the mismatch, then remove the fragment of the newly synthesized strand containing the error and ultimately resynthesize it correctly. In human cells, there are two ATPases protein complexes assigned to recognize mismatches: MuTSα and MuTSβ complexes. The first and most abundant is formed by MSH2 with MSH6 (MutS protein homolog 2 and 6). It preferentially recognizes the base-base mismatches and the small insertion/deletion mispairs. The second one is formed by MSH2 with MSH3 and it is charged to the recognition of larger insertions/deletions [64,65]. Once the mismatches have been recognized, another complex, MutL, links the MuTS, triggering the MMR downstream cascade [66,67]. Then, the proliferating cellular nuclear antigen (PCNA) interacts with MutL/MuTS complex introducing a nick to the mismatch region [68,69]. Exonuclease I (EXO1) catalyzes the excision of the newly synthesized DNA, removing the mismatch and the PCNA stimulates the DNA polymerase δ activation for the re-synthesis of the DNA strand. The remaining nick is then ligated by DNA ligase I and in this way the mismatch is corrected [70,71].
5.2. DNA Mismatch Repair in ALK-Positive Anaplastic Large Cell Lymphoma
Defects in the MMR mechanism increase the number of spontaneous mutations [72]. Indeed, MMR deficiencies have been associated to different diseases [62], such as hereditary non-polyposis colorectal cancer (Lynch syndrome) [73,74,75], as well as to resistance to chemotherapy drugs or abnormalities in meiosis [76,77]. Moreover, knockout mice deficient for MMR genes (MSH2, MSH6, MLH1, PMS2, EXO1) showed a high presence of microsatellite instability (MSI) and susceptibility to lymphoma development [77,78,79,80,81]. The fusion tyrosine kinase NPM-ALK also plays a role in the de-regulation of MMR mechanism in anaplastic large cell lymphoma. A proteome-wide study identified a large number of proteins interacting with NPM-ALK, including minichromosome maintenance complex component 6 (MCM6) and MSH2, which are both involved in DNA repair mechanisms [82]. Moreover, a direct interaction between NPM-ALK and MSH2, and the presence of microsatellite instability (MSI) in ALK-positive ALCL patients has been proven [83]. MSH2 is the key protein in the MuTS complexes. NPM-ALK interacts with MSH2, but not its normal partners in MuTS complexes (MSH6 or MSH3) [82]. In fact, subsequently, it has been shown that NPM-ALK phosphorylates MSH2 (at tyrosine 238) blocking the MSH2-MSH6 interaction (Figure 3B). MSH2 phosphorylated at Y238 is unable to bind MSH6, and then to recognize DNA mismatch, inducing loss of MMR activity in the presence of DNA damage [83,84]. The clinical significance of this finding needs further investigations, and restoring MMR mechanism strategy can be employed in the development of novel cancer treatment.
6. Conclusions
NPM-ALK clearly plays the main role in ALCL development modifying, not only the proliferation and survival pathways [15], but also acting on the genomic stability. The ALK translocation is able to indirectly regulate tumor suppressor p53 and directly p16INK4a and MSH2, acting in the DNA damage response and DNA repair pathways. Defects in these two mechanisms induce genomic instability and the accumulation of mutations, leading to higher cancer susceptibility. The standard first-line treatment of ALCL is the patient subjection to chemotherapy regimens. Nowadays, tyrosine kinase ALK inhibitors have been developed, such as Crizotinib, Ceritinib or Lorlatinib [85]. Despite the enormous efforts in the last years, a subgroup of patients still relapses and/or develops resistance to the treatment. Therefore, considerable effort needs to be undertaken to investigate new therapeutic strategies, such as nutlin-3a utilization. Targeting and restoring the normal condition of DDR and DNA repair pathways could turn out to be a new therapeutic strategy to fight cancer development. The phase I of several clinical trials using Nutlin-3a have been already completed in hematological neoplasm and solid tumors, including lymphoma (NCT00623870, NCT00559533). RG7112 is a small molecule derives from Nutlins and the first MDM2 inhibitors that are used in clinical trials. The results from a phase I study in patients with hematologic malignancies provide that MDM2 inhibitor has a sufficient clinical activity in restoring p53 function [86]. On the other hand, the trial highlights clinical adversity, such as hematological toxicity. In this regard, a new compound, RG7388, known as idasanutlin [87], is now under clinical studies. It is a second-generation MDM2 inhibitor and it needs a lower concentration to act respect RG7112. Moreover, a combination of MDM2 inhibitors with other agents, such as monoclonal antibodies, was tested with promising preliminary data [88].
As we described in this review, DNA damage response and DNA repair represent a complex machinery involving a wide number of players and an intricate network, therefore, further studies are necessary to investigate the role of other compounds in DNA damage response and DNA repair pathways. Moreover, the evolution of new high throughput techniques, such as next-generation sequencing, allows for investigating a broad spectrum of genes, and in particular, possible mutations that are involved in increasing genomic instability in the cell.
Acknowledgments
Cosimo Lobello, Andrea Janikova and Sarka Pospisilova are members of the European Research Initiative for ALK-Related Malignancies (www.erialcl.net). This project has received funding from the European Union’s Horizon 2020 Research and Innovation Programme under the [Marie Skłodowska-Curie grant agreement No. 675712; ALKATRAS] and from the Ministry of Education, Youth and Sports of the Czech Republic under the project CEITEC 2020 [LQ1601]. Publication reflects only the author’s view and that the Commission is not responsible for any use that may be made of the information it contains.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Niida, H.; Nakanishi, M. DNA damage checkpoints in mammals. Mutagenesis 2006, 21, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Skorski, T. Oncogenic tyrosine kinases and the DNA-damage response. Nat. Rev. Cancer 2002, 2, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.; Dugray, A.; AbdulKarim, B.; Marangoni, E.; Maggiorella, L.; Vaganay, S.; M’Kacher, R.; Rasy, S.D.; Eschwege, F.; Vainchenker, W.; et al. BCR-ABL down-regulates the DNA repair protein DNA-PKcs. Blood 2001, 97, 2084–2090. [Google Scholar] [CrossRef] [PubMed]
- Faderl, S.; Talpaz, M.; Estrov, Z.; O’Brien, S.; Kurzrock, R.; Kantarjian, H.M. The biology of chronic myeloid leukemia. N. Engl. J. Med. 1999, 341, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Benz-Lemoine, E.; Brizard, A.; Huret, J.L.; Babin, P.; Guilhot, F.; Couet, D.; Tanzer, J. Malignant histiocytosis: A specific t(2;5)(p23;q35) translocation? Review of the literature. Blood 1988, 72, 1045–1047. [Google Scholar] [PubMed]
- Morris, S.W.; Kirstein, M.N.; Valentine, M.B.; Dittmer, K.G.; Shapiro, D.N.; Saltman, D.L.; Look, A.T. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 1994, 263, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Mano, H. Alkoma: A cancer subtype with a shared target. Cancer Discov. 2012, 2, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Falini, B.; Pulford, K.; Pucciarini, A.; Carbone, A.; De Wolf-Peeters, C.; Cordell, J.; Fizzotti, M.; Santucci, A.; Pelicci, P.G.; Pileri, S.; et al. Lymphomas expressing ALK fusion protein(s) other than NPM-ALK. Blood 1999, 94, 3509–3515. [Google Scholar] [PubMed]
- Hernandez, L.; Bea, S.; Bellosillo, B.; Pinyol, M.; Falini, B.; Carbone, A.; Ott, G.; Rosenwald, A.; Fernandez, A.; Pulford, K.; et al. Diversity of genomic breakpoints in TFG-ALK translocations in anaplastic large cell lymphomas: Identification of a new TFG-ALK(XL) chimeric gene with transforming activity. Am. J. Pathol. 2002, 160, 1487–1494. [Google Scholar] [CrossRef]
- Lamant, L.; Dastugue, N.; Pulford, K.; Delsol, G.; Mariame, B. A new fusion gene tpm3-alk in anaplastic large cell lymphoma created by a (1;2)(q25;p23) translocation. Blood 1999, 93, 3088–3095. [Google Scholar] [PubMed]
- Colleoni, G.W.; Bridge, J.A.; Garicochea, B.; Liu, J.; Filippa, D.A.; Ladanyi, M. ATIC-ALK: A novel variant ALK gene fusion in anaplastic large cell lymphoma resulting from the recurrent cryptic chromosomal inversion, inv(2)(p23q35). Am. J. Pathol. 2000, 156, 781–789. [Google Scholar] [CrossRef]
- Zamo, A.; Chiarle, R.; Piva, R.; Howes, J.; Fan, Y.; Chilosi, M.; Levy, D.E.; Inghirami, G. Anaplastic lymphoma kinase (ALK) activates STAT3 and protects hematopoietic cells from cell death. Oncogene 2002, 21, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Slupianek, A.; Nieborowska-Skorska, M.; Hoser, G.; Morrione, A.; Majewski, M.; Xue, L.; Morris, S.W.; Wasik, M.A.; Skorski, T. Role of phosphatidylinositol 3-kinase-AKT pathway in nucleophosmin/anaplastic lymphoma kinase-mediated lymphomagenesis. Cancer Res. 2001, 61, 2194–2199. [Google Scholar] [PubMed]
- Bai, R.Y.; Dieter, P.; Peschel, C.; Morris, S.W.; Duyster, J. Nucleophosmin-anaplastic lymphoma kinase of large-cell anaplastic lymphoma is a constitutively active tyrosine kinase that utilizes phospholipase c-gamma to mediate its mitogenicity. Mol. Cell. Biol. 1998, 18, 6951–6961. [Google Scholar] [CrossRef] [PubMed]
- Chiarle, R.; Voena, C.; Ambrogio, C.; Piva, R.; Inghirami, G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat. Rev. Cancer 2008, 8, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Rouse, J.; Jackson, S.P. Interfaces between the detection, signaling, and repair of DNA damage. Science 2002, 297, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.H.; Zou, L. Checkpoint and coordinated cellular responses to DNA damage. Results Probl. Cell Differ. 2006, 42, 65–92. [Google Scholar] [PubMed]
- D’Amours, D.; Jackson, S.P. The mre11 complex: At the crossroads of DNA repair and checkpoint signalling. Nat. Rev. Mol. Cell Biol. 2002, 3, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Paull, T.T. ATM activation by DNA double-strand breaks through the MRE11-RAD50-NBS1 complex. Science 2005, 308, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Byun, T.S.; Pacek, M.; Yee, M.C.; Walter, J.C.; Cimprich, K.A. Functional uncoupling of mcm helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005, 19, 1040–1052. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Elledge, S.J. Sensing DNA damage through atrip recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Lukas, J. CHK1 and CHK2 kinases in checkpoint control and cancer. Cancer Cell 2003, 3, 421–429. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. Atm, atr, and DNA-PK: The trinity at the heart of the DNA damage response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Meulmeester, E.; Pereg, Y.; Shiloh, Y.; Jochemsen, A.G. ATM-mediated phosphorylations inhibit Mdmx/Mdm2 stabilization by HAUSP in favor of p53 activation. Cell Cycle 2005, 4, 1166–1170. [Google Scholar] [CrossRef] [PubMed]
- Stommel, J.M.; Wahl, G.M. Accelerated MDM2 auto-degradation induced by DNA-damage kinases is required for p53 activation. EMBO J. 2004, 23, 1547–1556. [Google Scholar] [CrossRef] [PubMed]
- Meek, D.W. Tumour suppression by p53: A role for the DNA damage response? Nat. Rev. Cancer 2009, 9, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Prives, C.; Hall, P.A. The p53 pathway. J. Pathol. 1999, 187, 112–126. [Google Scholar] [CrossRef]
- Joerger, A.C.; Fersht, A.R. The tumor suppressor p53: From structures to drug discovery. Cold Spring Harb. Perspect. Biol. 2010, 2, a000919. [Google Scholar] [CrossRef] [PubMed]
- Herrero, A.B.; Rojas, E.A.; Misiewicz-Krzeminska, I.; Krzeminski, P.; Gutierrez, N.C. Molecular mechanisms of p53 deregulation in cancer: An overview in multiple myeloma. Int. J. Mol. Sci. 2016, 17, 2003. [Google Scholar] [CrossRef] [PubMed]
- Bond, G.L.; Hu, W.; Bond, E.E.; Robins, H.; Lutzker, S.G.; Arva, N.C.; Bargonetti, J.; Bartel, F.; Taubert, H.; Wuerl, P.; et al. A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell 2004, 119, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Toledo, F.; Wahl, G.M. MDM2 and MDM4: p53 regulators as targets in anticancer therapy. Int. J. Biochem. Cell Biol. 2007, 39, 1476–1482. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Feng, Z.; Ma, L.; Wagner, J.; Rice, J.J.; Stolovitzky, G.; Levine, A.J. A single nucleotide polymorphism in the MDM2 gene disrupts the oscillation of p53 and MDM2 levels in cells. Cancer Res. 2007, 67, 2757–2765. [Google Scholar] [CrossRef] [PubMed]
- Whibley, C.; Pharoah, P.D.; Hollstein, M. p53 polymorphisms: Cancer implications. Nat. Rev. Cancer 2009, 9, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.B.; Schumacher, B. p53 in the DNA-damage-repair process. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Takakuwa, T.; Fujita, S.; Ham, M.F.; Luo, W.J.; Daibata, M.; Aozasa, K. Alterations of DNA damage-response genes ATM and ATR in pyothorax-associated lymphoma. Lab. Investig. J. Tech. Methods Pathol. 2005, 85, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Schaffner, C.; Idler, I.; Stilgenbauer, S.; Dohner, H.; Lichter, P. Mantle cell lymphoma is characterized by inactivation of the ATM gene. Proc. Natl. Acad. Sci. USA 2000, 97, 2773–2778. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Rasi, S.; Spina, V.; Bruscaggin, A.; Monti, S.; Ciardullo, C.; Deambrogi, C.; Khiabanian, H.; Serra, R.; Bertoni, F.; et al. Integrated mutational and cytogenetic analysis identifies new prognostic subgroups in chronic lymphocytic leukemia. Blood 2013, 121, 1403–1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, D.; Martinez, P.; Wade, R.; Hockley, S.; Oscier, D.; Matutes, E.; Dearden, C.E.; Richards, S.M.; Catovsky, D.; Morgan, G.J. Mutational status of the Tp53 gene as a predictor of response and survival in patients with chronic lymphocytic leukemia: Results from the LRF CLL4 trial. J. Clin. Oncol. 2011, 29, 2223–2229. [Google Scholar] [CrossRef] [PubMed]
- Te Raa, G.D.; Malcikova, J.; Pospisilova, S.; Trbusek, M.; Mraz, M.; Garff-Tavernier, M.L.; Merle-Beral, H.; Lin, K.; Pettitt, A.R.; Merkel, O.; et al. Overview of available p53 function tests in relation to Tp53 and ATM gene alterations and chemoresistance in chronic lymphocytic leukemia. Leuk. Lymphoma 2013, 54, 1849–1853. [Google Scholar] [CrossRef] [PubMed]
- Cesarman, E.; Inghirami, G.; Chadburn, A.; Knowles, D.M. High levels of p53 protein expression do not correlate with p53 gene mutations in anaplastic large cell lymphoma. Am. J. Pathol. 1993, 143, 845–856. [Google Scholar] [PubMed]
- Rassidakis, G.Z.; Thomaides, A.; Wang, S.; Jiang, Y.; Fourtouna, A.; Lai, R.; Medeiros, L.J. P53 gene mutations are uncommon but p53 is commonly expressed in anaplastic large-cell lymphoma. Leukemia 2005, 19, 1663–1669. [Google Scholar] [CrossRef] [PubMed]
- Boi, M.; Rinaldi, A.; Kwee, I.; Bonetti, P.; Todaro, M.; Tabbo, F.; Piva, R.; Rancoita, P.M.; Matolcsy, A.; Timar, B.; et al. PRDM1/BLIMP1 is commonly inactivated in anaplastic large T-cell lymphoma. Blood 2013, 122, 2683–2693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leventaki, V.; Drakos, E.; Medeiros, L.J.; Lim, M.S.; Elenitoba-Johnson, K.S.; Claret, F.X.; Rassidakis, G.Z. NPM-ALK oncogenic kinase promotes cell-cycle progression through activation of JNK/CJUN signaling in anaplastic large-cell lymphoma. Blood 2007, 110, 1621–1630. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.X.; Kerby, A.; McDuff, F.K.; Ye, H.; Turner, S.D. NPM-ALK inhibits the p53 tumor suppressor pathway in an MDM2 and JNK-dependent manner. Blood 2009, 113, 5217–5227. [Google Scholar] [CrossRef] [PubMed]
- McDuff, F.K.; Turner, S.D. Aberrant anaplastic lymphoma kinase activity induces a p53 and RB-dependent senescence-like arrest in the absence of detectable p53 stabilization. PLoS ONE 2011, 6, e17854. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.; et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005, 436, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Ventura, A.; Kirsch, D.G.; McLaughlin, M.E.; Tuveson, D.A.; Grimm, J.; Lintault, L.; Newman, J.; Reczek, E.E.; Weissleder, R.; Jacks, T. Restoration of p53 function leads to tumour regression in vivo. Nature 2007, 445, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [PubMed]
- Drakos, E.; Atsaves, V.; Schlette, E.; Li, J.; Papanastasi, I.; Rassidakis, G.Z.; Medeiros, L.J. The therapeutic potential of p53 reactivation by nutlin-3a in ALK+ anaplastic large cell lymphoma with wild-type or mutated p53. Leukemia 2009, 23, 2290–2299. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.; Blasco, M.A.; Serrano, M. Cellular senescence in cancer and aging. Cell 2007, 130, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.; Serrano, M. The power and the promise of oncogene-induced senescence markers. Nat. Rev. Cancer 2006, 6, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, G.I.; Edwards, C.D.; Kobzik, L.; Godleski, J.; Richards, W.; Sugarbaker, D.J.; Rollins, B.J. Reciprocal RB inactivation and p16ink4 expression in primary lung cancers and cell lines. Cancer Res. 1995, 55, 505–509. [Google Scholar] [PubMed]
- Martinelli, P.; Bonetti, P.; Sironi, C.; Pruneri, G.; Fumagalli, C.; Raviele, P.R.; Volorio, S.; Pileri, S.; Chiarle, R.; McDuff, F.K.; et al. The lymphoma-associated NPM-ALK oncogene elicits a P16INK4A/PRB-dependent tumor-suppressive pathway. Blood 2011, 117, 6617–6626. [Google Scholar] [CrossRef] [PubMed]
- Trotta, R.; Vignudelli, T.; Candini, O.; Intine, R.V.; Pecorari, L.; Guerzoni, C.; Santilli, G.; Byrom, M.W.; Goldoni, S.; Ford, L.P.; et al. BCR/ABL activates MDM2 mrna translation via the la antigen. Cancer Cell 2003, 3, 145–160. [Google Scholar] [CrossRef]
- Insinga, A.; Monestiroli, S.; Ronzoni, S.; Carbone, R.; Pearson, M.; Pruneri, G.; Viale, G.; Appella, E.; Pelicci, P.; Minucci, S. Impairment of p53 acetylation, stability and function by an oncogenic transcription factor. EMBO J. 2004, 23, 1144–1154. [Google Scholar] [CrossRef] [PubMed]
- Ceccon, M.; Merlo, M.E.B.; Mologni, L.; Poggio, T.; Varesio, L.M.; Menotti, M.; Bombelli, S.; Rigolio, R.; Manazza, A.D.; Di Giacomo, F.; et al. Excess of NPM-ALK oncogenic signaling promotes cellular apoptosis and drug dependency. Oncogene 2016, 35, 3854–3865. [Google Scholar] [CrossRef] [PubMed]
- Ceccon, M.; Mologni, L.; Giudici, G.; Piazza, R.; Pirola, A.; Fontana, D.; Gambacorti-Passerini, C. Treatment efficacy and resistance mechanisms using the second-generation ALK inhibitor ap26113 in human NPM-ALK-positive anaplastic large cell lymphoma. Mol. Cancer Res. 2015, 13, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Zdzalik, D.; Dymek, B.; Grygielewicz, P.; Gunerka, P.; Bujak, A.; Lamparska-Przybysz, M.; Wieczorek, M.; Dzwonek, K. Activating mutations in ALK kinase domain confer resistance to structurally unrelated ALK inhibitors in NPM-ALK-positive anaplastic large-cell lymphoma. J. Cancer Res. Clin. Oncol. 2014, 140, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Altieri, F.; Grillo, C.; Maceroni, M.; Chichiarelli, S. DNA damage and repair: From molecular mechanisms to health implications. Antioxid. Redox Signaling 2008, 10, 891–937. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T.; Barnes, D.E. Repair of endogenous DNA damage. Cold Spring Harb. Symp. Quant. Biol. 2000, 65, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Taylor, E.M.; Lehmann, A.R. Conservation of eukaryotic DNA repair mechanisms. Intern. J. Radiat. Biol. 1998, 74, 277–286. [Google Scholar]
- Modrich, P.; Lahue, R. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Ann. Rev. Biochem. 1996, 65, 101–133. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, T.A.; Erie, D.A. DNA mismatch repair. Ann. Rev. Biochem. 2005, 74, 681–710. [Google Scholar] [CrossRef] [PubMed]
- Acharya, S.; Wilson, T.; Gradia, S.; Kane, M.F.; Guerrette, S.; Marsischky, G.T.; Kolodner, R.; Fishel, R. hMSH2 forms specific mispair-binding complexes with hMSH3 and hMSH6. Proc. Natl. Acad. Sci. USA 1996, 93, 13629–13634. [Google Scholar] [CrossRef] [PubMed]
- Li, G.M. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008, 18, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Modrich, P. Mechanisms in eukaryotic mismatch repair. J. Biol. Chem. 2006, 281, 30305–30309. [Google Scholar] [CrossRef] [PubMed]
- Prolla, T.A.; Christie, D.M.; Liskay, R.M. Dual requirement in yeast DNA mismatch repair for MLH1 and PMS1, two homologs of the bacterial mutL gene. Mol. Cell. Biol. 1994, 14, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Presnell, S.R.; Yuan, F.; Zhang, Y.; Gu, L.; Li, G.M. Differential requirement for proliferating cell nuclear antigen in 5’ and 3’ nick-directed excision in human mismatch repair. J. Biol. Chem. 2004, 279, 16912–16917. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, F.C.; Jager, A.C.; Lutzen, A.; Bundgaard, J.R.; Rasmussen, L.J. Characterization of human exonuclease 1 in complex with mismatch repair proteins, subcellular localization and association with pcna. Oncogene 2004, 23, 1457–1468. [Google Scholar] [CrossRef] [PubMed]
- Tran, P.T.; Erdeniz, N.; Symington, L.S.; Liskay, R.M. EXO1-a multi-tasking eukaryotic nuclease. DNA Repair 2004, 3, 1549–1559. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Keijzers, G.; Rasmussen, L.J. DNA mismatch repair and its many roles in eukaryotic cells. Mutat. Res. 2017, 773, 174–187. [Google Scholar] [CrossRef] [PubMed]
- Tiraby, J.G.; Fox, M.S. Marker discrimination in transformation and mutation of pneumococcus. Proc. Natl. Acad. Sci. USA 1973, 70, 3541–3545. [Google Scholar] [CrossRef] [PubMed]
- Lynch, H.T.; de la Chapelle, A. Genetic susceptibility to non-polyposis colorectal cancer. J. Med. Genet. 1999, 36, 801–818. [Google Scholar] [PubMed]
- Hampel, H.; Frankel, W.L.; Martin, E.; Arnold, M.; Khanduja, K.; Kuebler, P.; Nakagawa, H.; Sotamaa, K.; Prior, T.W.; Westman, J.; et al. Screening for the lynch syndrome (hereditary nonpolyposis colorectal cancer). N. Engl. J. Med. 2005, 352, 1851–1860. [Google Scholar] [CrossRef] [PubMed]
- Barnetson, R.A.; Tenesa, A.; Farrington, S.M.; Nicholl, I.D.; Cetnarskyj, R.; Porteous, M.E.; Campbell, H.; Dunlop, M.G. Identification and survival of carriers of mutations in DNA mismatch-repair genes in colon cancer. N. Engl. J. Med. 2006, 354, 2751–2763. [Google Scholar] [CrossRef] [PubMed]
- Fink, D.; Nebel, S.; Norris, P.S.; Aebi, S.; Kim, H.K.; Haas, M.; Howell, S.B. The effect of different chemotherapeutic agents on the enrichment of DNA mismatch repair-deficient tumour cells. Br. J. Cancer 1998, 77, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Edelmann, W.; Yang, K.; Umar, A.; Heyer, J.; Lau, K.; Fan, K.; Liedtke, W.; Cohen, P.E.; Kane, M.F.; Lipford, J.R.; et al. Mutation in the mismatch repair gene msh6 causes cancer susceptibility. Cell 1997, 91, 467–477. [Google Scholar] [CrossRef]
- Reitmair, A.H.; Schmits, R.; Ewel, A.; Bapat, B.; Redston, M.; Mitri, A.; Waterhouse, P.; Mittrucker, H.W.; Wakeham, A.; Liu, B.; et al. MSH2 deficient mice are viable and susceptible to lymphoid tumours. Nat. Genet. 1995, 11, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.M.; Plug, A.W.; Prolla, T.A.; Bronner, C.E.; Harris, A.C.; Yao, X.; Christie, D.M.; Monell, C.; Arnheim, N.; Bradley, A.; et al. Involvement of mouse MLH1 in DNA mismatch repair and meiotic crossing over. Nat. Genet. 1996, 13, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Prolla, T.A.; Baker, S.M.; Harris, A.C.; Tsao, J.L.; Yao, X.; Bronner, C.E.; Zheng, B.; Gordon, M.; Reneker, J.; Arnheim, N.; et al. Tumour susceptibility and spontaneous mutation in mice deficient in MLH1, PMS1 and PMS2 DNA mismatch repair. Nat. Genet. 1998, 18, 276–279. [Google Scholar] [CrossRef] [PubMed]
- Wei, K.; Clark, A.B.; Wong, E.; Kane, M.F.; Mazur, D.J.; Parris, T.; Kolas, N.K.; Russell, R.; Hou, H., Jr.; Kneitz, B.; et al. Inactivation of exonuclease 1 in mice results in DNA mismatch repair defects, increased cancer susceptibility, and male and female sterility. Genes Dev. 2003, 17, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Wang, P.; Young, L.C.; Lai, R.; Li, L. Proteome-wide identification of novel binding partners to the oncogenic fusion gene protein, NPM-ALK, using tandem affinity purification and mass spectrometry. Am. J. Pathol. 2009, 174, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Young, L.C.; Bone, K.M.; Wang, P.; Wu, F.; Adam, B.A.; Hegazy, S.; Gelebart, P.; Holovati, J.; Li, L.; Andrew, S.E.; et al. Fusion tyrosine kinase NPM-ALK deregulates MSH2 and suppresses DNA mismatch repair function novel insights into a potent oncoprotein. Am. J. Pathol. 2011, 179, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Bone, K.M.; Wang, P.; Wu, F.; Wu, C.; Li, L.; Bacani, J.T.; Andrew, S.E.; Lai, R. Npm-alk mediates phosphorylation of msh2 at tyrosine 238, creating a functional deficiency in MSH2 and the loss of mismatch repair. Blood Cancer J. 2015, 5, e311. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Savooji, J.; Liu, D. Second- and third-generation ALK inhibitors for non-small cell lung cancer. J. Hematol. Oncol. 2016, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Andreeff, M.; Kelly, K.R.; Yee, K.; Assouline, S.; Strair, R.; Popplewell, L.; Bowen, D.; Martinelli, G.; Drummond, M.W.; Vyas, P.; et al. Results of the phase I trial of RG7112, a small-molecule MDM2 antagonist in leukemia. Clin. Cancer Res. 2016, 22, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Zhang, Z.; Liu, J.J.; Jiang, N.; Zhang, J.; Ross, T.M.; Chu, X.J.; Bartkovitz, D.; Podlaski, F.; Janson, C.; et al. Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J. Med. Chem. 2013, 56, 5979–5983. [Google Scholar] [CrossRef] [PubMed]
- Herting, F.; Herter, S.; Friess, T.; Muth, G.; Bacac, M.; Sulcova, J.; Umana, P.; Dangl, M.; Klein, C. Antitumour activity of the glycoengineered type II anti-CD20 antibody obinutuzumab (GA101) in combination with the MDM2-selective antagonist idasanutlin (RG7388). Eur. J. Haematol. 2016, 97, 461–470. [Google Scholar] [CrossRef] [PubMed]
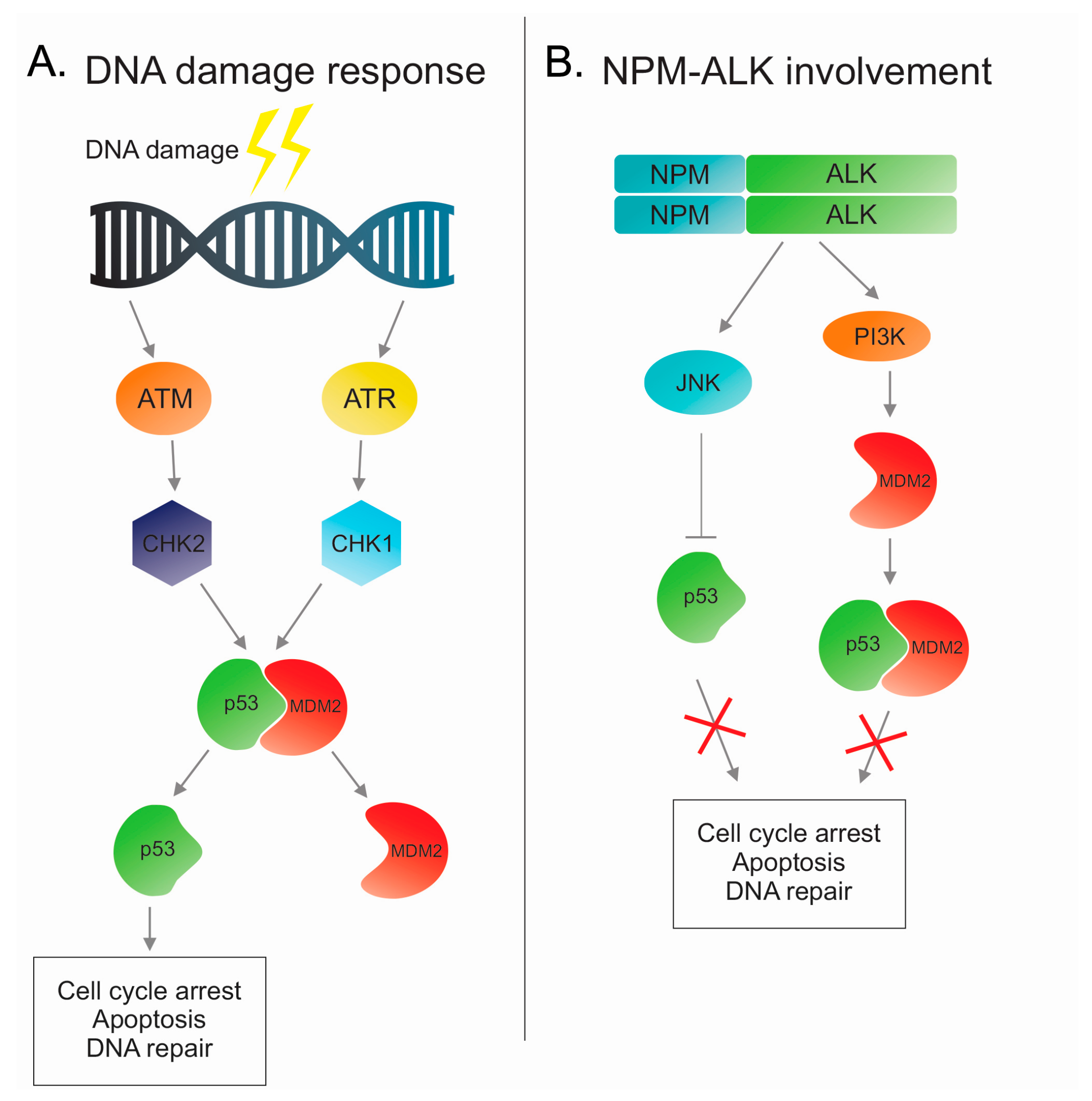
Figure 1.
Involvement of nucleophosmin (NPM)-anaplastic lymphoma kinase (ALK) in DNA damage response pathway. (A) A schematic overview of the DDR pathway with the stimulation of ataxia telangiectasia mutated (ATM) or ATM-Rad3-related (ATR) after DNA damage and the following cascade including p53 activation. (B) NPM-ALK activates (Jun-N-terminal kinase) JNK or phosphatidylinositol-3-kinase (PI3K) pathways and the subsequently inhibits the p53 pathway.
Figure 1.
Involvement of nucleophosmin (NPM)-anaplastic lymphoma kinase (ALK) in DNA damage response pathway. (A) A schematic overview of the DDR pathway with the stimulation of ataxia telangiectasia mutated (ATM) or ATM-Rad3-related (ATR) after DNA damage and the following cascade including p53 activation. (B) NPM-ALK activates (Jun-N-terminal kinase) JNK or phosphatidylinositol-3-kinase (PI3K) pathways and the subsequently inhibits the p53 pathway.

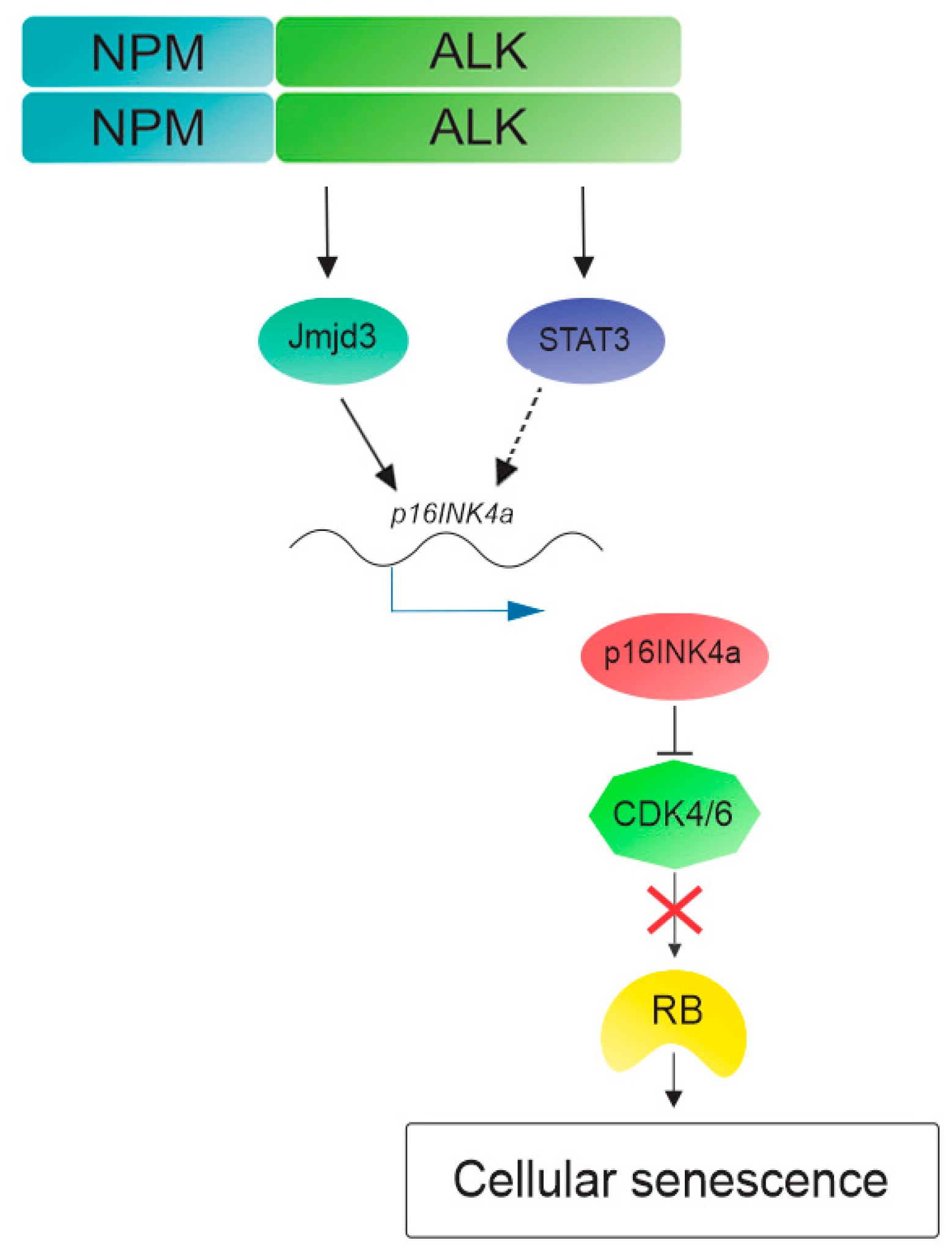
Figure 2.
Cellular senescence pathway activated by NPM-ALK. NPM-ALK promotes oncogenic-induced senescence through de-methylation of p16INK4a promoter performed by Jmjd3. Multiple mechanisms participate in p16INK4a activation, including signal transducer and activator of transcription 3 (STAT3). Then p16INK4a inhibits cyclin-dependent kinase 4 and 6 (CDK4/CDK6) allowing for retinoblastoma (RB) to induce cellular senescence and cell cycle arrest.
Figure 2.
Cellular senescence pathway activated by NPM-ALK. NPM-ALK promotes oncogenic-induced senescence through de-methylation of p16INK4a promoter performed by Jmjd3. Multiple mechanisms participate in p16INK4a activation, including signal transducer and activator of transcription 3 (STAT3). Then p16INK4a inhibits cyclin-dependent kinase 4 and 6 (CDK4/CDK6) allowing for retinoblastoma (RB) to induce cellular senescence and cell cycle arrest.
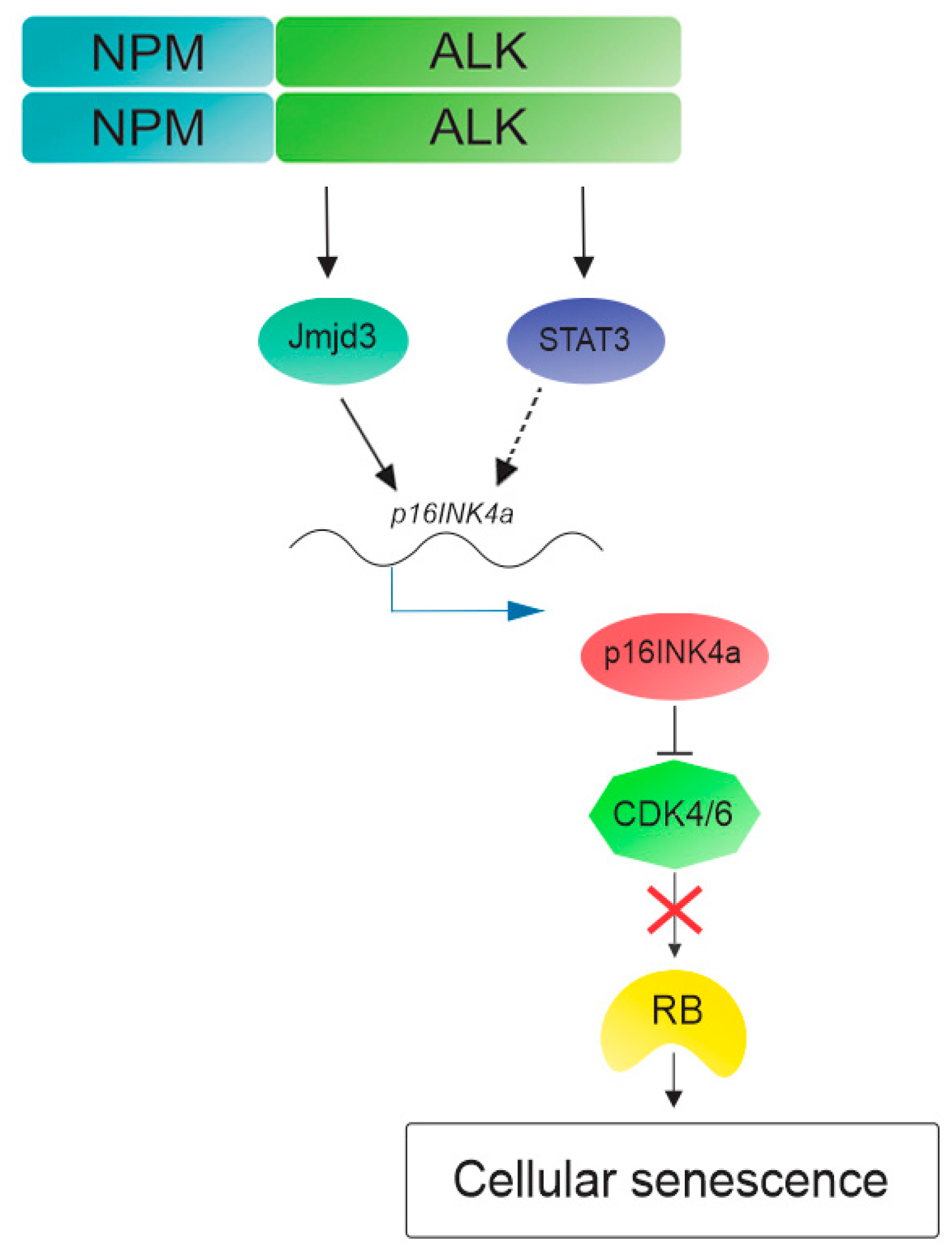
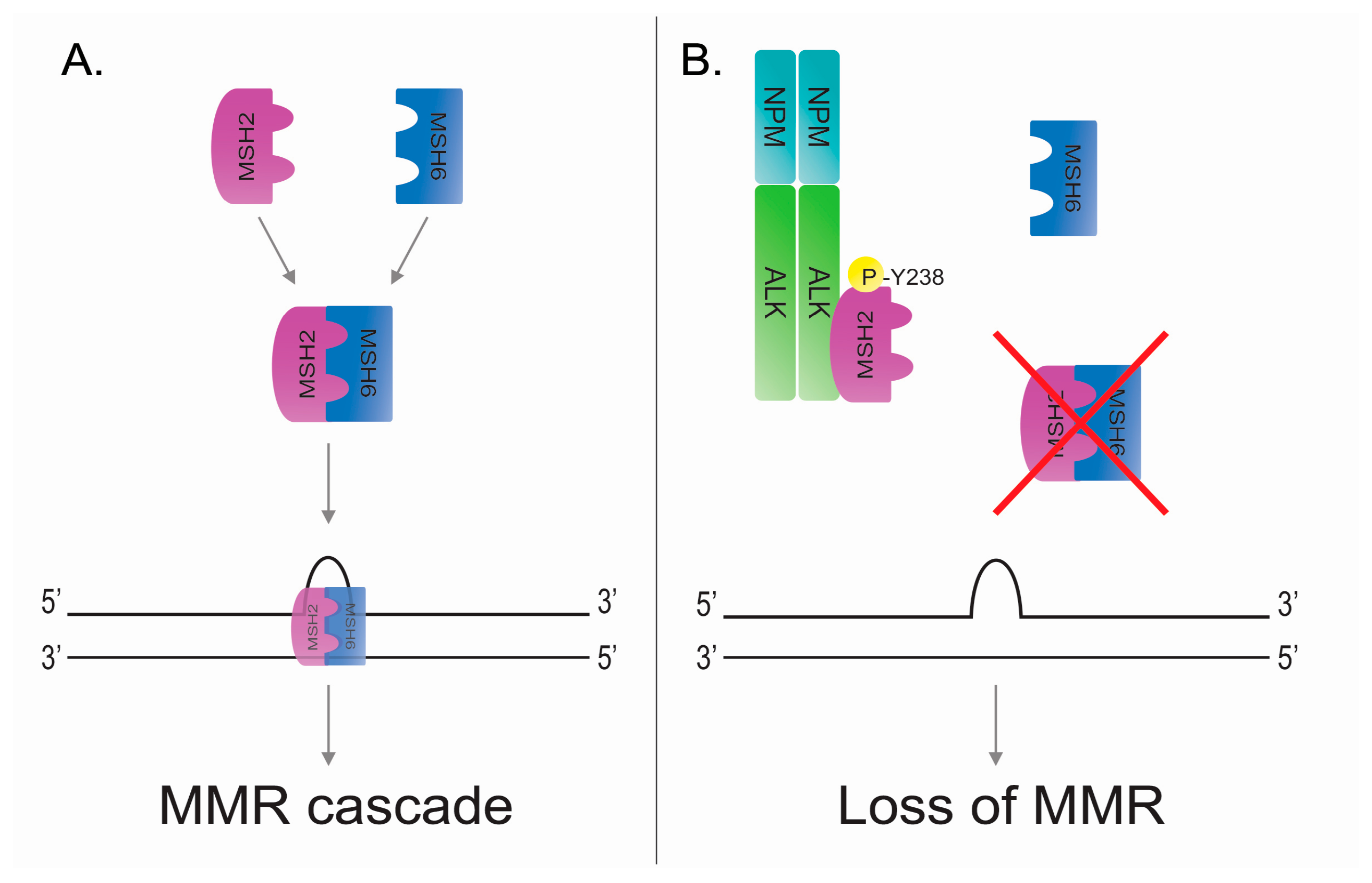
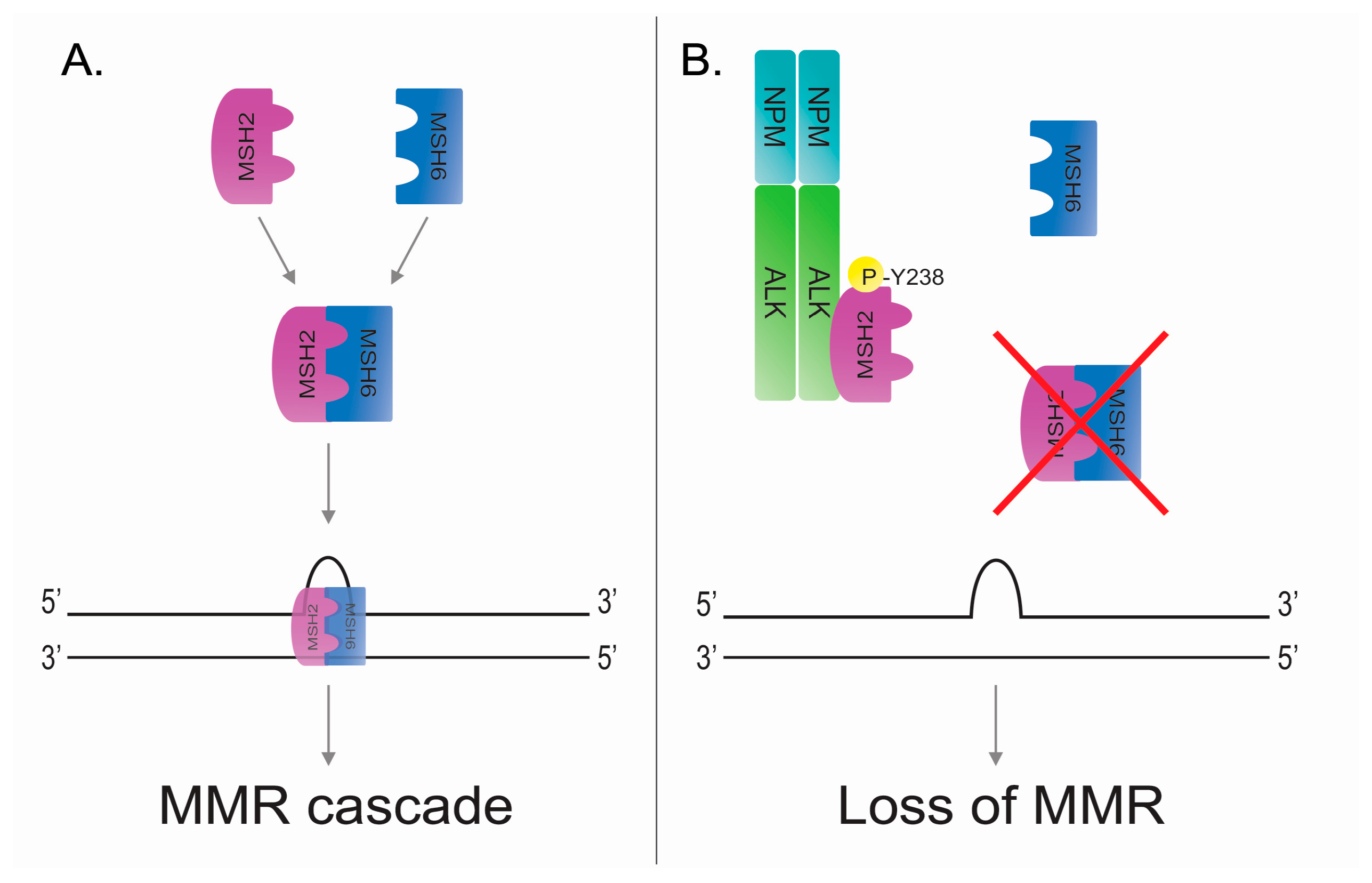
Figure 3.
NPM-ALK mediates the phosphorylation of MutS protein homolog 2 (MSH2) at tyrosine 238 leading to loss of DNA mismatch repair (MMR). (A) Physiologic activation of MMR after mismatch. MSH2 and MSH6 can interact forming a MuTSα complex and then translocating to the nucleus where the complex actives the MMR cascade. (B) Oncogenic tyrosine kinase NPM-ALK phosphorylates MSH2 at tyrosine 238 and it avoids the MSH2:MSH6 interaction and the normal activation of MMR mechanism in the presence of DNA mismatches.
Figure 3.
NPM-ALK mediates the phosphorylation of MutS protein homolog 2 (MSH2) at tyrosine 238 leading to loss of DNA mismatch repair (MMR). (A) Physiologic activation of MMR after mismatch. MSH2 and MSH6 can interact forming a MuTSα complex and then translocating to the nucleus where the complex actives the MMR cascade. (B) Oncogenic tyrosine kinase NPM-ALK phosphorylates MSH2 at tyrosine 238 and it avoids the MSH2:MSH6 interaction and the normal activation of MMR mechanism in the presence of DNA mismatches.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lobello, C.; Bikos, V.; Janikova, A.; Pospisilova, S. The Role of Oncogenic Tyrosine Kinase NPM-ALK in Genomic Instability. Cancers 2018, 10, 64. https://doi.org/10.3390/cancers10030064
AMA Style
Lobello C, Bikos V, Janikova A, Pospisilova S. The Role of Oncogenic Tyrosine Kinase NPM-ALK in Genomic Instability. Cancers. 2018; 10(3):64. https://doi.org/10.3390/cancers10030064
Chicago/Turabian StyleLobello, Cosimo, Vasilis Bikos, Andrea Janikova, and Sarka Pospisilova. 2018. "The Role of Oncogenic Tyrosine Kinase NPM-ALK in Genomic Instability" Cancers 10, no. 3: 64. https://doi.org/10.3390/cancers10030064
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.