Coming of Age for Autotaxin and Lysophosphatidate Signaling: Clinical Applications for Preventing, Detecting and Targeting Tumor-Promoting Inflammation



Abstract
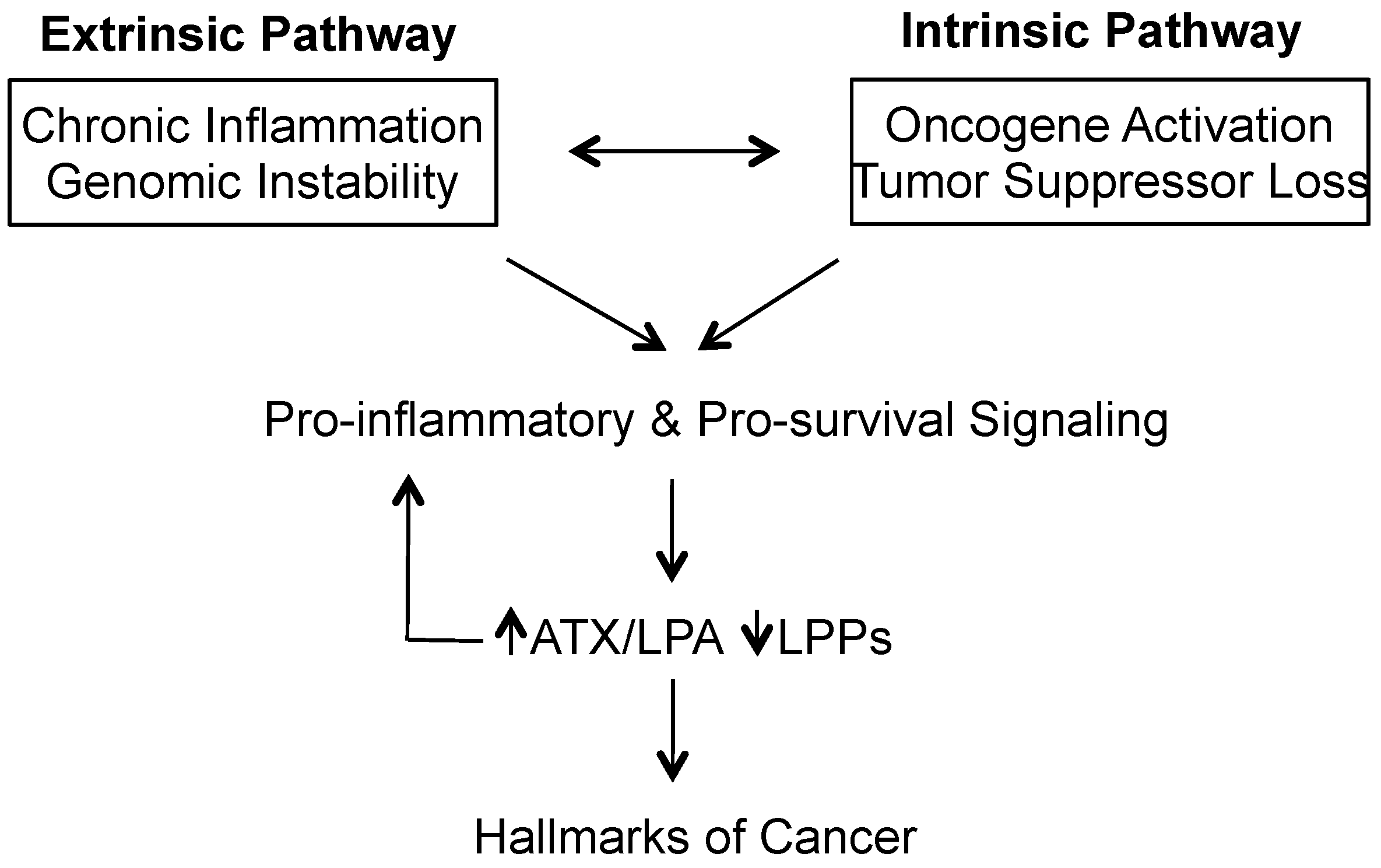
:1. Introduction—Cancer as the Ultimate Disease of Chronic Inflammation
2. Overview of LPA Signaling and Its Importance in Reproduction and Embryonic Development
3. LPA Signaling in Wound Healing and Immunity
4. Maladaptive LPA Signaling in Chronic Inflammatory Diseases
4.1. ATX and LPA in Inflamed Adipose Tissue and the Metabolic Syndrome
4.2. ATX and Neurological Disorders
4.3. ATX and Arthritis
4.4. ATX and LPA in Pulmonary Fibrosis and Asthma
4.5. ATX and LPA in Autoimmune and Retinal Diseases
4.6. ATX and LPA in Hepatic Diseases
4.7. Importance of the Site of ATX Production in Inflammatory Signaling
5. Recent Developments in LPA Signaling in Cancer Biology
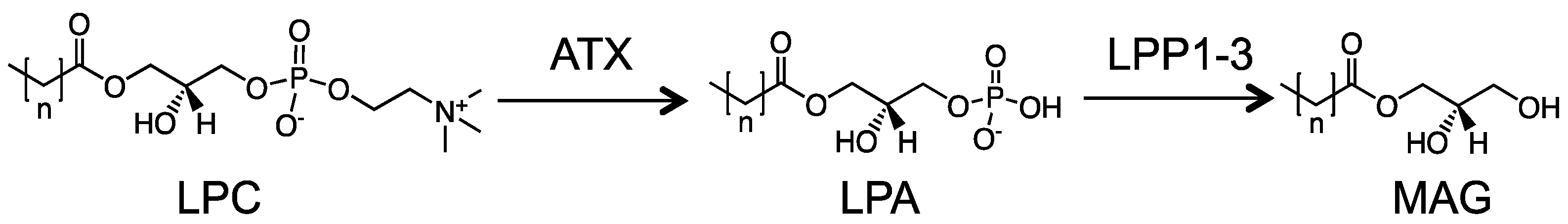
5.1. Mechanisms of ATX Overexpression in Cancers
5.2. LPA Signaling in Cancer Progression
6. The Roles of LPPs in Cancers
7. Pharmacological Targeting of LPA-Mediated Inflammation and Cancer Progression
7.1. Limiting Fibrosis through Potent ATX Inhibition
7.2. Improving Cancer Chemotherapy Effectiveness by Blocking LPA-Mediated Inflammatory Signaling
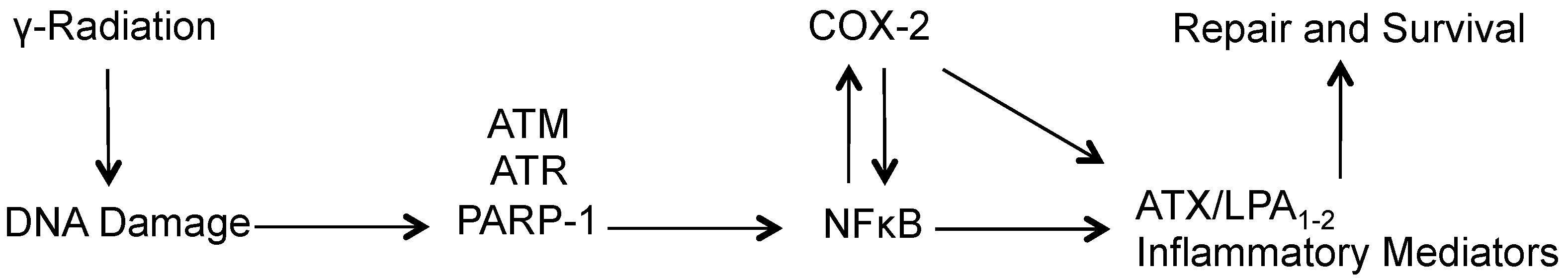
7.3. Sensitizing Tumors to Radiation Therapy by Blockage of LPA Signaling
7.4. Inhibitors of LPA Signaling Entering into Clinical Trials
8. Potential Applications for Inhibitors of LPA Signaling
9. Conclusions
Acknowledgments
Conflicts of Interest
References
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [PubMed]
- Mantovani, A.; Sica, A. Macrophages, innate immunity and cancer: Balance, tolerance, and diversity. Curr. Opin. Immunol. 2010, 22, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Zeligs, K.P.; Neuman, M.K.; Annunziata, C.M. Molecular pathways: The balance between cancer and the immune system challenges the therapeutic specificity of targeting nuclear factor-κb signaling for cancer treatment. Clin. Cancer Res. 2016, 22, 4302–4308. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Gkretsi, V.; Zacharia, L.C.; Stylianopoulos, T. Targeting inflammation to improve tumor drug delivery. Trends Cancer 2017, 3, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, J. Profile: Bert vogelstein. Cancer genetics with an edge. Science 2012, 337, 282–284. [Google Scholar] [CrossRef] [PubMed]
- Roxburgh, C.S.; McMillan, D.C. Cancer and systemic inflammation: Treat the tumour and treat the host. Br. J. Cancer 2014, 110, 1409–1412. [Google Scholar] [CrossRef] [PubMed]
- Brindley, D.N.; Benesch, M.G.K.; Murph, M.M. Autotaxin—An enzymatic augmenter of malignant progression linked to inflammation. In Melanoma—Current Clinical Management and Future Therapeutics; InTech Open: London, UK, 2015; pp. 297–324. [Google Scholar] [CrossRef]
- Benesch, M.G.; Tang, X.; Venkatraman, G.; Bekele, R.T.; Brindley, D.N. Recent advances in targeting the autotaxin-lysophosphatidate-lipid phosphate phosphatase axis in vivo. J. Biomed. Res. 2016, 30, 272–284. [Google Scholar] [PubMed]
- Benesch, M.G.K.; Yang, Z.; Tang, X.; Meng, G.; Brindley, D.N. Lysophosphatidate signaling: The tumor microenvironment’s new nemesis. Trends Cancer 2017, 3, 748–752. [Google Scholar] [CrossRef] [PubMed]
- Benesch, M.G.K.; Ko, Y.M.; McMullen, T.P.W.; Brindley, D.N. Autotaxin in the crosshairs: Taking aim at cancer and other inflammatory conditions. FEBS Lett. 2014, 588, 2712–2727. [Google Scholar] [CrossRef] [PubMed]
- Van Meeteren, L.A.; Ruurs, P.; Stortelers, C.; Bouwman, P.; van Rooijen, M.A.; Pradere, J.P.; Pettit, T.R.; Wakelam, M.J.; Saulnier-Blache, J.S.; Mummery, C.L.; et al. Autotaxin, a secreted lysophospholipase D, is essential for blood vessel formation during development. Mol. Cell. Biol. 2006, 26, 5015–5022. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Okudaira, S.; Kishi, Y.; Ohkawa, R.; Iseki, S.; Ota, M.; Noji, S.; Yatomi, Y.; Aoki, J.; Arai, H. Autotaxin stabilizes blood vessels and is required for embryonic vasculature by producing lysophosphatidic acid. J. Biol. Chem. 2006, 281, 25822–25830. [Google Scholar] [CrossRef] [PubMed]
- Gierse, J.; Thorarensen, A.; Beltey, K.; Bradshaw-Pierce, E.; Cortes-Burgos, L.; Hall, T.; Johnston, A.; Murphy, M.; Nemirovskiy, O.; Ogawa, S.; et al. A novel autotaxin inhibitor reduces lysophosphatidic acid levels in plasma and the site of inflammation. J. Pharmacol. Exp. Ther. 2010, 334, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Dusaulcy, R.; Rancoule, C.; Gres, S.; Wanecq, E.; Colom, A.; Guigne, C.; van Meeteren, L.A.; Moolenaar, W.H.; Valet, P.; Saulnier-Blache, J.S. Adipose-specific disruption of autotaxin enhances nutritional fattening and reduces plasma lysophosphatidic acid. J. Lipid Res. 2011, 52, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Quan, M.; Cui, J.J.; Feng, X.; Huang, Q. The critical role and potential target of the autotaxin/lysophosphatidate axis in pancreatic cancer. Tumour Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Oka, S.; Ota, R.; Shima, M.; Yamashita, A.; Sugiura, T. GPR35 is a novel lysophosphatidic acid receptor. Biochem. Biophys. Res. Commun. 2010, 395, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Tabata, K.; Baba, K.; Shiraishi, A.; Ito, M.; Fujita, N. The orphan GPCR GPR87 was deorphanized and shown to be a lysophosphatidic acid receptor. Biochem. Biophys. Res. Commun. 2007, 363, 861–866. [Google Scholar] [CrossRef] [PubMed]
- Ochiai, S.; Furuta, D.; Sugita, K.; Taniura, H.; Fujita, N. GPR87 mediates lysophosphatidic acid-induced colony dispersal in A431 cells. Eur. J. Pharmacol. 2013, 715, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Shiraishi, A.; Tabata, K.; Fujita, N. Identification of the orphan GPCR, P2Y(10) receptor as the sphingosine-1-phosphate and lysophosphatidic acid receptor. Biochem. Biophys. Res. Commun. 2008, 371, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Rai, V.; Toure, F.; Chitayat, S.; Pei, R.; Song, F.; Li, Q.; Zhang, J.; Rosario, R.; Ramasamy, R.; Chazin, W.J.; et al. Lysophosphatidic acid targets vascular and oncogenic pathways via rage signaling. J. Exp. Med. 2012, 209, 2339–2350. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Zhao, Y.Y.; Dewald, J.; Curtis, J.M.; Brindley, D.N. Tetracyclines increase lipid phosphate phosphatase expression on plasma membranes and turnover of plasma lysophosphatidate. J. Lipid Res. 2016, 57, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Benesch, M.G.; Brindley, D.N. Lipid phosphate phosphatases and their roles in mammalian physiology and pathology. J. Lipid Res. 2015, 56, 2048–2060. [Google Scholar] [CrossRef] [PubMed]
- Aikawa, S.; Hashimoto, T.; Kano, K.; Aoki, J. Lysophosphatidic acid as a lipid mediator with multiple biological actions. J. Biochem. 2015, 157, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Yung, Y.C.; Stoddard, N.C.; Chun, J. LPA receptor signaling: Pharmacology, physiology, and pathophysiology. J. Lipid Res. 2014, 55, 1192–1214. [Google Scholar] [CrossRef] [PubMed]
- Riaz, A.; Huang, Y.; Johansson, S. G-protein-coupled lysophosphatidic acid receptors and their regulation of AKT signaling. Int. J. Mol. Sci. 2016, 17, 215. [Google Scholar] [CrossRef] [PubMed]
- Mueller, P.; Ye, S.; Morris, A.; Smyth, S.S. Lysophospholipid mediators in the vasculature. Exp. Cell Res. 2015, 333, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Masse, K.; Bhamra, S.; Allsop, G.; Dale, N.; Jones, E.A. Ectophosphodiesterase/nucleotide phosphohydrolase (Enpp) nucleotidases: Cloning, conservation and developmental restriction. Int. J. Dev. Biol. 2010, 54, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Federico, L.; Jeong, K.J.; Vellano, C.P.; Mills, G.B. Autotaxin, a lysophospholipase D with pleomorphic effects in oncogenesis and cancer progression. J. Lipid Res. 2016, 57, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Nishimasu, H.; Okudaira, S.; Hama, K.; Mihara, E.; Dohmae, N.; Inoue, A.; Ishitani, R.; Takagi, J.; Aoki, J.; Nureki, O. Crystal structure of autotaxin and insight into GPCR activation by lipid mediators. Nat. Struct. Mol. Biol. 2011, 18, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Lyu, L.; Wang, B.; Xiong, C.; Zhang, X.; Zhang, X.; Zhang, J. Selective export of autotaxin from the endoplasmic reticulum. J. Biol. Chem. 2017, 292, 7011–7022. [Google Scholar] [CrossRef] [PubMed]
- Jansen, S.; Stefan, C.; Creemers, J.W.; Waelkens, E.; Van Eynde, A.; Stalmans, W.; Bollen, M. Proteolytic maturation and activation of autotaxin (NPP2), a secreted metastasis-enhancing lysophospholipase D. J. Cell Sci. 2005, 118, 3081–3089. [Google Scholar] [CrossRef] [PubMed]
- Tokumura, A.; Majima, E.; Kariya, Y.; Tominaga, K.; Kogure, K.; Yasuda, K.; Fukuzawa, K. Identification of human plasma lysophospholipase D, a lysophosphatidic acid-producing enzyme, as autotaxin, a multifunctional phosphodiesterase. J. Biol. Chem. 2002, 277, 39436–39442. [Google Scholar] [CrossRef] [PubMed]
- Umezu-Goto, M.; Kishi, Y.; Taira, A.; Hama, K.; Dohmae, N.; Takio, K.; Yamori, T.; Mills, G.B.; Inoue, K.; Aoki, J.; et al. Autotaxin has lysophospholipase D activity leading to tumor cell growth and motility by lysophosphatidic acid production. J. Cell Biol. 2002, 158, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Stracke, M.L.; Krutzsch, H.C.; Unsworth, E.J.; Arestad, A.; Cioce, V.; Schiffmann, E.; Liotta, L.A. Identification, purification, and partial sequence analysis of autotaxin, a novel motility-stimulating protein. J. Biol. Chem. 1992, 267, 2524–2529. [Google Scholar] [PubMed]
- Perrakis, A.; Moolenaar, W.H. Autotaxin: Structure-function and signaling. J. Lipid Res. 2014, 55, 1010–1018. [Google Scholar] [CrossRef] [PubMed]
- Tabchy, A.; Tigyi, G.; Mills, G.B. Location, location, location: A crystal-clear view of autotaxin saturating LPA receptors. Nat. Struct. Mol. Biol. 2011, 18, 117–118. [Google Scholar] [CrossRef] [PubMed]
- Aikawa, S.; Kano, K.; Inoue, A.; Aoki, J. Proliferation of mouse endometrial stromal cells in culture is highly sensitive to lysophosphatidic acid signaling. Biochem. Biophys. Res. Commun. 2017, 484, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Sinderewicz, E.; Grycmacher, K.; Boruszewska, D.; Kowalczyk-Zieba, I.; Staszkiewicz, J.; Slezak, T.; Woclawek-Potocka, I. Bovine ovarian follicular growth and development correlate with lysophosphatidic acid expression. Theriogenology 2017, 106, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sinderewicz, E.; Grycmacher, K.; Boruszewska, D.; Kowalczyk-Zieba, I.; Staszkiewicz, J.; Slezak, T.; Woclawek-Potocka, I. Expression of factors involved in apoptosis and cell survival is correlated with enzymes synthesizing lysophosphatidic acid and its receptors in granulosa cells originating from different types of bovine ovarian follicles. Reprod. Biol. Endocrinol. 2017, 15, 72. [Google Scholar] [CrossRef] [PubMed]
- Masuda, K.; Haruta, S.; Orino, K.; Kawaminami, M.; Kurusu, S. Autotaxin as a novel, tissue-remodeling-related factor in regressing corpora lutea of cycling rats. FEBS J. 2013, 280, 6600–6612. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, M.; Nagamatsu, T.; Schust, D.J.; Kawai-Iwasawa, Y.; Kawana, K.; Yamashita, T.; Osuga, Y.; Aoki, J.; Yatomi, Y.; Fujii, T. Placental autotaxin expression is diminished in women with pre-eclampsia. J. Obstet. Gynaecol. Res. 2015, 41, 1406–1411. [Google Scholar] [CrossRef] [PubMed]
- Erenel, H.; Yilmaz, N.; Cift, T.; Bulut, B.; Sozen, I.; Aslan Cetin, B.; Gezer, A.; Ekmekci, H.; Kaya, B.; Tuten, A. Maternal serum autotaxin levels in early- and late-onset preeclampsia. Hypertens. Pregnancy 2017, 36, 310–314. [Google Scholar] [CrossRef] [PubMed]
- Ferry, G.; Giganti, A.; Coge, F.; Bertaux, F.; Thiam, K.; Boutin, J.A. Functional invalidation of the autotaxin gene by a single amino acid mutation in mouse is lethal. FEBS Lett. 2007, 581, 3572–3578. [Google Scholar] [CrossRef] [PubMed]
- Fotopoulou, S.; Oikonomou, N.; Grigorieva, E.; Nikitopoulou, I.; Paparountas, T.; Thanassopoulou, A.; Zhao, Z.; Xu, Y.; Kontoyiannis, D.L.; Remboutsika, E.; et al. ATX expression and LPA signalling are vital for the development of the nervous system. Dev. Biol. 2010, 339, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Yuelling, L.W.; Waggener, C.T.; Afshari, F.S.; Lister, J.A.; Fuss, B. Autotaxin/ENPP2 regulates oligodendrocyte differentiation in vivo in the developing zebrafish hindbrain. Glia 2012, 60, 1605–1618. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, N.A.; Lister, J.A.; Fuss, B. The autotaxin-lysophosphatidic acid axis modulates histone acetylation and gene expression during oligodendrocyte differentiation. J. Neurosci. 2015, 35, 11399–11414. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.L.; Yao, W.L.; Tsao, K.C.; Houben, A.J.; Albers, H.M.; Ovaa, H.; Moolenaar, W.H.; Lee, S.J. Autotaxin/Lpar3 signaling regulates Kupffer’s vesicle formation and left-right asymmetry in zebrafish. Development 2012, 139, 4439–4448. [Google Scholar] [CrossRef] [PubMed]
- Yukiura, H.; Kano, K.; Kise, R.; Inoue, A.; Aoki, J. Autotaxin overexpression causes embryonic lethality and vascular defects. PLoS ONE 2015, 10, e0126734. [Google Scholar] [CrossRef] [PubMed]
- Escalante-Alcalde, D.; Hernandez, L.; Le Stunff, H.; Maeda, R.; Lee, H.S.; Cheng, G., Jr.; Sciorra, V.A.; Daar, I.; Spiegel, S.; Morris, A.J.; et al. The lipid phosphatase LPP3 regulates extra-embryonic vasculogenesis and axis patterning. Development 2003, 130, 4623–4637. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, I.; Baruah, J.; Lurie, E.E.; Wary, K.K. Endothelial lipid phosphate phosphatase-3 deficiency that disrupts the endothelial barrier function is a modifier of cardiovascular development. Cardiovasc. Res. 2016, 111, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Frisca, F.; Colquhoun, D.; Goldshmit, Y.; Anko, M.L.; Pebay, A.; Kaslin, J. Role of ectonucleotide pyrophosphatase/phosphodiesterase 2 in the midline axis formation of zebrafish. Sci. Rep. 2016, 6, 37678. [Google Scholar] [CrossRef] [PubMed]
- Nakanaga, K.; Hama, K.; Kano, K.; Sato, T.; Yukiura, H.; Inoue, A.; Saigusa, D.; Tokuyama, H.; Tomioka, Y.; Nishina, H.; et al. Overexpression of autotaxin, a lysophosphatidic acid-producing enzyme, enhances cardia bifida induced by hypo-sphingosine-1-phosphate signaling in zebrafish embryo. J. Biochem. 2014, 155, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Nakanaga, K.; Hama, K.; Aoki, J. Autotaxin—An LPA producing enzyme with diverse functions. J. Biochem. 2010, 148, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Knowlden, S.; Georas, S.N. The autotaxin-LPA axis emerges as a novel regulator of lymphocyte homing and inflammation. J. Immunol. 2014, 192, 851–857. [Google Scholar] [CrossRef] [PubMed]
- Ray, R.; Rai, V. Lysophosphatidic acid converts monocytes into macrophages in both mice and humans. Blood 2017, 129, 1177–1183. [Google Scholar] [CrossRef] [PubMed]
- Fulkerson, Z.; Wu, T.; Sunkara, M.; Kooi, C.V.; Morris, A.J.; Smyth, S.S. Binding of autotaxin to integrins localizes lysophosphatidic acid production to platelets and mammalian cells. J. Biol. Chem. 2011, 286, 34654–34663. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Guan, M.; Zhao, Z.; Zhang, J. Type I interferons function as autocrine and paracrine factors to induce autotaxin in response to TLR activation. PLoS ONE 2015, 10, e0136629. [Google Scholar] [CrossRef] [PubMed]
- Katakai, T.; Kondo, N.; Ueda, Y.; Kinashi, T. Autotaxin produced by stromal cells promotes LFA-1-independent and Rho-dependent interstitial T cell motility in the lymph node paracortex. J. Immunol. 2014, 193, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Knowlden, S.A.; Capece, T.; Popovic, M.; Chapman, T.J.; Rezaee, F.; Kim, M.; Georas, S.N. Regulation of T cell motility in vitro and in vivo by LPA and LPA2. PLoS ONE 2014, 9, e101655. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Kobayashi, D.; Aoi, K.; Sasaki, N.; Sugiura, Y.; Igarashi, H.; Tohya, K.; Inoue, A.; Hata, E.; Akahoshi, N.; et al. Fibroblastic reticular cell-derived lysophosphatidic acid regulates confined intranodal T-cell motility. eLife 2016, 5, e10561. [Google Scholar] [CrossRef] [PubMed]
- Kime, C.; Sakaki-Yumoto, M.; Goodrich, L.; Hayashi, Y.; Sami, S.; Derynck, R.; Asahi, M.; Panning, B.; Yamanaka, S.; Tomoda, K. Autotaxin-mediated lipid signaling intersects with LIF and BMP signaling to promote the naive pluripotency transcription factor program. Proc. Natl. Acad. Sci. USA 2016, 113, 12478–12483. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, K.; Brunkhorst, R.; de Bruin, N.; Mayer, C.A.; Haussler, A.; Ferreiros, N.; Schiffmann, S.; Parnham, M.J.; Tunaru, S.; Chun, J.; et al. Dysregulation of lysophosphatidic acids in multiple sclerosis and autoimmune encephalomyelitis. Acta Neuropathol. Commun. 2017, 5, 42. [Google Scholar] [CrossRef] [PubMed]
- Raddatz, B.B.; Hansmann, F.; Spitzbarth, I.; Kalkuhl, A.; Deschl, U.; Baumgartner, W.; Ulrich, R. Transcriptomic meta-analysis of multiple sclerosis and its experimental models. PLoS ONE 2014, 9, e86643. [Google Scholar] [CrossRef] [PubMed]
- Nogaroli, L.; Yuelling, L.M.; Dennis, J.; Gorse, K.; Payne, S.G.; Fuss, B. Lysophosphatidic acid can support the formation of membranous structures and an increase in MBP mRNA levels in differentiating oligodendrocytes. Neurochem. Res. 2009, 34, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Thirunavukkarasu, K.; Tan, B.; Swearingen, C.A.; Rocha, G.; Bui, H.H.; McCann, D.J.; Jones, S.B.; Norman, B.H.; Pfeifer, L.A.; Saha, J.K. Pharmacological characterization of a potent inhibitor of autotaxin in animal models of inflammatory bowel disease and multiple sclerosis. J. Pharmacol. Exp. Ther. 2016, 359, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, T.; Arima, N.; Kano, K.; Hama, K.; Itai, E.; Yukiura, H.; Kise, R.; Inoue, A.; Kim, S.H.; Solnica-Krezel, L.; et al. ATX-LPA1 axis contributes to proliferation of chondrocytes by regulating fibronectin assembly leading to proper cartilage formation. Sci. Rep. 2016, 6, 23433. [Google Scholar] [CrossRef] [PubMed]
- George, J.; Headen, K.V.; Ogunleye, A.O.; Perry, G.A.; Wilwerding, T.M.; Parrish, L.C.; McVaney, T.P.; Mattson, J.S.; Cerutis, D.R. Lysophosphatidic acid signals through specific lysophosphatidic acid receptor subtypes to control key regenerative responses of human gingival and periodontal ligament fibroblasts. J. Periodontol. 2009, 80, 1338–1347. [Google Scholar] [CrossRef] [PubMed]
- Cerutis, D.R.; Weston, M.D.; Ogunleye, A.O.; McVaney, T.P.; Miyamoto, T. Lysophosphatidic acid (LPA) 18:1 transcriptional regulation of primary human gingival fibroblasts. Genom. Data 2014, 2, 375–377. [Google Scholar] [CrossRef] [PubMed]
- Cerutis, D.R.; Weston, M.D.; Alnouti, Y.; Bathena, S.P.; Nunn, M.E.; Ogunleye, A.O.; McVaney, T.P.; Headen, K.V.; Miyamoto, T. A major human oral lysophosphatidic acid species, LPA 18:1, regulates novel genes in human gingival fibroblasts. J. Periodontol. 2015, 86, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Benesch, M.G.K.; Zhao, Y.Y.; Curtis, J.M.; McMullen, T.P.; Brindley, D.N. Regulation of autotaxin expression and secretion by lysophosphatidate and sphingosine 1-phosphate. J. Lipid Res. 2015, 56, 1134–1144. [Google Scholar] [CrossRef] [PubMed]
- Jansen, S.; Andries, M.; Vekemans, K.; Vanbilloen, H.; Verbruggen, A.; Bollen, M. Rapid clearance of the circulating metastatic factor autotaxin by the scavenger receptors of liver sinusoidal endothelial cells. Cancer Lett. 2009, 284, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Nikitopoulou, I.; Oikonomou, N.; Karouzakis, E.; Sevastou, I.; Nikolaidou-Katsaridou, N.; Zhao, Z.; Mersinias, V.; Armaka, M.; Xu, Y.; Masu, M.; et al. Autotaxin expression from synovial fibroblasts is essential for the pathogenesis of modeled arthritis. J. Exp. Med. 2012, 209, 925–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oikonomou, N.; Mouratis, M.A.; Tzouvelekis, A.; Kaffe, E.; Valavanis, C.; Vilaras, G.; Karameris, A.; Prestwich, G.D.; Bouros, D.; Aidinis, V. Pulmonary autotaxin expression contributes to the pathogenesis of pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2012, 47, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Kaffe, E.; Katsifa, A.; Xylourgidis, N.; Ninou, I.; Zannikou, M.; Harokopos, V.; Foka, P.; Dimitriadis, A.; Evangelou, K.; Moulas, A.N.; et al. Hepatocyte autotaxin expression promotes liver fibrosis and cancer. Hepatology 2017, 65, 1369–1383. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Cha, Y.J.; Koo, J.S. Adipocyte biology in breast cancer: From silent bystander to active facilitator. Prog. Lipid Res. 2017, 69, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Rancoule, C.; Dusaulcy, R.; Treguer, K.; Gres, S.; Guigne, C.; Quilliot, D.; Valet, P.; Saulnier-Blache, J.S. Depot-specific regulation of autotaxin with obesity in human adipose tissue. J. Physiol. Biochem. 2012, 68, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, R.; Okura, T.; Fujioka, Y.; Sumi, K.; Matsuzawa, K.; Izawa, S.; Ueta, E.; Kato, M.; Taniguchi, S.I.; Yamamoto, K. Serum fatty acid-binding protein 4 (FABP4) concentration is associated with insulin resistance in peripheral tissues, a clinical study. PLoS ONE 2017, 12, e0179737. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, K.; Kane, D.A.; Touaibia, M.; Kershaw, E.E.; Pulinilkunnil, T.; Kienesberger, P.C. Autotaxin is regulated by glucose and insulin in adipocytes. Endocrinology 2017, 158, 791–803. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Jaramillo, P.; Gomez-Arbelaez, D.; Lopez-Lopez, J.; Lopez-Lopez, C.; Martinez-Ortega, J.; Gomez-Rodriguez, A.; Triana-Cubillos, S. The role of leptin/adiponectin ratio in metabolic syndrome and diabetes. Horm. Mol. Biol. Clin. Investig. 2014, 18, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Stern, J.H.; Rutkowski, J.M.; Scherer, P.E. Adiponectin, leptin, and fatty acids in the maintenance of metabolic homeostasis through adipose tissue crosstalk. Cell Metab. 2016, 23, 770–784. [Google Scholar] [CrossRef] [PubMed]
- Fruhbeck, G.; Catalan, V.; Rodriguez, A.; Ramirez, B.; Becerril, S.; Salvador, J.; Portincasa, P.; Colina, I.; Gomez-Ambrosi, J. Involvement of the leptin-adiponectin axis in inflammation and oxidative stress in the metabolic syndrome. Sci. Rep. 2017, 7, 6619. [Google Scholar] [CrossRef] [PubMed]
- Reeves, V.L.; Trybula, J.S.; Wills, R.C.; Goodpaster, B.H.; Dube, J.J.; Kienesberger, P.C.; Kershaw, E.E. Serum Autotaxin/ENPP2 correlates with insulin resistance in older humans with obesity. Obesity 2015, 23, 2371–2376. [Google Scholar] [CrossRef] [PubMed]
- Federico, L.; Ren, H.; Mueller, P.A.; Wu, T.; Liu, S.; Popovic, J.; Blalock, E.M.; Sunkara, M.; Ovaa, H.; Albers, H.M.; et al. Autotaxin and its product lysophosphatidic acid suppress brown adipose differentiation and promote diet-induced obesity in mice. Mol. Endocrinol. 2012, 26, 786–797. [Google Scholar] [CrossRef] [PubMed]
- Fayyaz, S.; Japtok, L.; Schumacher, F.; Wigger, D.; Schulz, T.J.; Haubold, K.; Gulbins, E.; Voller, H.; Kleuser, B. Lysophosphatidic acid inhibits insulin signaling in primary rat hepatocytes via the LPA3 receptor subtype and is increased in obesity. Cell. Physiol. Biochem. 2017, 43, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Wang, R.; Song, J.; Guan, M.; Li, N.; Zhang, X.; Zhao, Z.; Zhang, J. Blocking gp130 signaling suppresses autotaxin expression in adipocytes and improves insulin sensitivity in diet-induced obesity. J. Lipid Res. 2017, 58, 2102–2113. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, S.; Nagasaki, M.; Okudaira, S.; Aoki, J.; Ohmori, T.; Ohkawa, R.; Nakamura, K.; Igarashi, K.; Yamashita, H.; Eto, K.; et al. ENPP2 contributes to adipose tissue expansion and insulin resistance in diet-induced obesity. Diabetes 2014, 63, 4154–4164. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Furuichi, K.; Toyama, T.; Yamahana, J.; Ohkawa, R.; Igarashi, K.; Aoki, J.; Kaneko, S.; Yatomi, Y.; Wada, T. Serum autotaxin levels are associated with proteinuria and kidney lesions in japanese type 2 diabetic patients with biopsy-proven diabetic nephropathy. Intern. Med. 2016, 55, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Valdes-Rives, S.A.; Gonzalez-Arenas, A. Autotaxin-lysophosphatidic acid: From inflammation to cancer development. Mediat. Inflamm. 2017, 2017, 9173090. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Wang, F.; Zhao, Z.; Wang, H.; Cong, X.; Chen, X. Lysophosphatidic acid is associated with atherosclerotic plaque instability by regulating NF-κb dependent matrix metalloproteinase-9 expression via LPA2 in macrophages. Front. Physiol. 2017, 8, 266. [Google Scholar] [CrossRef] [PubMed]
- Nsaibia, M.J.; Mahmut, A.; Boulanger, M.C.; Arsenault, B.J.; Bouchareb, R.; Simard, S.; Witztum, J.L.; Clavel, M.A.; Pibarot, P.; Bosse, Y.; et al. Autotaxin interacts with lipoprotein(a) and oxidized phospholipids in predicting the risk of calcific aortic valve stenosis in patients with coronary artery disease. J. Intern. Med. 2016, 280, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, P.; Arsenault, B.J.; Boulanger, M.C.; Bosse, Y.; Koschinsky, M.L. Pathobiology of Lp(a) in calcific aortic valve disease. Expert Rev. Cardiovasc. Ther. 2017, 15, 797–807. [Google Scholar] [CrossRef] [PubMed]
- Bouchareb, R.; Mahmut, A.; Nsaibia, M.J.; Boulanger, M.C.; Dahou, A.; Lepine, J.L.; Laflamme, M.H.; Hadji, F.; Couture, C.; Trahan, S.; et al. Autotaxin derived from lipoprotein(a) and valve interstitial cells promotes inflammation and mineralization of the aortic valve. Circulation 2015, 132, 677–690. [Google Scholar] [CrossRef] [PubMed]
- Smyth, S.S.; Mueller, P.; Yang, F.; Brandon, J.A.; Morris, A.J. Arguing the case for the autotaxin-lysophosphatidic acid-lipid phosphate phosphatase 3-signaling nexus in the development and complications of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Touat-Hamici, Z.; Weidmann, H.; Blum, Y.; Proust, C.; Durand, H.; Iannacci, F.; Codoni, V.; Gaignard, P.; Therond, P.; Civelek, M.; et al. Role of lipid phosphate phosphatase 3 in human aortic endothelial cell function. Cardiovasc. Res. 2016, 112, 702–713. [Google Scholar] [CrossRef] [PubMed]
- Panchatcharam, M.; Salous, A.K.; Brandon, J.; Miriyala, S.; Wheeler, J.; Patil, P.; Sunkara, M.; Morris, A.J.; Escalante-Alcalde, D.; Smyth, S.S. Mice with targeted inactivation of Ppap2b in endothelial and hematopoietic cells display enhanced vascular inflammation and permeability. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 837–845. [Google Scholar] [CrossRef] [PubMed]
- Parimisetty, A.; Dorsemans, A.C.; Awada, R.; Ravanan, P.; Diotel, N.; Lefebvre d’Hellencourt, C. Secret talk between adipose tissue and central nervous system via secreted factors-an emerging frontier in the neurodegenerative research. J. Neuroinflamm. 2016, 13, 67. [Google Scholar] [CrossRef] [PubMed]
- McLimans, K.E.; Willette, A.A. Autotaxin is related to metabolic dysfunction and predicts alzheimer’s disease outcomes. J. Alzheimers Dis. 2017, 56, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Velasco, M.; O’Sullivan, C.; Sheridan, G.K. Lysophosphatidic acid receptors (LPARs): Potential targets for the treatment of neuropathic pain. Neuropharmacology 2017, 113, 608–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDougall, J.J.; Albacete, S.; Schuelert, N.; Mitchell, P.G.; Lin, C.; Oskins, J.L.; Bui, H.H.; Chambers, M.G. Lysophosphatidic acid provides a missing link between osteoarthritis and joint neuropathic pain. Osteoarthr. Cartil. 2017, 25, 926–934. [Google Scholar] [CrossRef] [PubMed]
- Herr, D.R.; Ong, J.H.; Ong, W.Y. Potential therapeutic applications for inhibitors of autotaxin, a bioactive lipid-producing lysophospholipase D, in disorders affecting the nervous system. ACS Chem. Neurosci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Mabey, T.; Taleongpong, P.; Udomsinprasert, W.; Jirathanathornnukul, N.; Honsawek, S. Plasma and synovial fluid autotaxin correlate with severity in knee osteoarthritis. Clin. Chim. Acta 2015, 444, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Datta, P.; Zhang, Y.; Parousis, A.; Sharma, A.; Rossomacha, E.; Endisha, H.; Wu, B.; Kacprzak, I.; Mahomed, N.N.; Gandhi, R.; et al. High-fat diet-induced acceleration of osteoarthritis is associated with a distinct and sustained plasma metabolite signature. Sci. Rep. 2017, 7, 8205. [Google Scholar] [CrossRef] [PubMed]
- Orosa, B.; Garcia, S.; Conde, C. The autotaxin-lysophosphatidic acid pathway in pathogenesis of rheumatoid arthritis. Eur. J. Pharmacol. 2015, 765, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.; Wei, X.; Lu, S.; He, P. Autotaxin-LPA receptor axis in the pathogenesis of lung diseases. Int. J. Clin. Exp. Med. 2015, 8, 17117–17122. [Google Scholar] [PubMed]
- Tager, A.M.; LaCamera, P.; Shea, B.S.; Campanella, G.S.; Selman, M.; Zhao, Z.; Polosukhin, V.; Wain, J.; Karimi-Shah, B.A.; Kim, N.D.; et al. The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat. Med. 2008, 14, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Black, K.E.; Berdyshev, E.; Bain, G.; Castelino, F.V.; Shea, B.S.; Probst, C.K.; Fontaine, B.A.; Bronova, I.; Goulet, L.; Lagares, D.; et al. Autotaxin activity increases locally following lung injury, but is not required for pulmonary lysophosphatidic acid production or fibrosis. FASEB J. 2016, 30, 2435–2450. [Google Scholar] [CrossRef] [PubMed]
- Cao, P.; Aoki, Y.; Badri, L.; Walker, N.M.; Manning, C.M.; Lagstein, A.; Fearon, E.R.; Lama, V.N. Autocrine lysophosphatidic acid signaling activates β-catenin and promotes lung allograft fibrosis. J. Clin. Investig. 2017, 127, 1517–1530. [Google Scholar] [CrossRef] [PubMed]
- Nowak-Machen, M.; Lange, M.; Exley, M.; Wu, S.; Usheva, A.; Robson, S.C. Lysophosphatidic acid generation by pulmonary NKT cell ENPP-2/autotaxin exacerbates hyperoxic lung injury. Purinergic Signal. 2015, 11, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, S.J.; Park, G.Y.; Christman, J.W.; Nyenhuis, S.; Berdyshev, E.; Natarajan, V. Polyunsaturated lysophosphatidic acid as a potential asthma biomarker. Biomark. Med. 2016, 10, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Kim, D.; Lee, S.M.; Jun, H.S. Inhibition of lysophosphatidic acid receptor ameliorates Sjogren’s syndrome in NOD mice. Oncotarget 2017, 8, 27240–27251. [Google Scholar] [PubMed]
- Moisseiev, E.; Loewenstein, A. Diabetic macular edema: Emerging strategies and treatment algorithms. Dev. Ophthalmol. 2017, 60, 165–174. [Google Scholar] [PubMed]
- Dacheva, I.; Ullmer, C.; Ceglowska, K.; Nogoceke, E.; Hartmann, G.; Muller, S.; Rejdak, R.; Nowomiejska, K.; Reich, M.; Nobl, M.; et al. Lysophosphatidic acids and autotaxin in retinal vein occlusion. Retina 2016, 36, 2311–2318. [Google Scholar] [CrossRef] [PubMed]
- Udomsinprasert, W.; Honsawek, S.; Anomasiri, W.; Chongsrisawat, V.; Vejchapipat, P.; Poovorawan, Y. Serum autotaxin levels correlate with hepatic dysfunction and severity in postoperative biliary atresia. Biomarkers 2015, 20, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Udomsinprasert, W.; Kitkumthorn, N.; Mutirangura, A.; Chongsrisawat, V.; Poovorawan, Y.; Honsawek, S. Association between promoter hypomethylation and overexpression of autotaxin with outcome parameters in biliary atresia. PLoS ONE 2017, 12, e0169306. [Google Scholar] [CrossRef] [PubMed]
- Erstad, D.J.; Tager, A.M.; Hoshida, Y.; Fuchs, B.C. The autotaxin-lysophosphatidic acid pathway emerges as a therapeutic target to prevent liver cancer. Mol. Cell. Oncol. 2017, 4, e1311827. [Google Scholar] [CrossRef] [PubMed]
- Mazzocca, A.; Dituri, F.; De Santis, F.; Filannino, A.; Lopane, C.; Betz, R.C.; Li, Y.Y.; Mukaida, N.; Winter, P.; Tortorella, C.; et al. Lysophosphatidic acid receptor LPAR6 supports the tumorigenicity of hepatocellular carcinoma. Cancer Res. 2015, 75, 532–543. [Google Scholar] [CrossRef] [PubMed]
- Rachakonda, V.P.; Reeves, V.L.; Aljammal, J.; Wills, R.C.; Trybula, J.S.; DeLany, J.P.; Kienesberger, P.C.; Kershaw, E.E. Serum autotaxin is independently associated with hepatic steatosis in women with severe obesity. Obesity 2015, 23, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhang, W.; Evans, J.F.; Floreani, A.; Zou, Z.; Nishio, Y.; Qi, R.; Leung, P.S.; Bowlus, C.L.; Gershwin, M.E. Autotaxin, pruritus and primary biliary cholangitis (PBC). Autoimmun. Rev. 2016, 15, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, H.; Yatomi, Y. Autotaxin in liver fibrosis. Clin. Chim. Acta 2012, 413, 1817–1821. [Google Scholar] [CrossRef] [PubMed]
- Wunsch, E.; Krawczyk, M.; Milkiewicz, M.; Trottier, J.; Barbier, O.; Neurath, M.F.; Lammert, F.; Kremer, A.E.; Milkiewicz, P. Serum autotaxin is a marker of the severity of liver injury and overall survival in patients with cholestatic liver diseases. Sci. Rep. 2016, 6, 30847. [Google Scholar] [CrossRef] [PubMed]
- Ringelhan, M.; McKeating, J.A.; Protzer, U. Viral hepatitis and liver cancer. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372. [Google Scholar] [CrossRef]
- Ikeda, H.; Kobayashi, M.; Kumada, H.; Enooku, K.; Koike, K.; Kurano, M.; Sato, M.; Nojiri, T.; Kobayashi, T.; Ohkawa, R.; et al. Annals express: Performance of autotaxin as a serum marker of liver fibrosis. Ann. Clin. Biochem. 2017. [Google Scholar] [CrossRef]
- Joshita, S.; Ichikawa, Y.; Umemura, T.; Usami, Y.; Sugiura, A.; Shibata, S.; Yamazaki, T.; Fujimori, N.; Komatsu, M.; Matsumoto, A.; et al. Serum autotaxin is a useful liver fibrosis marker in patients with chronic hepatitis B virus infection. Hepatol. Res. 2018, 48, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Joshita, S.; Umemura, T.; Usami, Y.; Sugiura, A.; Fujimori, N.; Shibata, S.; Ichikawa, Y.; Komatsu, M.; Matsumoto, A.; et al. Association of serum autotaxin levels with liver fibrosis in patients with chronic hepatitis C. Sci. Rep. 2017, 7, 46705. [Google Scholar] [CrossRef] [PubMed]
- Farquhar, M.J.; Humphreys, I.S.; Rudge, S.A.; Wilson, G.K.; Bhattacharya, B.; Ciaccia, M.; Hu, K.; Zhang, Q.; Mailly, L.; Reynolds, G.M.; et al. Autotaxin-lysophosphatidic acid receptor signalling regulates hepatitis C virus replication. J. Hepatol. 2017, 66, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Kostadinova, L.; Shive, C.L.; Judge, C.; Zebrowski, E.; Compan, A.; Rife, K.; Hirsch, A.; Falck-Ytter, Y.; Schlatzer, D.M.; Li, X.; et al. During Hepatitis C Virus (HCV) infection and HCV-HIV coinfection, an elevated plasma level of autotaxin is associated with lysophosphatidic acid and markers of immune activation that normalize during interferon-free HCV therapy. J. Infect. Dis. 2016, 214, 1438–1448. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, N.; Friedman, S.L.; Goossens, N.; Hoshida, Y. Risk factors and prevention of hepatocellular carcinoma in the era of precision medicine. J. Hepatol. 2018, 68, 526–549. [Google Scholar] [CrossRef] [PubMed]
- Hausmann, J.; Kamtekar, S.; Christodoulou, E.; Day, J.E.; Wu, T.; Fulkerson, Z.; Albers, H.M.; van Meeteren, L.A.; Houben, A.J.; van Zeijl, L.; et al. Structural basis of substrate discrimination and integrin binding by autotaxin. Nat. Struct. Mol. Biol. 2011, 18, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, S.; Wang, W.; Ning, B.F.; Chen, F.; Shen, W.; Ding, J.; Chen, W.; Xie, W.F.; Zhang, X. Cancer-associated fibroblasts promote hepatocellular carcinoma metastasis through chemokine-activated hedgehog and TGF-β pathways. Cancer Lett. 2016, 379, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Benesch, M.G.K.; Tang, X.; Dewald, J.; Dong, W.F.; Mackey, J.R.; Hemmings, D.G.; McMullen, T.P.; Brindley, D.N. Tumor-induced inflammation in mammary adipose tissue stimulates a vicious cycle of autotaxin expression and breast cancer progression. FASEB J. 2015, 29, 3990–4000. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Kooi, C.V.; Shah, P.; Charnigo, R.; Huang, C.; Smyth, S.S.; Morris, A.J. Integrin-mediated cell surface recruitment of autotaxin promotes persistent directional cell migration. FASEB J. 2014, 28, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Barbayianni, E.; Kaffe, E.; Aidinis, V.; Kokotos, G. Autotaxin, a secreted lysophospholipase D, as a promising therapeutic target in chronic inflammation and cancer. Prog. Lipid Res. 2015, 58, 76–96. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Hogberg, J.; Stenius, U. ATM-activated autotaxin (ATX) propagates inflammation and DNA damage in lung epithelial cells; a new mode of action for silica-induced DNA damage? Carcinogenesis 2017, 38, 1196–1206. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Zhang, X.; Lyu, L.; Li, X.; Yao, S.; Zhang, J. Autotaxin expression is regulated at the post-transcriptional level by the RNA-binding proteins HuR and AUF1. J. Biol. Chem. 2016, 291, 25823–25836. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Zhou, Y.; Zheng, W.; Chambers, S.K. Hur expression in the nucleus correlates with high histological grade and poor disease-free survival in ovarian cancer. Aust. N. Z. J. Obstet. Gynaecol. 2009, 49, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Wang, B.; Bi, J.; Zhang, C.; Guo, Y.; Chu, H.; Liang, X.; Zhong, C.; Wang, J. Cytoplasmic HuR expression correlates with P-gp, HER-2 positivity, and poor outcome in breast cancer. Tumour Biol. 2013, 34, 2299–2308. [Google Scholar] [CrossRef] [PubMed]
- Benesch, M.G.K.; Ko, Y.M.; Tang, X.; Dewald, J.; Lopez-Campistrous, A.; Zhao, Y.Y.; Lai, R.; Curtis, J.M.; Brindley, D.N.; McMullen, T.P. Autotaxin is an inflammatory mediator and therapeutic target in thyroid cancer. Endocr. Relat. Cancer 2015, 22, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Baldan, F.; Mio, C.; Allegri, L.; Conzatti, K.; Toffoletto, B.; Puppin, C.; Radovic, S.; Vascotto, C.; Russo, D.; Di Loreto, C.; et al. Identification of tumorigenesis-related mRNAs associated with RNA-binding protein HuR in thyroid cancer cells. Oncotarget 2016, 7, 63388–63407. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Umezu-Goto, M.; Murph, M.; Lu, Y.; Liu, W.; Zhang, F.; Yu, S.; Stephens, L.C.; Cui, X.; Murrow, G.; et al. Expression of autotaxin and lysophosphatidic acid receptors increases mammary tumorigenesis, invasion, and metastases. Cancer Cell 2009, 15, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Benesch, M.G.K.; Tang, X.; Maeda, T.; Ohhata, A.; Zhao, Y.Y.; Kok, B.P.C.; Dewald, J.; Hitt, M.; Curtis, J.M.; McMullen, T.P.W.; et al. Inhibition of autotaxin delays breast tumor growth and lung metastasis in mice. FASEB J. 2014, 28, 2655–2666. [Google Scholar] [CrossRef] [PubMed]
- Volden, P.A.; Skor, M.N.; Johnson, M.B.; Singh, P.; Patel, F.N.; McClintock, M.K.; Brady, M.J.; Conzen, S.D. Mammary adipose tissue-derived lysophospholipids promote estrogen receptor-negative mammary epithelial cell proliferation. Cancer Prev. Res. 2016, 9, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, S.; Johnson, A.R.; Makowski, L. Obesity, metabolism and the microenvironment: Links to cancer. J. Carcinog. 2013, 12, 19. [Google Scholar] [CrossRef] [PubMed]
- De Pergola, G.; Silvestris, F. Obesity as a major risk factor for cancer. J. Obes. 2013, 2013, 291546. [Google Scholar] [CrossRef] [PubMed]
- Memet, I.; Tsalkidou, E.; Tsaroucha, A.K.; Lambropoulou, M.; Chatzaki, E.; Trypsianis, G.; Schizas, D.; Pitiakoudis, M.; Simopoulos, C. Autotaxin expression in hepatocellular carcinoma. J. Investig. Surg. 2017, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Enooku, K.; Uranbileg, B.; Ikeda, H.; Kurano, M.; Sato, M.; Kudo, H.; Maki, H.; Koike, K.; Hasegawa, K.; Kokudo, N.; et al. Higher LPA2 and LPA6 mRNA levels in hepatocellular carcinoma are associated with poorer differentiation, microvascular invasion and earlier recurrence with higher serum autotaxin levels. PLoS ONE 2016, 11, e0161825. [Google Scholar] [CrossRef] [PubMed]
- Lopane, C.; Agosti, P.; Gigante, I.; Sabbà, C.; Mazzocca, A. Implications of the lysophosphatidic acid signaling axis in liver cancer. Biochim. Biophys. Acta 2017, 1868, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Nakai, Y.; Ikeda, H.; Nakamura, K.; Kume, Y.; Fujishiro, M.; Sasahira, N.; Hirano, K.; Isayama, H.; Tada, M.; Kawabe, T.; et al. Specific increase in serum autotaxin activity in patients with pancreatic cancer. Clin. Biochem. 2011, 44, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Kadekar, S.; Silins, I.; Korhonen, A.; Dreij, K.; Al-Anati, L.; Hogberg, J.; Stenius, U. Exocrine pancreatic carcinogenesis and autotaxin expression. PLoS ONE 2012, 7, e43209. [Google Scholar] [CrossRef] [PubMed]
- Emoto, S.; Kurano, M.; Kano, K.; Matsusaki, K.; Yamashita, H.; Nishikawa, M.; Igarashi, K.; Ikeda, H.; Aoki, J.; Kitayama, J.; et al. Analysis of glycero-lysophospholipids in gastric cancerous ascites. J. Lipid Res. 2017, 58, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Muinao, T.; Deka Boruah, H.P.; Pal, M. Diagnostic and prognostic biomarkers in ovarian cancer and the potential roles of cancer stem cells—An updated review. Exp. Cell Res. 2018, 362, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Seo, E.J.; Kwon, Y.W.; Jang, I.H.; Kim, D.K.; Lee, S.I.; Choi, E.J.; Kim, K.H.; Suh, D.S.; Lee, J.H.; Choi, K.U.; et al. Autotaxin regulates maintenance of ovarian cancer stem cells through lysophosphatidic acid-mediated autocrine mechanism. Stem Cells 2016, 34, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Wasniewski, T.; Woclawek-Potocka, I.; Boruszewska, D.; Kowalczyk-Zieba, I.; Sinderewicz, E.; Grycmacher, K. The significance of the altered expression of lysophosphatidic acid receptors, autotaxin and phospholipase A2 as the potential biomarkers in type 1 endometrial cancer biology. Oncol. Rep. 2015, 34, 2760–2767. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Fukushima, K.; Fukushima, N.; Honoki, K.; Tsujiuchi, T. Enhanced cellular functions through induction of LPA2 by cisplatin in fibrosarcoma HT1080 cells. Mol. Cell. Biochem. 2017, 431, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Kanehira, M.; Fujiwara, T.; Nakajima, S.; Okitsu, Y.; Onishi, Y.; Fukuhara, N.; Ichinohasama, R.; Okada, Y.; Harigae, H. A lysophosphatidic acid receptors 1 and 3 axis governs cellular senescence of mesenchymal stromal cells and promotes growth and vascularization of multiple myeloma. Stem Cells 2017, 35, 739–753. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, S.; Mikami, S.; Sugino, H.; Yoshikawa, A.; Hashimoto, A.; Onodera, Y.; Furukawa, S.; Handa, H.; Oikawa, T.; Okada, Y.; et al. Lysophosphatidic acid activates Arf6 to promote the mesenchymal malignancy of renal cancer. Nat. Commun. 2016, 7, 10656. [Google Scholar] [CrossRef] [PubMed]
- Braeuer, R.R.; Zigler, M.; Kamiya, T.; Dobroff, A.S.; Huang, L.; Choi, W.; McConkey, D.J.; Shoshan, E.; Mobley, A.K.; Song, R.; et al. Galectin-3 contributes to melanoma growth and metastasis via regulation of NFAT1 and autotaxin. Cancer Res. 2012, 72, 5757–5766. [Google Scholar] [CrossRef] [PubMed]
- Shoshan, E.; Braeuer, R.R.; Kamiya, T.; Mobley, A.K.; Huang, L.; Vasquez, M.E.; Velazquez-Torres, G.; Chakravarti, N.; Ivan, C.; Prieto, V.; et al. NFAT1 directly regulates IL8 and MMP3 to promote melanoma tumor growth and metastasis. Cancer Res. 2016, 76, 3145–3155. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Fujiwara, Y.; Liu, J.; Yue, J.; Shimizu, Y.; Norman, D.D.; Wang, Y.; Tsukahara, R.; Szabo, E.; Patil, R.; et al. Autotaxin and LPA1 and LPA5 receptors exert disparate functions in tumor cells versus the host tissue microenvironment in melanoma invasion and metastasis. Mol. Cancer Res. 2015, 13, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Fujiwara, Y.; Tigyi, G.J. Uncovering unique roles of LPA receptors in the tumor microenvironment. Recept. Clin. Investig. 2015, 2. [Google Scholar] [CrossRef]
- Tang, X.; Benesch, M.G.; Dewald, J.; Zhao, Y.Y.; Patwardhan, N.; Santos, W.L.; Curtis, J.M.; McMullen, T.P.; Brindley, D.N. Lipid phosphate phosphatase-1 expression in cancer cells attenuates tumor growth and metastasis in mice. J. Lipid Res. 2014, 55, 2389–2400. [Google Scholar] [CrossRef] [PubMed]
- Tanyi, J.L.; Morris, A.J.; Wolf, J.K.; Fang, X.; Hasegawa, Y.; Lapushin, R.; Auersperg, N.; Sigal, Y.J.; Newman, R.A.; Felix, E.A.; et al. The human lipid phosphate phosphatase-3 decreases the growth, survival, and tumorigenesis of ovarian cancer cells: Validation of the lysophosphatidic acid signaling cascade as a target for therapy in ovarian cancer. Cancer Res. 2003, 63, 1073–1082. [Google Scholar] [PubMed]
- Mao, X.Y.; Lee, M.J.; Zhu, J.; Zhu, C.; Law, S.M.; Snijders, A.M. Genome-wide screen identifies a novel prognostic signature for breast cancer survival. Oncotarget 2017, 8, 14003–14016. [Google Scholar] [CrossRef] [PubMed]
- Morris, K.E.; Schang, L.M.; Brindley, D.N. Lipid phosphate phosphatase-2 activity regulates S-phase entry of the cell cycle in Rat2 fibroblasts. J. Biol. Chem. 2006, 281, 9297–9306. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, J.M.; Funes, J.M.; Henderson, S.; Wild, L.; Carey, N.; Boshoff, C. Genomics screen in transformed stem cells reveals RNASEH2A, PPAP2C, and ADARB1 as putative anticancer drug targets. Mol. Cancer Ther. 2009, 8, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Wang, X.; Zhao, Y.Y.; Curtis, J.M.; Brindley, D.N. Doxycycline attenuates breast cancer related inflammation by decreasing plasma lysophosphatidate concentrations and inhibiting NF-κb activation. Mol. Cancer 2017, 16, 36. [Google Scholar] [CrossRef] [PubMed]
- Nikolaou, A.; Kokotou, M.G.; Limnios, D.; Psarra, A.; Kokotos, G. Autotaxin inhibitors: A patent review (2012–2016). Expert Opin. Ther. Pat. 2017, 27, 815–829. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.; Cheasty, A.; Foxton, C.; Raynham, T.; Farooq, M.; Gutierrez, I.F.; Lejeune, A.; Pritchard, M.; Turnbull, A.; Pang, L.; et al. Discovery of potent inhibitors of the lysophospholipase autotaxin. Bioorg. Med. Chem. Lett. 2016, 26, 5403–5410. [Google Scholar] [CrossRef] [PubMed]
- Llona-Minguez, S.; Ghassemian, A.; Helleday, T. Lysophosphatidic acid receptor (LPAR) modulators: The current pharmacological toolbox. Prog. Lipid Res. 2015, 58, 51–75. [Google Scholar] [CrossRef] [PubMed]
- Katsifa, A.; Kaffe, E.; Nikolaidou-Katsaridou, N.; Economides, A.N.; Newbigging, S.; McKerlie, C.; Aidinis, V. The bulk of autotaxin activity is dispensable for adult mouse life. PLoS ONE 2015, 10, e0143083. [Google Scholar] [CrossRef] [PubMed]
- Castelino, F.V.; Bain, G.; Pace, V.A.; Black, K.E.; George, L.; Probst, C.K.; Goulet, L.; Lafyatis, R.; Tager, A.M. An autotaxin/lysophosphatidic acid/interleukin-6 amplification loop drives scleroderma fibrosis. Arthritis Rheumatol. 2016, 68, 2964–2974. [Google Scholar] [CrossRef] [PubMed]
- Castelino, F.V.; Seiders, J.; Bain, G.; Brooks, S.F.; King, C.D.; Swaney, J.S.; Lorrain, D.S.; Chun, J.; Luster, A.D.; Tager, A.M. Amelioration of dermal fibrosis by genetic deletion or pharmacologic antagonism of lysophosphatidic acid receptor 1 in a mouse model of scleroderma. Arthritis Rheum. 2011, 63, 1405–1415. [Google Scholar] [CrossRef] [PubMed]
- Sakkas, L.I. Spotlight on tocilizumab and its potential in the treatment of systemic sclerosis. Drug Des. Dev. Ther. 2016, 10, 2723–2728. [Google Scholar] [CrossRef] [PubMed]
- Bain, G.; Shannon, K.E.; Huang, F.; Darlington, J.; Goulet, L.; Prodanovich, P.; Ma, G.L.; Santini, A.M.; Stein, A.J.; Lonergan, D.; et al. Selective inhibition of autotaxin is efficacious in mouse models of liver fibrosis. J. Pharmacol. Exp. Ther. 2017, 360, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Wei, L.; Song, W.M.; Higashi, T.; Ghoshal, S.; Kim, R.S.; Bian, C.B.; Yamada, S.; Sun, X.; Venkatesh, A.; et al. Molecular liver cancer prevention in cirrhosis by organ transcriptome analysis and lysophosphatidic acid pathway inhibition. Cancer Cell 2016, 30, 879–890. [Google Scholar] [CrossRef] [PubMed]
- Mouratis, M.A.; Magkrioti, C.; Oikonomou, N.; Katsifa, A.; Prestwich, G.D.; Kaffe, E.; Aidinis, V. Autotaxin and endotoxin-induced acute lung injury. PLoS ONE 2015, 10, e0133619. [Google Scholar] [CrossRef] [PubMed]
- Thirunavukkarasu, K.; Swearingen, C.A.; Oskins, J.L.; Lin, C.; Bui, H.H.; Jones, S.B.; Pfeifer, L.A.; Norman, B.H.; Mitchell, P.G.; Chambers, M.G. Identification and pharmacological characterization of a novel inhibitor of autotaxin in rodent models of joint pain. Osteoarthr. Cartil. 2017, 25, 935–942. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Ikeda, H.; Miyakawa, S.; Futakawa, S.; Nonaka, Y.; Fujiwara, M.; Okudaira, S.; Kano, K.; Aoki, J.; Morita, J.; et al. Structural basis for specific inhibition of autotaxin by a DNA aptamer. Nat. Struct. Mol. Biol. 2016, 23, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Keune, W.J.; Hausmann, J.; Bolier, R.; Tolenaars, D.; Kremer, A.; Heidebrecht, T.; Joosten, R.P.; Sunkara, M.; Morris, A.J.; Matas-Rico, E.; et al. Steroid binding to autotaxin links bile salts and lysophosphatidic acid signalling. Nat. Commun. 2016, 7, 11248. [Google Scholar] [CrossRef] [PubMed]
- Venkatraman, G.; Benesch, M.G.; Tang, X.; Dewald, J.; McMullen, T.P.; Brindley, D.N. Lysophosphatidate signaling stabilizes Nrf2 and increases the expression of genes involved in drug resistance and oxidative stress responses: Implications for cancer treatment. FASEB J. 2015, 29, 772–785. [Google Scholar] [CrossRef] [PubMed]
- Bekele, R.T.; Venkatraman, G.; Liu, R.Z.; Tang, X.; Mi, S.; Benesch, M.G.; Mackey, J.R.; Godbout, R.; Curtis, J.M.; McMullen, T.P.; et al. Oxidative stress contributes to the tamoxifen-induced killing of breast cancer cells: Implications for tamoxifen therapy and resistance. Sci. Rep. 2016, 6, 21164. [Google Scholar] [CrossRef] [PubMed]
- Balogh, A.; Shimizu, Y.; Lee, S.C.; Norman, D.D.; Gangwar, R.; Bavaria, M.; Moon, C.; Shukla, P.; Rao, R.; Ray, R.; et al. The autotaxin-LPA2 GPCR axis is modulated by gamma-irradiation and facilitates DNA damage repair. Cell. Signal. 2015, 27, 1751–1762. [Google Scholar] [CrossRef] [PubMed]
- Jayakumar, S.; Pal, D.; Sandur, S.K. Nrf2 facilitates repair of radiation induced DNA damage through homologous recombination repair pathway in a ros independent manner in cancer cells. Mutat. Res. 2015, 779, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Sekhar, K.R.; Freeman, M.L. Nrf2 promotes survival following exposure to ionizing radiation. Free Radic. Biol. Med. 2015, 88, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Ye, W.; Shao, Q.; Zhang, M.; Liang, J. Nrf2 is a potential therapeutic target in radioresistance in human cancer. Crit. Rev. Oncol. Hematol. 2013, 88, 706–715. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Tang, X.; Yang, Y.; Benesch, M.G.K.; Marshall, A.; Murray, D.; Hemmings, D.G.; Wuest, F.; McMullen, T.P.W.; Brindley, D.N. Gamma-radiation causes an inflammatory wound healing response in adipose tissue involving autotaxin and lysophosphatidate signaling. FASEB J. 2017. [Google Scholar] [CrossRef]
- Schleicher, S.M.; Thotala, D.K.; Linkous, A.G.; Hu, R.; Leahy, K.M.; Yazlovitskaya, E.M.; Hallahan, D.E. Autotaxin and LPA receptors represent potential molecular targets for the radiosensitization of murine glioma through effects on tumor vasculature. PLoS ONE 2011, 6, e22182. [Google Scholar] [CrossRef] [PubMed]
- Bhave, S.R.; Dadey, D.Y.; Karvas, R.M.; Ferraro, D.J.; Kotipatruni, R.P.; Jaboin, J.J.; Hallahan, A.N.; Dewees, T.A.; Linkous, A.G.; Hallahan, D.E.; et al. Autotaxin inhibition with PF-8380 enhances the radiosensitivity of human and murine glioblastoma cell lines. Front. Oncol. 2013, 3, 236. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.R. Fifty years of progress in radiation therapy for breast cancer. Am. Soc. Clin. Oncol. Educ. Book 2014, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Xue, J.X.; Li, X.; Liu, D.S.; Ge, Y.; Ni, P.Y.; Deng, L.; Lu, Y.; Jiang, W. Blockade of lysophosphatidic acid receptors LPAR1/3 ameliorates lung fibrosis induced by irradiation. Biochem. Biophys. Res. Commun. 2011, 409, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Gan, L.; Li, X.; Li, J.; Qi, G.; Wu, Y.; Fu, X.; Mao, Q.; Ao, R.; Lang, J.; et al. Effects of lysophosphatidic acid and its receptors LPA(1/3) on radiation pneumonitis. Oncol. Rep. 2010, 24, 1515–1520. [Google Scholar] [PubMed]
- Desroy, N.; Housseman, C.; Bock, X.; Joncour, A.; Bienvenu, N.; Cherel, L.; Labeguere, V.; Rondet, E.; Peixoto, C.; Grassot, J.M.; et al. Discovery of 2-[[2-ethyl-6-[4-[2-(3-hydroxyazetidin-1-yl)-2-oxoethyl]piperazin-1-yl]-8-methyli midazo[1,2-a]pyridin-3-yl]methylamino]-4-(4-fluorophenyl)thiazole-5-carbonitrile (GLPG1690), a first-in-class autotaxin inhibitor undergoing clinical evaluation for the treatment of idiopathic pulmonary fibrosis. J. Med. Chem. 2017, 60, 3580–3590. [Google Scholar] [PubMed]
- Kihara, Y.; Mizuno, H.; Chun, J. Lysophospholipid receptors in drug discovery. Exp. Cell Res. 2015, 333, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Crack, P.J.; Zhang, M.; Morganti-Kossmann, M.C.; Morris, A.J.; Wojciak, J.M.; Fleming, J.K.; Karve, I.; Wright, D.; Sashindranath, M.; Goldshmit, Y.; et al. Anti-lysophosphatidic acid antibodies improve traumatic brain injury outcomes. J. Neuroinflamm. 2014, 11, 37. [Google Scholar] [CrossRef] [PubMed]
- Brindley, D.N.; Lin, F.T.; Tigyi, G.J. Role of the autotaxin-lysophosphatidate axis in cancer resistance to chemotherapy and radiotherapy. Biochim. Biophys. Acta 2013, 1831, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Rancoule, C.; Espenel, S.; Trone, J.-C.; Langrand-Escure, J.; Vallard, A.; Rehailia-Blanchard, A.; Hamrouni, A.E.M.; Xia, Y.; Guy, J.-B.; Ben-Mrad, M.; et al. Lysophosphatidic acid (LPA) as a pro-fibrotic and pro-oncogenic factor: A pivotal target to improve the radiotherapy therapeutic index. Oncotarget 2017, 8, 43543–43554. [Google Scholar] [CrossRef] [PubMed]
- Citrin, D.E.; Mitchell, J.B. Mechanisms of normal tissue injury from irradiation. Semin. Radiat. Oncol. 2017, 27, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Gangadhar, T.C.; Vonderheide, R.H. Mitigating the toxic effects of anticancer immunotherapy. Nat. Rev. Clin. Oncol. 2014, 11, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Yu, H.; Dong, S.; Zhong, Y.; Hu, S. Recognizing and managing on toxicities in cancer immunotherapy. Tumour Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Compound | Mechanism of Action | Clinical Stage | Clinical Indication | Company | Clinicaltrials.gov ID |
|---|---|---|---|---|---|
| GLPG1690 | ATX direct inhibition | Phase 2 | IPF | Galapagos NV (Mechelen, Belgium) | NCT02738801 |
| BMS-986020 | LPA1 antagonist | Phase 2 | IPF | Bristol-Myers Squibb (New York, NY, USA) | NCT01766817 |
| SAR100842 | LPA1/3 antagonist | Phase 2 | Systemic Sclerosis | Sanofi (Paris, France) | NCT01651143 |
| Lpathomab TM | LPA monoclonal antibody | Phase 1 | ___ | Lpath, Inc. (San Diego, CA, USA) | NCT02341508 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benesch, M.G.K.; MacIntyre, I.T.K.; McMullen, T.P.W.; Brindley, D.N. Coming of Age for Autotaxin and Lysophosphatidate Signaling: Clinical Applications for Preventing, Detecting and Targeting Tumor-Promoting Inflammation. Cancers 2018, 10, 73. https://doi.org/10.3390/cancers10030073
Benesch MGK, MacIntyre ITK, McMullen TPW, Brindley DN. Coming of Age for Autotaxin and Lysophosphatidate Signaling: Clinical Applications for Preventing, Detecting and Targeting Tumor-Promoting Inflammation. Cancers. 2018; 10(3):73. https://doi.org/10.3390/cancers10030073
Chicago/Turabian StyleBenesch, Matthew G.K., Iain T.K. MacIntyre, Todd P.W. McMullen, and David N. Brindley. 2018. "Coming of Age for Autotaxin and Lysophosphatidate Signaling: Clinical Applications for Preventing, Detecting and Targeting Tumor-Promoting Inflammation" Cancers 10, no. 3: 73. https://doi.org/10.3390/cancers10030073