Should We Keep Walking along the Trail for Pancreatic Cancer Treatment? Revisiting TNF-Related Apoptosis-Inducing Ligand for Anticancer Therapy

 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Pancreatic Cancer
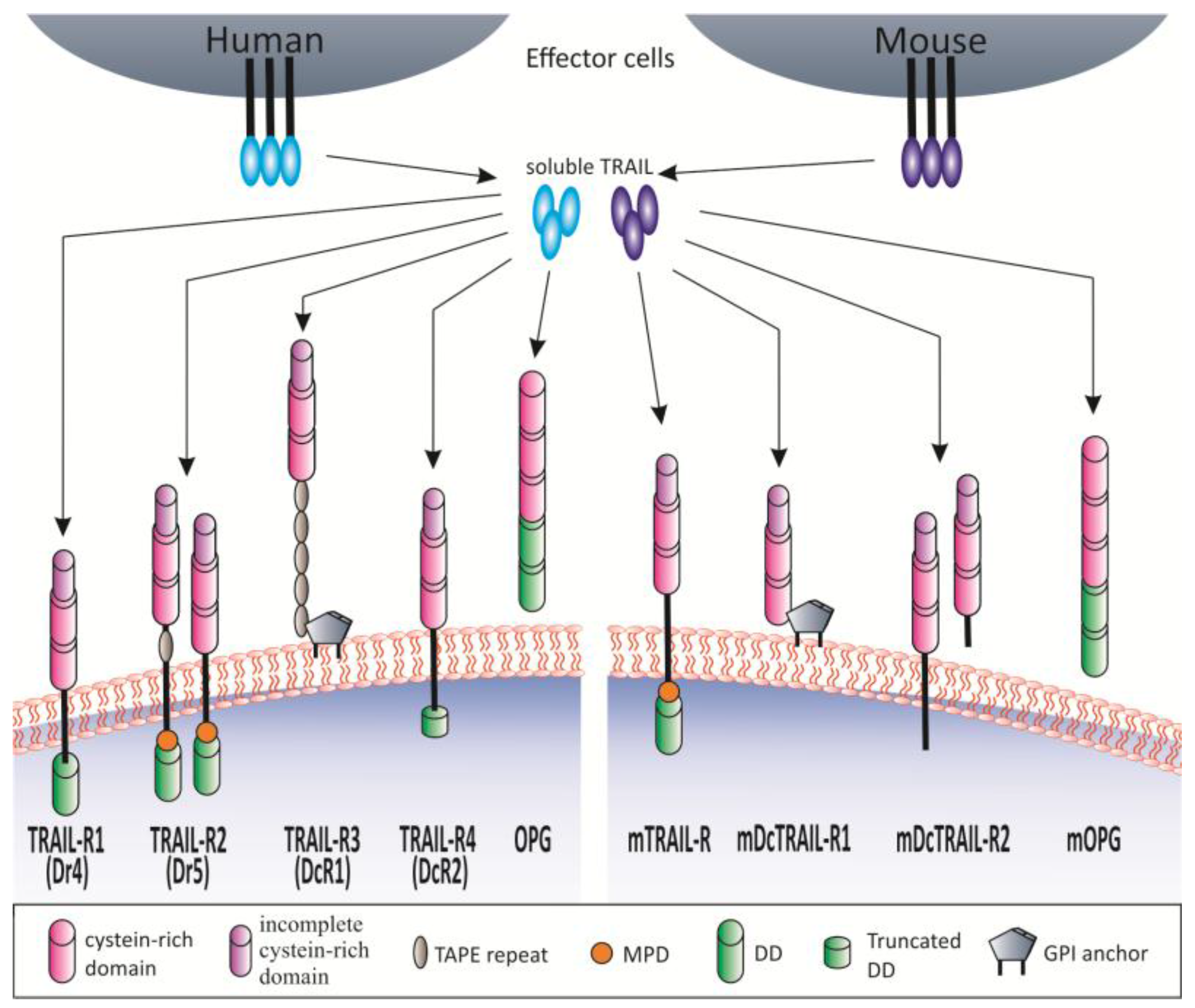
1.2. TRAIL and TRAIL Receptors
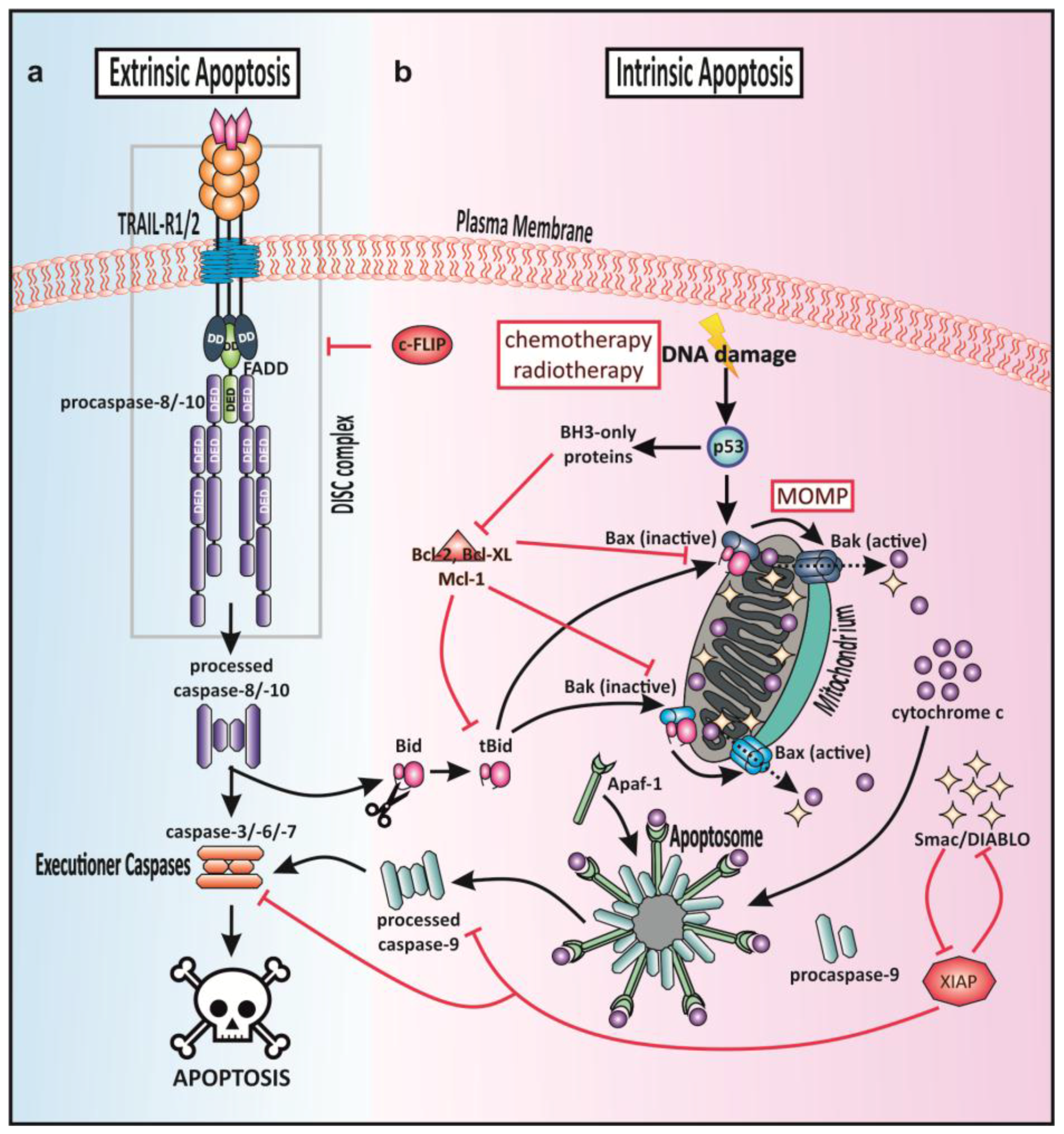
1.3. Apoptotic Signaling
1.4. TRAIL-Mediated Cell Death Signaling—Extrinsic Apoptosis
1.5. TRAIL-R Agonists in Clinical Trials
1.6. Recombinant TRAIL
1.7. TRAIL-R-Specific Agonistic Antibodies
1.8. Dysfunctional TRAIL-Induced Apoptosis in Pancreatic Cancer
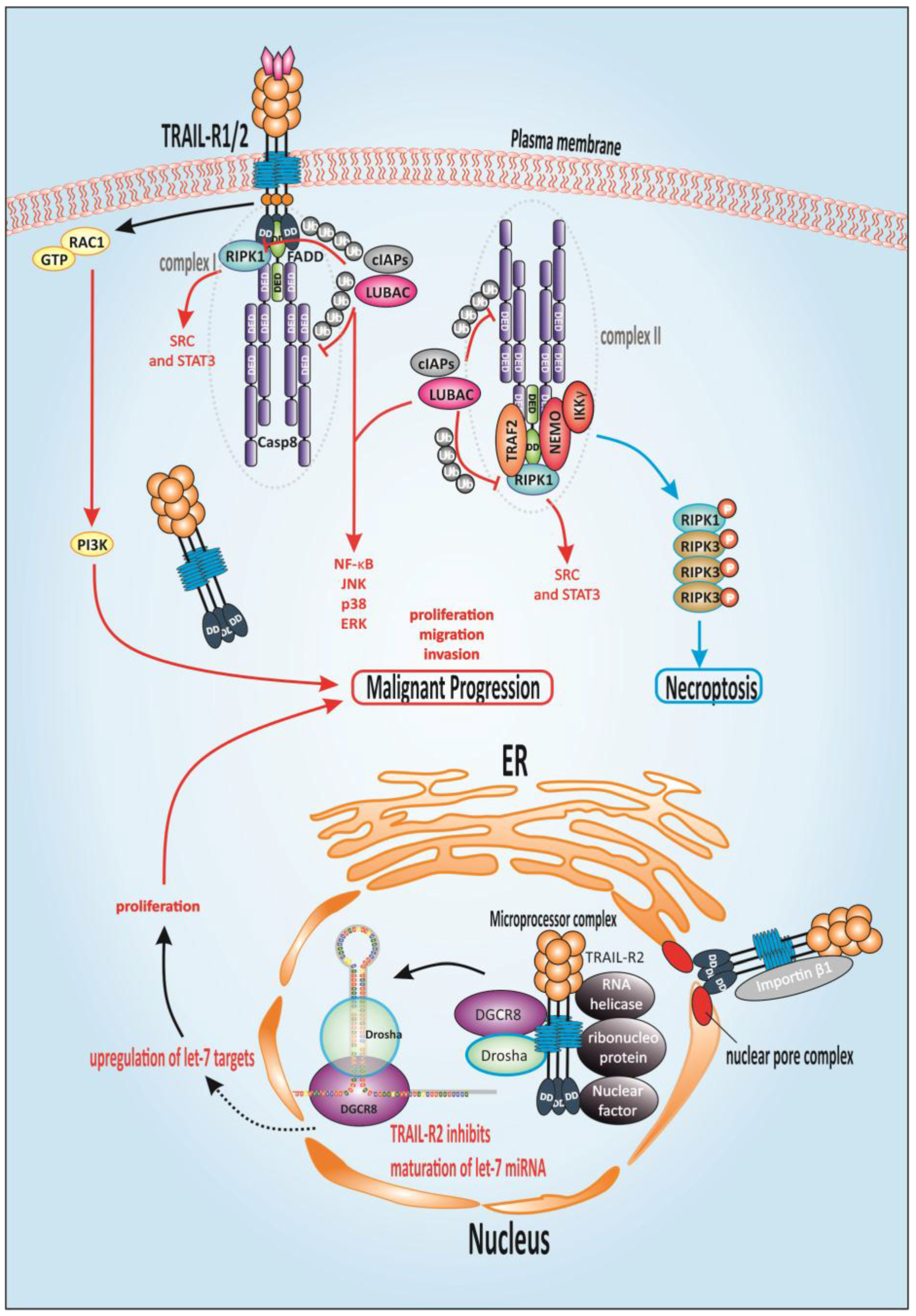
1.9. TRAIL-Mediated Non-Canonical Signaling
1.10. Alternative TRAIL-Mediated Death Signaling
2. Concluding Remarks and Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 5-FU | 5-fluorouracil |
| ADCC | antibody-dependent cellular cytotoxicity |
| AKT | protein kinase B |
| ATM | mutated in the disease ataxia telangiectasia |
| ATP | adenosine triphosphate |
| Apo2L | Apo-2 ligand |
| Bak | Bcl-2 antagonist or killer |
| Bax | Bcl-2-associated X protein |
| Bcl-2 | B-cell CLL/lymphoma 2 |
| Bcl-XL | B-cell lymphoma-extra-large |
| BH3 | Bcl-2 homology domain 3 |
| Bid | BH3-only protein |
| BITC | benzyl isothiocyanate |
| CDK9 | cyclin-dependent kinase 9 |
| c-FLIP | cellular (FADD-like IL-1β-converting enzyme)-inhibitory protein |
| cIAP | cellular IAP |
| c-Myc | avian myelocytomatosis virus oncogene cellular homolog |
| DcR | decoy receptor |
| CPT | circularly permuted TRAIL |
| DD | death domain |
| DDR | DNA damage response |
| DGCR8 | DiGeorge critical region8 |
| DR | death receptor |
| ER | endoplasmic reticulum |
| ERK | extracellular signal-regulated kinase |
| FADD | Fas-associated death domain |
| Fc | Fragment crystallizable |
| Flag | DYKDDDDK |
| FLICE | FADD-like IL-1β-converting enzyme |
| FOLFIRINOX | FOL-folinic acid, F-5-fluorouracil (5-FU), IRIN-irinotecan, OX-oxaliplatin |
| GALNT14 | N-acetylgalactosaminyltransferase-14 |
| GTP | guanosine-5′-triphosphate |
| HcAbs | heavy-chain-only antibodies |
| HDAC | histone deacetylase |
| HMGA2 | high-mobility group AT-hook 2 |
| IAP | inhibitor of apoptosis protein |
| IGF-R | insulin-like growth factor receptor |
| IgG | immunoglobulin |
| IGFR | insulin-like growth factor receptor |
| IKK | inhibitor of κB kinase |
| IMS | intermembrane space |
| IκB | inhibitor of κB |
| IZ | isoleucine zipper |
| JNK | c-JUN N-terminal kinase |
| KRAS | Kirsten rat sarcoma viral oncogene homolog |
| LUBAC | linear ubiquitin chain assembly complex |
| LZ | leucine zipper |
| mAb | monoclonal antibody |
| MAPK | mitogen-activated protein kinase |
| Mcl-1 | myeloid leukemia cell differentiation protein |
| memTRAIL | membrane-bound TRAIL |
| miRNA | micro RNA |
| MOM | mitochondrial outer membrane |
| MOMP | mitochondrial outer membrane permeabilization |
| MTD | membrane-proximal domain |
| nab | nanoparticle albumin-bound |
| NEMO | NF-κB essential modifier |
| NF-κB | nuclear factor kappa light chain enhancer of activated B cells |
| N-glyc | N-glycosylation |
| NIK | NF-κB-inducing kinase |
| nTRAIL | nuclear TRAIL |
| O-glyc | O-glycosylation |
| OPG | osteoprotegerin |
| p38 MAPK | p38 mitogen-activated protein kinase |
| PARP | poly-ADP ribose polymerase |
| PCD | programmed cell death |
| PDAC | pancreatic ductal adenocarcinoma |
| Rac1 | Ras-related C3 botulinum toxin substrate 1 |
| RANKL | receptor activator of nuclear factor κB ligand |
| Ras | Rat sarcoma |
| rhTRAIL | recombinant human TRAIL |
| RIPK | receptor-interacting serine/threonine protein kinase 1 |
| scTRAIL-RBD | single-chain-TRAIL-receptor-binding domain |
| Smac/DIABLO | second mitochondrial activator of caspases/direct inhibitor of apoptosis-binding protein with low pI |
| SR | superfamily |
| SRC | Rous sarcoma oncogene cellular homolog |
| STAT | signal transducer and activator of transcription |
| sTRAIL | soluble TRAIL |
| TNF | tumor necrosis factor |
| TAPE | threonine, alanine, proline, glutamine |
| TRAIL | TNF-related apoptosis-inducing ligand |
| TRAIL-R | Trail receptor |
| Ub | ubiquitin |
| VHH | heavy chain domain |
| XIAP | X-linked inhibitor of apoptosis protein |
References
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [PubMed]
- Westphal, S.; Kalthoff, H. Apoptosis: Targets in pancreatic cancer. Mol. Cancer 2003, 2, 6. [Google Scholar] [PubMed]
- Johnstone, R.W.; Ruefli, A.A.; Lowe, S.W. Apoptosis: A link between cancer genetics and chemotherapy. Cell 2002, 108, 153–164. [Google Scholar] [PubMed]
- Ashkenazi, A. Targeting death and decoy receptors of the tumour-necrosis factor superfamily. Nat. Rev. Cancer 2002, 2, 420–430. [Google Scholar] [PubMed]
- Wallace-Brodeur, R.R.; Lowe, S.W. Clinical implications of p53 mutations. Cell. Mol. Life Sci. 1999, 55, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Lemke, J.; von Karstedt, S.; Zinngrebe, J.; Walczak, H. Getting TRAIL back on track for cancer therapy. Cell Death Differ. 2014, 21, 1350–1364. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.K. Giovanni Battista Morgagni (1682–1771): Father of pathologic anatomy and pioneer of modern medicine. Anat. Sci. Int. 2017, 92, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Soerjomataram, I.; Ervik, M.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11 [online]. 2012; International Agency for Research on Cancer: Lyon, France, 2013. [Google Scholar]
- Hamacher, R.; Schmid, R.M.; Saur, D.; Schneider, G. Apoptotic pathways in pancreatic ductal adenocarcinoma. Mol. Cancer 2008, 7, 64. [Google Scholar] [PubMed]
- He, J.; Ahuja, N.; Makary, M.A.; Cameron, J.L.; Eckhauser, F.E.; Choti, M.A.; Hruban, R.H.; Pawlik, T.M.; Wolfgang, C.L. 2564 resected periampullary adenocarcinomas at a single institution: Trends over three decades. HPB (Oxford) 2014, 16, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Welsch, T.; Kleeff, J.; Friess, H. Molecular pathogenesis of pancreatic cancer: Advances and challenges. Curr. Mol. Med. 2007, 7, 504–521. [Google Scholar] [CrossRef] [PubMed]
- Lemke, J.; Schafer, D.; Sander, S.; Henne-Bruns, D.; Kornmann, M. Survival and prognostic factors in pancreatic and ampullary cancer. Anticancer Res. 2014, 34, 3011–3020. [Google Scholar] [PubMed]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, M. Pancreatic cancer. New Engl. J. Med. 2010, 362, 1605–1617. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A., 3rd; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef] [PubMed]
- Plunkett, W.; Huang, P.; Xu, Y.Z.; Heinemann, V.; Grunewald, R.; Gandhi, V. Gemcitabine: Metabolism, mechanisms of action, and self-potentiation. Semin. Oncol. 1995, 22 (Suppl. 11), 3–10. [Google Scholar] [PubMed]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. New Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. New Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Park, S.W. Systemic Chemotherapy in Advanced Pancreatic Cancer. Gut Liver 2016, 10, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Saung, M.T.; Zheng, L. Current Standards of Chemotherapy for Pancreatic Cancer. Clin. Ther. 2017, 39, 2125–2134. [Google Scholar] [CrossRef] [PubMed]
- Moran, R.G.; Keyomarsi, K. Biochemical rationale for the synergism of 5-fluorouracil and folinic acid. NCI Monogr. Publ. Natl. Cancer Inst. 1987, 5, 159–163. [Google Scholar]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Mathijssen, R.H.; Loos, W.J.; Verweij, J.; Sparreboom, A. Pharmacology of topoisomerase I inhibitors irinotecan (CPT-11) and topotecan. Curr. Cancer Drug Targets 2002, 2, 103–123. [Google Scholar] [PubMed]
- Raymond, E.; Faivre, S.; Woynarowski, J.M.; Chaney, S.G. Oxaliplatin: Mechanism of action and antineoplastic activity. Semin. Oncol. 1998, 25 (Suppl. 5), 4–12. [Google Scholar] [PubMed]
- Wani, M.C.; Taylor, H.L.; Wall, M.E.; Coggon, P.; McPhail, A.T. Plant antitumor agents. VI. The isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J. Am. Chem. Soc. 1971, 93, 2325–2327. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, R.M. Nab-paclitaxel: A limited improvement under the current therapeutic paradigm of pancreatic cancer. Expert Opin. Pharmacother. 2015, 16, 943–947. [Google Scholar] [CrossRef] [PubMed]
- Schiff, P.B.; Horwitz, S.B. Taxol stabilizes microtubules in mouse fibroblast cells. Proc. Natl. Acad. Sci. USA 1980, 77, 1561–1565. [Google Scholar] [CrossRef] [PubMed]
- Faris, J.E.; Blaszkowsky, L.S.; McDermott, S.; Guimaraes, A.R.; Szymonifka, J.; Huynh, M.A.; Ferrone, C.R.; Wargo, J.A.; Allen, J.N.; Dias, L.E.; et al. FOLFIRINOX in locally advanced pancreatic cancer: The Massachusetts General Hospital Cancer Center experience. Oncologist 2013, 18, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Gunturu, K.S.; Yao, X.; Cong, X.; Thumar, J.R.; Hochster, H.S.; Stein, S.M.; Lacy, J. FOLFIRINOX for locally advanced and metastatic pancreatic cancer: Single institution retrospective review of efficacy and toxicity. Med. Oncol. 2013, 30, 361. [Google Scholar] [CrossRef] [PubMed]
- Schniewind, B.; Christgen, M.; Kurdow, R.; Haye, S.; Kremer, B.; Kalthoff, H.; Ungefroren, H. Resistance of pancreatic cancer to gemcitabine treatment is dependent on mitochondria-mediated apoptosis. Int. J. Cancer 2004, 109, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Wiley, S.R.; Schooley, K.; Smolak, P.J.; Din, W.S.; Huang, C.P.; Nicholl, J.K.; Sutherland, G.R.; Smith, T.D.; Rauch, C.; Smith, C.A.; et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 1995, 3, 673–682. [Google Scholar] [CrossRef]
- Pitti, R.M.; Marsters, S.A.; Ruppert, S.; Donahue, C.J.; Moore, A.; Ashkenazi, A. Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J. Biol. Chem. 1996, 271, 12687–12690. [Google Scholar] [CrossRef] [PubMed]
- Walczak, H.; Miller, R.E.; Ariail, K.; Gliniak, B.; Griffith, T.S.; Kubin, M.; Chin, W.; Jones, J.; Woodward, A.; Le, T.; et al. Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat. Med. 1999, 5, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A.; Pai, R.C.; Fong, S.; Leung, S.; Lawrence, D.A.; Marsters, S.A.; Blackie, C.; Chang, L.; McMurtrey, A.E.; Hebert, A.; et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J. Clin. Investig. 1999, 104, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Pan, G.; Ni, J.; Wei, Y.F.; Yu, G.; Gentz, R.; Dixit, V.M. An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science 1997, 277, 815–818. [Google Scholar] [CrossRef] [PubMed]
- Walczak, H.; Degli-Esposti, M.A.; Johnson, R.S.; Smolak, P.J.; Waugh, J.Y.; Boiani, N.; Timour, M.S.; Gerhart, M.J.; Schooley, K.A.; Smith, C.A.; et al. TRAIL-R2: A novel apoptosis-mediating receptor for TRAIL. EMBO J. 1997, 16, 5386–5397. [Google Scholar] [CrossRef] [PubMed]
- Kawakubo, T.; Okamoto, K.; Iwata, J.; Shin, M.; Okamoto, Y.; Yasukochi, A.; Nakayama, K.I.; Kadowaki, T.; Tsukuba, T.; Yamamoto, K. Cathepsin E prevents tumor growth and metastasis by catalyzing the proteolytic release of soluble TRAIL from tumor cell surface. Cancer Res. 2007, 67, 10869–10878. [Google Scholar] [CrossRef] [PubMed]
- Hymowitz, S.G.; Christinger, H.W.; Fuh, G.; Ultsch, M.; O’Connell, M.; Kelley, R.F.; Ashkenazi, A.; de Vos, A.M. Triggering cell death: The crystal structure of Apo2L/TRAIL in a complex with death receptor 5. Mol. Cell 1999, 4, 563–571. [Google Scholar] [CrossRef]
- Mongkolsapaya, J.; Grimes, J.M.; Chen, N.; Xu, X.N.; Stuart, D.I.; Jones, E.Y.; Screaton, G.R. Structure of the TRAIL-DR5 complex reveals mechanisms conferring specificity in apoptotic initiation. Nat. Struct. Biol. 1999, 6, 1048–1053. [Google Scholar] [PubMed]
- Cha, S.S.; Kim, M.S.; Choi, Y.H.; Sung, B.J.; Shin, N.K.; Shin, H.C.; Sung, Y.C.; Oh, B.H. 2.8 A resolution crystal structure of human TRAIL, a cytokine with selective antitumor activity. Immunity 1999, 11, 253–261. [Google Scholar] [CrossRef]
- Hymowitz, S.G.; O’Connell, M.P.; Ultsch, M.H.; Hurst, A.; Totpal, K.; Ashkenazi, A.; de Vos, A.M.; Kelley, R.F. A unique zinc-binding site revealed by a high-resolution X-ray structure of homotrimeric Apo2L/TRAIL. Biochemistry 2000, 39, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Mariani, S.M.; Matiba, B.; Armandola, E.A.; Krammer, P.H. Interleukin 1β-converting enzyme related proteases/caspases are involved in TRAIL-induced apoptosis of myeloma and leukemia cells. J. Cell Biol. 1997, 137, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Gibellini, D.; Borderi, M.; De Crignis, E.; Cicola, R.; Vescini, F.; Caudarella, R.; Chiodo, F.; Re, M.C. RANKL/OPG/TRAIL plasma levels and bone mass loss evaluation in antiretroviral naive HIV-1-positive men. J. Med. Virol. 2007, 79, 1446–1454. [Google Scholar] [CrossRef] [PubMed]
- De Miguel, D.; Lemke, J.; Anel, A.; Walczak, H.; Martinez-Lostao, L. Onto better TRAILs for cancer treatment. Cell Death Differ. 2016, 23, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Von Karstedt, S.; Montinaro, A.; Walczak, H. Exploring the TRAILs less travelled: TRAIL in cancer biology and therapy. Nat. Rev. Cancer 2017, 17, 352–366. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, H.N.; Ashkenazi, A. Apo2L/TRAIL and its death and decoy receptors. Cell Death Differ. 2003, 10, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Emery, J.G.; McDonnell, P.; Burke, M.B.; Deen, K.C.; Lyn, S.; Silverman, C.; Dul, E.; Appelbaum, E.R.; Eichman, C.; DiPrinzio, R.; et al. Osteoprotegerin is a receptor for the cytotoxic ligand TRAIL. J. Biol. Chem. 1998, 273, 14363–14367. [Google Scholar] [CrossRef] [PubMed]
- Pan, G.; O’Rourke, K.; Chinnaiyan, A.M.; Gentz, R.; Ebner, R.; Ni, J.; Dixit, V.M. The receptor for the cytotoxic ligand TRAIL. Science 1997, 276, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, J.P.; Marsters, S.A.; Pitti, R.M.; Gurney, A.; Skubatch, M.; Baldwin, D.; Ramakrishnan, L.; Gray, C.L.; Baker, K.; Wood, W.I.; et al. Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science 1997, 277, 818–821. [Google Scholar] [CrossRef] [PubMed]
- Morizot, A.; Merino, D.; Lalaoui, N.; Jacquemin, G.; Granci, V.; Iessi, E.; Lanneau, D.; Bouyer, F.; Solary, E.; Chauffert, B.; et al. Chemotherapy overcomes TRAIL-R4-mediated TRAIL resistance at the DISC level. Cell Death Differ. 2011, 18, 700–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merino, D.; Lalaoui, N.; Morizot, A.; Schneider, P.; Solary, E.; Micheau, O. Differential inhibition of TRAIL-mediated DR5-DISC formation by decoy receptors 1 and 2. Mol. Cell. Biol. 2006, 26, 7046–7055. [Google Scholar] [CrossRef] [PubMed]
- Neumann, S.; Hasenauer, J.; Pollak, N.; Scheurich, P. Dominant negative effects of tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) receptor 4 on TRAIL receptor 1 signaling by formation of heteromeric complexes. J. Biol. Chem. 2014, 289, 16576–16587. [Google Scholar] [CrossRef] [PubMed]
- Vindrieux, D.; Reveiller, M.; Chantepie, J.; Yakoub, S.; Deschildre, C.; Ruffion, A.; Devonec, M.; Benahmed, M.; Grataroli, R. Down-regulation of DcR2 sensitizes androgen-dependent prostate cancer LNCaP cells to TRAIL-induced apoptosis. Cancer Cell Int. 2011, 11, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aydin, C.; Sanlioglu, A.D.; Karacay, B.; Ozbilim, G.; Dertsiz, L.; Ozbudak, O.; Akdis, C.A.; Sanlioglu, S. Decoy receptor-2 small interfering RNA (siRNA) strategy employing three different siRNA constructs in combination defeats adenovirus-transferred tumor necrosis factor-related apoptosis-inducing ligand resistance in lung cancer cells. Hum. Gene Ther. 2007, 18, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Simonet, W.S.; Lacey, D.L.; Dunstan, C.R.; Kelley, M.; Chang, M.S.; Luthy, R.; Nguyen, H.Q.; Wooden, S.; Bennett, L.; Boone, T.; et al. Osteoprotegerin: A novel secreted protein involved in the regulation of bone density. Cell 1997, 89, 309–319. [Google Scholar] [CrossRef]
- Takahashi, N.; Udagawa, N.; Suda, T. A new member of tumor necrosis factor ligand family, ODF/OPGL/TRANCE/RANKL, regulates osteoclast differentiation and function. Biochem. Biophys. Res. Commun. 1999, 256, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Truneh, A.; Sharma, S.; Silverman, C.; Khandekar, S.; Reddy, M.P.; Deen, K.C.; McLaughlin, M.M.; Srinivasula, S.M.; Livi, G.P.; Marshall, L.A.; et al. Temperature-sensitive differential affinity of TRAIL for its receptors. DR5 is the highest affinity receptor. J. Biol. Chem. 2000, 275, 23319–23325. [Google Scholar] [CrossRef] [PubMed]
- Lemke, J.; Noack, A.; Adam, D.; Tchikov, V.; Bertsch, U.; Roder, C.; Schutze, S.; Wajant, H.; Kalthoff, H.; Trauzold, A. TRAIL signaling is mediated by DR4 in pancreatic tumor cells despite the expression of functional DR5. J. Mol. Med. 2010, 88, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Stadel, D.; Mohr, A.; Ref, C.; MacFarlane, M.; Zhou, S.; Humphreys, R.; Bachem, M.; Cohen, G.; Moller, P.; Zwacka, R.M.; et al. TRAIL-induced apoptosis is preferentially mediated via TRAIL receptor 1 in pancreatic carcinoma cells and profoundly enhanced by XIAP inhibitors. Clin. Cancer Res. 2010, 16, 5734–5749. [Google Scholar] [CrossRef] [PubMed]
- MacFarlane, M.; Kohlhaas, S.L.; Sutcliffe, M.J.; Dyer, M.J.; Cohen, G.M. TRAIL receptor-selective mutants signal to apoptosis via TRAIL-R1 in primary lymphoid malignancies. Cancer Res. 2005, 65, 11265–11270. [Google Scholar] [CrossRef] [PubMed]
- MacFarlane, M.; Inoue, S.; Kohlhaas, S.L.; Majid, A.; Harper, N.; Kennedy, D.B.; Dyer, M.J.; Cohen, G.M. Chronic lymphocytic leukemic cells exhibit apoptotic signaling via TRAIL-R1. Cell Death Differ. 2005, 12, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Albarenque, S.M.; Cool, R.H.; Quax, W.J.; Mohr, A.; Zwacka, R.M. DR4 specific TRAIL variants are more efficacious than wild-type TRAIL in pancreatic cancer. Cancer Biol. Ther. 2014, 15, 1658–1666. [Google Scholar] [CrossRef] [PubMed]
- Mohr, A.; Yu, R.; Zwacka, R.M. TRAIL-receptor preferences in pancreatic cancer cells revisited: Both TRAIL-R1 and TRAIL-R2 have a licence to kill. BMC Cancer 2015, 15, 494. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, P.M.; Eby, M.; Jasmin, A.; Bookwalter, A.; Murray, J.; Hood, L. Death receptor 5, a new member of the TNFR family, and DR4 induce FADD-dependent apoptosis and activate the NF-κB pathway. Immunity 1997, 7, 821–830. [Google Scholar] [CrossRef]
- Wang, T.T.; Jeng, J. Coordinated regulation of two TRAIL-R2/KILLER/DR5 mRNA isoforms by DNA damaging agents, serum and 17β-estradiol in human breast cancer cells. Breast Cancer Res. Treat. 2000, 61, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.S.; Burns, T.F.; Zhan, Y.; Alnemri, E.S.; El-Deiry, W.S. Molecular cloning and functional analysis of the mouse homologue of the KILLER/DR5 tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) death receptor. Cancer Res. 1999, 59, 2770–2775. [Google Scholar] [PubMed]
- Schneider, P.; Olson, D.; Tardivel, A.; Browning, B.; Lugovskoy, A.; Gong, D.; Dobles, M.; Hertig, S.; Hofmann, K.; Van Vlijmen, H.; et al. Identification of a new murine tumor necrosis factor receptor locus that contains two novel murine receptors for tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). J. Biol. Chem. 2003, 278, 5444–5454. [Google Scholar] [CrossRef] [PubMed]
- Bossen, C.; Ingold, K.; Tardivel, A.; Bodmer, J.L.; Gaide, O.; Hertig, S.; Ambrose, C.; Tschopp, J.; Schneider, P. Interactions of tumor necrosis factor (TNF) and TNF receptor family members in the mouse and human. J. Biol. Chem. 2006, 281, 13964–13971. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, M.D.; Weil, M.; Raff, M.C. Programmed cell death in animal development. Cell 1997, 88, 347–354. [Google Scholar] [CrossRef]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Brune, W.; Andoniou, C.E. Die Another Day: Inhibition of Cell Death Pathways by Cytomegalovirus. Viruses 2017, 9, 249. [Google Scholar] [CrossRef] [PubMed]
- Brenner, D.; Mak, T.W. Mitochondrial cell death effectors. Curr. Opin. Cell Biol. 2009, 21, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Favaloro, B.; Allocati, N.; Graziano, V.; Di Ilio, C.; De Laurenzi, V. Role of apoptosis in disease. Aging (Albany NY) 2012, 4, 330–349. [Google Scholar] [CrossRef] [PubMed]
- Burke, P.J. Mitochondria, Bioenergetics and Apoptosis in Cancer. Trends Cancer 2017, 3, 857–870. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; El-Deiry, W.S. TRAIL and apoptosis induction by TNF-family death receptors. Oncogene 2003, 22, 8628–8633. [Google Scholar] [CrossRef] [PubMed]
- Neureiter, D.; Jager, T.; Ocker, M.; Kiesslich, T. Epigenetics and pancreatic cancer: Pathophysiology and novel treatment aspects. World J. Gastroenterol. 2014, 20, 7830–7848. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, E.F. The Toxins of William B. Coley and the Treatment of Bone and Soft-Tissue Sarcomas. Iowa Orthop. J. 2006, 26, 154–158. [Google Scholar] [PubMed]
- Carswell, E.A.; Old, L.J.; Kassel, R.L.; Green, S.; Fiore, N.; Williamson, B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc. Natl. Acad. Sci. USA 1975, 72, 3666–3670. [Google Scholar] [CrossRef] [PubMed]
- Roberts, N.J.; Zhou, S.; Diaz, L.A., Jr.; Holdhoff, M. Systemic use of tumor necrosis factor alpha as an anticancer agent. Oncotarget 2011, 2, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Trauth, B.C.; Klas, C.; Peters, A.M.; Matzku, S.; Moller, P.; Falk, W.; Debatin, K.M.; Krammer, P.H. Monoclonal antibody-mediated tumor regression by induction of apoptosis. Science 1989, 245, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Yonehara, S.; Ishii, A.; Yonehara, M. A cell-killing monoclonal antibody (anti-Fas) to a cell surface antigen co-downregulated with the receptor of tumor necrosis factor. J. Exp. Med. 1989, 169, 1747–1756. [Google Scholar] [CrossRef] [PubMed]
- Itoh, N.; Yonehara, S.; Ishii, A.; Yonehara, M.; Mizushima, S.; Sameshima, M.; Hase, A.; Seto, Y.; Nagata, S. The polypeptide encoded by the cDNA for human cell surface antigen Fas can mediate apoptosis. Cell 1991, 66, 233–243. [Google Scholar] [CrossRef]
- Oehm, A.; Behrmann, I.; Falk, W.; Pawlita, M.; Maier, G.; Klas, C.; Li-Weber, M.; Richards, S.; Dhein, J.; Trauth, B.C.; et al. Purification and molecular cloning of the APO-1 cell surface antigen, a member of the tumor necrosis factor/nerve growth factor receptor superfamily. Sequence identity with the Fas antigen. J. Biol. Chem. 1992, 267, 10709–10715. [Google Scholar] [PubMed]
- Ogasawara, J.; Watanabe-Fukunaga, R.; Adachi, M.; Matsuzawa, A.; Kasugai, T.; Kitamura, Y.; Itoh, N.; Suda, T.; Nagata, S. Lethal effect of the anti-Fas antibody in mice. Nature 1993, 364, 806–809. [Google Scholar] [CrossRef] [PubMed]
- Dickens, L.S.; Boyd, R.S.; Jukes-Jones, R.; Hughes, M.A.; Robinson, G.L.; Fairall, L.; Schwabe, J.W.; Cain, K.; Macfarlane, M. A death effector domain chain DISC model reveals a crucial role for caspase-8 chain assembly in mediating apoptotic cell death. Mol. Cell 2012, 47, 291–305. [Google Scholar] [CrossRef] [PubMed]
- Bodmer, J.L.; Holler, N.; Reynard, S.; Vinciguerra, P.; Schneider, P.; Juo, P.; Blenis, J.; Tschopp, J. TRAIL receptor-2 signals apoptosis through FADD and caspase-8. Nat. Cell Biol. 2000, 2, 241–243. [Google Scholar] [CrossRef] [PubMed]
- Schleich, K.; Buchbinder, J.H.; Pietkiewicz, S.; Kahne, T.; Warnken, U.; Ozturk, S.; Schnolzer, M.; Naumann, M.; Krammer, P.H.; Lavrik, I.N. Molecular architecture of the DED chains at the DISC: Regulation of procaspase-8 activation by short DED proteins c-FLIP and procaspase-8 prodomain. Cell Death Differ. 2016, 23, 681–694. [Google Scholar] [CrossRef] [PubMed]
- Horn, S.; Hughes, M.A.; Schilling, R.; Sticht, C.; Tenev, T.; Ploesser, M.; Meier, P.; Sprick, M.R.; MacFarlane, M.; Leverkus, M. Caspase-10 Negatively Regulates Caspase-8-Mediated Cell Death, Switching the Response to CD95L in Favor of NF-κB Activation and Cell Survival. Cell Rep. 2017, 19, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Hinshaw-Makepeace, J.; Huston, G.; Fortner, K.A.; Russell, J.Q.; Holoch, D.; Swain, S.; Budd, R.C. c-FLIP(S) reduces activation of caspase and NF-κB pathways and decreases T cell survival. Eur. J. Immunol. 2008, 38, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Dillon, C.P.; Oberst, A.; Weinlich, R.; Janke, L.J.; Kang, T.B.; Ben-Moshe, T.; Mak, T.W.; Wallach, D.; Green, D.R. Survival function of the FADD-CASPASE-8-cFLIP(L) complex. Cell Rep. 2012, 1, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Panayotova-Dimitrova, D.; Feoktistova, M.; Ploesser, M.; Kellert, B.; Hupe, M.; Horn, S.; Makarov, R.; Jensen, F.; Porubsky, S.; Schmieder, A.; et al. cFLIP Regulates Skin Homeostasis and Protects against TNF-Induced Keratinocyte Apoptosis. Cell Rep. 2013, 5, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.A.; Powley, I.R.; Jukes-Jones, R.; Horn, S.; Feoktistova, M.; Fairall, L.; Schwabe, J.W.R.; Leverkus, M.; Cain, K.; MacFarlane, M. Co-operative and Hierarchical Binding of c-FLIP and Caspase-8: A Unified Model Defines How c-FLIP Isoforms Differentially Control Cell Fate. Mol. Cell 2016, 61, 834–849. [Google Scholar] [CrossRef] [PubMed]
- Irmler, M.; Thome, M.; Hahne, M.; Schneider, P.; Hofmann, K.; Steiner, V.; Bodmer, J.L.; Schroter, M.; Burns, K.; Mattmann, C.; et al. Inhibition of death receptor signals by cellular FLIP. Nature 1997, 388, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, C.; Schmitz, I.; Krammer, P.H.; Peter, M.E. The role of c-FLIP in modulation of CD95-induced apoptosis. J. Biol. Chem. 1999, 274, 1541–1548. [Google Scholar] [CrossRef] [PubMed]
- Ozturk, S.; Schleich, K.; Lavrik, I.N. Cellular FLICE-like inhibitory proteins (c-FLIPs): Fine-tuners of life and death decisions. Exp. Cell Res. 2012, 318, 1324–1331. [Google Scholar] [CrossRef] [PubMed]
- Krueger, A.; Schmitz, I.; Baumann, S.; Krammer, P.H.; Kirchhoff, S. Cellular FLICE-inhibitory protein splice variants inhibit different steps of caspase-8 activation at the CD95 death-inducing signaling complex. J. Biol. Chem. 2001, 276, 20633–20640. [Google Scholar] [CrossRef] [PubMed]
- Vaux, D.L.; Silke, J. IAPs, RINGs and ubiquitylation. Nat. Rev. Mol. Cell Biol. 2005, 6, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Holcik, M.; Korneluk, R.G. XIAP, the guardian angel. Nat. Rev. Mol. Cell Biol. 2001, 2, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Jost, P.J.; Grabow, S.; Gray, D.; McKenzie, M.D.; Nachbur, U.; Huang, D.C.; Bouillet, P.; Thomas, H.E.; Borner, C.; Silke, J.; et al. XIAP discriminates between type I and type II FAS-induced apoptosis. Nature 2009, 460, 1035–1039. [Google Scholar] [CrossRef] [PubMed]
- Bratton, S.B.; Walker, G.; Srinivasula, S.M.; Sun, X.M.; Butterworth, M.; Alnemri, E.S.; Cohen, G.M. Recruitment, activation and retention of caspases-9 and -3 by Apaf-1 apoptosome and associated XIAP complexes. EMBO J. 2001, 20, 998–1009. [Google Scholar] [CrossRef] [PubMed]
- Deveraux, Q.L.; Roy, N.; Stennicke, H.R.; Van Arsdale, T.; Zhou, Q.; Srinivasula, S.M.; Alnemri, E.S.; Salvesen, G.S.; Reed, J.C. IAPs block apoptotic events induced by caspase-8 and cytochrome c by direct inhibition of distinct caspases. EMBO J. 1998, 17, 2215–2223. [Google Scholar] [CrossRef] [PubMed]
- Hinz, S.; Trauzold, A.; Boenicke, L.; Sandberg, C.; Beckmann, S.; Bayer, E.; Walczak, H.; Kalthoff, H.; Ungefroren, H. Bcl-XL protects pancreatic adenocarcinoma cells against CD95- and TRAIL-receptor-mediated apoptosis. Oncogene 2000, 19, 5477–5486. [Google Scholar] [CrossRef] [PubMed]
- Trauzold, A.; Wermann, H.; Arlt, A.; Schutze, S.; Schafer, H.; Oestern, S.; Roder, C.; Ungefroren, H.; Lampe, E.; Heinrich, M.; et al. CD95 and TRAIL receptor-mediated activation of protein kinase C and NF-κB contributes to apoptosis resistance in ductal pancreatic adenocarcinoma cells. Oncogene 2001, 20, 4258–4269. [Google Scholar] [CrossRef] [PubMed]
- Shamas-Din, A.; Kale, J.; Leber, B.; Andrews, D.W. Mechanisms of Action of Bcl-2 Family Proteins. Cold Spring Harb. Perspect. Biol. 2013, 5, a008714. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Zhang, J.; O’Neill, K.L.; Gurumurthy, C.B.; Quadros, R.M.; Tu, Y.; Luo, X. Cleavage by Caspase 8 and Mitochondrial Membrane Association Activate the BH3-only Protein Bid during TRAIL-induced Apoptosis. J. Biol. Chem. 2016, 291, 11843–11851. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Riedl, S.J.; Salvesen, G.S. The apoptosome: Signalling platform of cell death. Nat. Rev. Mol. Cell Biol. 2007, 8, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Llambi, F.; Moldoveanu, T.; Tait, S.W.; Bouchier-Hayes, L.; Temirov, J.; McCormick, L.L.; Dillon, C.P.; Green, D.R. A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Mol. Cell 2011, 44, 517–531. [Google Scholar] [CrossRef] [PubMed]
- Willis, S.N.; Chen, L.; Dewson, G.; Wei, A.; Naik, E.; Fletcher, J.I.; Adams, J.M.; Huang, D.C. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005, 19, 1294–1305. [Google Scholar] [CrossRef] [PubMed]
- Falschlehner, C.; Schaefer, U.; Walczak, H. Following TRAIL’s path in the immune system. Immunology 2009, 127, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Lin, Y.; Wu, X. TRAIL-induced apoptosis requires Bax-dependent mitochondrial release of Smac/DIABLO. Genes Dev. 2002, 16, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Ganten, T.M.; Koschny, R.; Sykora, J.; Schulze-Bergkamen, H.; Buchler, P.; Haas, T.L.; Schader, M.B.; Untergasser, A.; Stremmel, W.; Walczak, H. Preclinical differentiation between apparently safe and potentially hepatotoxic applications of TRAIL either alone or in combination with chemotherapeutic drugs. Clin. Cancer Res. 2006, 12, 2640–2646. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Eckhardt, S.G.; Kurzrock, R.; Ebbinghaus, S.; O’Dwyer, P.J.; Gordon, M.S.; Novotny, W.; Goldwasser, M.A.; Tohnya, T.M.; Lum, B.L.; et al. Phase I dose-escalation study of recombinant human Apo2L/TRAIL, a dual proapoptotic receptor agonist, in patients with advanced cancer. J. Clin. Oncol. 2010, 28, 2839–2846. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.C.; Mark, Z.; Zatloukal, P.; Szima, B.; Albert, I.; Juhasz, E.; Pujol, J.L.; Kozielski, J.; Baker, N.; Smethurst, D.; et al. Randomized phase II study of dulanermin in combination with paclitaxel, carboplatin, and bevacizumab in advanced non-small-cell lung cancer. J. Clin. Oncol. 2011, 29, 4442–4451. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.C.; Smit, E.; Khayat, D.; Besse, B.; Yang, X.; Hsu, C.P.; Reese, D.; Wiezorek, J.; Blackhall, F. Phase 1b study of dulanermin (recombinant human Apo2L/TRAIL) in combination with paclitaxel, carboplatin, and bevacizumab in patients with advanced non-squamous non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 1527–1533. [Google Scholar] [CrossRef] [PubMed]
- Yee, L.; Fanale, M.; Dimick, K.; Calvert, S.; Robins, C.; Ing, J.; Ling, J.; Novotny, W.; Ashkenazi, A.; Burris, H., III. A phase IB safety and pharmacokinetic (PK) study of recombinant human Apo2L/TRAIL in combination with rituximab in patients with low-grade non-Hodgkin lymphoma. J. Clin. Oncol. 2007, 25. [Google Scholar] [CrossRef]
- Wainberg, Z.A.; Messersmith, W.A.; Peddi, P.F.; Kapp, A.V.; Ashkenazi, A.; Royer-Joo, S.; Portera, C.C.; Kozloff, M.F. A phase 1B study of dulanermin in combination with modified FOLFOX6 plus bevacizumab in patients with metastatic colorectal cancer. Clin. Colorectal Cancer 2013, 12, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Yee, L.; Burris, H.A.; Kozloff, M.; Wainberg, Z.; Pao, M.; Skettino, S.; Novotny, W.; Durbin, B.; Weston, J.; Hurwitz, H. Phase Ib study of recombinant human Apo2L/TRAIL plus irinotecan and cetuximab or FOLFIRI in metastatic colorectal cancer (mCRC) patients (pts): Preliminary results. J. Clin. Oncol. 2009, 27. [Google Scholar] [CrossRef]
- Kasubhai, S.M.; Bendell, J.C.; Kozloff, M.; Kapp, A.V.; Ashkenazi, A.; Royer-Joo, S.; Portera, C.C. Phase Ib study of dulanermin combined with FOLFIRI (with or without bevacizumab [BV]) in previously treated patients (Pts) with metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2012, 30. [Google Scholar] [CrossRef]
- Quintavalle, C.; Condorelli, G. Dulanermin in cancer therapy: Still much to do. Transl. Lung Cancer Res. 2012, 1, 158–159. [Google Scholar] [PubMed]
- Kaplan-Lefko, P.J.; Graves, J.D.; Zoog, S.J.; Pan, Y.; Wall, J.; Branstetter, D.G.; Moriguchi, J.; Coxon, A.; Huard, J.N.; Xu, R.; et al. Conatumumab, a fully human agonist antibody to death receptor 5, induces apoptosis via caspase activation in multiple tumor types. Cancer Biol. Ther. 2010, 9, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Rosevear, H.M.; Lightfoot, A.J.; Griffith, T.S. Conatumumab, a fully human mAb against death receptor 5 for the treatment of cancer. Curr. Opin. Investig. Drugs 2010, 11, 688–698. [Google Scholar] [PubMed]
- Kindler, H.L.; Garbo, L.; Stephenson, J.; Wiezorek, J.; Sabin, T.; Hsu, M.; Civoli, F.; Richards, D. A phase Ib study to evaluate the safety and efficacy of AMG 655 in combination with gemcitabine (G) in patients (pts) with metastatic pancreatic cancer (PC). J. Clin. Oncol. 2009, 27. [Google Scholar] [CrossRef]
- Kindler, H.L.; Richards, D.A.; Stephenson, J.; Garbo, L.E.; Lima, C.S.R.; Safran, H.; Wiezorek, J.S.; Feigal, E.G.; Bray, S.; Fuchs, C. A placebo-controlled, randomized phase II study of conatumumab (C) or AMG 479 (A) or placebo (P) plus gemcitabine (G) in patients (pts) with metastatic pancreatic cancer (mPC). J. Clin. Oncol. 2010, 28. [Google Scholar] [CrossRef]
- Kindler, H.L.; Richards, D.A.; Garbo, L.E.; Garon, E.B.; Stephenson, J.J., Jr.; Rocha-Lima, C.M.; Safran, H.; Chan, D.; Kocs, D.M.; Galimi, F.; et al. A randomized, placebo-controlled phase 2 study of ganitumab (AMG 479) or conatumumab (AMG 655) in combination with gemcitabine in patients with metastatic pancreatic cancer. Ann. Oncol. 2012, 23, 2834–2842. [Google Scholar] [CrossRef] [PubMed]
- LoRusso, P.; Hong, D.; Heath, E.; Kurzrock, R.; Wang, D.; Hsu, M.; Goyal, L.; Wiezorek, J.; Storgard, C.; Herbst, R. First-in-human study of AMG 655, a pro-apoptotic TRAIL receptor-2 agonist, in adult patients with advanced solid tumors. J. Clin. Oncol. 2007, 25. [Google Scholar] [CrossRef]
- Beltran, P.J.; Mitchell, P.; Chung, Y.A.; Cajulis, E.; Lu, J.; Belmontes, B.; Ho, J.; Tsai, M.M.; Zhu, M.; Vonderfecht, S.; et al. AMG 479, a fully human anti-insulin-like growth factor receptor type I monoclonal antibody, inhibits the growth and survival of pancreatic carcinoma cells. Mol. Cancer Ther. 2009, 8, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Yada, A.; Yazawa, M.; Ishida, S.; Yoshida, H.; Ichikawa, K.; Kurakata, S.; Fujiwara, K. A novel humanized anti-human death receptor 5 antibody CS-1008 induces apoptosis in tumor cells without toxicity in hepatocytes. Ann. Oncol. 2008, 19, 1060–1067. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, K.; Liu, W.; Zhao, L.; Wang, Z.; Liu, D.; Ohtsuka, T.; Zhang, H.; Mountz, J.D.; Koopman, W.J.; Kimberly, R.P.; et al. Tumoricidal activity of a novel anti-human DR5 monoclonal antibody without hepatocyte cytotoxicity. Nat. Med. 2001, 7, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Buchsbaum, D.J.; Zhou, T.; Grizzle, W.E.; Oliver, P.G.; Hammond, C.J.; Zhang, S.; Carpenter, M.; LoBuglio, A.F. Antitumor efficacy of TRA-8 anti-DR5 monoclonal antibody alone or in combination with chemotherapy and/or radiation therapy in a human breast cancer model. Clin. Cancer Res. 2003, 9, 3731–3741. [Google Scholar] [PubMed]
- Straughn, J.M., Jr.; Oliver, P.G.; Zhou, T.; Wang, W.; Alvarez, R.D.; Grizzle, W.E.; Buchsbaum, D.J. Anti-tumor activity of TRA-8 anti-death receptor 5 (DR5) monoclonal antibody in combination with chemotherapy and radiation therapy in a cervical cancer model. Gynecol. Oncol. 2006, 101, 46–54. [Google Scholar] [CrossRef] [PubMed]
- DeRosier, L.C.; Huang, Z.Q.; Sellers, J.C.; Buchsbaum, D.J.; Vickers, S.M. Treatment with gemcitabine and TRA-8 anti-death receptor-5 mAb reduces pancreatic adenocarcinoma cell viability in vitro and growth in vivo. J. Gastrointest. Surg. 2006, 10, 1291–1300, discussion 1300. [Google Scholar] [CrossRef] [PubMed]
- Derosier, L.C.; Vickers, S.M.; Zinn, K.R.; Huang, Z.; Wang, W.; Grizzle, W.E.; Sellers, J.; Stockard, C.R., Jr.; Zhou, T.; Oliver, P.G.; et al. TRA-8 anti-DR5 monoclonal antibody and gemcitabine induce apoptosis and inhibit radiologically validated orthotopic pancreatic tumor growth. Mol. Cancer Ther. 2007, 6, 3198–3207. [Google Scholar] [CrossRef] [PubMed]
- Forero-Torres, A.; Shah, J.; Wood, T.; Posey, J.; Carlisle, R.; Copigneaux, C.; Luo, F.; Wojtowicz-Praga, S.; Percent, I.; Saleh, M. Phase I Trial of Weekly Tigatuzumab, an Agonistic Humanized Monoclonal Antibody Targeting Death Receptor 5 (DR5). Cancer Biother. Radiopharm. 2010, 25, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Forero-Torres, A.; Infante, J.R.; Waterhouse, D.; Wong, L.; Vickers, S.; Arrowsmith, E.; He, A.R.; Hart, L.; Trent, D.; Wade, J.; et al. Phase 2, multicenter, open-label study of tigatuzumab (CS-1008), a humanized monoclonal antibody targeting death receptor 5, in combination with gemcitabine in chemotherapy-naive patients with unresectable or metastatic pancreatic cancer. Cancer Med. 2013, 2, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Dhein, J.; Daniel, P.T.; Trauth, B.C.; Oehm, A.; Möller, P.; Krammer, P.H. Induction of apoptosis by monoclonal antibody anti-APO-1 class switch variants is dependent on cross-linking of APO-1 cell surface antigens. J. Immunol. 1992, 149, 3166–3173. [Google Scholar] [PubMed]
- Wajant, H.; Moosmayer, D.; Wuest, T.; Bartke, T.; Gerlach, E.; Schonherr, U.; Peters, N.; Scheurich, P.; Pfizenmaier, K. Differential activation of TRAIL-R1 and -2 by soluble and membrane TRAIL allows selective surface antigen-directed activation of TRAIL-R2 by a soluble TRAIL derivative. Oncogene 2001, 20, 4101–4106. [Google Scholar] [CrossRef] [PubMed]
- Thurber, G.M.; Schmidt, M.M.; Wittrup, K.D. Antibody tumor penetration: Transport opposed by systemic and antigen-mediated clearance. Adv. Drug Deliv. Rev. 2008, 60, 1421–1434. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog Signaling Enhances Delivery of Chemotherapy in a Mouse Model of Pancreatic Cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Koong, A.C.; Mehta, V.K.; Le, Q.T.; Fisher, G.A.; Terris, D.J.; Brown, J.M.; Bastidas, A.J.; Vierra, M. Pancreatic tumors show high levels of hypoxia. Int. J. Radiat. Oncol. Biol. Phys. 2000, 48, 919–922. [Google Scholar] [CrossRef]
- Wang, W.; Erbe, A.K.; Hank, J.A.; Morris, Z.S.; Sondel, P.M. NK Cell-Mediated Antibody-Dependent Cellular Cytotoxicity in Cancer Immunotherapy. Front. Immunol. 2015, 6, 368. [Google Scholar] [CrossRef] [PubMed]
- Wilson, N.S.; Yang, B.; Yang, A.; Loeser, S.; Marsters, S.; Lawrence, D.; Li, Y.; Pitti, R.; Totpal, K.; Yee, S.; et al. An Fcγ receptor-dependent mechanism drives antibody-mediated target-receptor signaling in cancer cells. Cancer Cell 2011, 19, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Graves, J.D.; Kordich, J.J.; Huang, T.H.; Piasecki, J.; Bush, T.L.; Sullivan, T.; Foltz, I.N.; Chang, W.; Douangpanya, H.; Dang, T.; et al. Apo2L/TRAIL and the death receptor 5 agonist antibody AMG 655 cooperate to promote receptor clustering and antitumor activity. Cancer Cell 2014, 26, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Tuthill, M.H.; Montinaro, A.; Zinngrebe, J.; Prieske, K.; Draber, P.; Prieske, S.; Newsom-Davis, T.; von Karstedt, S.; Graves, J.; Walczak, H. TRAIL-R2-specific antibodies and recombinant TRAIL can synergise to kill cancer cells. Oncogene 2015, 34, 2138–2144. [Google Scholar] [CrossRef] [PubMed]
- Todaro, M.; Lombardo, Y.; Francipane, M.G.; Alea, M.P.; Cammareri, P.; Iovino, F.; Di Stefano, A.B.; Di Bernardo, C.; Agrusa, A.; Condorelli, G.; et al. Apoptosis resistance in epithelial tumors is mediated by tumor-cell-derived interleukin-4. Cell Death Differ. 2008, 15, 762–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newsom-Davis, T.; Prieske, S.; Walczak, H. Is TRAIL the holy grail of cancer therapy? Apoptosis 2009, 14, 607–623. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, F.; Friess, H.; Kleeff, J.; Xu, Z.W.; Zimmermann, A.; Sheikh, M.S.; Büchler, M.W. Effects and expression of TRAIL and its apoptosis-promoting receptors in human pancreatic cancer. Cancer Lett. 2001, 163, 71–81. [Google Scholar] [CrossRef]
- Falschlehner, C.; Emmerich, C.H.; Gerlach, B.; Walczak, H. TRAIL signalling: Decisions between life and death. Int. J. Biochem. Cell Biol. 2007, 39, 1462–1475. [Google Scholar] [CrossRef] [PubMed]
- Trauzold, A.; Schmiedel, S.; Röder, C.; Tams, C.; Christgen, M.; Oestern, S.; Arlt, A.; Westphal, S.; Kapischke, M.; Ungefroren, H.; et al. Multiple and synergistic deregulations of apoptosis-controlling genes in pancreatic carcinoma cells. Br. J. Cancer 2003, 89, 1714–1721. [Google Scholar] [CrossRef] [PubMed]
- Trauzold, A.; Roder, C.; Sipos, B.; Karsten, K.; Arlt, A.; Jiang, P.; Martin-Subero, J.I.; Siegmund, D.; Muerkoster, S.; Pagerols-Raluy, L.; et al. CD95 and TRAF2 promote invasiveness of pancreatic cancer cells. FASEB J. 2005, 19, 620–622. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.W.; Punnoose, E.A.; Januario, T.; Lawrence, D.A.; Pitti, R.M.; Lancaster, K.; Lee, D.; von Goetz, M.; Yee, S.F.; Totpal, K.; et al. Death-receptor O-glycosylation controls tumor-cell sensitivity to the proapoptotic ligand Apo2L/TRAIL. Nat. Med. 2007, 13, 1070. [Google Scholar] [CrossRef] [PubMed]
- Dufour, F.; Rattier, T.; Shirley, S.; Picarda, G.; Constantinescu, A.A.; Morle, A.; Zakaria, A.B.; Marcion, G.; Causse, S.; Szegezdi, E.; et al. N-glycosylation of mouse TRAIL-R and human TRAIL-R1 enhances TRAIL-induced death. Cell Death Differ. 2017, 24, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Degli-Esposti, M.A.; Dougall, W.C.; Smolak, P.J.; Waugh, J.Y.; Smith, C.A.; Goodwin, R.G. The novel receptor TRAIL-R4 induces NF-κB and protects against TRAIL-mediated apoptosis, yet retains an incomplete death domain. Immunity 1997, 7, 813–820. [Google Scholar] [CrossRef]
- Braeuer, S.J.; Buneker, C.; Mohr, A.; Zwacka, R.M. Constitutively activated nuclear factor-κB, but not induced NF-κB, leads to TRAIL resistance by up-regulation of X-linked inhibitor of apoptosis protein in human cancer cells. Mol. Cancer Res. 2006, 4, 715–728. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Mayo, M.W.; Korneluk, R.G.; Goeddel, D.V.; Baldwin, A.S., Jr. NF-κB antiapoptosis: Induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science 1998, 281, 1680–1683. [Google Scholar] [CrossRef] [PubMed]
- Micheau, O.; Lens, S.; Gaide, O.; Alevizopoulos, K.; Tschopp, J. NF-κB signals induce the expression of c-FLIP. Mol. Cell. Biol. 2001, 21, 5299–5305. [Google Scholar] [CrossRef] [PubMed]
- Roux, J.; Hafner, M.; Bandara, S.; Sims, J.J.; Hudson, H.; Chai, D.; Sorger, P.K. Fractional killing arises from cell-to-cell variability in overcoming a caspase activity threshold. Mol. Syst. Biol. 2015, 11, 803. [Google Scholar] [CrossRef] [PubMed]
- Shlyakhtina, Y.; Pavet, V.; Gronemeyer, H. Dual role of DR5 in death and survival signaling leads to TRAIL resistance in cancer cells. Cell Death Dis. 2017, 8, e3025. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.; Thome, M.; Burns, K.; Bodmer, J.L.; Hofmann, K.; Kataoka, T.; Holler, N.; Tschopp, J. TRAIL receptors 1 (DR4) and 2 (DR5) signal FADD-dependent apoptosis and activate NF-κB. Immunity 1997, 7, 831–836. [Google Scholar] [CrossRef]
- Tran, S.E.; Holmstrom, T.H.; Ahonen, M.; Kahari, V.M.; Eriksson, J.E. MAPK/ERK overrides the apoptotic signaling from Fas, TNF, and TRAIL receptors. J. Biol. Chem. 2001, 276, 16484–16490. [Google Scholar] [CrossRef] [PubMed]
- Morel, J.; Audo, R.; Hahne, M.; Combe, B. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) induces rheumatoid arthritis synovial fibroblast proliferation through mitogen-activated protein kinases and phosphatidylinositol 3-kinase/Akt. J. Biol. Chem. 2005, 280, 15709–15718. [Google Scholar] [CrossRef] [PubMed]
- Azijli, K.; Weyhenmeyer, B.; Peters, G.J.; de Jong, S.; Kruyt, F.A. Non-canonical kinase signaling by the death ligand TRAIL in cancer cells: Discord in the death receptor family. Cell Death Differ. 2013, 20, 858–868. [Google Scholar] [CrossRef] [PubMed]
- Azijli, K.; Yuvaraj, S.; Peppelenbosch, M.P.; Wurdinger, T.; Dekker, H.; Joore, J.; van Dijk, E.; Quax, W.J.; Peters, G.J.; de Jong, S.; et al. Kinome profiling of non-canonical TRAIL signaling reveals RIP1-Src-STAT3-dependent invasion in resistant non-small cell lung cancer cells. J. Cell Sci. 2012, 125 Pt 19, 4651–4661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Zhou, J.Y.; Wei, W.Z.; Wu, G.S. Activation of the Akt survival pathway contributes to TRAIL resistance in cancer cells. PLoS ONE 2010, 5, e10226. [Google Scholar] [CrossRef] [PubMed]
- Lalaoui, N.; Morlé, A.; Mérino, D.; Jacquemin, G.; Iessi, E.; Morizot, A.; Shirley, S.; Robert, B.; Solary, E.; Garrido, C.; et al. TRAIL-R4 Promotes Tumor Growth and Resistance to Apoptosis in Cervical Carcinoma HeLa Cells through AKT. PLoS ONE 2011, 6, e19679. [Google Scholar] [CrossRef] [PubMed]
- Lafont, E.; Kantari-Mimoun, C.; Draber, P.; De Miguel, D.; Hartwig, T.; Reichert, M.; Kupka, S.; Shimizu, Y.; Taraborrelli, L.; Spit, M.; et al. The linear ubiquitin chain assembly complex regulates TRAIL-induced gene activation and cell death. EMBO J. 2017, 36, 1147–1166. [Google Scholar] [CrossRef] [PubMed]
- Ishimura, N.; Isomoto, H.; Bronk, S.F.; Gores, G.J. Trail induces cell migration and invasion in apoptosis-resistant cholangiocarcinoma cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G129–G136. [Google Scholar] [CrossRef] [PubMed]
- Trauzold, A.; Siegmund, D.; Schniewind, B.; Sipos, B.; Egberts, J.; Zorenkov, D.; Emme, D.; Roder, C.; Kalthoff, H.; Wajant, H. TRAIL promotes metastasis of human pancreatic ductal adenocarcinoma. Oncogene 2006, 25, 7434–7439. [Google Scholar] [CrossRef] [PubMed]
- Von Karstedt, S.; Conti, A.; Nobis, M.; Montinaro, A.; Hartwig, T.; Lemke, J.; Legler, K.; Annewanter, F.; Campbell, A.D.; Taraborrelli, L.; et al. Cancer cell-autonomous TRAIL-R signaling promotes KRAS-driven cancer progression, invasion, and metastasis. Cancer Cell 2015, 27, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Hoogwater, F.J.; Nijkamp, M.W.; Smakman, N.; Steller, E.J.; Emmink, B.L.; Westendorp, B.F.; Raats, D.A.; Sprick, M.R.; Schaefer, U.; Van Houdt, W.J.; et al. Oncogenic K-Ras turns death receptors into metastasis-promoting receptors in human and mouse colorectal cancer cells. Gastroenterology 2010, 138, 2357–2367. [Google Scholar] [CrossRef] [PubMed]
- Azijli, K.; Yuvaraj, S.; van Roosmalen, I.; Flach, K.; Giovannetti, E.; Peters, G.J.; de Jong, S.; Kruyt, F.A. MAPK p38 and JNK have opposing activities on TRAIL-induced apoptosis activation in NSCLC H460 cells that involves RIP1 and caspase-8 and is mediated by Mcl-1. Apoptosis 2013, 18, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Karl, I.; Jossberger-Werner, M.; Schmidt, N.; Horn, S.; Goebeler, M.; Leverkus, M.; Wajant, H.; Giner, T. TRAF2 inhibits TRAIL- and CD95L-induced apoptosis and necroptosis. Cell Death Dis. 2014, 5, e1444. [Google Scholar] [CrossRef] [PubMed]
- Varfolomeev, E.; Maecker, H.; Sharp, D.; Lawrence, D.; Renz, M.; Vucic, D.; Ashkenazi, A. Molecular determinants of kinase pathway activation by Apo2 ligand/tumor necrosis factor-related apoptosis-inducing ligand. J. Biol. Chem. 2005, 280, 40599–40608. [Google Scholar] [CrossRef] [PubMed]
- Hacker, H.; Karin, M. Regulation and function of IKK and IKK-related kinases. Sci. STKE 2006, 2006, re13. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, M.J.; Milutinovic, S.; Dickson, K.M.; Ho, W.C.; Boudreault, A.; Durkin, J.; Gillard, J.W.; Jaquith, J.B.; Morris, S.J.; Barker, P.A. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol. Cell 2008, 30, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Rahighi, S.; Ikeda, F.; Kawasaki, M.; Akutsu, M.; Suzuki, N.; Kato, R.; Kensche, T.; Uejima, T.; Bloor, S.; Komander, D.; et al. Specific recognition of linear ubiquitin chains by NEMO is important for NF-κB activation. Cell 2009, 136, 1098–1109. [Google Scholar] [PubMed]
- Ganten, T.M.; Sykora, J.; Koschny, R.; Batke, E.; Aulmann, S.; Mansmann, U.; Stremmel, W.; Sinn, H.P.; Walczak, H. Prognostic significance of tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) receptor expression in patients with breast cancer. J. Mol. Med. 2009, 87, 995–1007. [Google Scholar] [CrossRef] [PubMed]
- Macher-Goeppinger, S.; Aulmann, S.; Tagscherer, K.E.; Wagener, N.; Haferkamp, A.; Penzel, R.; Brauckhoff, A.; Hohenfellner, M.; Sykora, J.; Walczak, H.; et al. Prognostic value of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and TRAIL receptors in renal cell cancer. Clin. Cancer Res. 2009, 15, 650–659. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Williams, J.L.; Greenwood, S.L.; Baker, P.N.; Aplin, J.D.; Crocker, I.P. A placental protective role for trophoblast-derived TNF-related apoptosis-inducing ligand (TRAIL). Placenta 2009, 30, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Siegmund, D.; Klose, S.; Zhou, D.; Baumann, B.; Roder, C.; Kalthoff, H.; Wajant, H.; Trauzold, A. Role of caspases in CD95L- and TRAIL-induced non-apoptotic signalling in pancreatic tumour cells. Cell. Signal. 2007, 19, 1172–1184. [Google Scholar] [CrossRef] [PubMed]
- Bertsch, U.; Röder, C.; Kalthoff, H.; Trauzold, A. Compartmentalization of TNF-related apoptosis-inducing ligand (TRAIL) death receptor functions: Emerging role of nuclear TRAIL-R2. Cell Death Dis. 2014, 5, e1390. [Google Scholar] [CrossRef] [PubMed]
- Boyerinas, B.; Park, S.M.; Hau, A.; Murmann, A.E.; Peter, M.E. The role of let-7 in cell differentiation and cancer. Endocr.-Relat. Cancer 2010, 17, F19–F36. [Google Scholar] [CrossRef] [PubMed]
- Barh, D.; Malhotra, R.; Ravi, B.; Sindhurani, P. MicroRNA let-7: An emerging next-generation cancer therapeutic. Curr. Oncol. 2010, 17, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Haselmann, V.; Kurz, A.; Bertsch, U.; Hubner, S.; Olempska-Muller, M.; Fritsch, J.; Hasler, R.; Pickl, A.; Fritsche, H.; Annewanter, F.; et al. Nuclear death receptor TRAIL-R2 inhibits maturation of let-7 and promotes proliferation of pancreatic and other tumor cells. Gastroenterology 2014, 146, 278–290. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, Y.; Steller, H. Live to die another way: Modes of programmed cell death and the signals emanating from dying cells. Nat. Rev. Mol. Cell Biol. 2015, 16, 329. [Google Scholar] [CrossRef] [PubMed]
- Jouan-Lanhouet, S.; Arshad, M.I.; Piquet-Pellorce, C.; Martin-Chouly, C.; Le Moigne-Muller, G.; Van Herreweghe, F.; Takahashi, N.; Sergent, O.; Lagadic-Gossmann, D.; Vandenabeele, P.; et al. TRAIL induces necroptosis involving RIPK1/RIPK3-dependent PARP-1 activation. Cell Death Differ. 2012, 19, 2003. [Google Scholar] [CrossRef] [PubMed]
- Meylan, E.; Tschopp, J. The RIP kinases: Crucial integrators of cellular stress. Trends Biochem. Sci. 2005, 30, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Oberst, A.; Dillon, C.P.; Weinlich, R.; McCormick, L.L.; Fitzgerald, P.; Pop, C.; Hakem, R.; Salvesen, G.S.; Green, D.R. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 2011, 471, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.; Peng, W.; Liu, Y.; Yan, C.; Maki, J.; Degterev, A.; Yuan, J.; Shi, Y. Structural basis of RIP1 inhibition by necrostatins. Structure 2013, 21, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Seifert, L.; Werba, G.; Tiwari, S.; Giao Ly, N.N.; Alothman, S.; Alqunaibit, D.; Avanzi, A.; Barilla, R.; Daley, D.; Greco, S.H.; et al. The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature 2016, 532, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Lemke, J.; von Karstedt, S.; Abd El Hay, M.; Conti, A.; Arce, F.; Montinaro, A.; Papenfuss, K.; El-Bahrawy, M.A.; Walczak, H. Selective CDK9 inhibition overcomes TRAIL resistance by concomitant suppression of cFlip and Mcl-1. Cell Death Differ. 2014, 21, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yang, X.; Xu, T.; Kong, Q.; Zhang, Y.; Shen, Y.; Wei, Y.; Wang, G.; Chang, K. Overcoming resistance to TRAIL-induced apoptosis in solid tumor cells by simultaneously targeting death receptors, c-FLIP and IAPs. Int. J. Oncol. 2016, 49, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Wang, Y.; Wang, X. Tagged and untagged TRAIL show different activity against tumor cells. Oncol. Lett. 2012, 4, 1301–1304. [Google Scholar] [CrossRef] [PubMed]
- Holland, P.M. Targeting Apo2L/TRAIL receptors by soluble Apo2L/TRAIL. Cancer Lett. 2013, 332, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Huet, H.A.; Growney, J.D.; Johnson, J.A.; Li, J.; Bilic, S.; Ostrom, L.; Zafari, M.; Kowal, C.; Yang, G.; Royo, A.; et al. Multivalent nanobodies targeting death receptor 5 elicit superior tumor cell killing through efficient caspase induction. mAbs 2014, 6, 1560–1570. [Google Scholar] [CrossRef] [PubMed]
- Van Audenhove, I.; Gettemans, J. Nanobodies as Versatile Tools to Understand, Diagnose, Visualize and Treat Cancer. EBioMedicine 2016, 8, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, K.P.; Isaacs, R.; Bilic, S.; Kentsch, K.; Huet, H.A.; Hofmann, M.; Rasco, D.; Kundamal, N.; Tang, Z.; Cooksey, J.; et al. Unexpected hepatotoxicity in a phase I study of TAS266, a novel tetravalent agonistic Nanobody(R) targeting the DR5 receptor. Cancer Chemother. Pharmacol. 2015, 75, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Gieffers, C.; Kluge, M.; Merz, C.; Sykora, J.; Thiemann, M.; Schaal, R.; Fischer, C.; Branschadel, M.; Abhari, B.A.; Hohenberger, P.; et al. APG350 induces superior clustering of TRAIL receptors and shows therapeutic antitumor efficacy independent of cross-linking via Fcgamma receptors. Mol. Cancer Ther. 2013, 12, 2735–2747. [Google Scholar] [CrossRef] [PubMed]
- Leng, Y.; Hou, J.; Jin, J.; Zhang, M.; Ke, X.; Jiang, B.; Pan, L.; Yang, L.; Zhou, F.; Wang, J.; et al. Circularly permuted TRAIL plus thalidomide and dexamethasone versus thalidomide and dexamethasone for relapsed/refractory multiple myeloma: A phase 2 study. Cancer Chemother. Pharmacol. 2017, 79, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Geng, C.; Hou, J.; Zhao, Y.; Ke, X.; Wang, Z.; Qiu, L.; Xi, H.; Wang, F.; Wei, N.; Liu, Y.; et al. A multicenter, open-label phase II study of recombinant CPT (Circularly Permuted TRAIL) plus thalidomide in patients with relapsed and refractory multiple myeloma. Am. J. Hematol. 2014, 89, 1037–1042. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Qiu, L.; Hou, J.; Zhang, X.; Ke, X.; Wang, Z. Phase Ib study of recombinant circularly permuted TRAIL (CPT) in relapsed or refractory multiple myeloma patients. Blood 2012, 120, 1857. [Google Scholar]
- Fang, F.; Wang, A.P.; Yang, S.F. Antitumor activity of a novel recombinant mutant human tumor necrosis factor-related apoptosis-inducing ligand. Acta Pharmacol. Sin. 2005, 26, 1373–1381. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wen, X.Z.; Bu, Z.D.; Cheng, X.J.; Xing, X.F.; Wang, X.H.; Zhang, L.H.; Guo, T.; Du, H.; Hu, Y.; et al. Paclitaxel enhances tumoricidal potential of TRAIL via inhibition of MAPK in resistant gastric cancer cells. Oncol. Rep. 2016, 35, 3009–3017. [Google Scholar] [CrossRef] [PubMed]
- Jaworska, D.; Szliszka, E. Targeting Apoptotic Activity Against Prostate Cancer Stem Cells. Int. J. Mol. Sci. 2017, 18, 1648. [Google Scholar] [CrossRef] [PubMed]
- Min, S.Y.; Byeon, H.J.; Lee, C.; Seo, J.; Lee, E.S.; Shin, B.S.; Choi, H.G.; Lee, K.C.; Youn, Y.S. Facile one-pot formulation of TRAIL-embedded paclitaxel-bound albumin nanoparticles for the treatment of pancreatic cancer. Int. J. Pharm. 2015, 494, 506–515. [Google Scholar] [CrossRef] [PubMed]
- Wicker, C.A.; Sahu, R.P.; Kulkarni-Datar, K.; Srivastava, S.K.; Brown, T.L. BITC Sensitizes Pancreatic Adenocarcinomas to TRAIL-induced Apoptosis. Cancer Growth Metastasis 2010, 2009, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Henrich, C.J.; Brooks, A.D.; Erickson, K.L.; Thomas, C.L.; Bokesch, H.R.; Tewary, P.; Thompson, C.R.; Pompei, R.J.; Gustafson, K.R.; McMahon, J.B.; et al. Withanolide E sensitizes renal carcinoma cells to TRAIL-induced apoptosis by increasing cFLIP degradation. Cell Death Dis. 2015, 6, e1666. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Lee, D.H.; Kim, J.L.; Kim, B.R.; Na, Y.J.; Jo, M.J.; Jeong, Y.A.; Lee, S.Y.; Lee, S.I.; Lee, Y.Y.; et al. Metformin enhances TRAIL-induced apoptosis by Mcl-1 degradation via Mule in colorectal cancer cells. Oncotarget 2016, 7, 59503–59518. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, M.Z.; Romero, M.A.; Attar, R.; Javed, Z.; Farooqi, A.A. TRAIL and Bortezomib: Killing cancer with two stones. Asian Pac. J. Cancer Prev. 2015, 16, 1671–1674. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Park, S.B.; Kim, S.A.; Kwon, S.K.; Cha, H.; Lee, D.Y.; Ro, S.; Cho, J.M.; Song, S.Y. A novel HDAC inhibitor, CG200745, inhibits pancreatic cancer cell growth and overcomes gemcitabine resistance. Sci. Rep. 2017, 7, 41615. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, T.; Wakimoto, N.; Yin, D.; Gery, S.; Kawamata, N.; Takai, N.; Komatsu, N.; Chumakov, A.; Imai, Y.; Koeffler, H.P. Histone deacetylase inhibitor, suberoylanilide hydroxamic acid (Vorinostat, SAHA) profoundly inhibits the growth of human pancreatic cancer cells. Int. J. Cancer 2007, 121, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Arhoma, A.; Chantry, A.D.; Haywood-Small, S.L.; Cross, N.A. SAHA-induced TRAIL-sensitisation of Multiple Myeloma cells is enhanced in 3D cell culture. Exp. Cell Res. 2017, 360, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Feng, X.; Han, H.; Guo, S.; Wang, G. Synergistic effects of combined treatment with histone deacetylase inhibitor suberoylanilide hydroxamic acid and TRAIL on human breast cancer cells. Sci. Rep. 2016, 6, 28004. [Google Scholar] [CrossRef] [PubMed]
- Hari, Y.; Harashima, N.; Tajima, Y.; Harada, M. Bcl-xL inhibition by molecular-targeting drugs sensitizes human pancreatic cancer cells to TRAIL. Oncotarget 2015, 6, 41902–41915. [Google Scholar] [CrossRef] [PubMed]
- Perimenis, P.; Galaris, A.; Voulgari, A.; Prassa, M.; Pintzas, A. IAP antagonists Birinapant and AT-406 efficiently synergise with either TRAIL, BRAF, or BCL-2 inhibitors to sensitise BRAFV600E colorectal tumour cells to apoptosis. BMC Cancer 2016, 16, 624. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.S.; Wang, G.F.; Zhao, Z.Q.; Liang, Y.; Wang, H.B.; Wu, M.Y.; Min, P.; Chen, L.Z.; Feng, Q.S.; Bei, J.X.; et al. Smac mimetics in combination with TRAIL selectively target cancer stem cells in nasopharyngeal carcinoma. Mol. Cancer Ther. 2013, 12, 1728–1737. [Google Scholar] [CrossRef] [PubMed]
- Stern, H.M.; Padilla, M.; Wagner, K.; Amler, L.; Ashkenazi, A. Development of immunohistochemistry assays to assess GALNT14 and FUT3/6 in clinical trials of dulanermin and drozitumab. Clin. Cancer Res. 2010, 16, 1587–1596. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kretz, A.-L.; Von Karstedt, S.; Hillenbrand, A.; Henne-Bruns, D.; Knippschild, U.; Trauzold, A.; Lemke, J. Should We Keep Walking along the Trail for Pancreatic Cancer Treatment? Revisiting TNF-Related Apoptosis-Inducing Ligand for Anticancer Therapy. Cancers 2018, 10, 77. https://doi.org/10.3390/cancers10030077
Kretz A-L, Von Karstedt S, Hillenbrand A, Henne-Bruns D, Knippschild U, Trauzold A, Lemke J. Should We Keep Walking along the Trail for Pancreatic Cancer Treatment? Revisiting TNF-Related Apoptosis-Inducing Ligand for Anticancer Therapy. Cancers. 2018; 10(3):77. https://doi.org/10.3390/cancers10030077
Chicago/Turabian StyleKretz, Anna-Laura, Silvia Von Karstedt, Andreas Hillenbrand, Doris Henne-Bruns, Uwe Knippschild, Anna Trauzold, and Johannes Lemke. 2018. "Should We Keep Walking along the Trail for Pancreatic Cancer Treatment? Revisiting TNF-Related Apoptosis-Inducing Ligand for Anticancer Therapy" Cancers 10, no. 3: 77. https://doi.org/10.3390/cancers10030077