Role of Pattern Recognition Receptors in KSHV Infection


1
Department of Microbiology and Immunology, University of Nevada, Reno, School of Medicine, 1664 N, Virginia Street, MS 320, Reno, NV 89557, USA
2
Department of Internal Medicine, UTMB Galveston, League City Campus, 301 University Blvd, Galveston, TX 77555, USA
*
Author to whom correspondence should be addressed.
Cancers 2018, 10(3), 85; https://doi.org/10.3390/cancers10030085
Submission received: 27 January 2018
/
Revised: 12 March 2018
/
Accepted: 16 March 2018
/
Published: 20 March 2018
(This article belongs to the Special Issue Cancer Biomarkers)
Abstract
:Kaposi’s sarcoma-associated herpesvirus or Human herpesvirus-8 (KSHV/HHV-8), an oncogenic human herpesvirus and the leading cause of cancer in HIV-infected individuals, is a major public health concern with recurring reports of epidemics on a global level. The early detection of KSHV virus and subsequent activation of the antiviral immune response by the host’s immune system are crucial to prevent KSHV infection. The host’s immune system is an evolutionary conserved system that provides the most important line of defense against invading microbial pathogens, including viruses. Viruses are initially detected by the cells of the host innate immune system, which evoke concerted antiviral responses via the secretion of interferons (IFNs) and inflammatory cytokines/chemokines for elimination of the invaders. Type I IFN and cytokine gene expression are regulated by multiple intracellular signaling pathways that are activated by germline-encoded host sensors, i.e., pattern recognition receptors (PRRs) that recognize a conserved set of ligands, known as ‘pathogen-associated molecular patterns (PAMPs)’. On the contrary, persistent and dysregulated signaling of PRRs promotes numerous tumor-causing inflammatory events in various human cancers. Being an integral component of the mammalian innate immune response and due to their constitutive activation in tumor cells, targeting PRRs appears to be an effective strategy for tumor prevention and/or treatment. Cellular PRRs are known to respond to KSHV infection, and KSHV has been shown to be armed with an array of strategies to selectively inhibit cellular PRR-based immune sensing to its benefit. In particular, KSHV has acquired specific immunomodulatory genes to effectively subvert PRR responses during the early stages of primary infection, lytic reactivation and latency, for a successful establishment of a life-long persistent infection. The current review aims to comprehensively summarize the latest advances in our knowledge of role of PRRs in KSHV infections.
1. Introduction
Pathogens such as bacteria, fungi, viruses, and parasites (protozoa and worms) have long been implicated in several human malignancies, with viruses alone being identified as etiological agents of about 15–25% of different human cancers, worldwide [1,2,3]. Viruses, in general, are metabolically inert but can grow rapidly inside the host cell and kill the infected cell releasing progeny virions. Some viruses, such as tumor causing viruses or oncoviruses, on the other hand, may mediate transformation of an infected host cell into a cancer cell and may establish long-term persistent infection while maintaining low levels of viral replication. These oncoviruses either integrate into the host genome or persist as an episome and express viral oncogenes to instigate abnormal cellular proliferation and multistep tumor development (tumorigenesis). Tumor virus infections are generally asymptomatic however, it is the persistent nature of the latent and the lytic infection that triggers their oncogenic potential and stimulates tumor formation in their hosts. Presently, seven tumor-causing DNA/RNA viruses are known to exist and cause cancers in humans. They include Hepatitis B and C Viruses (HBV and HCV), Human Papillomavirus (HPV, Type 16 and 18), gammaherpesviruses namely, Epstein-Barr Virus (EBV) and Kaposi’s Sarcoma-associated Herpesvirus (KSHV), recently identified, Merkel Cell Polyomavirus (MCPyV) and Human T-cell lymphotrophic virus-1 (HTLV-1) [4,5]. Tumorigenesis is a multistep process, and although these seven oncogenic viruses follow diverse strategies and cause different human cancers, yet, as proposed by Weinburg et al. tumors in humans display some key biological characteristics, including the evasion of growth suppressors, apoptosis, induction of angiogenesis, invasion, metastasis, tumor-promoting inflammation, and evasion of immune destruction, to name a few [6,7]. These characteristics, often called “hallmarks of cancer”, are acquired by mammalian cells to transform, survive, proliferate and become tumorigenic.
Decades of intensive tumor virus and associated cancer research have made it increasingly clear that the interplay between host-intrinsic immunity and virus-mediated tumorigenesis is complex. The host immune system is a surveillance system that, via numerous innate immune and adaptive immune responses, protects the host against the intruding pathogens, tissue damage and metabolic disorders [8,9]. The main components of the innate immune system (i.e., macrophages, dendritic cells, and natural killer cells) play a pivotal role in modulating viral infection and determining disease severity and outcome by triggering (i) activation of type I interferon (IFN-α/β) signaling, (ii) activation of multiple cellular cytokines and chemokines, and subsequent (iii) initiation of host adaptive (antigen-specific) immune signals [10]. Hence, it is quintessential for invading viruses to efficiently antagonize cellular immune responses and immune checkpoints, to secure viral infection.
A growing body of information from clinical and epidemiological studies, along with studies using animal models of human cancer, provides evolving insights into the immune system as an important barrier to tumor formation and progression [11]. Research has also shed light on the involvement of immune suppression, and the role of immunosuppressive lymphoid and myeloid cells in the development of virus-induced tumors. For instance, a spontaneous tumor formation occurs in mice genetically deficient in IFN-γ, as compared to wild-type mice [12,13]. In addition, immune cell-deficient mice demonstrated increased susceptibility towards cell proliferation and tumor formation [11]. Further, severe immunosuppressed patients and individuals are reported to display higher incidence for viral oncogenicity [14,15,16]. KSHV/HHV-8-linked Kaposi’s Sarcoma (KS) and EBV-positive B-cell lymphomas are the most prominent tumors under these perturbed immune conditions [17,18,19].
2. KSHV Infections, Diagnosis and Treatment
2.1. KSHV Genome and Associated Malignancies
KSHV/HHV-8 is an oncogenic, large, enveloped, double-stranded (ds) DNA virus, that has been classified as class I carcinogen by International Agency for Research on Cancer (IARC). KSHV has been shown to effectively infect and persist in mammalian B-lymphocytes/endothelial cells (of different lineage) and can induce multiple sarcomas and lymphomas in the infected hosts. KSHV infection is unequivocally linked to KS, a leading cancer among untreated HIV-positive individuals or immune compromised organ transplant recipients [20,21]. The discovery of KSHV in 1994 from KS lesions played an instrumental role for our understanding of this disease [22]. KS is an unusual, aggressive, low-grade mesenchymal tumor of highly proliferative endothelial cells, characterized by infiltrative red/brown/purple lesions on the skin, mucosal surfaces, lymph nodes, and internal organs (such as lungs and digestive tracts) [21,23]. KS has been classified into four distinct epidemiological-clinical forms, i.e., classic/sporadic KS, a relatively aggressive endemic/African KS, an iatrogenic KS/post-transplant KS, and acquired immune deficiency syndrome (AIDS)-related epidemic KS [24,25,26,27]. Also, KSHV latent viral genome and gene products have been detected in all these forms of KS. KSHV has been characterized as a lymphotropic oncogenic herpesvirus, due to its association with two distinct B-cell lymphoproliferative diseases, namely, primary effusion lymphoma [20], and multicentric Castleman’s disease (MCD)-linked plasmablastic lymphoma [28]. PEL, also referred to as body cavity-based lymphoma (BCBL), is a B-cell non-Hodgkin’s lymphoma predominantly found in AIDS patients with compromised immune status, and is characterized by lymphomatous effusion tumor growth in body cavities. KSHV-MCD, on the other hand, is a rare but rapidly progressing polyclonal B-cell lymphoproliferative disorder which is characterized by vascular endothelial proliferation of the lymph nodes and lymphoid tissues in the KSHV/HIV-infected patients [29,30].
The infection with KSHV begins with the attachment of viral envelope glycoproteins to several host membrane receptors that are present on the cell surface of target cells [31]. Upon entry, the viral capsid is released in the cytoplasm and transported to the nuclear periphery followed by release of the viral genome into the nucleus [31,32,33]. Inside the nucleus, KSHV linear dsDNA is circularized and chromatinized to persist as a latent “minichromosome” or “episome”, which transcribes and replicates along with the host chromosome in proliferating infected cells for efficient long-term infection [31,32,33]. KSHV can exhibit either a latent, non-productive or a lytic, productive infection. However, like other gammaherpesviruses, KSHV prefers a prolonged latency phase with a restricted latent transcriptional program. The stringent default latency period is reversible, and changes in the host cell environment can reactivate the silent virus towards the lytic cycle, wherein tightly controlled and coordinated expression of multiple viral lytic genes allow viral genome amplification, virion assembly, and progeny virus production [34]. KSHV also displays a transient but abortive lytic cycle gene program following de novo infection, as some lytic transcripts have been detected in transcriptome analyses of KSHV-infected cells [35,36]. Indeed, KSHV can support different transcription profiles in lymphatic endothelial cells and blood vascular endothelial cells [37]. These findings imply that the lytic phase also plays a central role in the stable maintenance of the viral genome, an essential pre-requisite for the persistent infection and development of KSHV-induced tumors [38].
2.2. KS Diagnostic Markers and Therapeutic Approaches
Differential diagnosis of malignant, soft-tissue KS vascular tumors has been difficult, especially in the presence of other benign/malignant/non-vascular soft-tissue neoplasms. Immuno-histochemical staining and PCR detection of KSHV viral genome/DNA sequences in KS tissue are two major tools for diagnosis and clinical evaluation of KSHV-associated malignancies. Histologically, KS tumors are characterized by the presence of KSHV latency-associated nuclear antigen (LANA) [39] positive elongated spindle-like, poorly differentiated endothelial cells, and inflammatory infiltrates involving monocytes, B and T cells [40]. Studies have demonstrated that low peripheral blood CD4 lymphocyte count is a significant marker associated with AIDS-related KS [41]. As KS lesions harbor KSHV in its latent form, expression of KSHV latency transcripts, including major nuclear latency protein LANA (OR73), vFLIP (ORF71 or K13), vCyclin (ORF72), Kaposins (K12), and a cluster of viral microRNAs (miRNAs), serve as useful biomarkers of KS tumorigenesis [42,43,44,45]. LANA has been shown to dysregulate the p53 and pRb tumor suppressors in KSHV-infected cells [46,47]. Since KSHV can display both latent/lytic gene activity during KS infection, some KSHV lytic gene transcripts, such as vGPCR (viral G-protein coupled receptor), vIL-6 (viral Interleukin-6), vBcl2 homolog, vMIPs (viral macrophage inflammatory protein), and vIRF-1 and -3 (viral IFN regulatory factor), have also been detected in KS and PEL-tumor isolates [48,49,50]. Overexpression of vascular endothelial cell markers including CD36, CD138/syndecan-1 and factor XIII has been found in several KSHV latency models (Reviewed in [51,52,53]). Studies of KS tumors have found over-expression of selective lymphatic and blood vessel endothelial markers, such as LYVE-1 (Lymphatic vessel endothelial receptor-1), VEGFR-3 (Vascular endothelial growth factor receptor 3 precursor), PDPN (podoplanin), CXCR4 (Chemokine C-X-C motif receptor 4), DLL4 (Delta like protein 4), and CXCL12/SDF1 (Stromal cell derived factor 1) [54,55,56]. Rosado and colleagues have demonstrated the use of monoclonal antibodies directed against CD31, CD34, D2-40, and FLI1 (Friend leukemia integration 1 transcription factor) as specific and selective markers of endothelial differentiation in different clinical subtypes and tumor stages of KS [57]. Based on current available data, higher levels of inflammatory cytokines and pro-angiogenic growth factors, such as IFN-γ, interleukin 1/6 (IL-1/6), oncostatin M, TNF-α/β (tumor necrosis factor-α/β) and bFGFs (basic fibroblast growth factor) that favor the development of tumors, have been reported in HIV-positive KSHV-infected patients [58,59,60]. Detailed information on current biomarkers in KSHV-linked malignancies is summarized in Figure 1 and Table 1.
Survival of patients with KS has remained poor due to a combination of poor prognosis and lack of effective treatment therapies. The current therapeutic approaches available for the treatment of KS patients include combined antiretroviral therapy (cART), cytotoxic chemotherapy (i.e., liposomal polyethylene glycol doxorubicin-Doxil), radiotherapy, and surgery [102,103]. Though cART improves KS, cART-initiated regression of KS has been linked to immune reconstitution inflammatory syndrome (IRIS) of varying severity in some HIV-infected patients [104]. Standard cytotoxic chemotherapy is effective; however, access to this treatment of choice is limited and rarely leads to complete responses in advanced KS [105]. Though a number of nucleoside analog-based antiviral drugs, such as ganciclovir, acyclovir, foscarnet, and cidofovir have been reported to induce KS regression, these drugs have significant toxicities and fewer case reports suggest reduced KS incidence [106,107,108,109]. Several novel drugs including cepharanthine, and diethyldithiocarbamate (NF-κB inhibitors), bevacizumab (angiogenesis inhibitors), sorafenib and imatinib (receptor tyrosine kinase inhibitors), and rapamycin (mTOR inhibitors), are currently being developed for the treatment of KSHV infection; further investigations are however required to determine their efficacy and tolerability [110,111,112,113,114]. Although several therapeutic approaches are showing promising advancement in the treatment of KSHV-linked disorders in the infected patients, unfortunately, there is no definite cure for KSHV infections. Hence, there is a growing need to devise newer virus-targeted therapies without the toxicity of chemotherapy for the prevention/early treatment of KSHV-positive malignancies. In this regard, a detailed understanding of interplay between KSHV and the infected host’s immune system could assist in the development of KSHV therapeutics.
3. KSHV Inhibition of PRR-Dependent Immune Responses
Innate immunity is the first and most rapid line of host surveillance against intruding microbial pathogens. Host cell mediated intracellular discrimination of harmless “self” (cellular DNA/RNA) and harmful “non-self” (viral DNA/RNA) is mediated through germline-encoded proteins, known as pattern recognition receptors (PRRs). PRRs are located either on the cell surface, or within distinct cellular compartments of the cytosol, and respond to microbial molecular marks, known as pathogen-associated molecular patterns (PAMPs) (Figure 2) [115,116,117]. PAMPs are essential functional components of pathogens, such as microbial nucleic acids (dsDNA/dsRNA/single stranded (ss)RNA/5′-triphosphate RNA), lipopolysaccharide (LPS), lipoproteins, envelope glycoproteins, and peptidoglycans [117]. Sensing viral PAMPs, present in the incoming virus or released during viral replication, activate innate immune signals by cellular PRRs to produce type I interferon (IFNs) and multiple antiviral cytokines, that result in the robust expression of several antiviral proteins to promote elimination of invading viruses. Transient yet robust antiviral inflammatory cytokine activation not only recruits immune cells to the site of viral infection, but also stimulates antigen-specific adaptive immune responses to control the spread of the virus.
There are four major families of PRRs including Toll-like receptors (TLRs), nucleotide oligomerization domain (NOD)-like receptors (NLRs), retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs) and intracellular DNA sensors (e.g., cGAS, IFI16) (Reviewed in [117,118,119]) that recognize virus-associated nucleic acids in the endosomes, nucleus, and cytoplasm of the infected cells and initiate multiple immunomodulatory and inflammatory pathways through a complex yet well-coordinated signaling system. The TLR family of receptors are membrane-bound receptors whereas RLRs and NLRs are cytosolic receptors [115]. During herpesvirus infection, the viral genome and viral latency/lytic transcripts (ssRNA/dsRNA) often serve as immunogenic molecular ligands to cellular PRRs (Reviewed in [114]).
3.1. TLRs in KSHV Infection
Members of the TLR family are the most extensively studied and characterized PRRs (Reviewed in [120]). TLRs are type-I transmembrane glycoproteins, and are expressed by macrophages, neutrophils, Dendritic Cells, Natural Killer cells, epithelial and endothelial cells. The mammalian TLR family comprises of 10 members, designated as TLRs 1–10. In comparison to humans, 12 TLRs have been identified in mice numbered as TLR 1–9 and TLR 11–13 (TLR 10 is identified as a pseudogene) [121]. All TLRs shares a common structural organization and consist of an ectodomain containing leucine-rich repeats (LRRs), a transmembrane domain and a cytoplasmic Toll/Interleukin-1 (IL-1) receptor signaling domain [122]. Cell-surface expressed TLRs (TLR-1, -2, -4, -5, -6 and -10) mainly recognize pathogenic wall components, including lipids, proteins and polysaccharides, whereas endosomal TLRs (TLR-3, -7, -8, and -9) sense microbial nuclei acids, such as, viral dsDNA and ssRNA [123]. In addition, TLRs can also sense endogenous danger-associated molecular patterns, such as host alarmins and inflammatory mediators [124]. Following viral infection, TLRs predominantly employ either of two pathways: the MyD88-dependent pathway or the TRIF-dependent pathway, via different TIR-domain containing adaptor proteins, such as MyD88, Mal (MyD88 adaptor-like protein), TIRAP (TIR-associated protein), TRIF and TRAM [125]. Recruitment of adaptor proteins brings about the activation of IRF-3/7 transcription factors, which culminates in the production of IFNs-α/β and chemokines including chemokine C-C motif ligands (CCLs) and C-X-C motif ligands (CXCLs) [126]. More and more evidence suggests the involvement of TLRs in multiple inflammation-driven diseases, along with pro- and anti-inflammatory cytokines, growth factors, and tumor suppressors [126]. Various TLR-based immune adjuvants have been validated for multiple clinical applications, including cancer immunotherapy [127].
KSHV has been shown to be recognized by TLRs throughout its lifecycle (Figure 3). The Damania’s group showed that KSHV infection of human monocytes upregulates TLR-3, IFN-β, CCL2, and CXCL10 transcripts [128,129]. However, KSHV-encoded vIRFs (viral interferon regulatory factors-1/2/3) inhibit TLR-3-mediated activation of IFN-responsive promoters and suppress TLR-3 and CXCL10 gene expression [129]. KSHV encodes four viral IRFs (vIRFs 1-4), that show homology with cellular IRFs, and are negative regulators of cellular IFN immune responses [92,129]. KSHV vIRF-1 (K9) and vIRF-2 (K11/K11.1) repress IRF-3-mediated IFN-β production upon TLR-3 activation, though via different mechanisms [92]. According to a recent report by Jacobs et al. [130], vIRF-1 interacts with the cellular ISG15 E3 ligase HERC5 and decreases global ISGylation associated with TLR-3 activation. Previous studies have shown that vIRF-2 reduced the activation of the ISRE (IFN-induced interferon-response element) promoter through de-regulation of ISGF-3 (IFN-stimulated gene factor-3) [131]. KSHV infection of endothelial cells downregulates TLR-4 signaling by vGPCR-mediated ERK activation and vIRF-1, which, in turn, suppresses expression of TNF-α, IL-1β, IL-6 and IFN-β [132]. In THP-1 monocytes, KSHV infection results in inhibition of TLR-2/-4 signaling [133]. Furthermore, TLRs also play an important role during reactivation of KSHV from latency as TLR-7/-8 activation with a TLR-8 ligand, i.e., ss-polyuridine (a synthetic ssRNA homolog), was shown to trigger KSHV reactivation in latently infected B lymphocytes [134]. In plasmacytoid dendritic cells, TLR-9 has been reported to recognize KSHV, and lead to upregulation of CD83, CD86 and elevated IFN-α production [135].
The immediate-early lytic protein, RTA, which jump-starts the entire lytic replication cascade, has been shown to utilize several different strategies to inhibit TLR-mediated signaling, and consequently block inflammatory cytokines and type I IFN production. RTA has been found to regulate IFN production by targeting IRF-7, TRIF, TLR-2, and TLR-4 receptors [136]. RTA was also recently reported to down regulate MyD88 expression through the ubiquitin (Ub)-proteasomal degradation of MyD88, thereby blocking the TLR-4 signaling mediated IFN production and NF-κB activity [137]. KSHV-encoded miRNAs, miR-K12-9 and miR-K12-5 are found to regulate TLR/IL-1R signaling by suppressing IRAK1 and MyD88 protein expression [138]. As reported by Yang et al. IRAK1 kinase is constitutively phosphorylated and essential for PEL survival in culture and IRAK1-driven TLR signaling is a driving force for the PEL growth [139].
3.2. Intracellular NLRs, RLRs, and Cytosolic DNA Receptors in KSHV Infection
NLRs are key sensors of intracellular pathogens in the host cell cytoplasm (Reviewed in [140,141]). The NLR family comprises of more than 23 cytoplasmic receptor proteins with a central nucleotide-binding domain (NOD), C-terminal leucine-rich repeats (LRRs), and an N-terminal protein binding motif, composed of a caspase-recruitment domain (CARD), or a pyrin domain (PYD), or a baculovirus inhibitor of apoptosis protein repeat (BIR) domain [115,141]. NLRs can activate the formation of large multi-scaffold protein complexes called inflammasomes, that are composed of an adaptor protein called ASC (apoptosis-associated speck-like protein containing a CARD) [142], pro-caspase-1, and an oligomer of a particular NLR. To date, several families of NLR inflammasomes have been identified, including NLRX1, NLRP1, NLRP3, and NLRC4. Among these, NLRP3 is the most studied and best characterized NLR family member [143]. Oligomerization of NLRs and their adaptor through CARD-CARD interactions is functionally important for regulation and activation of pro-inflammatory caspase 1, which results in the cleavage and maturation of pro-IL-1β and pro-IL-18 to their active forms IL-1β and IL-18, respectively [142,144]. Increased expression of proinflammatory mediators IL-1β and IL-18 has been observed to induce pyroptosis, defined as a caspase 1-mediated programmed cell death [145]. NOD1 and NOD2, the two predominant members of non-inflammasome NLRs are shown to activate IRF-3/7, NF-κB and MAPK signaling pathways in response to different PAMPs [146,147].
KSHV ORF63, a viral homolog of cellular NLRP1, reduces caspase-1 activity and lowers the production of “alarm” pro-inflammatory cytokines (Figure 4), IL-1β and IL-18, by altering the interaction between NLRP1 and pro-caspase 1 through its association with NLRP1 oligomerization domains. Additionally, ORF63 inhibits NOD2 and NLRP3 inflammasome responses to suppress NLR-mediated signaling [148]. Recently, the ubiquitously expressed NLRX1, a negative regulator of the type I IFN response has been shown to play an important role in KSHV reactivation from latency by inhibiting MAVS-dependent IFN-I signaling [149].
RLRs are long recognized as intracellular viral dsRNA sensors in the cytoplasm (reviewed in [150]). However, there are reports of their involvement in the sensing of DNA viruses, expanding their repertoire as a cytoplasmic viral nucleic acid sensor [151]. The RLR family of proteins currently consists of three known members, namely RIG-I/DDX58, the melanoma differentiation-associated gene 5, MDA5/IFIH1, and the regulatory homologue laboratory of genetics protein 2, LGP2/DHX58 [152]. These RLR proteins are widely expressed in different cell types and are characterized by a central DExH/D-box RNA helicase domain and a C-terminal domain (CTD). In addition, RIG-I and MDA5 have tandem N-terminal CARD domains that activate signaling cascades to induce antiviral pro-inflammatory as well as type-I IFN-stimulated genes (ISGs). RIG-I primarily recognizes short dsRNA with an adjacent 5′- tri- or di- phosphate moiety, whereas MDA5 binds to internal sites in the long dsRNA with no end specificity [153]. In the absence of dsRNA, RIG-I is maintained in an inactive conformation where the CARD domains are bound by the RIG-I C-terminal repressor domains and cannot interact with the membrane-bound mitochondrial antiviral signaling adaptor protein, MAVS/IPS-I/VISA/CARDIF [154]. However, upon dsRNA binding, RIG-I undergoes conformational rearrangements in favor of CARD-CARD interactions between RIG-I and MAVS, [155,156] leading to activation of kinases that activate NF-kB and IRF-3/7 and in turn, induce the promoter for type I IFNs (IFN-α/β) [10,157,158]. In contrast to RIG-I, dsRNA binding triggers the helicase domains of MDA5 to wrap around dsRNA leading to a 20° rotation of MDA5 CTD. The rotation of MDA5-CTD promotes ATP-dependent MDA5 cooperative filament formation on dsRNA and induces oligomerization of MDA5 CARDs, which in turn, forms a scaffold for oligomerization of MAVS CARD [153,159,160]. LGP2, that lacks CARD domains for signaling, is thought to influence IFNs production by regulating the activity of RIG-I and MDA5. Though the exact function of LGP2 in viral infections is still unclear, it is proposed that LGP2 inhibits IFN induction by sequestering PAMPs from RIG-I [161]. A study by Childs et al. reported that LGP2 plays an important role in sensitizing MDA5 to activation by dsRNA [162].
In comparison to TLR and NLR signaling, little is known about the role of RLR signaling during KSHV infection. West et al. showed that both RIG-I and MAVS play a role in IFN-β production following KSHV infection and can suppress KSHV replication and/or reactivation. Nevertheless, KSHV-encoded deubiquitinase (DUB), ORF64, has been shown to inhibit RIG-I signaling and activation of IFN-β via the prevention of ubiquitination of RIG-I [163]. Hence, RIG-I or MDA5 could be further exploited as promising oncogenic targets.
Cytosolic DNA sensors belong to the most recently identified members of the PRR family, and hence, lack full characterization among known PRRs. To date, several intracellular DNA sensors have been identified. DNA-dependent activator of IFN-regulatory factors (DAI), absent in melanoma 2 (AIM2), gamma interferon-inducible protein 16 (IFI16), and cyclic guanosine monophosphate-adenosine monophosphate (cyclic GMP-AMP synthase; cGAS) are the major cytosolic DNA sensors involved in innate immune responses, following viral infection [164,165,166,167,168,169]. However, unlike, TLR and NLR signaling, not much is known about the viral sensing by the cGAS-STING pathway. Upon binding of cytosolic DNA, cGAS dimerizes and generates cyclic-GMP-AMP (cGAMP), which binds to a deep pocket in the adaptor protein STING/MITA/MPYS/ERIS located in the ER membrane [170]. STING acts as a scaffold to recruit IFN-inducing proteins, namely TBK-1 (TNFR-associated NF-kB kinase (TANK)-binding kinase-1) and transcription factor IRF-3, leading to production and secretion of IFNs [171,172]. IFI16, a recently characterized receptor for dual sensing of nuclear and cytosolic viral DNA, has been identified to play a key role in the activation of STING/TBK1/IRF-3 signaling, required for induction of several IFNs, ISGs, and inflammasomes [173,174].
IFI16, a cytosolic DNA receptor, has been recognized as a stimulator of innate immunity upon KSHV infection. KSHV DNA has been shown to activate IFI16, resulting in the formation of IFI16-ASC-procaspase-1-inflammasome assembly to activate caspase-1 and IL-1β (Figure 4) [175,176]. The major products of inflammasome activation, IL-1β and IL-18, are pro-tumorigenic and shown to play an important role in tumor-mediated angiogenesis, hence blocking their function may suppress tumor progression. A recent study identified BRCA1 as an important constituent of IFI16-ASC-procaspase-1-inflammasome assembly that promotes induction of inflammasome and IFN-β responses during de novo KSHV infection [177]. Roy et al., for the first time, has recently demonstrated the role of IFI16 in maintenance of KSHV latency in KSHV-infected PEL cell lines, by functioning as a transcriptional repressor for viral lytic promoters [178]. Cytoplasmic isoforms of KSHV LANA recruit and antagonize the function of cGAS to inhibit cGAS-STING mediated restriction of KSHV lytic replication and promote reactivation from latency [179]. KSHV ORF52, a gammaherpesvirus specific virion tegument protein, disrupts the cGAS signaling pathway by inhibiting the enzymatic activity of cGAS and, as a consequence, cGAMP synthesis [180]. KSHV vIRF1 has been reported to block cGAS-STING-mediated antiviral immunity in endothelial cells by de-stabilizing TBK1 binding to STING [181]. Since intracellular DNA receptors play diverse yet critical roles during de novo infection, as well as in KSHV reactivation from latency, KSHV-encoded cGAS/STING/TBK-1 inhibitors may provide targets for developing diagnostic approaches for KS-mediated tumorigenesis.
4. Conclusions and Future Perspectives
It has been well established that viral immune evasion strategies play a critical role in virus-induced tumor escape from the host’s surveillance machinery. Accumulating evidence has indicated the involvement of nearly all known PPRs in diseases associated with each of the seven oncogenic viruses. Activation of PRR-mediated signaling, through interaction between PRRs and viral PAMPs at the surface of the cell, in the endocytic compartments, in the cytoplasm, or in the nucleus, promotes a diverse range of inflammatory, innate and adaptive immune responses. Hence targeting these cellular immune sensors, immunomediators and associated pathways represents a promising cancer prevention/treatment approach.
Interestingly, prolonged and dysregulated activation of PRRs reroutes the protective PRR-triggered pathways, key transcription factors and specialized cells of the immune system, causing chronic inflammation that facilitates tumor progression [182]. It has been proven that abnormal expression of PRRs favors tumorigenesis via release of various proliferative, anti-apoptotic, and angiogenic factors that contribute to the tumor microenvironment [183]. Mounting evidence inextricably suggests the association of nearly all known members of PRR families with increased tumor incidence and disease severity in several human malignancies (Table 2) [184,185]. Several tumor models and hematological malignancies display aberrant expression of TLRs, in particular TLR-4 and TLR-9 [186,187]. For instance, the upregulation of TLR-4, expressed on head and neck squamous cell carcinoma, has been found to favor tumor growth and development [188]. Involvement of TLR-4/MyD88 signaling in epithelial ovarian cells is shown to promote paclitaxel chemoresistance as well as tumor progression [189]. TLR activation, when combined with inflammatory responses, causes inflammation-driven tumor progression. Elevated expression of major inflammasome activation products, IL-1β and IL-18, have been implicated in gastrointestinal and colitis-associated cancer [190].
The vast majority of studies are providing mounting evidence for the PRR involvement in KSHV recognition (via KSHV-PAMPs) and KS carcinogenesis [114]. Similar to other oncoviruses, KSHV has achieved a balance with its host to limit its recognition and ensure its long-term survival following infection. However, the exact knowledge of these intricate molecular mechanisms and the full spectrum of KSHV-encoded proteins that antagonize PRRs remains incompletely understood. Further studies to shed light into the important structure-function analyses of viral PAMP-host PRR interaction and context-specific relevance of KSHV-triggered PRRs modulation during KS tumorigenesis may stimulate the development of novel prophylactic and therapeutic approaches against KSHV-positive malignancies, and may contribute to long term clinical benefits.
Acknowledgments
We apologize to the authors whose important work could not be cited here due to the limits on reference numbers per journal policy. We thank members of Verma lab for stimulating discussions and critical review of the manuscript. This work is supported by Public Health Service Grants CA174459 and AI105000 from National Institute of Health (NIH).
Author Contributions
Timsy Uppal, Roni Sarkar and Ranjit Dhelaria conceived and wrote this article. Timsy Uppal and Subhash C. Verma did the final editing.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bouvard, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A review of human carcinogens—Part B: Biological agents. Lancet Oncol. 2009, 10, 321–322. [Google Scholar] [CrossRef]
- De Martel, C.; Ferlay, J.; Franceschi, S.; Vignat, J.; Bray, F.; Forman, D.; Plummer, M. Global burden of cancers attributable to infections in 2008: A review and synthetic analysis. Lancet Oncol. 2012, 13, 607–615. [Google Scholar] [CrossRef]
- Zur Hausen, H. The search for infectious causes of human cancers: Where and why. Virology 2009, 392, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zur Hausen, H. Oncogenic DNA viruses. Oncogene 2001, 20, 7820–7823. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Kaul, R.; Murakami, M.; Robertson, E.S. Tumor viruses and cancer biology: Modulating signaling pathways for therapeutic intervention. Cancer Biol. Ther. 2010, 10, 961–978. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Bonilla, F.A.; Oettgen, H.C. Adaptive immunity. J. Allergy Clin. Immunol. 2010, 125, 33S–40S. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B. Innate immunity: An overview. Mol. Immunol. 2004, 40, 845–859. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.J.; Takeuchi, O.; Akira, S. IPS-1, an adaptor triggering RIG-I- and MDA5-mediated type I interferon induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.; Emi, M.; Tanabe, K. Cancer immunoediting from immune surveillance to immune escape. Immunology 2007, 121, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Street, S.E.; Trapani, J.A.; MacGregor, D.; Smyth, M.J. Suppression of lymphoma and epithelial malignancies effected by interferon gamma. J. Exp. Med. 2002, 196, 129–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smyth, M.J.; Thia, K.Y.; Street, S.E.; MacGregor, D.; Godfrey, D.I.; Trapani, J.A. Perforin-mediated cytotoxicity is critical for surveillance of spontaneous lymphoma. J. Exp. Med. 2000, 192, 755–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Page, A.R.; Hansen, A.E.; Good, R.A. Occurrence of leukemia and lymphoma in patients with agammaglobulinemia. Blood 1963, 21, 197–206. [Google Scholar] [PubMed]
- Boder, E.; Sedgwick, R.P. Ataxia-telangiectasia; a familial syndrome of progressive cerebellar ataxia, oculocutaneous telangiectasia and frequent pulmonary infection. Pediatrics 1958, 21, 526–554. [Google Scholar] [PubMed]
- Gatti, R.A.; Good, R.A. Occurrence of malignancy in immunodeficiency diseases. A literature review. Cancer 1971, 28, 89–98. [Google Scholar] [CrossRef]
- Rabkin, C.S.; Janz, S.; Lash, A.; Coleman, A.E.; Musaba, E.; Liotta, L.; Biggar, R.J.; Zhuang, Z. Monoclonal origin of multicentric Kaposi’s sarcoma lesions. N. Engl. J. Med. 1997, 336, 988–993. [Google Scholar] [CrossRef] [PubMed]
- Masood, R.; Cai, J.; Law, R.; Gill, P. AIDS-associated Kaposi’s sarcoma pathogenesis, clinical features, and treatment. Curr. Opin. Oncol. 1993, 5, 831–834. [Google Scholar] [CrossRef] [PubMed]
- Allen, U.; Alfieri, C.; Preiksaitis, J.; Humar, A.; Moore, D.; Tapiero, B.; Tellier, R.; Green, M.; Davies, D.; Hébert, D.; et al. Epstein-Barr virus infection in transplant recipients: Summary of a workshop on surveillance, prevention and treatment. Can. J. Infect. Dis. 2002, 13, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Cesarman, E.; Chang, Y.; Moore, P.S.; Said, J.W.; Knowles, D.M. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 1995, 332, 1186–1191. [Google Scholar] [CrossRef] [PubMed]
- Gbabe, O.F.; Okwundu, C.I.; Dedicoat, M.; Freeman, E.E. Treatment of severe or progressive Kaposi’s sarcoma in HIV-infected adults. Cochrane Database Syst. Rev. 2014, CD003256. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Cesarman, E.; Pessin, M.S.; Lee, F.; Culpepper, J.; Knowles, D.M.; Moore, P.S. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 1994, 266, 1865–1869. [Google Scholar] [CrossRef] [PubMed]
- Cornali, E.; Zietz, C.; Benelli, R.; Weninger, W.; Masiello, L.; Breier, G.; Tschachler, E.; Albini, A.; Sturzl, M. Vascular endothelial growth factor regulates angiogenesis and vascular permeability in Kaposi’s sarcoma. Am. J. Pathol. 1996, 149, 1851–1869. [Google Scholar] [PubMed]
- Hengge, U.R.; Ruzicka, T.; Tyring, S.K.; Stuschke, M.; Roggendorf, M.; Schwartz, R.A.; Seeber, S. Update on Kaposi’s sarcoma and other HHV8 associated diseases. Part 1: Epidemiology, environmental predispositions, clinical manifestations, and therapy. Lancet Infect. Dis. 2002, 2, 281–292. [Google Scholar] [CrossRef]
- Borkovic, S.P.; Schwartz, R.A. Kaposi’s sarcoma presenting in the homosexual man—A new and striking phenomenon! Ariz. Med. 1981, 38, 902–904. [Google Scholar] [PubMed]
- Gottlieb, G.J.; Ragaz, A.; Vogel, J.V.; Friedman-Kien, A.; Rywlin, A.M.; Weiner, E.A.; Ackerman, A.B. A preliminary communication on extensively disseminated Kaposi’s sarcoma in young homosexual men. Am. J. Dermatopathol. 1981, 3, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Hymes, K.B.; Cheung, T.; Greene, J.B.; Prose, N.S.; Marcus, A.; Ballard, H.; William, D.C.; Laubenstein, L.J. Kaposi’s sarcoma in homosexual men-a report of eight cases. Lancet 1981, 2, 598–600. [Google Scholar] [CrossRef]
- Soulier, J.; Grollet, L.; Oksenhendler, E.; Cacoub, P.; Cazals-Hatem, D.; Babinet, P.; d’Agay, M.F.; Clauvel, J.P.; Raphael, M.; Degos, L.; et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric castleman’s disease. Blood 1995, 86, 1276–1280. [Google Scholar] [PubMed]
- Nador, R.G.; Cesarman, E.; Chadburn, A.; Dawson, D.B.; Ansari, M.Q.; Sald, J.; Knowles, D.M. Primary effusion lymphoma: A distinct clinicopathologic entity associated with the Kaposi’s sarcoma-associated herpes virus. Blood 1996, 88, 645–656. [Google Scholar] [PubMed]
- Saeed-Abdul-Rahman, I.; Al-Amri, A.M. Castleman disease. Korean J. Hematol. 2012, 47, 163–177. [Google Scholar] [CrossRef] [PubMed]
- Chandran, B. Early events in Kaposi’s sarcoma-associated herpesvirus infection of target cells. J. Virol. 2010, 84, 2188–2199. [Google Scholar] [CrossRef] [PubMed]
- Akula, S.M.; Pramod, N.P.; Wang, F.Z.; Chandran, B. Human herpesvirus 8 envelope-associated glycoprotein B interacts with heparan sulfate-like moieties. Virology 2001, 284, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.Z.; Akula, S.M.; Pramod, N.P.; Zeng, L.; Chandran, B. Human herpesvirus 8 envelope glycoprotein K8.1A interaction with the target cells involves heparan sulfate. J. Virol. 2001, 75, 7517–7527. [Google Scholar] [CrossRef] [PubMed]
- Greene, W.; Kuhne, K.; Ye, F.; Chen, J.; Zhou, F.; Lei, X.; Gao, S.J. Molecular biology of KSHV in relation to AIDS-associated oncogenesis. Cancer Treat. Res. 2007, 133, 69–127. [Google Scholar] [PubMed]
- Krishnan, H.H.; Naranatt, P.P.; Smith, M.S.; Zeng, L.; Bloomer, C.; Chandran, B. Concurrent expression of latent and a limited number of lytic genes with immune modulation and antiapoptotic function by Kaposi’s sarcoma-associated herpesvirus early during infection of primary endothelial and fibroblast cells and subsequent decline of lytic gene expression. J. Virol. 2004, 78, 3601–3620. [Google Scholar] [PubMed]
- Purushothaman, P.; Thakker, S.; Verma, S.C. Transcriptome analysis of Kaposi’s sarcoma-associated herpesvirus during de novo primary infection of human B and endothelial cells. J. Virol. 2015, 89, 3093–3111. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.; Ganem, D. A unique herpesviral transcriptional program in KSHV-infected lymphatic endothelial cells leads to mTORC1 activation and rapamycin sensitivity. Cell Host Microbe 2013, 13, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Grundhoff, A.; Ganem, D. Inefficient establishment of KSHV latency suggests an additional role for continued lytic replication in Kaposi sarcoma pathogenesis. J. Clin. Investig. 2004, 113, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.W.; Trotter, M.W.; Lagos, D.; Bourboulia, D.; Henderson, S.; Makinen, T.; Elliman, S.; Flanagan, A.M.; Alitalo, K.; Boshoff, C. Kaposi sarcoma herpesvirus-induced cellular reprogramming contributes to the lymphatic endothelial gene expression in Kaposi sarcoma. Nat. Genet. 2004, 36, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Gessain, A.; Duprez, R. Spindle cells and their role in Kaposi’s sarcoma. Int. J. Biochem. Cell Biol. 2005, 37, 2457–2465. [Google Scholar] [CrossRef] [PubMed]
- Tedeschi, R.; Enbom, M.; Bidoli, E.; Linde, A.; De Paoli, P.; Dillner, J. Viral load of human herpesvirus 8 in peripheral blood of human immunodeficiency virus-infected patients with Kaposi’s sarcoma. J. Clin. Microbiol. 2001, 39, 4269–4273. [Google Scholar] [CrossRef] [PubMed]
- Ballestas, M.E.; Chatis, P.A.; Kaye, K.M. Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science 1999, 284, 641–644. [Google Scholar] [CrossRef] [PubMed]
- Pearce, M.; Matsumura, S.; Wilson, A.C. Transcripts encoding K12, v-FLIP, v-Cyclin, and the microRNA cluster of Kaposi’s sarcoma-associated herpesvirus originate from a common promoter. J. Virol. 2005, 79, 14457–14464. [Google Scholar] [CrossRef] [PubMed]
- Rainbow, L.; Platt, G.M.; Simpson, G.R.; Sarid, R.; Gao, S.J.; Stoiber, H.; Herrington, C.S.; Moore, P.S.; Schulz, T.F. The 222- to 234-kilodalton latent nuclear protein (LNA) of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) is encoded by ORF73 and is a component of the latency-associated nuclear antigen. J. Virol. 1997, 71, 5915–5921. [Google Scholar] [PubMed]
- Samols, M.A.; Hu, J.; Skalsky, R.L.; Renne, R. Cloning and identification of a microRNA cluster within the latency-associated region of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2005, 79, 9301–9305. [Google Scholar] [CrossRef] [PubMed]
- Friborg, J., Jr.; Kong, W.; Hottiger, M.O.; Nabel, G.J. p53 inhibition by the LANA protein of KSHV protects against cell death. Nature 1999, 402, 889–894. [Google Scholar] [CrossRef] [PubMed]
- Radkov, S.A.; Kellam, P.; Boshoff, C. The latent nuclear antigen of Kaposi sarcoma-associated herpesvirus targets the retinoblastoma-E2F pathway and with the oncogene Hras transforms primary rat cells. Nat. Med. 2000, 6, 1121–1127. [Google Scholar] [CrossRef] [PubMed]
- Wen, K.W.; Damania, B. Kaposi sarcoma-associated herpesvirus (KSHV): Molecular biology and oncogenesis. Cancer Lett. 2010, 289, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Pulitzer, M. Molecular diagnosis of infection-related cancers in dermatopathology. Semin. Cutan. Med. Surg. 2012, 31, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Yarchoan, R.; Wyvill, K.; Okamoto, S.; Little, R.F.; Tosato, G. Detection of viral interleukin-6 in Kaposi sarcoma-associated herpesvirus-linked disorders. Blood 2001, 97, 2173–2176. [Google Scholar] [CrossRef] [PubMed]
- Ensoli, B.; Sturzl, M. Kaposi’s sarcoma: A result of the interplay among inflammatory cytokines, angiogenic factors and viral agents. Cytokine Growth Factor Rev. 1998, 9, 63–83. [Google Scholar] [CrossRef]
- Ensoli, B.; Sgadari, C.; Barillari, G.; Sirianni, M.C.; Sturzl, M.; Monini, P. Biology of Kaposi’s sarcoma. Eur. J. Cancer 2001, 37, 1251–1269. [Google Scholar] [CrossRef]
- Carbone, A.; Cilia, A.M.; Gloghini, A.; Capello, D.; Fassone, L.; Perin, T.; Rossi, D.; Canzonieri, V.; De Paoli, P.; Vaccher, E.; et al. Characterization of a novel HHV-8-positive cell line reveals implications for the pathogenesis and cell cycle control of primary effusion lymphoma. Leukemia 2000, 14, 1301–1309. [Google Scholar] [CrossRef] [PubMed]
- Carroll, P.A.; Brazeau, E.; Lagunoff, M. Kaposi’s sarcoma-associated herpesvirus infection of blood endothelial cells induces lymphatic differentiation. Virology 2004, 328, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Weninger, W.; Partanen, T.A.; Breiteneder-Geleff, S.; Mayer, C.; Kowalski, H.; Mildner, M.; Pammer, J.; Sturzl, M.; Kerjaschki, D.; Alitalo, K.; et al. Expression of vascular endothelial growth factor receptor-3 and podoplanin suggests a lymphatic endothelial cell origin of Kaposi’s sarcoma tumor cells. Lab. Investig. 1999, 79, 243–251. [Google Scholar] [PubMed]
- Marchio, S.; Primo, L.; Pagano, M.; Palestro, G.; Albini, A.; Veikkola, T.; Cascone, I.; Alitalo, K.; Bussolino, F. Vascular endothelial growth factor-C stimulates the migration and proliferation of Kaposi’s sarcoma cells. J. Boil. Chem. 1999, 274, 27617–27622. [Google Scholar] [CrossRef]
- Rosado, F.G.; Itani, D.M.; Coffin, C.M.; Cates, J.M. Utility of immunohistochemical staining with FLI1, D2-40, CD31, and CD34 in the diagnosis of acquired immunodeficiency syndrome-related and non-acquired immunodeficiency syndrome-related Kaposi sarcoma. Arch. Pathol. Lab. Med. 2012, 136, 301–304. [Google Scholar] [CrossRef] [PubMed]
- Faris, M.; Ensoli, B.; Kokot, N.; Nel, A.E. Inflammatory cytokines induce the expression of basic fibroblast growth factor (bFGF) isoforms required for the growth of Kaposi’s sarcoma and endothelial cells through the activation of AP-1 response elements in the bFGF promoter. AIDS 1998, 12, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.X.; Antakly, T.; Cadotte, M.; Kachra, Z.; Kunkel, L.; Masood, R.; Gill, P. Expression and cytokine regulation of glucocorticoid receptors in Kaposi’s sarcoma. Am. J. Pathol. 1996, 148, 1999–2008. [Google Scholar] [PubMed]
- Cai, J.; Gill, P.S.; Masood, R.; Chandrasoma, P.; Jung, B.; Law, R.E.; Radka, S.F. Oncostatin-M is an autocrine growth factor in Kaposi’s sarcoma. Am. J. Pathol. 1994, 145, 74–79. [Google Scholar] [PubMed]
- Liu, Z.; Fang, Q.; Zhou, S.; Minhas, V.; Wood, C.; He, N.; Zhang, T. Seroprevalence of Kaposi’s sarcoma-associated herpesvirus among HIV-infected uygurs in Xinjiang, china. J. Med. Virol. 2017, 89, 1629–1635. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, J.; Cope, A.; Gill, J.; Bourboulia, D.; Hayes, P.; Imami, N.; Kubo, T.; Marcelin, A.; Calvez, V.; Weiss, R.; et al. Identification of Kaposi’s sarcoma-associated herpesvirus (KSHV)-specific cytotoxic T-lymphocyte epitopes and evaluation of reconstitution of KSHV-specific responses in human immunodeficiency virus type 1-infected patients receiving highly active antiretroviral therapy. J. Virol. 2002, 76, 2634–2640. [Google Scholar] [PubMed]
- Pereira, P.F.; Cuzzi, T.; Galhardo, M.C. Immunohistochemical detection of the latent nuclear antigen-1 of the human herpesvirus type 8 to differentiate cutaneous epidemic Kaposi sarcoma and its histological simulators. An. Bras. Dermatol. 2013, 88, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Pantanowitz, L.; Dezube, B.J.; Pinkus, G.S.; Tahan, S.R. Histological characterization of regression in acquired immunodeficiency syndrome-related Kaposi’s sarcoma. J. Cutan. Pathol. 2004, 31, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Cheuk, W.; Wong, K.O.; Wong, C.S.; Dinkel, J.E.; Ben-Dor, D.; Chan, J.K. Immunostaining for human herpesvirus 8 latent nuclear antigen-1 helps distinguish Kaposi sarcoma from its mimickers. Am. J. Clin. Pathol. 2004, 121, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Long, E.; Ilie, M.; Hofman, V.; Havet, K.; Selva, E.; Butori, C.; Lacour, J.P.; Nelson, A.M.; Cathomas, G.; Hofman, P. LANA-1, Bcl-2, Mcl-1 and HIF-1α protein expression in HIV-associated Kaposi sarcoma. Virchows Arch. 2009, 455, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Ojala, P.M.; Tiainen, M.; Salven, P.; Veikkola, T.; Castanos-Velez, E.; Sarid, R.; Biberfeld, P.; Makela, T.P. Kaposi’s sarcoma-associated herpesvirus-encoded v-cyclin triggers apoptosis in cells with high levels of cyclin-dependent kinase 6. Cancer Res. 1999, 59, 4984–4989. [Google Scholar] [PubMed]
- Nagata, N.; Igari, T.; Shimbo, T.; Sekine, K.; Akiyama, J.; Hamada, Y.; Yazaki, H.; Ohmagari, N.; Teruya, K.; Oka, S.; et al. Diagnostic value of endothelial markers and HHV-8 staining in gastrointestinal Kaposi sarcoma and its difference in endoscopic tumor staging. World J. Gastroenterol. 2013, 19, 3608–3614. [Google Scholar] [CrossRef] [PubMed]
- Russell Jones, R.; Orchard, G.; Zelger, B.; Wilson Jones, E. Immunostaining for CD31 and CD34 in Kaposi sarcoma. J. Clin. Pathol. 1995, 48, 1011–1016. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.M.; Goldblum, J.R.; Hsi, E.D. Immunohistochemical detection of human herpes virus-8 latent nuclear antigen-1 is useful in the diagnosis of Kaposi sarcoma. Mod. Pathol. 2004, 17, 456–460. [Google Scholar] [CrossRef] [PubMed]
- Pantanowitz, L.; Schwartz, E.J.; Dezube, B.J.; Kohler, S.; Dorfman, R.F.; Tahan, S.R. C-kit (CD117) expression in AIDS-related, classic, and African endemic Kaposi sarcoma. Appl. Immunohistochem. Mol. Morphol. 2005, 13, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Moses, A.V.; Jarvis, M.A.; Raggo, C.; Bell, Y.C.; Ruhl, R.; Luukkonen, B.G.; Griffith, D.J.; Wait, C.L.; Druker, B.J.; Heinrich, M.C.; et al. Kaposi’s sarcoma-associated herpesvirus-induced upregulation of the c-kit proto-oncogene, as identified by gene expression profiling, is essential for the transformation of endothelial cells. J. Virol. 2002, 76, 8383–8399. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, M.M.; Biddolph, S.; Lucas, S.B.; Howells, D.D.; Picton, S.; McGee, J.O.; Silva, I.; Uhlmann, V.; Luttich, K.; O’Leary, J.J. Cyclin D1 expression and HHV8 in Kaposi sarcoma. J. Clin. Pathol. 1999, 52, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Koopal, S.; Furuhjelm, J.H.; Jarviluoma, A.; Jaamaa, S.; Pyakurel, P.; Pussinen, C.; Wirzenius, M.; Biberfeld, P.; Alitalo, K.; Laiho, M.; et al. Viral oncogene-induced DNA damage response is activated in Kaposi sarcoma tumorigenesis. PLoS Pathog. 2007, 3, 1348–1360. [Google Scholar] [CrossRef] [PubMed]
- Si, H.; Robertson, E.S. Kaposi’s sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen induces chromosomal instability through inhibition of p53 function. J. Virol. 2006, 80, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Petre, C.E.; Sin, S.H.; Dittmer, D.P. Functional p53 signaling in Kaposi’s sarcoma-associated herpesvirus lymphomas: Implications for therapy. J. Virol. 2007, 81, 1912–1922. [Google Scholar] [CrossRef] [PubMed]
- Sharma-Walia, N.; Chandran, K.; Patel, K.; Veettil, M.V.; Marginean, A. The Kaposi’s sarcoma-associated herpesvirus (KSHV)-induced 5-lipoxygenase-leukotriene B4 cascade plays key roles in KSHV latency, monocyte recruitment, and lipogenesis. J. Virol. 2014, 88, 2131–2156. [Google Scholar] [CrossRef] [PubMed]
- Boshoff, C.; Schulz, T.F.; Kennedy, M.M.; Graham, A.K.; Fisher, C.; Thomas, A.; McGee, J.O.; Weiss, R.A.; O’Leary, J.J. Kaposi’s sarcoma-associated herpesvirus infects endothelial and spindle cells. Nat. Med. 1995, 1, 1274–1278. [Google Scholar] [CrossRef] [PubMed]
- Sturzl, M.; Hohenadl, C.; Zietz, C.; Castanos-Velez, E.; Wunderlich, A.; Ascherl, G.; Biberfeld, P.; Monini, P.; Browning, P.J.; Ensoli, B. Expression of K13/v-FLIP gene of human herpesvirus 8 and apoptosis in Kaposi’s sarcoma spindle cells. J. Natl. Cancer Inst. 1999, 91, 1725–1733. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, S.; Pise-Masison, C.A.; Brady, J.N.; Tosato, G. Gene regulation and functional alterations induced by Kaposi’s sarcoma-associated herpesvirus-encoded ORFK13/vFLIP in endothelial cells. J. Virol. 2009, 83, 2140–2153. [Google Scholar] [CrossRef] [PubMed]
- Horenstein, M.G.; Cesarman, E.; Wang, X.; Linkov, I.; Prieto, V.G.; Louie, D.C. Cyclin D1 and retinoblastoma protein expression in Kaposi’s sarcoma. J. Cutan. Pathol. 1997, 24, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Borah, S.; Verma, S.C.; Robertson, E.S. ORF73 of herpesvirus saimiri, a viral homolog of Kaposi’s sarcoma-associated herpesvirus, modulates the two cellular tumor suppressor proteins p53 and pRb. J. Virol. 2004, 78, 10336–10347. [Google Scholar] [CrossRef] [PubMed]
- Kuwabara, H.; Nagai, M.; Shibanushi, T.; Ohmori, M.; Kawakami, K.; Asakura, H. CD138-positive and Kaposi’s sarcoma-associated herpesvirus (KSHV)-negative B-cell lymphoma with serosal spreading of the body cavity and lymphadenopathy: An autopsy case. Hum. Pathol. 2000, 31, 1171–1175. [Google Scholar] [CrossRef] [PubMed]
- Deloose, S.T.; Smit, L.A.; Pals, F.T.; Kersten, M.J.; van Noesel, C.J.; Pals, S.T. High incidence of Kaposi sarcoma-associated herpesvirus infection in HIV-related solid immunoblastic/plasmablastic diffuse large B-cell lymphoma. Leukemia 2005, 19, 851–855. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Lu, S.; Zhang, Z.; Gonzalez, C.M.; Damania, B.; Cullen, B.R. Kaposi’s sarcoma-associated herpesvirus expresses an array of viral microRNAs in latently infected cells. Proc. Natl. Acad. Sci. USA 2005, 102, 5570–5575. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, W.; Gao, S.J.; Lu, C. KSHV microRNAs: Tricks of the devil. Trends Microbiol. 2017, 25, 648–661. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Xu, Y.; Mo, D.; Huang, P.; Sun, R.; Huang, L.; Pan, S.; Xu, J. Kaposi’s sarcoma-associated herpesvirus (KSHV) vIL-6 promotes cell proliferation and migration by upregulating DNMT1 via STAT3 activation. PLoS ONE 2014, 9, e93478. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Harada, J.N.; Brown, H.J.; Deng, H.; Song, M.J.; Wu, T.T.; Kato-Stankiewicz, J.; Nelson, C.G.; Vieira, J.; Tamanoi, F.; et al. Systematic identification of cellular signals reactivating Kaposi sarcoma-associated herpesvirus. PLoS Pathog. 2007, 3, e44. [Google Scholar] [CrossRef] [PubMed]
- Umbach, J.L.; Cullen, B.R. In-depth analysis of Kaposi’s sarcoma-associated herpesvirus microRNA expression provides insights into the mammalian microRNA-processing machinery. J. Virol. 2010, 84, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Arai, E.; Kuramochi, A.; Tsuchida, T.; Tsuneyoshi, M.; Kage, M.; Fukunaga, M.; Ito, T.; Tada, T.; Izumi, M.; Shimizu, K.; et al. Usefulness of D2-40 immunohistochemistry for differentiation between kaposiform hemangioendothelioma and tufted angioma. J. Cutan. Pathol. 2006, 33, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Kahn, H.J.; Bailey, D.; Marks, A. Monoclonal antibody D2-40, a new marker of lymphatic endothelium, reacts with Kaposi’s sarcoma and a subset of angiosarcomas. Mod. Pathol. 2002, 15, 434–440. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.R.; Damania, B. The viral interferon regulatory factors of KSHV: Immunosuppressors or oncogenes? Front. Immunol. 2011, 2, 19. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.R.; Gregory, S.M.; West, J.A.; Wollish, A.C.; Bennett, C.L.; Blackbourn, D.J.; Heise, M.T.; Damania, B. The viral interferon regulatory factors of Kaposi’s sarcoma-associated herpesvirus differ in their inhibition of interferon activation mediated by toll-like receptor 3. J. Virol. 2013, 87, 798–806. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.C.; Robertson, E.S. Molecular biology and pathogenesis of Kaposi sarcoma-associated herpesvirus. FEMS Microbiol. Lett. 2003, 222, 155–163. [Google Scholar] [CrossRef]
- Gutierrez, K.D.; Morris, V.A.; Wu, D.; Barcy, S.; Lagunoff, M. Ets-1 is required for the activation of VEGFR3 during latent Kaposi’s sarcoma-associated herpesvirus infection of endothelial cells. J. Virol. 2013, 87, 6758–6768. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.K.; Foreman, K.; Shin, J.W.; Hirakawa, S.; Curry, C.L.; Sage, D.R.; Libermann, T.; Dezube, B.J.; Fingeroth, J.D.; Detmar, M. Lymphatic reprogramming of blood vascular endothelium by Kaposi sarcoma-associated herpesvirus. Nat. Genet. 2004, 36, 683–685. [Google Scholar] [CrossRef] [PubMed]
- Bottero, V.; Sharma-Walia, N.; Kerur, N.; Paul, A.G.; Sadagopan, S.; Cannon, M.; Chandran, B. Kaposi sarcoma-associated herpes virus (KSHV) G protein-coupled receptor (vGPCR) activates the ORF50 lytic switch promoter: A potential positive feedback loop for sustained ORF50 gene expression. Virology 2009, 392, 34–51. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Verma, S.C.; Lu, J.; Robertson, E.S. Molecular biology of Kaposi’s sarcoma-associated herpesvirus and related oncogenesis. Adv. Virus Res. 2010, 78, 87–142. [Google Scholar] [PubMed]
- Naranatt, P.P.; Krishnan, H.H.; Svojanovsky, S.R.; Bloomer, C.; Mathur, S.; Chandran, B. Host gene induction and transcriptional reprogramming in Kaposi’s sarcoma-associated herpesvirus (KSHV/HHV-8)-infected endothelial, fibroblast, and B cells: Insights into modulation events early during infection. Cancer Res. 2004, 64, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Amaral, M.C.; Miles, S.; Kumar, G.; Nel, A.E. Oncostatin-M stimulates tyrosine protein phosphorylation in parallel with the activation of p42MAPK/ERK-2 in Kaposi’s cells. Evidence that this pathway is important in Kaposi cell growth. J. Clin. Investig. 1993, 92, 848–857. [Google Scholar] [CrossRef] [PubMed]
- Simonart, T.; Van Vooren, J.P. Interleukin-1 β increases the BCL-2/BAX ratio in Kaposi’s sarcoma cells. Cytokine 2002, 19, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, D.; Richards, K.; Damania, B. Treatment of Kaposi sarcoma-associated herpesvirus-associated cancers. Front. Microbiol. 2012, 3, 141. [Google Scholar] [CrossRef] [PubMed]
- Yanik, E.L.; Napravnik, S.; Cole, S.R.; Achenbach, C.J.; Gopal, S.; Olshan, A.; Dittmer, D.P.; Kitahata, M.M.; Mugavero, M.J.; Saag, M.; et al. Incidence and timing of cancer in HIV-infected individuals following initiation of combination antiretroviral therapy. Clin. Infect. Dis. 2013, 57, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Walker, N.F.; Scriven, J.; Meintjes, G.; Wilkinson, R.J. Immune reconstitution inflammatory syndrome in HIV-infected patients. HIV/AIDS 2015, 7, 49–64. [Google Scholar] [PubMed]
- Hosseinipour, M.C.; Sweet, K.M.; Xiong, J.; Namarika, D.; Mwafongo, A.; Nyirenda, M.; Chiwoko, L.; Kamwendo, D.; Hoffman, I.; Lee, J.; et al. Viral profiling identifies multiple subtypes of Kaposi’s sarcoma. mBio 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Glesby, M.J.; Hoover, D.R.; Weng, S.; Graham, N.M.; Phair, J.P.; Detels, R.; Ho, M.; Saah, A.J. Use of antiherpes drugs and the risk of Kaposi’s sarcoma: Data from the multicenter AIDS cohort study. J. Infect. Dis. 1996, 173, 1477–1480. [Google Scholar] [CrossRef] [PubMed]
- Mazzi, R.; Parisi, S.G.; Sarmati, L.; Uccella, I.; Nicastri, E.; Carolo, G.; Gatti, F.; Concia, E.; Andreoni, M. Efficacy of cidofovir on human herpesvirus 8 viraemia and Kaposi’s sarcoma progression in two patients with AIDS. AIDS 2001, 15, 2061–2062. [Google Scholar] [CrossRef] [PubMed]
- Bossini, N.; Sandrini, S.; Setti, G.; Luppi, M.; Maiorca, P.; Maffei, C.; Cancarini, G. Successful treatment with liposomal doxorubicin and foscarnet in a patient with widespread Kaposi’s sarcoma and human herpes virus 8-related, serious hemophagocytic syndrome, after renal transplantation. G. Ital. Nefrol. 2005, 22, 281–286. [Google Scholar] [PubMed]
- Sergerie, Y.; Boivin, G. Evaluation of susceptibility of human herpesvirus 8 to antiviral drugs by quantitative real-time PCR. J. Clin. Microbiol. 2003, 41, 3897–3900. [Google Scholar] [CrossRef] [PubMed]
- Takahashi-Makise, N.; Suzu, S.; Hiyoshi, M.; Ohsugi, T.; Katano, H.; Umezawa, K.; Okada, S. Biscoclaurine alkaloid cepharanthine inhibits the growth of primary effusion lymphoma in vitro and in vivo and induces apoptosis via suppression of the NF-κB pathway. Int. J. Cancer 2009, 125, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- Matsuno, T.; Kariya, R.; Yano, S.; Morino-Koga, S.; Taura, M.; Suico, M.A.; Shimauchi, Y.; Matsuyama, S.; Okamoto, Y.; Shuto, T.; et al. Diethyldithiocarbamate induces apoptosis in HHV-8-infected primary effusion lymphoma cells via inhibition of the NF-κB pathway. Int. J. Oncol. 2012, 40, 1071–1078. [Google Scholar] [PubMed]
- Uldrick, T.S.; Goncalves, P.H.; Wyvill, K.M.; Peer, C.J.; Bernstein, W.; Aleman, K.; Polizzotto, M.N.; Venzon, D.; Steinberg, S.M.; Marshall, V.; et al. A phase Ib study of sorafenib (BAY 43-9006) in patients with Kaposi sarcoma. Oncologist 2017, 22. [Google Scholar] [CrossRef] [PubMed]
- Koon, H.B.; Krown, S.E.; Lee, J.Y.; Honda, K.; Rapisuwon, S.; Wang, Z.; Aboulafia, D.; Reid, E.G.; Rudek, M.A.; Dezube, B.J.; et al. Phase II trial of imatinib in AIDS-associated Kaposi’s sarcoma: AIDS malignancy consortium protocol 042. J. Clin. Oncol. 2014, 32, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, D.P.; Damania, B. Kaposi sarcoma-associated herpesvirus: Immunobiology, oncogenesis, and therapy. J. Clin. Investig. 2016, 126, 3165–3175. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.R.; Kaminski, J.J.; Kurt-Jones, E.A.; Fitzgerald, K.A. Pattern recognition receptors and the innate immune response to viral infection. Viruses 2011, 3, 920–940. [Google Scholar] [CrossRef] [PubMed]
- Brubaker, S.W.; Bonham, K.S.; Zanoni, I.; Kagan, J.C. Innate immune pattern recognition: A cell biological perspective. Annu. Rev. Immunol. 2015, 33, 257–290. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. TLR-mediated innate immune recognition. Semin. Immunol. 2007, 19, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, S.; Fujiwara, N.; Ido, A.; Oono, M.; Takeuchi, Y.; Tateno, M.; Suzuki, K.; Takahashi, R.; Tooyama, I.; Taniguchi, N.; et al. Induction of protective immunity by vaccination with wild-type apo superoxide dismutase 1 in mutant SOD1 transgenic mice. J. Neuropathol. Exp. Neurol. 2010, 69, 1044–1056. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, A.; Pierce, S.K. How location governs toll-like receptor signaling. Traffic 2009, 10, 621–628. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.; Bowie, A.G. The family of five: TIR-domain-containing adaptors in toll-like receptor signalling. Nat. Rev. Immunol. 2007, 7, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Srikrishna, G.; Freeze, H.H. Endogenous damage-associated molecular pattern molecules at the crossroads of inflammation and cancer. Neoplasia 2009, 11, 615–628. [Google Scholar] [CrossRef] [PubMed]
- De Nardo, D. Toll-like receptors: Activation, signalling and transcriptional modulation. Cytokine 2015, 74, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Li, T.T.; Ogino, S.; Qian, Z.R. Toll-like receptor signaling in colorectal cancer: Carcinogenesis to cancer therapy. World J. Gastroenterol. 2014, 20, 17699–17708. [Google Scholar] [CrossRef] [PubMed]
- Steinhagen, F.; Kinjo, T.; Bode, C.; Klinman, D.M. TLR-based immune adjuvants. Vaccine 2011, 29, 3341–3355. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.R.; Amatya, R.; Jung, J.U. Multi-step regulation of innate immune signaling by Kaposi’s sarcoma-associated herpesvirus. Virus Res. 2015, 209, 39–44. [Google Scholar] [CrossRef] [PubMed]
- West, J.; Damania, B. Upregulation of the TLR3 pathway by Kaposi’s sarcoma-associated herpesvirus during primary infection. J. Virol. 2008, 82, 5440–5449. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.R.; Stopford, C.M.; West, J.A.; Bennett, C.L.; Giffin, L.; Damania, B. Kaposi’s sarcoma-associated herpesvirus viral interferon regulatory factor 1 interacts with a member of the interferon-stimulated gene 15 pathway. J. Virol. 2015, 89, 11572–11583. [Google Scholar] [CrossRef] [PubMed]
- Mutocheluh, M.; Hindle, L.; Areste, C.; Chanas, S.A.; Butler, L.M.; Lowry, K.; Shah, K.; Evans, D.J.; Blackbourn, D.J. Kaposi’s sarcoma-associated herpesvirus viral interferon regulatory factor-2 inhibits type 1 interferon signalling by targeting interferon-stimulated gene factor-3. J. Gen. Virol. 2011, 92, 2394–2398. [Google Scholar] [CrossRef] [PubMed]
- Lagos, D.; Vart, R.J.; Gratrix, F.; Westrop, S.J.; Emuss, V.; Wong, P.-P.; Robey, R.; Imami, N.; Bower, M.; Gotch, F.; et al. Toll-like receptor 4 mediates innate immunity to Kaposi sarcoma herpesvirus. Cell Host Microbe 2008, 4, 470–483. [Google Scholar] [CrossRef] [PubMed]
- Bussey, K.A.; Reimer, E.; Todt, H.; Denker, B.; Gallo, A.; Konrad, A.; Ottinger, M.; Adler, H.; Stürzl, M.; Brune, W.; et al. The gammaherpesviruses Kaposi’s sarcoma-associated herpesvirus and murine gammaherpesvirus 68 modulate the toll-like receptor-induced proinflammatory cytokine response. J. Virol. 2014, 88, 9245–9259. [Google Scholar] [CrossRef] [PubMed]
- Gregory, S.M.; West, J.A.; Dillon, P.J.; Hilscher, C.; Dittmer, D.P.; Damania, B. Toll-like receptor signaling controls reactivation of KSHV from latency. Proc. Natl. Acad. Sci. USA 2009, 106, 11725–11730. [Google Scholar] [CrossRef] [PubMed]
- West, J.A.; Gregory, S.M.; Sivaraman, V.; Su, L.; Damania, B. Activation of plasmacytoid dendritic cells by Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2011, 85, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, H.; Gubbels, R.; Ehlers, E.; Meyer, F.; Waterbury, T.; Lin, R.; Zhang, L. Kaposi sarcoma-associated herpesvirus degrades cellular toll-interleukin-1 receptor domain-containing adaptor-inducing β-interferon (TRIF). J. Boil. Chem. 2011, 286, 7865–7872. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Liang, D.; Sun, R.; Jia, B.; Xia, T.; Xiao, H.; Lan, K. Kaposi’s sarcoma-associated herpesvirus-encoded replication and transcription activator impairs innate immunity via ubiquitin-mediated degradation of myeloid differentiation factor 88. J. Virol. 2015, 89, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Abend, J.R.; Ramalingam, D.; Kieffer-Kwon, P.; Uldrick, T.S.; Yarchoan, R.; Ziegelbauer, J.M. Kaposi’s sarcoma-associated herpesvirus microRNAs target IRAK1 and MyD88, two components of the toll-like receptor/interleukin-1R signaling cascade, to reduce inflammatory-cytokine expression. J. Virol. 2012, 86, 11663–11674. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Chen, W.; Xiong, J.; Sherrod, C.J.; Henry, D.H.; Dittmer, D.P. Interleukin 1 receptor-associated kinase 1 (IRAK1) mutation is a common, essential driver for Kaposi sarcoma herpesvirus lymphoma. Proc. Natl. Acad. Sci. USA 2014, 111, E4762–E4768. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.K.; Wen, H.; Ting, J.P. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu. Rev. Immunol. 2011, 29, 707–735. [Google Scholar] [CrossRef] [PubMed]
- Monie, T.P. NLR activation takes a direct route. Trends Biochem. Sci. 2013, 38, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proil-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Sharma, D.; Kanneganti, T.-D. The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. J. Cell Biol. 2016, 213, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Vajjhala, P.R.; Ve, T.; Bentham, A.; Stacey, K.J.; Kobe, B. The molecular mechanisms of signaling by cooperative assembly formation in innate immunity pathways. Mol. Immunol. 2017, 86, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Coutermarsh-Ott, S.; Eden, K.; Allen, I.C. Beyond the inflammasome: Regulatory nod-like receptor modulation of the host immune response following virus exposure. J. Gen. Virol. 2016, 97, 825–838. [Google Scholar] [CrossRef] [PubMed]
- Lupfer, C.; Kanneganti, T.D. The expanding role of NLRs in antiviral immunity. Immunol. Rev. 2013, 255, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Gregory, S.M.; Davis, B.K.; West, J.A.; Taxman, D.J.; Matsuzawa, S.; Reed, J.C.; Ting, J.P.; Damania, B. Discovery of a viral NLR homolog that inhibits the inflammasome. Science 2011, 331, 330–334. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Hopcraft, S.E.; Yang, F.; Petrucelli, A.; Guo, H.; Ting, J.P.; Dittmer, D.P.; Damania, B. NLRX1 negatively modulates type I IFN to facilitate KSHV reactivation from latency. PLoS Pathog. 2017, 13, e1006350. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int. Immunol. 2009, 21, 317–337. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.H.; Macmillan, J.B.; Chen, Z.J. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 2009, 138, 576–591. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Fujita, T. Structural mechanism of RNA recognition by the RIG-I-like receptors. Immunity 2008, 29, 178–181. [Google Scholar] [CrossRef] [PubMed]
- Reikine, S.; Nguyen, J.B.; Modis, Y. Pattern recognition and signaling mechanisms of RIG-I and MDA5. Front. Immunol. 2014, 5, 342. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Ramanathan, A.; Miller, M.T.; Tang, G.Q.; Gale, M., Jr.; Patel, S.S.; Marcotrigiano, J. Structural basis of RNA recognition and activation by innate immune receptor RIG-I. Nature 2011, 479, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.; Eisenacher, K.; Kirchhofer, A.; Brzozka, K.; Lammens, A.; Lammens, K.; Fujita, T.; Conzelmann, K.K.; Krug, A.; Hopfner, K.P. The C-terminal regulatory domain is the RNA 5′-triphosphate sensor of RIG-I. Mol. Cell 2008, 29, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Takahasi, K.; Yoneyama, M.; Nishihori, T.; Hirai, R.; Kumeta, H.; Narita, R.; Gale, M., Jr.; Inagaki, F.; Fujita, T. Nonself RNA-sensing mechanism of RIG-I helicase and activation of antiviral immune responses. Mol. Cell 2008, 29, 428–440. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of mavs, a mitochondrial antiviral signaling protein that activates NF-κB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Wang, Y.; Han, K.; Li, L.; Zhai, Z.; Shu, H. VISA is an adapter protein required for virus-triggered IFN-β signaling. Mol. Cell 2005, 19, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Berke, I.C.; Modis, Y. MDA5 cooperatively forms dimers and ATP-sensitive filaments upon binding double-stranded RNA. EMBO J. 2012, 31, 1714–1726. [Google Scholar] [CrossRef] [PubMed]
- Peisley, A.; Lin, C.; Wu, B.; Orme-Johnson, M.; Liu, M.; Walz, T.; Hur, S. Cooperative assembly and dynamic disassembly of MDA5 filaments for viral dsRNA recognition. Proc. Natl. Acad. Sci. USA 2011, 108, 21010–21015. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Kikuchi, M.; Matsumoto, K.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Foy, E.; Loo, Y.M.; Gale, M., Jr.; Akira, S.; et al. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J. Immunol. 2005, 175, 2851–2858. [Google Scholar] [CrossRef] [PubMed]
- Childs, K.S.; Randall, R.E.; Goodbourn, S. LGP2 plays a critical role in sensitizing MDA-5 to activation by double-stranded RNA. PLoS ONE 2013, 8, e64202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inn, K.S.; Lee, S.H.; Rathbun, J.Y.; Wong, L.Y.; Toth, Z.; Machida, K.; Ou, J.H.; Jung, J.U. Inhibition of RIG-I-mediated signaling by Kaposi’s sarcoma-associated herpesvirus-encoded deubiquitinase ORF64. J. Virol. 2011, 85, 10899–10904. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, A.; Wang, Z.; Choi, M.K.; Yanai, H.; Negishi, H.; Ban, T.; Lu, Y.; Miyagishi, M.; Kodama, T.; Honda, K.; et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 2007, 448, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Unterholzner, L.; Keating, S.E.; Baran, M.; Horan, K.A.; Jensen, S.B.; Sharma, S.; Sirois, C.M.; Jin, T.; Latz, E.; Xiao, T.S.; et al. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010, 11, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Brennan, K.; Bowie, A.G. Activation of host pattern recognition receptors by viruses. Curr. Opin. Microbial. 2010, 13, 503–507. [Google Scholar] [CrossRef] [PubMed]
- Nakaya, Y.; Lilue, J.; Stavrou, S.; Moran, E.A.; Ross, S.R. AIM2-like receptors positively and negatively regulate the interferon response induced by cytosolic DNA. mBio 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Chen, Y.; Wang, H.; Wang, R. Mechanisms and pathways of innate immune activation and regulation in health and cancer. Hum. Vaccines Immunother. 2014, 10, 3270–3285. [Google Scholar] [CrossRef] [PubMed]
- Ablasser, A.; Goldeck, M.; Cavlar, T.; Deimling, T.; Witte, G.; Rohl, I.; Hopfner, K.P.; Ludwig, J.; Hornung, V. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates sting. Nature 2013, 498, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Burdette, D.L.; Vance, R.E. Sting and the innate immune response to nucleic acids in the cytosol. Nat. Immunol. 2013, 14, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Vance, R.E. Cytosolic DNA sensing: The field narrows. Immunity 2016, 45, 227–228. [Google Scholar] [CrossRef] [PubMed]
- Christensen, M.H.; Paludan, S.R. Viral evasion of DNA-stimulated innate immune responses. Cell. Mol. Immunol. 2017, 14, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Diner, B.A.; Lum, K.K.; Cristea, I.M. The emerging role of nuclear viral DNA sensors. J. Boil. Chem. 2015, 290, 26412–26421. [Google Scholar] [CrossRef] [PubMed]
- Kerur, N.; Veettil, M.V.; Sharma-Walia, N.; Bottero, V.; Sadagopan, S.; Otageri, P.; Chandran, B. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi sarcoma-associated herpesvirus infection. Cell Host Microbe 2011, 9, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.V.; Kerur, N.; Bottero, V.; Dutta, S.; Chakraborty, S.; Ansari, M.A.; Paudel, N.; Chikoti, L.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus latency in endothelial and B cells activates gamma interferon-inducible protein 16-mediated inflammasomes. J. Virol. 2013, 87, 4417–4431. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Dutta, S.; Veettil, M.V.; Roy, A.; Ansari, M.A.; Iqbal, J.; Chikoti, L.; Kumar, B.; Johnson, K.E.; Chandran, B. BRCA1 regulates IFI16 mediated nuclear innate sensing of herpes viral DNA and subsequent induction of the innate inflammasome and interferon-β responses. PLoS Pathog. 2015, 11, e1005030. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Dutta, D.; Iqbal, J.; Pisano, G.; Gjyshi, O.; Ansari, M.A.; Kumar, B.; Chandran, B. Nuclear innate immune DNA sensor IFI16 is degraded during lytic reactivation of Kaposi’s sarcoma-associated herpesvirus (KSHV): Role of IFI16 in maintenance of KSHV latency. J. Virol. 2016, 90, 8822–8841. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Chan, B.; Samarina, N.; Abere, B.; Weidner-Glunde, M.; Buch, A.; Pich, A.; Brinkmann, M.M.; Schulz, T.F. Cytoplasmic isoforms of Kaposi sarcoma herpesvirus LANA recruit and antagonize the innate immune DNA sensor cGAS. Proc. Natl. Acad. Sci. USA 2016, 113, E1034–E1043. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.J.; Li, W.; Shao, Y.; Avey, D.; Fu, B.; Gillen, J.; Hand, T.; Ma, S.; Liu, X.; Miley, W.; et al. Inhibition of cGAS DNA sensing by a herpesvirus virion protein. Cell Host Microbe 2015, 18, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Jacobs, S.R.; West, J.A.; Stopford, C.; Zhang, Z.; Davis, Z.; Barber, G.N.; Glaunsinger, B.A.; Dittmer, D.P.; Damania, B. Modulation of the cGAS-sting DNA sensing pathway by gammaherpesviruses. Proc. Natl. Acad. Sci. USA 2015, 112, E4306–E4315. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.; Singh, S.; Anang, V.; Bhatt, A.N.; Natarajan, K.; Dwarakanath, B.S. Pattern recognition receptors in cancer progression and metastasis. Cancer Growth Metastasis 2015, 8, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Ridnour, L.A.; Cheng, R.Y.; Switzer, C.H.; Heinecke, J.L.; Ambs, S.; Glynn, S.; Young, H.A.; Trinchieri, G.; Wink, D.A. Molecular pathways: Toll-like receptors in the tumor microenvironment—Poor prognosis or new therapeutic opportunity. Clin. Cancer Res. 2013, 19, 1340–1346. [Google Scholar] [CrossRef] [PubMed]
- Conroy, H.; Marshall, N.A.; Mills, K.H. TLR ligand suppression or enhancement of Treg cells? A double-edged sword in immunity to tumours. Oncogene 2008, 27, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Goto, Y.; Narita, N.; Hoon, D.S. Cancer cells expressing toll-like receptors and the tumor microenvironment. Cancer Microenviron. 2009, 2 (Suppl. S1), 205–214. [Google Scholar] [CrossRef] [PubMed]
- Jarnicki, A.; Putoczki, T.; Ernst, M. Stat3: Linking inflammation to epithelial cancer—More than a “gut” feeling? Cell Div. 2010, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Salcedo, R.; Cataisson, C.; Hasan, U.; Yuspa, S.H.; Trinchieri, G. MyD88 and its divergent toll in carcinogenesis. Trends Immunol. 2013, 34, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Szczepanski, M.J.; Czystowska, M.; Szajnik, M.; Harasymczuk, M.; Boyiadzis, M.; Kruk-Zagajewska, A.; Szyfter, W.; Zeromski, J.; Whiteside, T.L. Triggering of toll-like receptor 4 expressed on human head and neck squamous cell carcinoma promotes tumor development and protects the tumor from immune attack. Cancer Res. 2009, 69, 3105–3113. [Google Scholar] [CrossRef] [PubMed]
- Kelly, M.G.; Alvero, A.B.; Chen, R.; Silasi, D.A.; Abrahams, V.M.; Chan, S.; Visintin, I.; Rutherford, T.; Mor, G. TLR-4 signaling promotes tumor growth and paclitaxel chemoresistance in ovarian cancer. Cancer Res. 2006, 66, 3859–3868. [Google Scholar] [CrossRef] [PubMed]
- Kolb, R.; Liu, G.-H.; Janowski, A.M.; Sutterwala, F.S.; Zhang, W. Inflammasomes in cancer: A double-edged sword. Protein Cell 2014, 5, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Lowe, E.L.; Crother, T.R.; Rabizadeh, S.; Hu, B.; Wang, H.; Chen, S.; Shimada, K.; Wong, M.H.; Michelsen, K.S.; Arditi, M. Toll-like receptor 2 signaling protects mice from tumor development in a mouse model of colitis-induced cancer. PLoS ONE 2010, 5, e13027. [Google Scholar] [CrossRef] [PubMed]
- Slattery, M.L.; Herrick, J.S.; Bondurant, K.L.; Wolff, R.K. Toll-like receptor genes and their association with colon and rectal cancer development and prognosis. Int. J. Cancer 2012, 130, 2974–2980. [Google Scholar] [CrossRef] [PubMed]
- Isaza-Correa, J.M.; Liang, Z.; van den Berg, A.; Diepstra, A.; Visser, L. Toll-like receptors in the pathogenesis of human B cell malignancies. J. Hematol. Oncol. 2014, 7, 57. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Liu, Q.; Wang, L.; Chen, W.; Li, N.; Cao, X. TLR4 signaling promotes immune escape of human lung cancer cells by inducing immunosuppressive cytokines and apoptosis resistance. Mol. Immunol. 2007, 44, 2850–2859. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, M.R.; Conejo-Garcia, J.R. TLR5 signaling, commensal microbiota and systemic tumor promoting inflammation: The three parcae of malignant progression. Oncoimmunology 2015, 4, e1021542. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Zhu, S.; Qiao, Y.; Liu, Y.; Chen, W.; Zhao, G.; Chen, J. Recent advances in the role of toll-like receptors and TLR agonists in immunotherapy for human glioma. Protein Cell 2014, 5, 899–911. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.; Kozhaya, L.; Martiniuk, F.; Meng, T.C.; Chiriboga, L.; Liebes, L.; Hochman, T.; Shuman, N.; Axelrod, D.; Speyer, J.; et al. Topical TLR7 agonist imiquimod can induce immune-mediated rejection of skin metastases in patients with breast cancer. Clin. Cancer Res. 2012, 18, 6748–6757. [Google Scholar] [CrossRef] [PubMed]
- Kaczanowska, S.; Joseph, A.M.; Davila, E. TLR agonists: Our best frenemy in cancer immunotherapy. J. Leukocyte Biol. 2013, 93, 847–863. [Google Scholar] [CrossRef] [PubMed]
- Luddy, K.A.; Robertson-Tessi, M.; Tafreshi, N.K.; Soliman, H.; Morse, D.L. The role of toll-like receptors in colorectal cancer progression: Evidence for epithelial to leucocytic transition. Front. Immunol. 2014, 5, 429. [Google Scholar] [CrossRef] [PubMed]
- Sandholm, J.; Selander, K.S. Toll-like receptor 9 in breast cancer. Front. Immunol. 2014, 5, 330. [Google Scholar] [CrossRef] [PubMed]
- Arvaniti, E.; Ntoufa, S.; Papakonstantinou, N.; Touloumenidou, T.; Laoutaris, N.; Anagnostopoulos, A.; Lamnissou, K.; Caligaris-Cappio, F.; Stamatopoulos, K.; Ghia, P.; et al. Toll-like receptor signaling pathway in chronic lymphocytic leukemia: Distinct gene expression profiles of potential pathogenic significance in specific subsets of patients. Haematologica 2011, 96, 1644–1652. [Google Scholar] [CrossRef] [PubMed]
- Duewell, P.; Beller, E.; Kirchleitner, S.V.; Adunka, T.; Bourhis, H.; Siveke, J.; Mayr, D.; Kobold, S.; Endres, S.; Schnurr, M. Targeted activation of melanoma differentiation-associated protein 5 (MDA5) for immunotherapy of pancreatic carcinoma. Oncoimmunology 2015, 4, e1029698. [Google Scholar] [CrossRef] [PubMed]
- Sallets, A.; Kardosh, A.; Robinson, S.; Levy, R. In-situ vaccination using sting agonists combined with immune-modulating antibodies to treat lymphoma. Blood 2017, 130 (Suppl. S1), 4102. [Google Scholar]
Figure 1.
Summary of significant cancer diagnosis markers detected in Kaposi’s sarcoma-associated herpesvirus (KSHV)-associated neoplasms. These biomarkers can be categorized into virus specific markers (KSHV genome or specific proteins), cellular markers, cellular oncogenes, and inflammatory cytokines/growth factors.
Figure 1.
Summary of significant cancer diagnosis markers detected in Kaposi’s sarcoma-associated herpesvirus (KSHV)-associated neoplasms. These biomarkers can be categorized into virus specific markers (KSHV genome or specific proteins), cellular markers, cellular oncogenes, and inflammatory cytokines/growth factors.

Figure 2.
Schematic representation of the key principles involved in innate immune recognition by host pattern recognition receptors (PRRs). Upon viral infection, viral pathogen-associated molecular patterns (PAMPs), such as the viral genome or gene products are recognized by several cellular PRRs present on the cell membrane, in the cytoplasm or within intracellular compartments. Sensing of PAMPs by PRR triggers PRR-mediated signaling pathways and activates antiviral responses, leading to subversion of the infection. Abnormal activation of PRRs may also trigger auto-immunity. Abbreviations: TLRs, Toll-like receptors; NLRs, nucleotide oligomerization domain (NOD)-like receptors; RLRs, retinoic acid-inducible gene I (RIG-I)-like receptors; IFN-α/β, type-I interferons α/β; CCLs/CXCLs, chemokine ligands; IL-1β/IL-18, inflammatory cytokines.
Figure 2.
Schematic representation of the key principles involved in innate immune recognition by host pattern recognition receptors (PRRs). Upon viral infection, viral pathogen-associated molecular patterns (PAMPs), such as the viral genome or gene products are recognized by several cellular PRRs present on the cell membrane, in the cytoplasm or within intracellular compartments. Sensing of PAMPs by PRR triggers PRR-mediated signaling pathways and activates antiviral responses, leading to subversion of the infection. Abnormal activation of PRRs may also trigger auto-immunity. Abbreviations: TLRs, Toll-like receptors; NLRs, nucleotide oligomerization domain (NOD)-like receptors; RLRs, retinoic acid-inducible gene I (RIG-I)-like receptors; IFN-α/β, type-I interferons α/β; CCLs/CXCLs, chemokine ligands; IL-1β/IL-18, inflammatory cytokines.
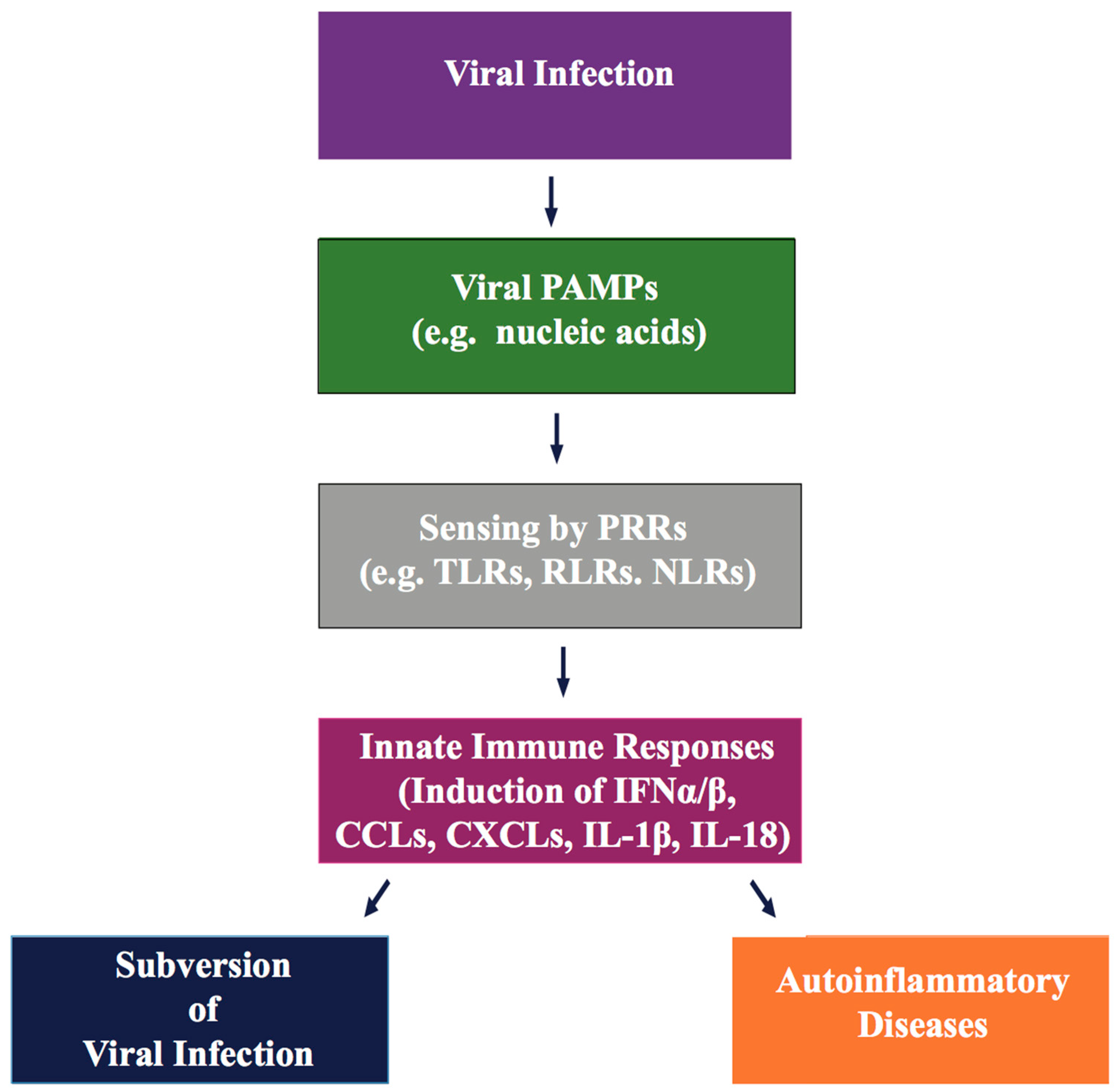
Figure 3.
KSHV regulation of the TLR signaling pathway. Upon KSHV infection, TLRs that are expressed either on the cell surface (TLR-2 and 4) or in endosomes (TLR-3, 4, 7, 8, and 9) recruit TIR-domain containing MyD88 and TRIF adaptor molecules. The MyD88 and TRIF molecules then recruit downstream molecules, culminating in the activation of transcription factors such as IRF3/7 and NF-κB, which regulate the production of type I IFNs and inflammatory cytokines.
Figure 3.
KSHV regulation of the TLR signaling pathway. Upon KSHV infection, TLRs that are expressed either on the cell surface (TLR-2 and 4) or in endosomes (TLR-3, 4, 7, 8, and 9) recruit TIR-domain containing MyD88 and TRIF adaptor molecules. The MyD88 and TRIF molecules then recruit downstream molecules, culminating in the activation of transcription factors such as IRF3/7 and NF-κB, which regulate the production of type I IFNs and inflammatory cytokines.
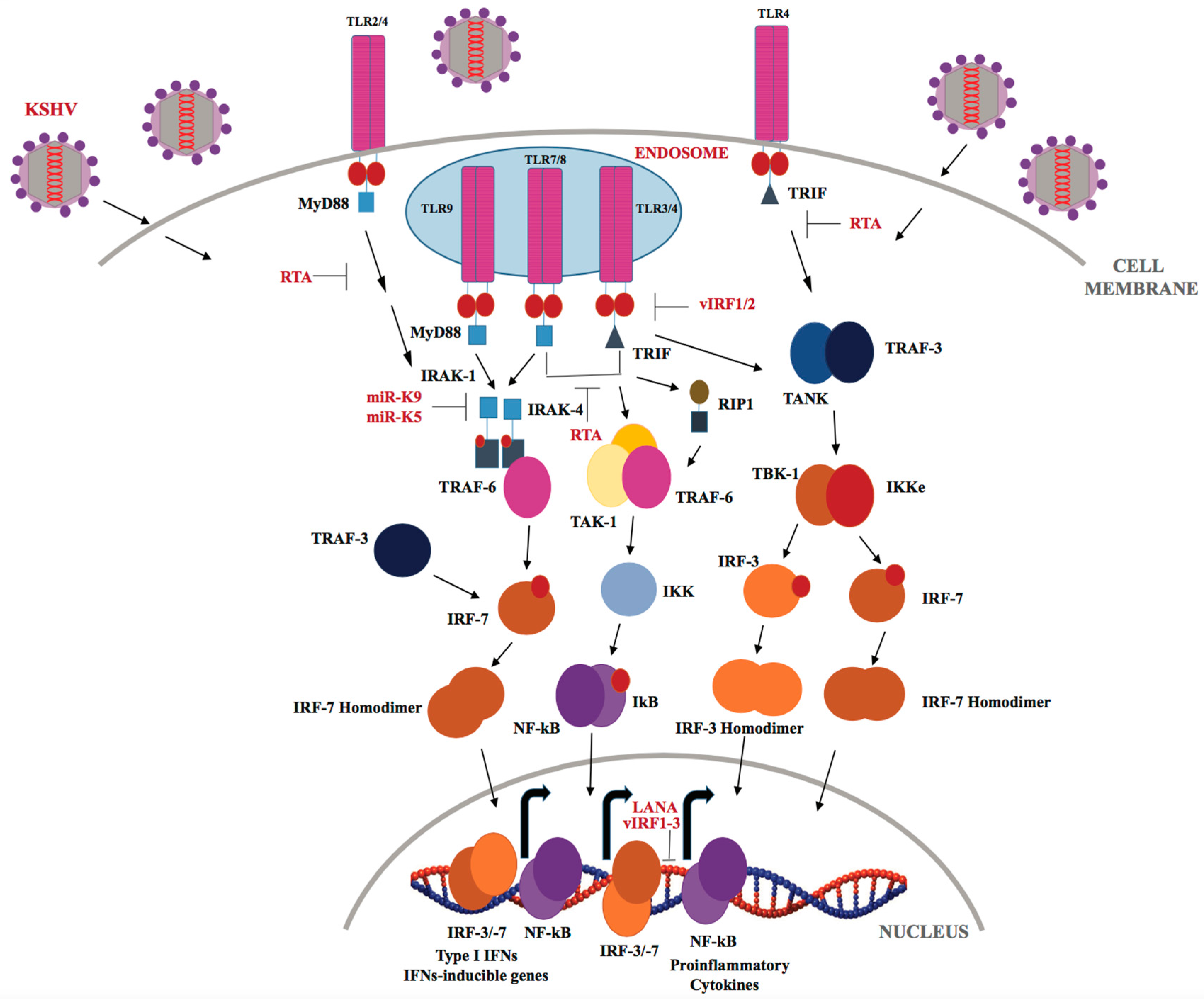
Figure 4.
KSHV regulation of NLR, RLR, and cGAS-STING signaling pathways. Following KSHV entry into the host cell, the capsid extrudes KSHV dsDNA in the cytoplasm which is sensed by cGAS resulting in the STING/TBK1/IRF3 signaling and type I IFNs production. KSHV dsDNA is transcribed into dsRNA polymerase III and sensed by RIG-I and MDA-5, leading to activation and polymerization of MAVS and activation of NF-κB. KSHV also controls the inflammatory responses via NLRP-1/3-dependent caspase-1 activation and inhibition of IL-1β and IL-18 production.
Figure 4.
KSHV regulation of NLR, RLR, and cGAS-STING signaling pathways. Following KSHV entry into the host cell, the capsid extrudes KSHV dsDNA in the cytoplasm which is sensed by cGAS resulting in the STING/TBK1/IRF3 signaling and type I IFNs production. KSHV dsDNA is transcribed into dsRNA polymerase III and sensed by RIG-I and MDA-5, leading to activation and polymerization of MAVS and activation of NF-κB. KSHV also controls the inflammatory responses via NLRP-1/3-dependent caspase-1 activation and inhibition of IL-1β and IL-18 production.


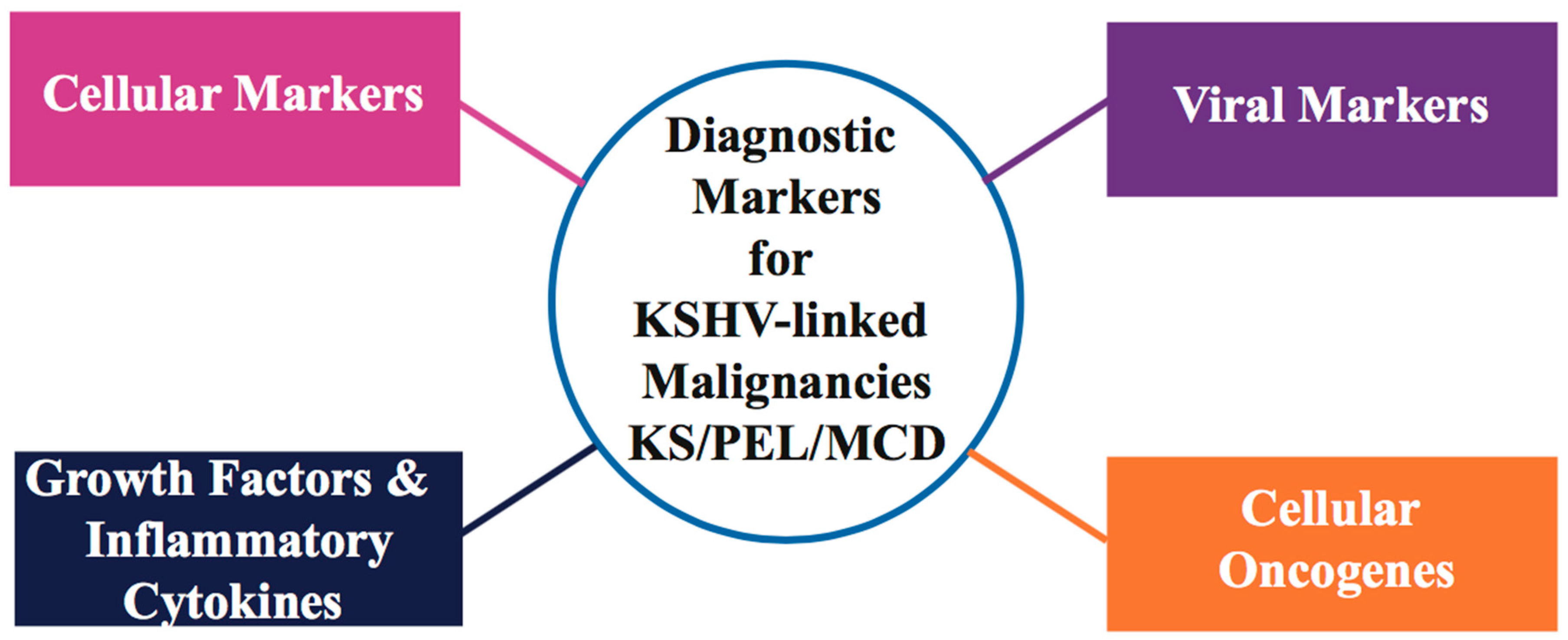{kind=link}
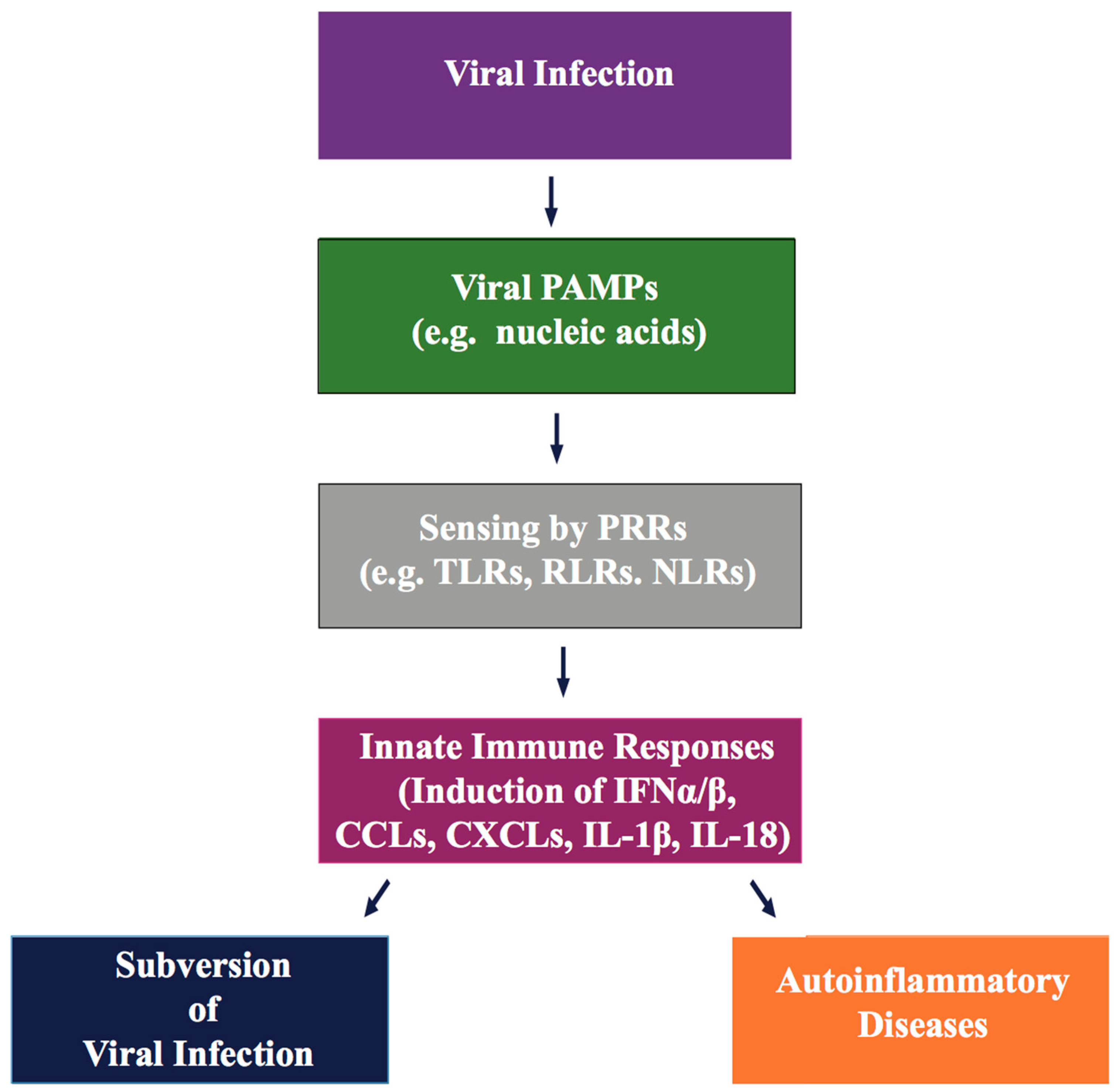{kind=link}
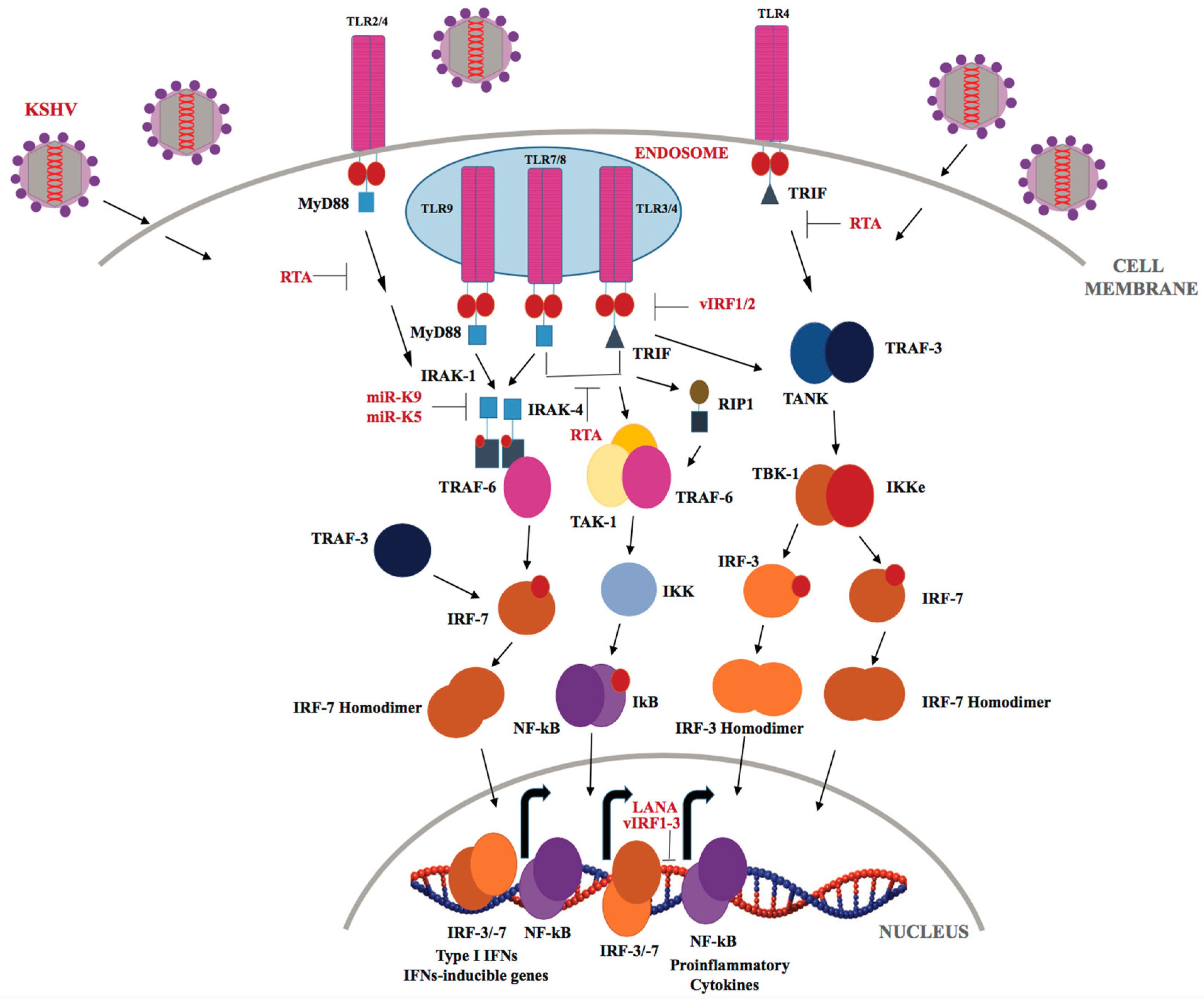{kind=link}
{kind=link}
Table 1.
Summary of current biomarkers in acquired immune deficiency syndrome (AIDS)-associated KS.
| Cellular Markers | Viral Markers/Proteins | Cellular Oncogenes | Growth Factors/Inflammatory Cytokines |
|---|---|---|---|
| CD4 Count [41,61,62] | KSHV Virus [63,64,65] | bcl-2 [49,66,67] | CD31 [57,64,68,69] |
| LANA/ORF73 [64,65,66,70] | c-kit [71,72] | CD34 [57,68,69] | |
| vCyclin/ORF72 [67,73,74] | p53 [46,75,76] | CD36 [51,77,78] | |
| vFLIP/ORF71 [49,74,79,80] | pRb [47,81,82] | CD138/Syndecan-1 [83,84] | |
| K12/Kaposins [49,85,86] | FLI1 [57,87,88] | ||
| miRNAs [48,85,86,89] | D2-40 [57,90,91] | ||
| vIRFs [48,92,93,94] | Podoplanin [54,55,95] | ||
| vIL-6 [49,50,59] | VEGFR3 [39,56,95,96] | ||
| vGPCR/ORF74 [48,97] | LYVE-1 [39,54,95,96] | ||
| TNF-α [58,59] | |||
| bFGF [39,58,98,99] | |||
| Oncostatin M [58,60,100] | |||
| vIL-6 [49,50,59] | |||
| IL-I [58,59,101] |
Table 2.
Summary of key PRRs and associated human cancers.
| PRRs | Cellular Localization | PAMPs | Associated Tumors |
|---|---|---|---|
| TLR-1/2 | Plasma membrane | Lipoprotein | Colon cancer [191] |
| TLR-3 | Endosome | Double stranded (ds)RNA | Melanoma, Colorectal adenoma, low-grade B-cell Lymphoma, Solid tumors [192,193] |
| TLR-4 | Plasma membrane | Lipopolysaccharides | Lung cancer, Non-Hodgkin’s lymphoma [194] |
| TLR-5 | Plasma membrane | Flagellin | Advanced/Metastatic solid tumors [195] |
| TLR-7/8 | Endosome | Single stranded (ss)RNA | Ovarian cancer, Solid tumors [196,197,198] |
| TLR-9 | Endosome | CpG-DNA | Colorectal cancer, Breast cancer, Chronic Lymphocytic Leukemia [199,200,201] |
| MDA5 | Cytoplasm | Long dsRNA | Solid tumors [202] |
| STING | Cytoplasm | dsDNA | Advanced/Metastatic solid tumors [203] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Uppal, T.; Sarkar, R.; Dhelaria, R.; Verma, S.C. Role of Pattern Recognition Receptors in KSHV Infection. Cancers 2018, 10, 85. https://doi.org/10.3390/cancers10030085
AMA Style
Uppal T, Sarkar R, Dhelaria R, Verma SC. Role of Pattern Recognition Receptors in KSHV Infection. Cancers. 2018; 10(3):85. https://doi.org/10.3390/cancers10030085
Chicago/Turabian StyleUppal, Timsy, Roni Sarkar, Ranjit Dhelaria, and Subhash C. Verma. 2018. "Role of Pattern Recognition Receptors in KSHV Infection" Cancers 10, no. 3: 85. https://doi.org/10.3390/cancers10030085
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.