YAP and TAZ in Lung Cancer: Oncogenic Role and Clinical Targeting



1
Oncogenomic and Epigenetic Unit, Molecular Medicine Area Regina Elena National Cancer Institute, via Elio Chianesi 53, 00144 Rome, Italy
2
Molecular Chemoprevention Group, Molecular Medicine Area Regina Elena National Cancer Institute, via Elio Chianesi 53 00144 Rome, Italy
*
Author to whom correspondence should be addressed.
Cancers 2018, 10(5), 137; https://doi.org/10.3390/cancers10050137
Submission received: 30 March 2018
/
Revised: 1 May 2018
/
Accepted: 2 May 2018
/
Published: 6 May 2018
(This article belongs to the Special Issue Hippo Pathway in Cancer, towards Realization of the Hippo-Targeted Therapy)
Abstract
:Lung cancer is the leading cause of cancer death in the world and there is no current treatment able to efficiently treat the disease as the tumor is often diagnosed at an advanced stage. Moreover, cancer cells are often resistant or acquire resistance to the treatment. Further knowledge of the mechanisms driving lung tumorigenesis, aggressiveness, metastasization, and resistance to treatments could provide new tools for detecting the disease at an earlier stage and for a better response to therapy. In this scenario, Yes Associated Protein (YAP) and Trascriptional Coactivator with PDZ-binding motif (TAZ), the final effectors of the Hippo signaling transduction pathway, are emerging as promising therapeutic targets. Here, we will discuss the most recent advances made in YAP and TAZ biology in lung cancer and, more importantly, on the newly discovered mechanisms of YAP and TAZ inhibition in lung cancer as well as their clinical implications.
1. Introduction
Among solid tumors, lung cancer is the first cause of cancer death worldwide with a 5-year survival rate lower than 20%. Nearly 80–85% of lung cancers are non-small cell lung cancer (NSCLC), which include adenocarcinoma (LAC), lung squamous cell carcinoma (LSCC), and large-cell carcinoma [1]. One of the reasons for this short survival is the lack of clear symptoms occurring until advanced stages of the disease. Many diagnoses are given when the cancer has already progressed beyond a localized state. Moreover, in some patients, cancer cells are often resistant or acquire resistance to therapy. Finally, in 10% to 25% of lung cancer patients, brain metastases can occur and are associated with unfavorable prognosis and loss of cognitive functions [2]. Up until now, current treatment based on surgery, radiation, chemotherapy, laser therapy, and photodynamic therapy associated with palliative care only increase the overall survival and quality of life of patients, but NSCLC still remains one of the most aggressive malignant tumors with the lowest survival rate [3,4,5]. This observation spurred the development of new technology for the early detection of lung cancer in people who are at very high risk [6,7,8] as well as the development of targeted therapies for patients with known mutations driving lung cancer. In this scenario, the characterization of early molecular biomarkers can provide a useful tool for early diagnosis while a deeper understanding of the mechanisms driving lung transformation, metastasization, and resistance to therapy could provide new therapeutic targets.
2. The Hippo Pathway
The Hippo signaling transduction pathway controls animal organ development, growth, and regeneration upon injury, and its dysregulation is often involved in tumorigenesis [9,10,11]. Cell contact, cell polarity, and metabolic and mechanical signals, which change during organ development and growth to properly orchestrate these processes, regulate the activity of the Hippo pathway core components, consisting of a cascade of kinases (MST1/2 and LATS1/2) with adaptor proteins (SAV and MOB) whose final targets are the transcriptional coactivators Yes Associated Protein (YAP) and Trascriptional Coactivator with PDZ-binding motif (TAZ) [11,12,13]. When the Hippo pathway is turned on, YAP and TAZ are phosphorylated by LATS1/2, promoting their cytoplasmic sequestration and proteasome-mediated degradation. When the Hippo pathway is turned off, YAP and TAZ are dephosphorylated and translocated to the nucleus where they are able to activate target genes in association with different transcription factors ultimately regulating cell growth, metabolism, proliferation, migration, invasion, or cell death [12,14,15]. Recent evidence added new complexity to this simple model, adding new players and regulators to the Hippo pathway core kinases [16]. For example, two groups of MAP4Ks (mitogen-activated protein kinase)—MAP4K1/2/3/5 and MAP4K4/6/8—were recently discovered acting in parallel with MST1/2 to phosphorylate and activate LATS1/2 [17,18]. Moreover, Hippo core kinases may be involved in the regulation of proliferation and the cell cycle independently of YAP/TAZ function [19,20,21] and, vice versa, YAP and TAZ are not exclusively regulated by the Hippo pathway core kinases. In fact, YAP and TAZ may undergo different post-translational modifications beyond Ser127 phosphorylation and can interact with several different protein partners, resulting in different activation states and/or subcellular localization of YAP/TAZ [22,23,24,25,26,27,28,29,30]. Additionally, YAP/TAZ and other Hippo pathway components crosstalk with several other signaling pathways, such as EGFR, Wnt, TGF-β, and Notch, involved in development and cell proliferation (reviewed in [31,32]). Finally, YAP and TAZ are often treated as single proteins, but they are two distinct proteins which exert both overlapping and exclusive functions [9,33,34,35,36].
3. Regulation of the Hippo Pathway
Physiological organ development and growth or regeneration upon injury are the final results of specific patterns of cell growth, proliferation, migration, commitment, differentiation, senescence, or apoptosis. These, in turn, are orchestrated by the mechanical and biochemical stimuli originating from intrinsic cell machineries and from the extracellular matrix (ECM). These stimuli regulate proteins involved in mechanotransduction, cell junction, cell polarity, G-protein-coupled receptor (GPCR) signaling, receptor tyrosine kinase (RTK) mitogenic signaling, and metabolism which, in turn, regulate the core Hippo pathway and subsequent YAP and TAZ nuclear activity [11,12,13]. In general, low mechanical stress (which can be experienced by cells grown on a soft extracellular substrate or at a high cell density), serum deprivation, low glucose and nutrients, as well as diffusible signals that inhibit cell proliferation and metabolism are activators of the Hippo pathway core kinases and thus inhibit YAP/TAZ nuclear activity. Conversely, low cell density, stiff extracellular substrate or high mechanical stress transduced to the cytoskeleton, mitogenic signals, inflammation, and high nutrient uptake activate nuclear YAP and TAZ [11,12]. The nature and the position of the post-translational modifications of YAP and/or TAZ, their subcellular localization, their total abundance, their specific interactions with other proteins, and their crosstalk with other signaling pathways will eventually determine their final biological outcome. This, in turn, depends on (1) the signals coming from the extracellular environment (upstream signaling) and (2) the cell type which transduces these signals (downstream signaling).
4. YAP and TAZ are Pro-Oncogenic in Solid Tumors
During physiologic embryogenesis and development, YAP and TAZ cooperate with transcriptional complexes in the regulation of cell stemness, cell growth, specification and differentiation, while in the adult, YAP and TAZ are required for proliferation of adult progenitors/stem cells and for the regeneration of damaged tissues [37,38]. These functions are elicited through the regulation of different transcriptional programs in a context-dependent manner. The dysregulation of YAP and TAZ function in adult cells has shown both protumorigenic and pro-apoptotic effects in different experimental systems and conditions.
Generally, in hematologic malignancies, YAP is either deleted or downregulated and lower YAP expression correlates with poorer prognosis and shorter survival of patients [39]. In contrast, in many solid cancers, YAP and TAZ behave as oncogenes and are upregulated or hyperactivated compared to normal tissues, and a higher YAP/TAZ level or activity correlates with poorer prognosis and shorter patient survival [9,10,11].
In the context of solid cancers, YAP and TAZ transcriptionally activate genes involved in cell pluripotency and stemness, proliferation, migration, and invasiveness, and associate with oncogenic transcription factors like TEAD1-4, Smad1, 2/3, KLF5, AP-1, β-catenin, TBX5, ERG, Erb-B4 and mutp53 [29,40,41,42,43,44,45,46,47,48,49,50,51]. Recent lines of evidence have shown YAP/TAZ’s role also in transcriptional repression of tumor suppressors [52]. Frequently, higher YAP/TAZ expression levels can be observed in tumoral compared to matched nontumoral tissues in different cancer types. However, increased activity of YAP and/or TAZ in solid cancer is rarely caused by genomic amplification of YAP/TAZ loci. Instead, the main mechanisms that increase YAP and TAZ oncogenic activity in vivo are the modifications of the extracellular and intracellular signals occurring during tumorigenesis, for example, the formation of a hypoxic environment, the disruption of the apico-basal cell polarization in epithelial tissues, the presence of inflammation and cell/tissue damage signals, and increased mechanical stress induced by a higher production of cellular and extracellular constituents by cancer cells [37,53,54,55,56]. Additionally, several metabolic alterations occurring in most tumors increase YAP/TAZ oncogenic function. For example, cancer cells preferentially metabolize glucose through glycolysis, producing less energy (ATP) but promoting anabolism, cell growth, and proliferation [57] compared to normal cells which preferentially use oxidative phosphorylation to produce ATP. Glycolysis has been shown to activate and sustain the protumorigenic functions of YAP and TAZ [58,59,60]. Also, the increase of lipid biosynthesis is required to sustain the increased tissue growth during tumorigenesis [61,62]. Among the pathways involved in lipid biosynthesis, the mevalonate pathway and the synthesis of monounsaturated fatty acids have been shown to sustain YAP/TAZ oncogenic function [63,64,65] and will be discussed later in detail. All these mechanisms are described in Figure 1.
5. Hippo Pathway in Lung Cancer
In physiological conditions, YAP and TAZ are required for normal lung development and regeneration after injury [66,67]. In a mouse model of developing lung, the apical polarity protein Crb3 inhibits YAP/TAZ nuclear activity by sequestering them at the apical plasma membrane together with LATS1/2 in proximal airway epithelial cells. This inhibits proliferation and promotes differentiation. Conversely, in undifferentiated basal progenitor cells, where Crb3 levels are undetectable, YAP/TAZ are mainly nuclear and promote cell stemness and proliferation. Downregulation of Crb3 in proximal airway epithelium increases YAP/TAZ nuclear localization and cellular proliferation, leading to morphological changes which resemble a premalignant epithelium [68]. This suggests that the subcellular localization of YAP and TAZ is fine-tuned during lung development, when a balance between nuclear and cytoplasmic YAP and TAZ is necessary to support normal organ development and patterning.
Interestingly, in adults, YAP and TAZ are not required for normal tissue homeostasis, while numerous experimental results show that nuclear YAP and TAZ are required to drive lung tumor formation, survival, stemness, progression, metastasization, and resistance to therapy, in particular in non-small cell lung cancer (NSCLC) that will be thoroughly described in the following sections. Conversely and interestingly, in a specific subset of small cell lung cancer cell lines and patients, the loss of YAP1 has been shown to be associated with an increase of neuroendocrine markers and a poorer prognosis [69,70] similar to what has been observed in haematologic malignancies. However, much of the literature reports evidence of the oncogenic role of YAP and TAZ in NSCLC which will be the focus of our review; meanwhile, we will keep in mind the opposite role of YAP and TAZ observed in SCLC compared to NSCLC that become clinically relevant when NSCLC is transformed into SCLC and needs different therapeutic treatments [71].
In NSCLC, YAP overexpression has been associated with development, progression, and poor prognosis [72,73,74,75,76,77] similar to TAZ [78,79,80], and an epidemiological study showed that a germline YAP mutation resulting in its oncogenic hyperactivation is associated with the occurrence of lung cancer [81]. In vivo studies in mouse models of lung adenocarcinoma showed that the genetic loss of YAP reduces the number of tumor masses experimentally induced in mice, while the knock-down of YAP or TAZ in human NSCLC cells impairs tumor formation after injection into nude mice [82,83]. TAZ overexpression in normal bronchial epithelial cells was found to be sufficient to promote tumor formation when injected into nude mice in one study [80] and not in another study which used a different cell line [78], while overexpression of YAP alone was not able to induce de novo tumor formation but only to promote progression of tumors driven by Kirsten RAt Sarcoma (KRAS) or Liver Kinase B1 (LKB1) mutations in mice [82,83]. Moreover, YAP and TAZ staining was stronger in metastatic versus nonmetastatic experimentally induced tumors in mice [82,83,84].
When translating these results into clinics, an increase of the YAP protein was observed in the nucleus and not in the cytoplasm in lung cancer specimens compared to the matched normal tissues [76]. In another study, the presence of higher levels of nuclear YAP was associated with poorer prognosis while, conversely, higher levels of cytoplasmic YAP were associated with lower histologic grade and TNM stage in NSCLC [75]. This indicates that in different cellular compartments, YAP and/or TAZ may exert completely different functions. In particular, this study clearly reinforces the idea that the oncogenic function of YAP and TAZ is exerted mainly in the nucleus through the transcriptional regulation of genes involved in tumorigenesis, while in the cytoplasm, YAP and TAZ are prevented from activating pro-proliferative genes. In agreement with this hypothesis, in different cohorts of lung cancer patients, higher expression of genes belonging to the YAP/TAZ signature correlates with poorer prognosis [80,82].
The increase of YAP and TAZ nuclear activity can be induced by an increase of signals, receptors, or transducers which positively regulate YAP/TAZ activity or by a decrease in signals or molecules that negatively regulate YAP/TAZ oncogenic function. This depends on one hand on the acquired genetic and epigenetic alterations of the genes coding for YAP/TAZ regulators in cancer cells, and on the other hand on the mechanical and biochemical changes occurring in the tumor microenvironment to sustain tumor growth. For example, the oncogenic ABL1 and 2 kinases, shown to be overexpressed or hyperactivated in NSCLC [85,86,87,88] and involved in tumor growth and metastasization in experimental models of lung cancer [27,87,88], have been recently demonstrated to act in part through the activation of TAZ and β-catenin by inhibiting their binding to β-TrCP, thus increasing their stability. In mouse models, ABL1 and 2 inactivation reduced the metastasization of NSCLC to the bone and brain, impinging on the expression of a subset of TAZ/β-catenin oncogenic target genes. In clinical lung cancer patient data, high expression levels of ABL1 and ABL2 and concomitant high transcription of TAZ/β-catenin targets correlate with shorter survival [27]. Also, several RTKs including EGFR or their downstream transducers/effectors like Src/Ras/Raf/MEK and PI3K/AKT/mTOR are hyperactivated and contribute to bad prognosis in lung cancer [89,90,91,92,93,94,95,96]. EGFR signaling crosstalks with Wnt-βcatenin signaling [97], GPCR signaling [98,99], and Hippo signaling [100]. It participates in the positive regulation of YAP and TAZ oncogenic function in lung cancer [101] and can synergize with YAP and TAZ in lung tumorigenesis and tumor growth and invasion [102].
Among YAP/TAZ endogenous inhibitors, it has been shown that LATS2 is mutated or more frequently downregulated in NSCLC through multiple mechanisms such as promoter hypermethylation, or recruitment of repressive epigenetic complexes including long noncoding RNAs and repressive Enhancer of Zeste Homolog 2 (EZH2) [103,104,105,106,107]. Higher LATS2 levels in lung cancer patients contribute to better prognosis [108] while forced overexpression of LATS2 in vitro reduces tumorigenicity of NSCLC [104]. Similarly, LATS1 is downregulated in 60% of NSCLC cancers while its high level contributes to good prognosis and negatively regulates oncogenic YAP in NSCLC [105,109,110]. AMOT, a scaffold protein that sequesters YAP and TAZ into the cytoplasm, inhibiting their nuclear function, has been shown to decrease lung cancer progression in vitro while its knock-down increases lung cancer growth and metastasis in vivo, and is significantly reduced in lung cancer specimens [111]. Also, MST1 kinase has been shown to inhibit NSCLC growth in vitro and in vivo [112]. Moreover, VGLL4, an inhibitor of YAP–TEAD interaction, inhibits cancer progression through YAP inactivation and is downregulated in lung adenocarcinoma [113]. Ras-association domain family 1 isoform A (RASSF1A), which activates MST1/2 and LATS1 in the presence of DNA damage or other stress signals [114,115,116], is epigenetically inactivated on its promoter in 40% of primary NSCLC [117,118,119]. Importantly, a significant hypermethylation as well as somatic loss of etherozigosity (LOH) of the RASSF1A locus has been observed also in SCLC [118,119]. This inactivation is associated with poor prognosis [119,120]. Mechanistically, it has been shown that RASSF1A inactivation in normal bronchial epithelial cells contributes to cell invasion and migration in part through the activation of nuclear YAP [121]. LKB1 has been recently shown to suppress YAP/TAZ nuclear function through either MST1 activation [84] or Hippo-independent pathways [122] in response to metabolic stress. LKB1 inactivation is involved in tumorigenesis [123], it is the third most frequently mutated gene in adenocarcinoma [124], and somatic mutations of LKB1 have been reported in 20–30% of NSCLC and derivative cell lines [124,125,126,127,128]. Mutations in LKB1 gene often occur together with KRAS-activating mutations, the most frequent oncogenic mutations found in NSCLC [128]. In mouse lung cancer models, LKB1 downregulation does not drive tumorigenesis by itself but it induces a more aggressive phenotype in KRAS-driven tumors, suggesting a role in lung cancer progression and metastasis. Accordingly, in NSCLC cell lines, LKB1 represses the expression of pro-metastatic genes [126] or pro-survival genes [83], and its lower expression in patients correlates with advanced disease [128]. A recent study showed that the interference of RhoA, which transduces mechanical cytoskeletal forces to YAP/TAZ, inhibited proliferation and metastasization in a cellular model of NSCLC [129]. A recent study found that Ski inhibits YAP and mainly TAZ in lung because it induces their phosphorylation mediated by LATS1/2, thus inducing their proteasome-mediated degradation but not their cytoplasmic translocation. In 56% of NSCLC tissues, the Ski promoter is hypermethylated and its downregulation is associated with poorer prognosis [130].
A recent study by Noto and coworkers showed that YAP and TAZ are required for the maintenance of stemness properties in lung cancer cell cultures [64]. Before this report, the role of YAP and TAZ in cancer stem cells was investigated in detail only in breast cancer, hepatocarcinoma, and esophageal cancer [35,131,132,133]. Moreover, the authors showed that stearoyl-CoA-desaturase 1 (SCD1), involved in the conversion of saturated into monounsaturated fatty acids, is important for the expression, nuclear localization, and transcriptional activity of YAP and TAZ, in part by acting through the Wnt-β-catenin pathway. Accordingly, the high co-expression of SCD1, β-catenin, and YAP/TAZ has a negative prognostic value in adenocarcinoma lung cancer patients [64]. This is the first report showing a correlation between an increased biosynthetic lipid metabolism driven by tumorigenesis and a hyperactivation of oncogenic YAP/TAZ in NSCLC. Before this study, this had been described only in other cancer types [134]. This report couples with our recent report showing that pharmacological inhibition of the mevalonate pathway inhibits YAP/TAZ oncogenic function also in NSCLC models [135] (see below). Later on, another work confirmed a role for TAZ in promoting cell stemness and tumorigenesis in lung through the upregulation of ALDH1A [51].
YAP overexpression has been associated with increased resistance of NSCLC to current therapies because YAP provides parallel survival input to treatments [136,137,138]. All these observations underlie an important oncogenic role of YAP/TAZ as well as their upstream regulators in lung cancer (Table 1 and Figure 2).
6. YAP and mutP53 Can Synergize in Lung Cancer
P53 is one of the most studied tumor suppressors and its coding gene, TP53, is the most commonly mutated in human cancer [139]. More than 50% of tumors arise from mutations of the wild-type TP53 gene sequence. These mutational events may lead to loss of tumor suppressor function of p53 or may generate alternative functional proteins with oncogenic properties. The functional mutated forms of p53 protein (mutp53), including those with certain amino acid substitutions (R175, G245, R248, R249, R273, and R282), are more stable, interact with protein partners different from those of wild-type p53, and lose the original DNA-binding specificity, thus targeting genes promoting oncogenic activities such as invasion, migration, tissue remodelling, angiogenesis, stem cell expansion, proliferation, survival, and increased chemo-resistance [140,141]. Immunohistochemistry is employed in the clinical setting in order to assess the presence of mutp53 in cancer tissues, as mutp53 can easily be detected, being much more stable than the wild-type counterpart. Moreover, in several tumors including lung cancer, the accumulation of mutp53 may trigger an autoimmune response against p53 (p53Abs) [142]. In lung cancer, p53 overexpression is detected at an early stage, also in pre-neoplastic lesions, and the presence of p53 antibodies is observed many months before the lung cancer diagnosis in heavy smokers [143]. Two meta-analysis studies showed that the presence of p53 staining is an independent prognostic factor in lung cancer [144,145]. Moreover, a recent systematic review with meta-analysis showed that p53Abs are more abundant in the blood of patients affected by several types of cancers, including lung cancer, than in healthy controls [146] and together with other markers, p53Abs can be used for lung cancer detection [147]. A recent study showed that the presence of p53Abs was significantly associated with poorly differentiated and higher-grade tumors and was predictive for shorter survival in NSCLC patients [148]. A search conducted in the cBioPortal for Cancer Genomics website using data derived from The Cancer Genome Atlas [149] shows that mutp53 is present in nearly 85% of small cell lung cancer and in 60% of non-small cell lung carcinoma (Figure 3).
All these lines of evidence indicate that mutp53 plays a pivotal role in lung tumorigenesis and can be a valid diagnostic and prognostic factor in lung cancer. Strikingly, in our laboratory it has been shown that mutp53 is able to form a complex with the transcriptional coactivator YAP and the transcription factor NFY on pro-oncogenic genes such as cyclin A, cylin B, and CDK1 [48]. This has been observed in different models of human cancers (metastatic breast cancer, head and neck and pancreatic cancer cell lines) bearing different mutp53 isoforms. Moreover, treatment with inhibitors of the mevalonate pathway reduced tumor growth in vivo and reduced the expression of mutp53/YAP oncogenic targets in vitro by impairing the recruitment of the YAP/mutp53/NFY transcriptional complex onto their promoter [48].
Based on these lines of evidence, it will be interesting in the future to investigate any synergism between YAP and mutp53 function in this context. Consistently, tissues from NSCLC patients with higher expression and nuclear localization of YAP showed a higher level of cyclinA [77]. In the study mentioned, the status of TP53 was not investigated, but YAP may cooperate with mutp53 on the promoter of cyclinA and other cell-cycle-related genes.
YAP and mutp53 can synergize not only as transcriptional regulators of common oncogenic targets. In fact, mutp53 can support tumor growth both when expressed in cancer cells and also in the stroma surrounding tumors [150,151,152]. The same has been observed for YAP, which has been shown to be overexpressed in Cancer-Associated Fibroblasts (CAFs), promoting matrix stiffening and tumor growth [153]. Moreover, the Hippo and p53 signaling pathways cooperate to regulate cholesterol/lipid levels [134] which are in turn regulators of YAP. Accordingly, pharmacological inhibition of pathways involved in lipid biosynthesis inhibits YAP/TAZ oncogenic function [63,64,154,155]. In cancer cells, lipid and cholesterol biosynthesis are upregulated and drive tumor malignancy [156] partly through the upregulation of the sterol regulatory element binding protein (SREBP) transcription factors [134]. While wild-type p53 and LATS2 cooperate in inhibiting SREBP expression, mutp53 has been shown to sustain the expression of SREBP in breast cancer cells, suggesting an additional layer of positive regulation of YAP by mutp53, through the upregulation of lipid metabolism [156]. This can be investigated also in lung cancer where lipid metabolism is showing an emerging role [65,157]. Interestingly, when we interrogated 1144 samples from lung cancer patients collected in the cBioportal website database (Pan-Lung Cancer, TCGA Nat. Genet. 2016), we found that YAP amplification or LATS2 deletion co-occur with oncogenic driver mutations of the TP53 gene (Figure 4), suggesting a possible cooperation between mutp53 and the overexpression or hyperactivation of YAP in lung cancer. Interestingly, while YAP1 and TP53 alterations co-occur in lung cancer, TAZ and TP53 alterations are mutually exclusive. This is in agreement with a work from our laboratory where a transcriptional cooperation on the promoter of cell-cycle-regulating genes has been observed between mutp53 and YAP, but not between mutp53 with TAZ, highlighting a difference between YAP and TAZ [48]. Based on this observation, a detailed analysis of the TP53 mutational status combined with the activity of YAP (the latter not only determined by gene amplification but also by gene expression and protein activity) in relation to stage/grade of the tumor and prognosis of lung cancer patients can be a starting point for future studies headed in this direction.
7. An Emerging Role of YAP/TAZ in microRNA Regulation in NSCLC
Long lists of YAP/TAZ pro-oncogenic target genes have been published from studies on different experimental systems and conditions in order to dissect the mechanisms through which YAP/TAZ could elicit their oncogenic role [41,49,82,159,160,161,162]. However, not much is known about YAP/TAZ-regulated microRNAs, with only two reports in MCF10A cells or in human pulmonary arterial adventitial fibroblasts [163,164] and two other reports showing that YAP/TAZ can influence miRNA biogenesis by modulating the activity of specific subunits of the miRNA processing machinery [165,166] independently of YAP/TAZ function as transcriptional coactivators.
With the exception of these studies, evidence of a direct transcriptional role of YAP/TAZ in microRNA regulation in the tumor context and on a large scale have never been investigated. This lack of knowledge is relevant, since altered miRNA expression can contribute to tumorigenesis by inappropriately modulating critical genes involved in tumor cell biology. Our recent study is the first report of a list of microRNAs specifically regulated by YAP and TAZ in NSCLC [135]. We focused on a cluster of three oncogenic microRNAs (miR-25, 93, and 106b) located in the intron 13 of MCM7 (Minichromosome Maintenance complex 7) gene. Both the coding MCM7 transcript and the hosted microRNAs are oncogenic, and they are upregulated in lung cancer tissues and show a prognostic role [167,168,169,170,171,172,173]. We found that MCM7 transcript and the hosted microRNAs form a bi-oncogenic locus [174] that is positively regulated by YAP/TAZ as a single transcriptional unit [135]. We showed that in NSCLC cell lines, MCM7 and hosted miRs repress a list of cancer-related genes, and focused on the p21 cell cycle inhibitor. Two previous works showed YAP as a negative regulator of the p21 gene at a transcriptional level [175,176], while we elucidated a new layer of p21 inhibition mediated by YAP/TAZ through a post-transcriptional mechanism [135]. Moreover, these findings hold great potential for translational medicine as microRNAs are frequently used as molecular biomarkers of diagnostic and prognostic relevance in human cancers, for patient stratification and for monitoring the response of cancer patients to treatment. It will be important in the future to characterize the whole transcriptome under YAP/TAZ modulation in addition to microRNAs because competing endogenous RNAs, long noncoding RNAs, and enhancer-associated RNAs are emerging for their role in development and cancer, including lung cancer [177,178]). In agreement with this hypothesis, YAP/TAZ have recently been globally mapped at enhancers much more than in the proximity of transcription start sites [49].
8. YAP/TAZ as Therapeutic Targets in NSCLC: State of the Art
Current therapy for lung cancer involves platinum-based chemotherapy, while radiation is used for the treatment of locally advanced disease, concurrently with platinum chemotherapy. These therapies, however, show strong side effects in patients because they are highly cytotoxic also for healthy cells. In order to overcome these side effects, increased effort is being put towards targeted therapy according to the biomarker status of the tumor. The best characterized driver mutations for lung cancer are KRAS, EGFR, ALK, ROS1, BRAF, and MET. These mutations induce upregulation of hyperactivation of the encoded receptors or kinases belonging to different signalings (RAS/RAF/MEK/ERK, JAK/STAT, SRC/STAT, PI3K/AKT/mTOR) which are responsible for the aberrant proliferation and survival of cancer cells. Thus, inhibition of these signaling pathways through employing small molecule inhibitors of kinases or monoclonal antibodies against receptors may be a good therapy for patients whose driver oncogenic mutations are known. Antagonists of the epidermal growth factor receptor (EGFR), ALK-ROS1 inhibitors, and RAF-MEK inhibitors have been successfully used in NSCLC clinical trials and approved by the FDA, while the employment of KRAS inhibitors in clinics needs further trials [179,180]. Promising results have been obtained with EGFR inhibitors, which after several randomized phase 3 trials have been approved as first-line treatment in advanced NSCLC patients positive for EGFR mutations, showing higher progression-free survival and lower toxicity when compared to standard chemotherapy [181]. However, in Western populations, only 20% of patients with metastatic lung adenocarcinoma can be stratified for treatment with current targeted therapies with median responses of approximately a year [182,183]; thus, cytotoxic chemotherapy remains the elected therapy for first-line treatment of advanced disease. Often, the emergence of resistance to chemotherapy, radiotherapy, or targeted therapies reduces their efficacy after an initial response [184,185,186]. Resistance to therapies may involve the acquisition of new specific mutations by cancer cells that re-activate the signaling pathways targeted by the therapy through some alternative mechanisms. Thus, it is important to dissect the molecular mechanisms regulating not only lung tumor formation, progression, and metastasization, but also the mechanisms responsible for the primary and acquired resistance to therapy of cancer cells in order to overcome them, for example, by inhibiting the bypass pathways conferring resistance. Interestingly, as discussed below, all the driver genes for lung cancer listed above along with their downstream pathways have been shown to regulate YAP and TAZ at different levels in several contexts, including lung cancer. Thus, inhibition of YAP and TAZ downstream of these pathways may provide a valid therapeutic opportunity in addition to current targeted therapies.
The clinical targeting of YAP and TAZ in cancer is attractive because in most adult organs, and in the absence of any injury, they seem to be unnecessary for normal tissue homeostasis, while they appear to be required for tumor survival, growth, metastasization, and resistance to several treatments. Thus, their inhibition may have beneficial effects on cancer therapy without gross side effects on normal cells and healthy tissues. In line with this, several recent reports provided evidence that YAP and TAZ pharmacological inhibition reduces their oncogenic function in NSCLC models (see below), and, importantly, synergizes with current treatments which inhibit the main oncogenic pathways driving NSCLC (see Section 9).
A recent study showed that norcantharidin (NCTD) inhibits cell growth, migration, and invasiveness while enhancing apoptosis and senescence in NSCLC cells, partly through a specific downregulation of an aberrantly activated YAP signaling observed both in NSCLC cell lines and tumor tissues [76]. NCTD was already applied in treatment of breast cancer, liver cancer, gallbladder carcinoma, prostate cancer, mantle cell lymphoma, hepatocellular carcinoma, leukemia, and colon cancer with few side effects [187,188,189,190,191,192,193,194,195]. The study by Guo et al. shows the NCTD role in the inhibition of YAP-driven oncogenic pathways and suggests the possibility to also use this drug in lung cancer therapy. Another recent work showed that rottlerin, a natural polyphenolic compound derived from Mallotus philipinensis, exhibits antitumor activity in NSCLC cells (inhibits cancer cell proliferation, migration, and invasion, and enhances cell cycle arrest and apoptosis) partly through the inhibition of oncogenic TAZ [196], supporting the intriguing possibility to use natural compounds in order to inhibit oncogenic YAP and/or TAZ function. Other possible therapies against oncogenic YAP and TAZ may be represented by compounds able to activate their endogenous inhibitors. For example, LKB1, which inhibits YAP and TAZ in response to energy stress [84,122], has been shown to be induced and activated by Honokiol (HNK), a bioactive molecule from Magnolia grandiflora, in breast cancer experimental models [197]. Honokiol was able to inhibit breast tumorigenesis in mice in an LKB1-dependent manner [197]. Honokiol antitumor effects have been experimentally tested in different types of cancer, including lung cancer, where it was shown to play an anti-metastatic role through the inhibition of pro-metastatic proteins, among which was β-catenin, known to synergize with YAP/TAZ and to be a target itself of YAP and TAZ [198,199,200]. Finally, the following GPCRs (GPR87, CMKOR1, LGR4, FZD10, and P2RY11) were observed to be overexpressed in NSCLC patients [201] and some of their natural ligands were shown to activate oncogenic YAP and TAZ function [202]. This suggests that pharmacological inhibition of these GPCRs could represent another mechanism through which the pro-oncogenic function of YAP and TAZ can be inhibited in NSCLC [203]. All these observations need to be translated into clinics.
9. Synergism between YAP/TAZ Inhibition and Current Therapies in NSCLC Experimental Models
Recent studies have shown that in NSCLC, the hyperactivation of YAP may confer increased patient resistance to several treatments including cisplatin, radiation therapy, EGFR inhibitors, and BRAF or MEK inhibitors [102,136,137,138,204].
In those studies, YAP was overexpressed in cells that acquired resistance to the mentioned therapies, while knock-down of YAP and TAZ or treatment with the YAP/TAZ pharmacologic inhibitor verteporfin enhanced the response to cisplatin, radiation therapy, and EGFR inhibitors [136,137,138,204]. The mechanisms at the basis of resistance can be multiple and are described below.
Chemo-radiotherapy: Platinum-based chemotherapeutic drugs crosslink with the purine bases on the DNA, interfering with DNA repair mechanisms, eventually causing DNA damage, and inducing apoptosis in cancer cells. Pemetrexed inhibits purine and pyrimidine synthesis, interfering with cell proliferation and DNA damage repair, while radiotherapy causes DNA damage and cell death via the generation of free radicals. It has been proposed that YAP and TAZ may be involved in the protection of genomic stability in different models, including lung cancer [64,205,206] and that YAP stimulates nucleotide biosynthesis in liver cancer [207]. This suggests that YAP- and TAZ-overexpressing cells can overcome the DNA damage and the inhibition of DNA synthesis induced by chemotherapy. YAP and TAZ can also reduce sensitivity to DNA damaging agents by upregulating signaling which bypass cell cycle checkpoints induced by DNA damage, or by upregulating pro-survival and pro-proliferative signaling, typically hyperactivated in cells with stemness properties, where YAP/TAZ are often overexpressed and whose number increases over the course of radiotherapy and chemotherapy to repopulate the tumor mass [208]. It has also been shown that hyperactivation of β-catenin signaling can increase chemoresistance and radioresistance in different cancer types [209,210,211], and a synergism between β-catenin and YAP/TAZ has been described in cancer, including lung cancer [64,212,213,214], suggesting a possible role of YAP and TAZ in chemoresistance and radioresistance through its crosstalk with β-catenin signaling that needs to be further investigated in lung cancer. All the mechanisms described above can explain how cancer cells overexpressing YAP and/or TAZ can bypass the inhibitory effect of cytotoxic chemotherapy and radiotherapy.
Resistance to EGFR inhibitors: EGFR receptors, activated by the EGF ligands, induce three main pro-survival signaling pathways: SRC/JAK/STAT signaling, RAS/RAF/MEK/ERK signaling, and PI3K/AKT/mTOR signaling.
YAP has been shown to be overexpressed and more active in NSCLC specimens bearing higher resistance to EGFR inhibitors, and to mediate this resistance through several mechanisms [102,136,204,215]. One study shows that YAP overexpression increases Erbb3 [204]; through the activation of the Erbb3 signaling, NSCLC cells can reactivate ERK1/2 also in the presence of EGFR inhibitors [216,217]. In another study, YAP has been shown to activate AXL tyrosine kinase which induces PI3K/AKT signaling, providing a parallel survival input in the presence of EGFR inhibitors [215,218]. Finally, a recent study has shown that YAP works in parallel with RAF and MEK in the regulation of the antiapoptotic gene BCL-xL, thus promoting resistance to RAF and MEK inhibitors in BRAF mutant NSCLC.
In this scenario, YAP/TAZ inhibition may have a therapeutic effect when combined with other therapies already used for lung cancer treatment. Strikingly, in the abovementioned studies, YAP is pharmacologically inhibited with verteporfin, which disrupts its interaction with the TEAD transcription factor. This is important because verteporfin is already used in clinics for neovascular macular degeneration and opens up the opportunity to also use this drug in improving the response to current treatments in lung cancer.
Growing evidence shows that statins—inhibitors of the mevalonate (MVA) pathway, conventionally used in the treatment of hypercholesterolemia in cardiovascular disorders—also inhibit YAP-/TAZ-mediated transcriptional activation of pro-oncogenic genes and reduce breast tumor growth in mice xenografts [48,63,155]. Statins and similar compounds showed a promising role in the prevention and treatment of different cancer types in some preclinical and epidemiological studies, while in other studies the therapeutic efficacy and safety of statin was questionable (reviewed in [219]). Moreover, evidence of a therapeutic role of statin in lung cancer is still lacking. In vitro, we previously showed that treatment of NSCLC cells with the statin cerivastatin partially recapitulated the tumor suppressive effects observed upon YAP/TAZ interference, while treatment with MVA reversed the cerivastatin effect [135]. This result reinforces previous experimental works where statins showed anticancer effects in NSCLC cell lines [220,221,222] and in mouse models of lung cancer [222], linking the anti-oncogenic potential of statins to the inhibition of YAP and TAZ. At a clinical level, two recent prospective studies on a large population-based cohort of lung cancer patients showed that patients who used statins prior to the cancer diagnosis had reduced rates of cancer-specific mortality [223,224] and provided some evidence (to be confirmed with clinical trials) that statins may increase the survival of patients when prescribed at the time of lung cancer diagnosis [224]. A more recent prospective study showed that statin treatment increased survival in a large nationwide (Taiwan) population-based cohort of patients with lung cancer concomitantly receiving EGFR-TKI therapy [225], in line with previous case-control studies [226]. At a molecular level, statins were shown to act synergistically with conventional EGFR inhibitors in models of breast cancer and head and neck squamous cell carcinomas (HNSCC) through the inhibition of EGFR autophosphorylation [227]. In models of NSCLC, statins counteract the resistance to EGFR-TKI through the suppression of AKT/β-catenin signaling [228]. Moreover, as mentioned above, statins inhibit the oncogenic function of YAP, and YAP inhibition increases sensitivity to EGFR-TKI. Overall, these studies expand the potential therapeutic application of inhibitors of the mevalonate pathway in NSCLC, where early-phase clinical trials are still ongoing employing statins in combination with the current therapies (ClinicalTrials.gov Identifier: NCT00966472, NCT00452634, NCT00433498, NCT00452244, NCT01441349 (see the website http://clinicaltrials.gov)). All the possible therapeutic strategies targeting oncogenic YAP and TAZ and their upstream regulators are schematically represented in Figure 5.
10. Immune Evasion
Cancer cells often acquire the ability to evade the host immune response, even in the presence of an intact immune system, through multiple mechanisms. The dissection of the mechanisms driving immune evasion is under investigation with the aim to make the tumor more sensitive to the host immune response [229]. Several lines of evidence have suggested that YAP can be involved in acquired immune tolerance in different types of cancer cells. For example, in murine models of prostate cancer, YAP1 has been shown to promote the transcription of Cxcl5, a chemochine which attracts myeloid-derived suppressor cells (MDSCs) to the tumor microenvironment, reducing the immune anti-tumor response, and the knock-down of YAP1 reduced MDSCs in the intratumoral population of immune cells, impairing tumor progression [230]. The increased presence of MDSCs has been observed in patients with various solid tumors, including lung cancer, where a higher number of MDSCs correlates with tumor progression and poor prognosis [231,232,233]. Promising preclinical studies showed that pharmacological inhibition of MDSCs contributes to reducing tumor growth, progression, and metastasis, and increases the response to chemotherapy and radiotherapy in lung cancer models. However, these studies need to be translated into clinics (reviewed in [234]). Another study showed that in mouse epicardial cells, YAP and TAZ recruit a specific subset of suppressive CD4+ Tregs to injured myocardium after myocardial infarction. However, functional evidence of a role of YAP and TAZ in immunosuppression in vivo in lung cancer is still scarce. Two recent studies have shown that YAP transcriptionally upregulates PD-L1 (programmed cell death-ligand 1) in NSCLC cells [235,236]. PD-L1 is a ligand which binds to the PD1 receptor, an immune checkpoint receptor involved in tumor immune escape [237], and its overexpression is prognostic in lung cancer and increases cancer cell proliferation and chemoresistance [238,239]. PD-L1 has been widely studied in the few last years as a novel therapeutic target in lung cancer patients, and PD1/PD-L1 inhibitors are employed in several clinical trials in comparison or in combination with current therapies.
In August 2017, in a randomized, pivotal, phase 3 study of KEYNOTE-024 (ClinicalTrials.gov Identifier: NCT02142738), pembrolizumab, a PD1 inhibitor, was approved by FDA as a first-line therapy for advanced metastatic (stage IV) NSCLC, according to PD-L1 expression status of the tumor, giving superior progression-free survival in comparison to platinum-based therapy. Importantly, the 305 enrolled patients were found positive to programmed cell death ligand 1 (PD-L1) (in 50% of tumor cells or more) and lacked any EGFR or ALK aberrations. In another recent phase III clinical trial (Pacific), the PD-L1 inhibitor durvalumab (Imfinzi) was approved by FDA for the treatment of patients with locally advanced, unresectable stage III NSCLC who had not progressed following chemoradiotherapy. In that trial (where 713 patients were enrolled), durvalumab improved the median progression-free survival by 11.2 months compared to placebo, irrespective of PDL1 expression before chemoradiotherapy (ClinicalTrials.gov Identifier: NCT02125461). Other clinical trials are still ongoing for employing a PD-L1 blockade in combination with stereotactic radiation therapy (SRT) in stage I–IIIA NSCLC patients who are planned to undergo surgical resection of lung cancer (ClinicalTrials.gov Identifier: NCT03217071) or in combination with standard chemotherapy or EGFR-targeted therapies. Future studies are needed in order to demonstrate any possible role of YAP and TAZ in the regulation of immune evasion in human lung cancer through PDL1, Cxcl5, or other mechanisms.
11. Concluding Remarks
The Hippo pathway regulates several physiological processes and is implicated in numerous diseases including cancer. This makes it an exciting field of investigation. Knowledge on the mechanisms regulating and regulated by this pathway is continuously expanding, opening a wide range of possible players which can be therapeutically targeted in clinics. The oncogenic role of YAP and TAZ in lung cancer has been widely demonstrated.
However, in general, YAP and/or TAZ activation alone is not sufficient to drive tumorigenesis, but it promotes tumor growth and aggressiveness when it occurs concurrently with other oncogenic modifications of tumor cells and/or the tumor microenvironment, which may vary in different contexts. Moreover, it is important to keep in mind that YAP and TAZ, even if paralogues, can exert completely different, and sometimes opposite, functions in the same context or tissue. For example, in contrast to what has been widely shown for YAP (increased YAP expression and activation in NSCLC specimens resistant to EGFR-targeted therapy), Noguchi and coworkers showed that TAZ is overexpressed in cells that are more sensitive to EGFR inhibitors [80]. Thus, further knowledge is still required in order to understand controversial experimental findings and to design the correct therapy for each case, when combining current therapies with inhibitors of YAP and/or TAZ oncogenic activity in YAP- and /or TAZ-positive lung cancer specimens, in the different genetic and epigenetic backgrounds of lung cancer.
Acknowledgments
This research was funded by AIRC, grant number 20613.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Yokota, J.; Kohno, T. Molecular footprints of human lung cancer progression. Cancer Sci. 2004, 95, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Eichler, A.F.; Chung, E.; Kodack, D.P.; Loeffler, J.S.; Fukumura, D.; Jain, R.K. The biology of brain metastases-translation to new therapies. Nat. Rev. Clin. Oncol. 2011, 8, 344–356. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Siegel, R.L.; Jemal, A. Lung cancer statistics. Adv. Exp. Med. Biol. 2016, 893, 1–19. [Google Scholar] [PubMed]
- Back, A.L.; Park, E.R.; Greer, J.A.; Jackson, V.A.; Jacobsen, J.C.; Gallagher, E.R.; Temel, J.S. Clinician roles in early integrated palliative care for patients with advanced cancer: A qualitative study. J. Palliat. Med. 2014, 17, 1244–1248. [Google Scholar] [CrossRef] [PubMed]
- Greer, J.A.; Jackson, V.A.; Meier, D.E.; Temel, J.S. Early integration of palliative care services with standard oncology care for patients with advanced cancer. CA Cancer J. Clin. 2013, 63, 349–363. [Google Scholar] [CrossRef] [PubMed]
- Begnaud, A.; Hall, T.; Allen, T. Lung cancer screening with low-dose CT: Implementation amid changing public policy at one health care system. Am. Soc. Clin. Oncol. Educ. Book 2016, 35, e468–e475. [Google Scholar] [CrossRef] [PubMed]
- Eberth, J.M.; Sercy, E. Implementation of lung cancer screening in the United States: Changing trends based on a survey of society of thoracic radiology members. J. Thorac. Imaging 2015, 30, W60–W62. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, J.H.; Ashraf, H. Implementation and organization of lung cancer screening. Ann. Transl. Med. 2016, 4, 152. [Google Scholar] [CrossRef] [PubMed]
- Moroishi, T.; Hansen, C.G.; Guan, K.L. The emerging roles of YAP and TAZ in cancer. Nat. Rev. Cancer 2015, 15, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the roots of cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef] [PubMed]
- Panciera, T.; Azzolin, L.; Cordenonsi, M.; Piccolo, S. Mechanobiology of YAP and TAZ in physiology and disease. Nat. Rev. Mol. Cell. Biol. 2017, 18, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Moroishi, T.; Guan, K.L. Mechanisms of Hippo pathway regulation. Genes Dev. 2016, 30, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Santinon, G.; Pocaterra, A.; Dupont, S. Control of YAP/TAZ activity by metabolic and nutrient-sensing pathways. Trends Cell Biol. 2016, 26, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The biology of YAP/TAZ: Hippo signaling and beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zhang, N.; Xie, R.; Wang, W.; Cai, J.; Choi, K.S.; David, K.K.; Huang, B.; Yabuta, N.; Nojima, H.; et al. Homeostatic control of Hippo signaling activity revealed by an endogenous activating mutation in YAP. Genes Dev. 2015, 29, 1285–1297. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.Q.; Zhou, Z.C.; Tian, W. Research progresses on the molecular structure of the Hippo signaling pathway components. Yi Chuan 2017, 39, 659–674. [Google Scholar] [PubMed]
- Zheng, Y.; Wang, W.; Liu, B.; Deng, H.; Uster, E.; Pan, D. Identification of Happyhour/MAP4K as Alternative Hpo/Mst-like Kinases in the Hippo Kinase Cascade. Dev. Cell 2015, 34, 642–655. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Moroishi, T.; Mottier-Pavie, V.; Plouffe, S.W.; Hansen, C.G.; Hong, A.W.; Park, H.W.; Mo, J.S.; Lu, W.; Lu, S.; et al. MAP4K family kinases act in parallel to MST1/2 to activate LATS1/2 in the Hippo pathway. Nat. Commun. 2015, 6, 8357. [Google Scholar] [CrossRef] [PubMed]
- Maejima, Y.; Kyoi, S.; Zhai, P.; Liu, T.; Li, H.; Ivessa, A.; Sciarretta, S.; Del Re, D.P.; Zablocki, D.K.; Hsu, C.P.; et al. MST1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat. Med. 2013, 19, 1478–1488. [Google Scholar] [CrossRef] [PubMed]
- Furth, N.; Aylon, Y. The LATS1 and LATS2 tumor suppressors: Beyond the Hippo pathway. Cell Death Differ. 2017, 24, 1488–1501. [Google Scholar] [CrossRef] [PubMed]
- Thompson, B.J.; Sahai, E. MST kinases in development and disease. J. Cell Biol. 2015, 210, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Oudhoff, M.J.; Freeman, S.A.; Couzens, A.L.; Antignano, F.; Kuznetsova, E.; Min, P.H.; Northrop, J.P.; Lehnertz, B.; Barsyte-Lovejoy, D.; Vedadi, M.; et al. Control of the Hippo pathway by Set7-dependent methylation of YAP. Dev. Cell 2013, 26, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Mao, B.; Hu, F.; Cheng, J.; Wang, P.; Xu, M.; Yuan, F.; Meng, S.; Wang, Y.; Yuan, Z.; Bi, W. SIRT1 regulates YAP2-mediated cell proliferation and chemoresistance in hepatocellular carcinoma. Oncogene 2014, 33, 1468–1474. [Google Scholar] [CrossRef] [PubMed]
- Hata, S.; Hirayama, J.; Kajiho, H.; Nakagawa, K.; Hata, Y.; Katada, T.; Furutani-Seiki, M.; Nishina, H. A novel acetylation cycle of transcription co-activator Yes-associated protein that is downstream of Hippo pathway is triggered in response to SN2 alkylating agents. J. Biol. Chem. 2012, 287, 22089–22098. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, V.; Gudmundsdottir, K.; Luong, P.; Leung, K.Y.; Knebel, A.; Basu, S. JNK phosphorylates Yes-associated protein (YAP) to regulate apoptosis. Cell Death Dis. 2010, 1, e29. [Google Scholar] [CrossRef] [PubMed]
- Keshet, R.; Adler, J.; Ricardo Lax, I.; Shanzer, M.; Porat, Z.; Reuven, N.; Shaul, Y. c-Abl antagonizes the YAP oncogenic function. Cell Death Differ. 2015, 22, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.J.; Rouse, C.; Xu, X.; Wang, J.; Onaitis, M.W.; Pendergast, A.M. Inactivation of ABL kinases suppresses non-small cell lung cancer metastasis. JCI Insight 2016, 1, e89647. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Totty, N.F.; Irwin, M.S.; Sudol, M.; Downward, J. Akt phosphorylates the Yes-associated protein, YAP, to induce interaction with 14-3-3 and attenuation of p73-mediated apoptosis. Mol. Cell 2003, 11, 11–23. [Google Scholar] [CrossRef]
- Rosenbluh, J.; Nijhawan, D.; Cox, A.G.; Li, X.; Neal, J.T.; Schafer, E.J.; Zack, T.I.; Wang, X.; Tsherniak, A.; Schinzel, A.C.; et al. β-catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell 2012, 151, 1457–1473. [Google Scholar] [CrossRef] [PubMed]
- Kohli, P.; Bartram, M.P.; Habbig, S.; Pahmeyer, C.; Lamkemeyer, T.; Benzing, T.; Schermer, B.; Rinschen, M.M. Label-free quantitative proteomic analysis of the YAP/TAZ interactome. Am. J. Physiol. Cell Physiol. 2014, 306, C805–C818. [Google Scholar] [CrossRef] [PubMed]
- Barry, E.R.; Camargo, F.D. The Hippo superhighway: Signaling crossroads converging on the Hippo/YAP pathway in stem cells and development. Curr. Opin. Cell Biol. 2013, 25, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Hansen, C.G.; Moroishi, T.; Guan, K.L. YAP and TAZ: A nexus for Hippo signaling and beyond. Trends Cell Biol. 2015, 25, 499–513. [Google Scholar] [CrossRef] [PubMed]
- Finch-Edmondson, M.L.; Strauss, R.P.; Passman, A.M.; Sudol, M.; Yeoh, G.C.; Callus, B.A. TAZ protein accumulation is negatively regulated by YAP abundance in mammalian cells. J. Biol. Chem. 2015, 290, 27928–27938. [Google Scholar] [CrossRef] [PubMed]
- Kaan, H.Y.K.; Chan, S.W.; Tan, S.K.J.; Guo, F.; Lim, C.J.; Hong, W.; Song, H. Crystal structure of TAZ-TEAD complex reveals a distinct interaction mode from that of YAP-TEAD complex. Sci. Rep. 2017, 7, 2035. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Higashi, T.; Yokoyama, N.; Kaida, T.; Sakamoto, K.; Fukushima, Y.; Ishimoto, T.; Kuroki, H.; Nitta, H.; Hashimoto, D.; et al. An imbalance in TAZ and YAP expression in hepatocellular carcinoma confers cancer stem cell-like behaviors contributing to disease progression. Cancer Res. 2015, 75, 4985–4997. [Google Scholar] [CrossRef] [PubMed]
- Hau, J.C.; Erdmann, D.; Mesrouze, Y.; Furet, P.; Fontana, P.; Zimmermann, C.; Schmelzle, T.; Hofmann, F.; Chene, P. The TEAD4-YAP/TAZ protein-protein interaction: Expected similarities and unexpected differences. Chembiochem 2013, 14, 1218–1225. [Google Scholar] [CrossRef] [PubMed]
- Elbediwy, A.; Vincent-Mistiaen, Z.I.; Spencer-Dene, B.; Stone, R.K.; Boeing, S.; Wculek, S.K.; Cordero, J.; Tan, E.H.; Ridgway, R.; Brunton, V.G.; et al. Integrin signalling regulates YAP and TAZ to control skin homeostasis. Development 2016, 143, 1674–1687. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yu, A.; Yu, F.X. The Hippo pathway in tissue homeostasis and regeneration. Protein Cell 2017, 8, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Cottini, F.; Hideshima, T.; Xu, C.; Sattler, M.; Dori, M.; Agnelli, L.; ten Hacken, E.; Bertilaccio, M.T.; Antonini, E.; Neri, A.; et al. Rescue of Hippo coactivator YAP1 triggers DNA damage-induced apoptosis in hematological cancers. Nat. Med. 2014, 20, 599–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Liu, Y.; Zheng, Y.; Dong, J.; Pan, D. The TEAD/TEF family protein scalloped mediates transcriptional output of the Hippo growth-regulatory pathway. Dev. Cell 2008, 14, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Ye, X.; Yu, J.; Li, L.; Li, W.; Li, S.; Yu, J.; Lin, J.D.; Wang, C.Y.; Chinnaiyan, A.M.; et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008, 22, 1962–1971. [Google Scholar] [CrossRef] [PubMed]
- Shao, D.D.; Xue, W.; Krall, E.B.; Bhutkar, A.; Piccioni, F.; Wang, X.; Schinzel, A.C.; Sood, S.; Rosenbluh, J.; Kim, J.W.; et al. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell 2014, 158, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, A.; Kaneko, K.J.; Shu, H.; Zhao, Y.; DePamphilis, M.L. TEAD/TEF transcription factors utilize the activation domain of YAP65, a Src/Yes-associated protein localized in the cytoplasm. Genes Dev. 2001, 15, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Komuro, A.; Nagai, M.; Navin, N.E.; Sudol, M. WW domain-containing protein YAP associates with Erbb-4 and acts as a co-transcriptional activator for the carboxyl-terminal fragment of Erbb-4 that translocates to the nucleus. J. Biol. Chem. 2003, 278, 33334–33341. [Google Scholar] [CrossRef] [PubMed]
- Haskins, J.W.; Nguyen, D.X.; Stern, D.F. Neuregulin 1-activated Erbb4 interacts with YAP to induce Hippo pathway target genes and promote cell migration. Sci. Signal. 2014, 7, ra116. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, W.M., Jr.; Hong, J.H.; Yaffe, M.B.; Farrance, I.K. The transcriptional co-activator TAZ interacts differentially with transcriptional enhancer factor-1 (TEF-1) family members. Biochem. J. 2005, 388, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Nakagawa, M.; Olson, E.N.; Nakagawa, O. A WW domain protein TAZ is a critical coactivator for TBX5, a transcription factor implicated in Holt-Oram syndrome. Proc. Natl. Acad. Sci. USA 2005, 102, 18034–18039. [Google Scholar] [CrossRef] [PubMed]
- Di Agostino, S.; Sorrentino, G.; Ingallina, E.; Valenti, F.; Ferraiuolo, M.; Bicciato, S.; Piazza, S.; Strano, S.; Del Sal, G.; Blandino, G. YAP enhances the pro-proliferative transcriptional activity of mutant p53 proteins. EMBO Rep. 2016, 17, 188–201. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Forcato, M.; Battilana, G.; Azzolin, L.; Quaranta, E.; Bodega, B.; Rosato, A.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol. 2015, 17, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Tretiakova, M.S.; Silvis, M.R.; Lucas, J.; Klezovitch, O.; Coleman, I.; Bolouri, H.; Kutyavin, V.I.; Morrissey, C.; True, L.D.; et al. Erg activates the YAP1 transcriptional program and induces the development of age-related prostate tumors. Cancer Cell 2015, 27, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Alharbi, A.; Shan, H.; Hao, Y.; Snetsinger, B.; Rauh, M.J.; Yang, X. TAZ induces lung cancer stem cell properties and tumorigenesis by up-regulating ALDH1A1. Oncotarget 2017, 8, 38426–38443. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Kim, T.; Johnson, R.L.; Lim, D.S. Transcriptional co-repressor function of the Hippo pathway transducers YAP and TAZ. Cell Rep. 2015, 11, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Chen, Y.; Chen, L.; Cheng, H.; Mu, C.; Li, J.; Gao, R.; Zhou, C.; Cao, L.; Liu, J.; et al. Hypoxia regulates Hippo signalling through the SIAH2 ubiquitin E3 ligase. Nat. Cell Biol. 2015, 17, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.Y.; Zhuang, L.H.; Wang, D.D.; Zhou, T.Y.; Chang, L.L.; Gai, R.H.; Zhu, D.F.; Yang, B.; Zhu, H.; He, Q.J. Nuclear translocation and activation of YAP by hypoxia contributes to the chemoresistance of SN38 in hepatocellular carcinoma cells. Oncotarget 2016, 7, 6933–6947. [Google Scholar] [CrossRef] [PubMed]
- Low, B.C.; Pan, C.Q.; Shivashankar, G.V.; Bershadsky, A.; Sudol, M.; Sheetz, M. YAP/TAZ as mechanosensors and mechanotransducers in regulating organ size and tumor growth. FEBS Lett. 2014, 588, 2663–2670. [Google Scholar] [CrossRef] [PubMed]
- Pesic, M.; Greten, F.R. Inflammation and cancer: Tissue regeneration gone awry. Curr. Opin. Cell Biol. 2016, 43, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Enzo, E.; Santinon, G.; Pocaterra, A.; Aragona, M.; Bresolin, S.; Forcato, M.; Grifoni, D.; Pession, A.; Zanconato, F.; Guzzo, G.; et al. Aerobic glycolysis tunes YAP/TAZ transcriptional activity. EMBO J. 2015, 34, 1349–1370. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xiao, Z.D.; Li, X.; Aziz, K.E.; Gan, B.; Johnson, R.L.; Chen, J. AMPK modulates Hippo pathway activity to regulate energy homeostasis. Nat. Cell Biol. 2015, 17, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Mo, J.S.; Meng, Z.; Kim, Y.C.; Park, H.W.; Hansen, C.G.; Kim, S.; Lim, D.S.; Guan, K.L. Cellular energy stress induces AMPK-mediated regulation of YAP and the Hippo pathway. Nat. Cell Biol. 2015, 17, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Cheng, C.; Tan, Z.; Li, N.; Tang, M.; Yang, L.; Cao, Y. Emerging roles of lipid metabolism in cancer metastasis. Mol. Cancer 2017, 16, 76. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, G.; Ruggeri, N.; Specchia, V.; Cordenonsi, M.; Mano, M.; Dupont, S.; Manfrin, A.; Ingallina, E.; Sommaggio, R.; Piazza, S.; et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat. Cell Biol. 2014, 16, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Noto, A.; De Vitis, C.; Pisanu, M.E.; Roscilli, G.; Ricci, G.; Catizone, A.; Sorrentino, G.; Chianese, G.; Taglialatela-Scafati, O.; Trisciuoglio, D.; et al. Stearoyl-CoA-desaturase 1 regulates lung cancer stemness via stabilization and nuclear localization of YAP/TAZ. Oncogene 2017, 36, 4573–4584. [Google Scholar] [CrossRef] [PubMed]
- Mancini, R.; Noto, A.; Pisanu, M.E.; De Vitis, C.; Maugeri-Sacca, M.; Ciliberto, G. Metabolic features of cancer stem cells: The emerging role of lipid metabolism. Oncogene 2018. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.L.; Zhao, W.Y.; Zhang, W.J.; Song, H.; Ji, H.B.; Tang, N. Hippo signaling pathway in lung development, regeneration, and diseases. Yi Chuan 2017, 39, 597–606. [Google Scholar] [PubMed]
- Dai, Y.; Jablons, D.; You, L. Hippo pathway in lung development. J. Thorac. Dis. 2017, 9, 2246–2250. [Google Scholar] [CrossRef] [PubMed]
- Szymaniak, A.D.; Mahoney, J.E.; Cardoso, W.V.; Varelas, X. Crumbs3-mediated polarity directs airway epithelial cell fate through the Hippo pathway effector YAP. Dev. Cell 2015, 34, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Matsubara, D.; Tanaka, I.; Makiya, K.; Tanei, Z.I.; Kumagai, Y.; Shiu, S.J.; Nakaoka, H.J.; Ishikawa, S.; Isagawa, T.; et al. Loss of YAP1 defines neuroendocrine differentiation of lung tumors. Cancer Sci. 2016, 107, 1527–1538. [Google Scholar] [CrossRef] [PubMed]
- Horie, M.; Saito, A.; Ohshima, M.; Suzuki, H.I.; Nagase, T. YAP and TAZ modulate cell phenotype in a subset of small cell lung cancer. Cancer Sci. 2016, 107, 1755–1766. [Google Scholar] [CrossRef] [PubMed]
- Oser, M.G.; Niederst, M.J.; Sequist, L.V.; Engelman, J.A. Transformation from non-small-cell lung cancer to small-cell lung cancer: Molecular drivers and cells of origin. Lancet Oncol. 2015, 16, e165–e172. [Google Scholar] [CrossRef]
- Su, L.L.; Ma, W.X.; Yuan, J.F.; Shao, Y.; Xiao, W.; Jiang, S.J. Expression of Yes-associated protein in non-small cell lung cancer and its relationship with clinical pathological factors. Chin. Med. J. 2012, 125, 4003–4008. [Google Scholar] [PubMed]
- Wang, Y.; Dong, Q.; Zhang, Q.; Li, Z.; Wang, E.; Qiu, X. Overexpression of Yes-associated protein contributes to progression and poor prognosis of non-small-cell lung cancer. Cancer Sci. 2010, 101, 1279–1285. [Google Scholar] [CrossRef] [PubMed]
- Lorenzetto, E.; Brenca, M.; Boeri, M.; Verri, C.; Piccinin, E.; Gasparini, P.; Facchinetti, F.; Rossi, S.; Salvatore, G.; Massimino, M.; et al. YAP1 acts as oncogenic target of 11q22 amplification in multiple cancer subtypes. Oncotarget 2014, 5, 2608–2621. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Kang, D.W.; Long, L.Z.; Huang, S.M.; Yeo, M.K.; Yi, E.S.; Kim, K.H. Differential expression of Yes-associated protein is correlated with expression of cell cycle markers and pathologic TNM staging in non-small-cell lung carcinoma. Hum. Pathol. 2011, 42, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Wu, Y.; Yang, L.; Du, J.; Gong, K.; Chen, W.; Dai, J.; Li, X.; Xi, S. Repression of YAP by NCTD disrupts NSCLC progression. Oncotarget 2017, 8, 2307–2319. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.L.; Han, F.F.; Peng, X.H.; Chen, X.; Luan, C.Y.; Han, R.C.; Xu, W.G.; Guo, X.J. Yes-associated protein 1 promotes adenocarcinoma growth and metastasis through activation of the receptor tyrosine kinase AXL. Int. J. Immunopathol. Pharmacol. 2012, 25, 989–1001. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Hao, Y.; Liu, N.; Raptis, L.; Tsao, M.S.; Yang, X. TAZ is a novel oncogene in non-small cell lung cancer. Oncogene 2011, 30, 2181–2186. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Zhang, L.; He, C.S.; Hou, J.H.; Lin, S.X.; Hu, Z.H.; Xu, F.; Zhao, H.Y. Prognostic significance of TAZ expression in resected non-small cell lung cancer. J. Thorac. Oncol. 2012, 7, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, S.; Saito, A.; Horie, M.; Mikami, Y.; Suzuki, H.I.; Morishita, Y.; Ohshima, M.; Abiko, Y.; Mattsson, J.S.; Konig, H.; et al. An integrative analysis of the tumorigenic role of TAZ in human non-small cell lung cancer. Clin. Cancer Res. 2014, 20, 4660–4672. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Y.; Yu, S.L.; Ho, B.C.; Su, K.Y.; Hsu, Y.C.; Chang, C.S.; Li, Y.C.; Yang, S.Y.; Hsu, P.Y.; Ho, H.; et al. R331W missense mutation of oncogene YAP1 is a germline risk allele for lung adenocarcinoma with medical actionability. J. Clin. Oncol. 2015, 33, 2303–2310. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.N.; Curtis, S.J.; Fillmore, C.M.; Rowbotham, S.P.; Mohseni, M.; Wagner, D.E.; Beede, A.M.; Montoro, D.T.; Sinkevicius, K.W.; Walton, Z.E.; et al. Tumor-propagating cells and YAP/TAZ activity contribute to lung tumor progression and metastasis. EMBO J. 2014, 33, 468–481. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Gao, Y.; Li, F.; Tong, X.; Ren, Y.; Han, X.; Yao, S.; Long, F.; Yang, Z.; Fan, H.; et al. YAP promotes malignant progression of LKB1-deficient lung adenocarcinoma through downstream regulation of survivin. Cancer Res. 2015, 75, 4450–4457. [Google Scholar] [CrossRef] [PubMed]
- Mohseni, M.; Sun, J.; Lau, A.; Curtis, S.; Goldsmith, J.; Fox, V.L.; Wei, C.; Frazier, M.; Samson, O.; Wong, K.K.; et al. A genetic screen identifies an LKB1-mark signalling axis controlling the Hippo-YAP pathway. Nat. Cell Biol. 2014, 16, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Greuber, E.K.; Smith-Pearson, P.; Wang, J.; Pendergast, A.M. Role of ABL family kinases in cancer: From leukaemia to solid tumours. Nat. Rev. Cancer 2013, 13, 559–571. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar]
- Sos, M.L.; Michel, K.; Zander, T.; Weiss, J.; Frommolt, P.; Peifer, M.; Li, D.; Ullrich, R.; Koker, M.; Fischer, F.; et al. Predicting drug susceptibility of non-small cell lung cancers based on genetic lesions. J. Clin. Investig. 2009, 119, 1727–1740. [Google Scholar] [CrossRef] [PubMed]
- Testoni, E.; Stephenson, N.L.; Torres-Ayuso, P.; Marusiak, A.A.; Trotter, E.W.; Hudson, A.; Hodgkinson, C.L.; Morrow, C.J.; Dive, C.; Brognard, J. Somatically mutated ABL1 is an actionable and essential NSCLC survival gene. EMBO Mol. Med. 2016, 8, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.H.; Chou, H.C.; Fu, Y.N.; Yeh, C.L.; Cheng, H.W.; Chang, I.C.; Liu, K.J.; Chang, G.C.; Tsai, T.F.; Tsai, S.F.; et al. EGFR over-expression in non-small cell lung cancers harboring EGFR mutations is associated with marked down-regulation of CD82. Biochim. Biophys. Acta 2015, 1852, 1540–1549. [Google Scholar] [CrossRef] [PubMed]
- Bethune, G.; Bethune, D.; Ridgway, N.; Xu, Z. Epidermal growth factor receptor (EGFR) in lung cancer: An overview and update. J. Thorac. Dis. 2010, 2, 48–51. [Google Scholar] [PubMed]
- Formisano, L.; D’Amato, V.; Servetto, A.; Brillante, S.; Raimondo, L.; Di Mauro, C.; Marciano, R.; Orsini, R.C.; Cosconati, S.; Randazzo, A.; et al. Src inhibitors act through different mechanisms in non-small cell lung cancer models depending on EGFR and RAS mutational status. Oncotarget 2015, 6, 26090–26103. [Google Scholar] [CrossRef] [PubMed]
- Rothschild, S.I.; Gautschi, O.; Haura, E.B.; Johnson, F.M. Src inhibitors in lung cancer: Current status and future directions. Clin. Lung Cancer 2010, 11, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Giaccone, G.; Zucali, P.A. Src as a potential therapeutic target in non-small-cell lung cancer. Ann. Oncol. 2008, 19, 1219–1223. [Google Scholar] [CrossRef] [PubMed]
- Westcott, P.M.; To, M.D. The genetics and biology of KRAS in lung cancer. Chin. J. Cancer 2013, 32, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Rolfo, C.; Caparica, R. B-RAF mutation in non-small cell lung cancer: The sleeping beauty is waking up. Transl. Lung Cancer Res. 2016, 5, 367–369. [Google Scholar] [CrossRef] [PubMed]
- Stinchcombe, T.E.; Johnson, G.L. MEK inhibition in non-small cell lung cancer. Lung Cancer 2014, 86, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Li, C. Convergence between WNT-β-catenin and EGFR signaling in cancer. Mol. Cancer 2010, 9, 236. [Google Scholar] [CrossRef] [PubMed]
- Moody, T.W.; Nuche-Berenguer, B.; Nakamura, T.; Jensen, R.T. EGFR transactivation by peptide g protein-coupled receptors in cancer. Curr. Drug Targets 2016, 17, 520–528. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z. Transactivation of epidermal growth factor receptor by G protein-coupled receptors: Recent progress, challenges and future research. Int. J. Mol. Sci. 2016, 17, 95. [Google Scholar] [CrossRef] [PubMed]
- Reddy, B.V.; Irvine, K.D. Regulation of Hippo signaling by EGFR-MAPK signaling through AJUBA family proteins. Dev. Cell 2013, 24, 459–471. [Google Scholar] [CrossRef] [PubMed]
- You, B.; Yang, Y.L.; Xu, Z.; Dai, Y.; Liu, S.; Mao, J.H.; Tetsu, O.; Li, H.; Jablons, D.M.; You, L. Inhibition of ERK1/2 down-regulates the Hippo/YAP signaling pathway in human NSCLC cells. Oncotarget 2015, 6, 4357–4368. [Google Scholar] [CrossRef] [PubMed]
- McGowan, M.; Kleinberg, L.; Halvorsen, A.R.; Helland, A.; Brustugun, O.T. NSCLC depend upon YAP expression and nuclear localization after acquiring resistance to EGFR inhibitors. Genes Cancer 2017, 8, 497–504. [Google Scholar] [PubMed]
- Strazisar, M.; Mlakar, V.; Glavac, D. LATS2 tumour specific mutations and down-regulation of the gene in non-small cell carcinoma. Lung Cancer 2009, 64, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Yao, F.; Liu, H.; Li, Z.; Zhong, C.; Fang, W. Down-regulation of LATS2 in non-small cell lung cancer promoted the growth and motility of cancer cells. Tumour. Biol. 2015, 36, 2049–2057. [Google Scholar] [CrossRef] [PubMed]
- Malik, S.A.; Khan, M.S.; Dar, M.; Hussain, M.U.; Shah, M.A.; Shafi, S.M.; Mudassar, S. Molecular alterations and expression dynamics of LATS1 and LATS2 genes in non-small-cell lung carcinoma. Pathol. Oncol. Res. 2018, 24, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Sun, M.; Liu, G.J.; Wei, C.C.; Zhang, E.B.; Kong, R.; Xu, T.P.; Huang, M.D.; Wang, Z.X. Long noncoding RNA PVT1 promotes non-small cell lung cancer cell proliferation through epigenetically regulating LATS2 expression. Mol. Cancer Ther. 2016, 15, 1082–1094. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Sun, M.; Zang, C.; Ma, P.; He, J.; Zhang, M.; Huang, Z.; Ding, Y.; Shu, Y. Upregulated long non-coding RNA AGAP2-AS1 represses LATS2 and KLF2 expression through interacting with EZH2 and LSD1 in non-small-cell lung cancer cells. Cell Death Dis. 2016, 7, e2225. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.Y.; Sit, K.Y.; Sihoe, A.D.; Suen, W.S.; Au, W.K.; Tang, X.; Ma, E.S.; Chan, W.K.; Wistuba, I.I.; Minna, J.D.; et al. Aberrant large tumor suppressor 2 (LATS2) gene expression correlates with EGFR mutation and survival in lung adenocarcinomas. Lung Cancer 2014, 85, 282–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, X.Y.; Zhang, X.P.; Wu, J.H.; Qiu, X.S.; Wang, E.H. Expression of LATS1 contributes to good prognosis and can negatively regulate YAP oncoprotein in non-small-cell lung cancer. Tumour Biol. 2014, 35, 6435–6443. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Bachman, J.; Lai, Z.C. Evidence for a tumor suppressor role for the large tumor suppressor genes LATS1 and LATS2 in human cancer. Genetics 2013, 195, 1193–1196. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.L.; Hung, J.Y.; Chou, S.H.; Huang, M.S.; Tsai, M.J.; Lin, Y.S.; Chiang, S.Y.; Ho, Y.W.; Wu, C.Y.; Kuo, P.L. Angiomotin decreases lung cancer progression by sequestering oncogenic YAP/TAZ and decreasing Cyr61 expression. Oncogene 2015, 34, 4056–4068. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.M.; Liu, W.W.; Liu, C.J.; Wen, C.; Lu, H.F.; Wan, F.S. MST1 overexpression inhibited the growth of human non-small cell lung cancer in vitro and in vivo. Cancer Gene Ther. 2013, 20, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Gao, Y.; Li, P.; Shi, Z.; Guo, T.; Li, F.; Han, X.; Feng, Y.; Zheng, C.; Wang, Z.; et al. VGLL4 functions as a new tumor suppressor in lung cancer by negatively regulating the YAP-TEAD transcriptional complex. Cell Res. 2014, 24, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Matallanas, D.; Romano, D.; Yee, K.; Meissl, K.; Kucerova, L.; Piazzolla, D.; Baccarini, M.; Vass, J.K.; Kolch, W.; O’Neill, E. RASSF1A elicits apoptosis through an MST2 pathway directing proapoptotic transcription by the p73 tumor suppressor protein. Mol. Cell 2007, 27, 962–975. [Google Scholar] [CrossRef] [PubMed]
- Scrace, S.F.; O’Neill, E. Rassf signalling and DNA damage: Monitoring the integrity of the genome? Mol. Biol. Int. 2012, 2012, 141732. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Zhang, X.; Pfeifer, G.P. The tumor suppressor RASSF1A prevents dephosphorylation of the mammalian STE20-like kinases MST1 and MST2. J. Biol. Chem. 2011, 286, 6253–6261. [Google Scholar] [CrossRef] [PubMed]
- Dammann, R.; Li, C.; Yoon, J.H.; Chin, P.L.; Bates, S.; Pfeifer, G.P. Epigenetic inactivation of a RAS association domain family protein from the lung tumour suppressor locus 3p21.3. Nat. Genet. 2000, 25, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Agathanggelou, A.; Honorio, S.; Macartney, D.P.; Martinez, A.; Dallol, A.; Rader, J.; Fullwood, P.; Chauhan, A.; Walker, R.; Shaw, J.A.; et al. Methylation associated inactivation of RASSF1A from region 3p21.3 in lung, breast and ovarian tumours. Oncogene 2001, 20, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Burbee, D.G.; Forgacs, E.; Zochbauer-Muller, S.; Shivakumar, L.; Fong, K.; Gao, B.; Randle, D.; Kondo, M.; Virmani, A.; Bader, S.; et al. Epigenetic inactivation of RASSF1A in lung and breast cancers and malignant phenotype suppression. J. Natl. Cancer Inst. 2001, 93, 691–699. [Google Scholar] [CrossRef] [PubMed]
- De Fraipont, F.; Levallet, G.; Creveuil, C.; Bergot, E.; Beau-Faller, M.; Mounawar, M.; Richard, N.; Antoine, M.; Rouquette, I.; Favrot, M.C.; et al. An apoptosis methylation prognostic signature for early lung cancer in the IFCT-0002 trial. Clin. Cancer Res. 2012, 18, 2976–2986. [Google Scholar] [CrossRef] [PubMed]
- Dubois, F.; Keller, M.; Calvayrac, O.; Soncin, F.; Hoa, L.; Hergovich, A.; Parrini, M.C.; Mazieres, J.; Vaisse-Lesteven, M.; Camonis, J.; et al. RASSF1A suppresses the invasion and metastatic potential of human non-small cell lung cancer cells by inhibiting YAP activation through the GEF-H1/RHOB pathway. Cancer Res. 2016, 76, 1627–1640. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.B.; Babcock, J.T.; Wells, C.D.; Quilliam, L.A. LKB1 tumor suppressor regulates AMP kinase/mTOR-independent cell growth and proliferation via the phosphorylation of YAP. Oncogene 2013, 32, 4100–4109. [Google Scholar] [CrossRef] [PubMed]
- Momcilovic, M.; Shackelford, D.B. Targeting LKB1 in cancer—Exposing and exploiting vulnerabilities. Br. J. Cancer 2015, 113, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Getz, G.; Wheeler, D.A.; Mardis, E.R.; McLellan, M.D.; Cibulskis, K.; Sougnez, C.; Greulich, H.; Muzny, D.M.; Morgan, M.B.; et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008, 455, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Carretero, J.; Medina, P.P.; Pio, R.; Montuenga, L.M.; Sanchez-Cespedes, M. Novel and natural knockout lung cancer cell lines for the LKB1/STK11 tumor suppressor gene. Oncogene 2004, 23, 4037–4040. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Ramsey, M.R.; Hayes, D.N.; Fan, C.; McNamara, K.; Kozlowski, P.; Torrice, C.; Wu, M.C.; Shimamura, T.; Perera, S.A.; et al. LKB1 modulates lung cancer differentiation and metastasis. Nature 2007, 448, 807–810. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Cespedes, M.; Parrella, P.; Esteller, M.; Nomoto, S.; Trink, B.; Engles, J.M.; Westra, W.H.; Herman, J.G.; Sidransky, D. Inactivation of LKB1/STK11 is a common event in adenocarcinomas of the lung. Cancer Res. 2002, 62, 3659–3662. [Google Scholar] [PubMed]
- Matsumoto, S.; Iwakawa, R.; Takahashi, K.; Kohno, T.; Nakanishi, Y.; Matsuno, Y.; Suzuki, K.; Nakamoto, M.; Shimizu, E.; Minna, J.D.; et al. Prevalence and specificity of LKB1 genetic alterations in lung cancers. Oncogene 2007, 26, 5911–5918. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zheng, F.; Zhang, S.; Lu, J. Loss of RhoA expression prevents proliferation and metastasis of SPCA1 lung cancer cells in vitro. Biomed. Pharmacother. 2015, 69, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Wu, X.; Zhang, J.; Zhang, J.; Li, X. Ski regulates Smads and TAZ signaling to suppress lung cancer progression. Mol. Carcinog. 2017, 56, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- Bartucci, M.; Dattilo, R.; Moriconi, C.; Pagliuca, A.; Mottolese, M.; Federici, G.; Benedetto, A.D.; Todaro, M.; Stassi, G.; Sperati, F.; et al. TAZ is required for metastatic activity and chemoresistance of breast cancer stem cells. Oncogene 2015, 34, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Cordenonsi, M.; Zanconato, F.; Azzolin, L.; Forcato, M.; Rosato, A.; Frasson, C.; Inui, M.; Montagner, M.; Parenti, A.R.; Poletti, A.; et al. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell 2011, 147, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Ajani, J.A.; Honjo, S.; Maru, D.M.; Chen, Q.; Scott, A.W.; Heallen, T.R.; Xiao, L.; Hofstetter, W.L.; Weston, B.; et al. Hippo coactivator YAP1 upregulates SOX9 and endows esophageal cancer cells with stem-like properties. Cancer Res. 2014, 74, 4170–4182. [Google Scholar] [CrossRef] [PubMed]
- Aylon, Y.; Oren, M. The Hippo pathway, p53 and cholesterol. Cell Cycle 2016, 15, 2248–2255. [Google Scholar] [CrossRef] [PubMed]
- Lo Sardo, F.; Forcato, M.; Sacconi, A.; Capaci, V.; Zanconato, F.; di Agostino, S.; Del Sal, G.; Pandolfi, P.P.; Strano, S.; Bicciato, S.; et al. MCM7 and its hosted miR-25, 93 and 106b cluster elicit YAP/TAZ oncogenic activity in lung cancer. Carcinogenesis 2017, 38, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Zhang, Z.; Rodriguez-Barrueco, R.; Borczuk, A.; Liu, H.; Yu, J.; Silva, J.M.; Cheng, S.K.; Perez-Soler, R.; Halmos, B. Functional genomics screen identifies YAP1 as a key determinant to enhance treatment sensitivity in lung cancer cells. Oncotarget 2016, 7, 28976–28988. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Park, H.S.; Lee, D.; Yoo, G.; Kim, T.; Jeon, H.; Yeo, M.K.; Lee, C.S.; Moon, J.Y.; Jung, S.S.; et al. Hippo pathway effector YAP inhibition restores the sensitivity of EGFR-TKI in lung adenocarcinoma having primary or acquired EGFR-TKI resistance. Biochem. Biophys. Res. Commun. 2016, 474, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Sabnis, A.J.; Chan, E.; Olivas, V.; Cade, L.; Pazarentzos, E.; Asthana, S.; Neel, D.; Yan, J.J.; Lu, X.; et al. The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat. Genet. 2015, 47, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Vousden, K.H. P53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Soussi, T. P53 antibodies in the sera of patients with various types of cancer: A review. Cancer Res. 2000, 60, 1777–1788. [Google Scholar] [PubMed]
- Lubin, R.; Zalcman, G.; Bouchet, L.; Tredanel, J.; Legros, Y.; Cazals, D.; Hirsch, A.; Soussi, T. Serum p53 antibodies as early markers of lung cancer. Nat. Med. 1995, 1, 701–702. [Google Scholar] [CrossRef] [PubMed]
- Mitsudomi, T.; Hamajima, N.; Ogawa, M.; Takahashi, T. Prognostic significance of p53 alterations in patients with non-small cell lung cancer: A meta-analysis. Clin. Cancer Res. 2000, 6, 4055–4063. [Google Scholar] [PubMed]
- Steels, E.; Paesmans, M.; Berghmans, T.; Branle, F.; Lemaitre, F.; Mascaux, C.; Meert, A.P.; Vallot, F.; Lafitte, J.J.; Sculier, J.P. Role of p53 as a prognostic factor for survival in lung cancer: A systematic review of the literature with a meta-analysis. Eur. Respir. J. 2001, 18, 705–719. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xu, Z.; Yu, L.; Chen, M.; Li, K. Assessment of the potential diagnostic value of serum p53 antibody for cancer: A meta-analysis. PLoS ONE 2014, 9, e99255. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Kim, Y.; Lee, J.H.; Lee, E.Y.; Kim, H.S. Usefulness of serum anti-p53 antibody assay for lung cancer diagnosis. Arch. Pathol. Lab. Med. 2011, 135, 1570–1575. [Google Scholar] [CrossRef] [PubMed]
- Mattioni, M.; Soddu, S.; Prodosmo, A.; Visca, P.; Conti, S.; Alessandrini, G.; Facciolo, F.; Strigari, L. Prognostic role of serum p53 antibodies in lung cancer. BMC Cancer 2015, 15, 148. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narendran, A.; Ganjavi, H.; Morson, N.; Connor, A.; Barlow, J.W.; Keystone, E.; Malkin, D.; Freedman, M.H. Mutant p53 in bone marrow stromal cells increases VEGF expression and supports leukemia cell growth. Exp. Hematol. 2003, 31, 693–701. [Google Scholar] [CrossRef]
- Madar, S.; Harel, E.; Goldstein, I.; Stein, Y.; Kogan-Sakin, I.; Kamer, I.; Solomon, H.; Dekel, E.; Tal, P.; Goldfinger, N.; et al. Mutant p53 attenuates the anti-tumorigenic activity of fibroblasts-secreted interferon beta. PLoS ONE 2013, 8, e61353. [Google Scholar] [CrossRef] [PubMed]
- Patocs, A.; Zhang, L.; Xu, Y.; Weber, F.; Caldes, T.; Mutter, G.L.; Platzer, P.; Eng, C. Breast-cancer stromal cells with TP53 mutations and nodal metastases. N. Engl. J. Med. 2007, 357, 2543–2551. [Google Scholar] [CrossRef] [PubMed]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wu, Y.; Wang, H.; Zhang, Y.; Mei, L.; Fang, X.; Zhang, X.; Zhang, F.; Chen, H.; Liu, Y.; et al. Interplay of mevalonate and Hippo pathways regulates RHAMM transcription via YAP to modulate breast cancer cell motility. Proc. Natl. Acad. Sci. USA 2014, 111, E89–E98. [Google Scholar] [CrossRef] [PubMed]
- Taccioli, C.; Sorrentino, G.; Zannini, A.; Caroli, J.; Beneventano, D.; Anderlucci, L.; Lolli, M.; Bicciato, S.; Del Sal, G. MDP, a database linking drug response data to genomic information, identifies dasatinib and statins as a combinatorial strategy to inhibit YAP/TAZ in cancer cells. Oncotarget 2015, 6, 38854–38865. [Google Scholar] [CrossRef] [PubMed]
- Freed-Pastor, W.A.; Mizuno, H.; Zhao, X.; Langerod, A.; Moon, S.H.; Rodriguez-Barrueco, R.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia, A.; et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012, 148, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Merino Salvador, M.; Gomez de Cedron, M.; Merino Rubio, J.; Falagan Martinez, S.; Sanchez Martinez, R.; Casado, E.; Ramirez de Molina, A.; Sereno, M. Lipid metabolism and lung cancer. Crit. Rev. Oncol. Hematol. 2017, 112, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.D.; Alexandrov, A.; Kim, J.; Wala, J.; Berger, A.H.; Pedamallu, C.S.; Shukla, S.A.; Guo, G.; Brooks, A.N.; Murray, B.A.; et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 2016, 48, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Beyer, T.A.; Weiss, A.; Khomchuk, Y.; Huang, K.; Ogunjimi, A.A.; Varelas, X.; Wrana, J.L. Switch enhancers interpret TGF-β and Hippo signaling to control cell fate in human embryonic stem cells. Cell Rep. 2013, 5, 1611–1624. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, T.; Murakami, H.; Fujii, M.; Ishiguro, F.; Tanaka, I.; Kondo, Y.; Akatsuka, S.; Toyokuni, S.; Yokoi, K.; Osada, H.; et al. YAP induces malignant mesothelioma cell proliferation by upregulating transcription of cell cycle-promoting genes. Oncogene 2012, 31, 5117–5122. [Google Scholar] [CrossRef] [PubMed]
- Lian, I.; Kim, J.; Okazawa, H.; Zhao, J.; Zhao, B.; Yu, J.; Chinnaiyan, A.; Israel, M.A.; Goldstein, L.S.; Abujarour, R.; et al. The role of YAP transcription coactivator in regulating stem cell self-renewal and differentiation. Genes Dev. 2010, 24, 1106–1118. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Stanger, B.Z. YAP regulates s-phase entry in endothelial cells. PLoS ONE 2015, 10, e0117522. [Google Scholar] [CrossRef] [PubMed]
- Tumaneng, K.; Schlegelmilch, K.; Russell, R.C.; Yimlamai, D.; Basnet, H.; Mahadevan, N.; Fitamant, J.; Bardeesy, N.; Camargo, F.D.; Guan, K.L. YAP mediates crosstalk between the Hippo and PI(3)K-TOR pathways by suppressing PTEN via miR-29. Nat. Cell Biol. 2012, 14, 1322–1329. [Google Scholar] [CrossRef] [PubMed]
- Bertero, T.; Oldham, W.M.; Cottrill, K.A.; Pisano, S.; Vanderpool, R.R.; Yu, Q.; Zhao, J.; Tai, Y.; Tang, Y.; Zhang, Y.Y.; et al. Vascular stiffness mechanoactivates YAP/TAZ-dependent glutaminolysis to drive pulmonary hypertension. J. Clin. Investig. 2016, 126, 3313–3335. [Google Scholar] [CrossRef] [PubMed]
- Chaulk, S.G.; Lattanzi, V.J.; Hiemer, S.E.; Fahlman, R.P.; Varelas, X. The Hippo pathway effectors TAZ/YAP regulate dicer expression and microRNA biogenesis through Let-7. J. Biol. Chem. 2014, 289, 1886–1891. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.; Triboulet, R.; Mohseni, M.; Schlegelmilch, K.; Shrestha, K.; Camargo, F.D.; Gregory, R.I. Hippo signaling regulates microprocessor and links cell-density-dependent miRNA biogenesis to cancer. Cell 2014, 156, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Ivanovska, I.; Ball, A.S.; Diaz, R.L.; Magnus, J.F.; Kibukawa, M.; Schelter, J.M.; Kobayashi, S.V.; Lim, L.; Burchard, J.; Jackson, A.L.; et al. Micrornas in the miR-106b family regulate p21/CDKN1A and promote cell cycle progression. Mol. Cell Biol. 2008, 28, 2167–2174. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Inoue, I. Conditional expression of MCM7 increases tumor growth without altering DNA replication activity. FEBS Lett. 2003, 553, 213–217. [Google Scholar] [CrossRef]
- Ren, B.; Yu, G.; Tseng, G.C.; Cieply, K.; Gavel, T.; Nelson, J.; Michalopoulos, G.; Yu, Y.P.; Luo, J.H. MCM7 amplification and overexpression are associated with prostate cancer progression. Oncogene 2006, 25, 1090–1098. [Google Scholar] [CrossRef] [PubMed]
- Erkan, E.P.; Strobel, T.; Lewandrowski, G.; Tannous, B.; Madlener, S.; Czech, T.; Saydam, N.; Saydam, O. Depletion of minichromosome maintenance protein 7 inhibits glioblastoma multiforme tumor growth in vivo. Oncogene 2014, 33, 4778–4785. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, S.; Shomori, K.; Nishihara, K.; Yamaga, K.; Nosaka, K.; Araki, K.; Haruki, T.; Taniguchi, Y.; Nakamura, H.; Ito, H. Expression of minichromosome maintenance 7 (MCM7) in small lung adenocarcinomas (pT1): Prognostic implication. Lung Cancer 2009, 65, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Toyokawa, G.; Masuda, K.; Daigo, Y.; Cho, H.S.; Yoshimatsu, M.; Takawa, M.; Hayami, S.; Maejima, K.; Chino, M.; Field, H.I.; et al. Minichromosome maintenance protein 7 is a potential therapeutic target in human cancer and a novel prognostic marker of non-small cell lung cancer. Mol. Cancer 2011, 10, 65. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Z.; Jiang, Y.Y.; Hao, J.J.; Lu, S.S.; Zhang, T.T.; Shang, L.; Cao, J.; Song, X.; Wang, B.S.; Cai, Y.; et al. Prognostic significance of MCM7 expression in the bronchial brushings of patients with non-small cell lung cancer (NSCLC). Lung Cancer 2012, 77, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Poliseno, L.; Salmena, L.; Riccardi, L.; Fornari, A.; Song, M.S.; Hobbs, R.M.; Sportoletti, P.; Varmeh, S.; Egia, A.; Fedele, G.; et al. Identification of the miR-106b~25 microRNA cluster as a proto-oncogenic PTEN-targeting intron that cooperates with its host gene MCM7 in transformation. Sci. Signal. 2010, 3, ra29. [Google Scholar] [CrossRef] [PubMed]
- Vitolo, M.I.; Anglin, I.E.; Mahoney, W.M., Jr.; Renoud, K.J.; Gartenhaus, R.B.; Bachman, K.E.; Passaniti, A. The RUNX2 transcription factor cooperates with the Yes-associated protein, YAP65, to promote cell transformation. Cancer Biol. Ther. 2007, 6, 856–863. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, T.; Imoto, I.; Matsui, T.; Kozaki, K.; Haruki, S.; Sudol, M.; Shimada, Y.; Tsuda, H.; Kawano, T.; Inazawa, J. YAP is a candidate oncogene for esophageal squamous cell carcinoma. Carcinogenesis 2011, 32, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Yang, J.J.; Zhou, X.; Deng, Z.Y.; Shi, K.H.; Li, J. Emerging role of long noncoding RNAs in lung cancer: Current status and future prospects. Respir. Med. 2016, 110, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Kunz, M.; Wolf, B.; Schulze, H.; Atlan, D.; Walles, T.; Walles, H.; Dandekar, T. Non-coding RNAs in lung cancer: Contribution of bioinformatics analysis to the development of non-invasive diagnostic tools. Genes 2016, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Mayekar, M.K.; Bivona, T.G. Current landscape of targeted therapy in lung cancer. Clin. Pharmacol. Ther. 2017, 102, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Tomasini, P.; Walia, P.; Labbe, C.; Jao, K.; Leighl, N.B. Targeting the KRAS pathway in non-small cell lung cancer. Oncologist 2016, 21, 1450–1460. [Google Scholar] [CrossRef] [PubMed]
- Tiefenbacher, A.; Pirker, R. EGFR tyrosine kinase inhibitors as first-line therapy in advanced EGFR mutation-positive non-small cell lung cancer: Strategies to improve clinical outcome. J. Thorac. Dis. 2017, 9, 4208–4211. [Google Scholar] [CrossRef] [PubMed]
- Heist, R.S.; Engelman, J.A. Snapshot: Non-small cell lung cancer. Cancer Cell 2012, 21, 448.e442. [Google Scholar] [CrossRef] [PubMed]
- Komiya, T.; Perez, R.P.; Erickson, K.D.; Huang, C.H. Systematic analysis of design and stratification for phase III trials in first-line advanced non-small cell lung cancer. Thorac. Cancer 2016, 7, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Maione, P.; Sacco, P.C.; Sgambato, A.; Casaluce, F.; Rossi, A.; Gridelli, C. Overcoming resistance to targeted therapies in NSCLC: Current approaches and clinical application. Ther. Adv. Med. Oncol. 2015, 7, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.J.; Shaw, A.T. Resisting resistance: Targeted therapies in lung cancer. Trends Cancer 2016, 2, 350–364. [Google Scholar] [CrossRef] [PubMed]
- Morgillo, F.; Della Corte, C.M.; Fasano, M.; Ciardiello, F. Mechanisms of resistance to EGFR-targeted drugs: Lung cancer. ESMO Open 2016, 1, e000060. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.F.; Chen, Y.J.; Chou, C.H.; Wang, F.W.; Kuo, C.D. Norcantharidin induces cell cycle arrest and inhibits progression of human leukemic Jurkat T cells through mitogen-activated protein kinase-mediated regulation of interleukin-2 production. Toxicol. In Vitro 2011, 25, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.Z.; Zhao, Z.M.; Fu, J.Y.; Chen, C.Q.; Sun, W. Norcantharidin inhibits growth of human gallbladder carcinoma xenografted tumors in nude mice by inducing apoptosis and blocking the cell cycle in vivo. Hepatobiliary Pancreat. Dis. Int. 2010, 9, 414–422. [Google Scholar] [PubMed]
- Chen, Y.J.; Chang, W.M.; Liu, Y.W.; Lee, C.Y.; Jang, Y.H.; Kuo, C.D.; Liao, H.F. A small-molecule metastasis inhibitor, norcantharidin, downregulates matrix metalloproteinase-9 expression by inhibiting Sp1 transcriptional activity in colorectal cancer cells. Chem. Biol. Interact. 2009, 181, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Li, Y.; Du, H.; Fang, J.; Song, X.; Zhang, J. The synthetic compound norcantharidin induced apoptosis in mantle cell lymphoma in vivo and in vitro through the PI3K-Akt-NF-κB signaling pathway. Evid. Based Complement. Alternat. Med. 2013, 2013, 461487. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Li, G.; Ma, X.; Wang, Y.; Liu, G.; Feng, L.; Zhao, Y.; Zhang, G.; Wu, Y.; Ye, X.; et al. Norcantharidin enhances ABT-737-induced apoptosis in hepatocellular carcinoma cells by transcriptional repression of Mcl-1. Cell. Signal. 2012, 24, 1803–1809. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.H.; Yang, Y.Y.; Huang, Y.F.; Chow, K.C.; Chen, M.F. Induction of apoptosis in human Hep3B hepatoma cells by norcantharidin through a p53 independent pathway via TRAIL/DR5 signal transduction. Chin. J. Integr. Med. 2012, 18, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Tsai, Y.M.; Kuo, C.D.; Ku, K.L.; Shie, H.S.; Liao, H.F. Norcantharidin is a small-molecule synthetic compound with anti-angiogenesis effect. Life Sci. 2009, 85, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, W.; Zhang, W.Z.; Ge, C.Y.; Zhang, J.T.; Liu, Z.Y.; Fan, Y.Z. Inhibition of tumor vasculogenic mimicry and prolongation of host survival in highly aggressive gallbladder cancers by norcantharidin via blocking the ephrin type a receptor 2/focal adhesion kinase/paxillin signaling pathway. PLoS ONE 2014, 9, e96982. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.T.; Fan, Y.Z.; Chen, C.Q.; Zhao, Z.M.; Sun, W. Norcantharidin: A potential antiangiogenic agent for gallbladder cancers in vitro and in vivo. Int. J. Oncol. 2012, 40, 1501–1514. [Google Scholar] [PubMed]
- Zhao, Z.; Zheng, N.; Wang, L.; Hou, Y.; Zhou, X.; Wang, Z. Rottlerin exhibits antitumor activity via down-regulation of TAZ in non-small cell lung cancer. Oncotarget 2017, 8, 7827–7838. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Nagalingam, A.; Muniraj, N.; Bonner, M.Y.; Mistriotis, P.; Afthinos, A.; Kuppusamy, P.; Lanoue, D.; Cho, S.; Korangath, P.; et al. Activation of tumor suppressor LKB1 by honokiol abrogates cancer stem-like phenotype in breast cancer via inhibition of oncogenic Stat3. Oncogene 2017, 36, 5709–5721. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.Q.; Qiao, X.R.; Su, L.; Chen, S.Z. Honokiol inhibits EMT-mediated motility and migration of human non-small cell lung cancer cells in vitro by targeting c-FLIP. Acta Pharmacol. Sin. 2016, 37, 1574–1586. [Google Scholar] [CrossRef] [PubMed]
- Singh, T.; Katiyar, S.K. Honokiol inhibits non-small cell lung cancer cell migration by targeting PGE(2)-mediated activation of β-catenin signaling. PLoS ONE 2013, 8, e60749. [Google Scholar]
- Pan, J.; Lee, Y.; Zhang, Q.; Xiong, D.; Wan, T.C.; Wang, Y.; You, M. Honokiol decreases lung cancer metastasis through inhibition of the Stat3 signaling pathway. Cancer Prev. Res. 2017, 10, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Gugger, M.; White, R.; Song, S.; Waser, B.; Cescato, R.; Riviere, P.; Reubi, J.C. GPR87 is an overexpressed G-protein coupled receptor in squamous cell carcinoma of the lung. Dis. Markers 2008, 24, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.X.; Zhao, B.; Panupinthu, N.; Jewell, J.L.; Lian, I.; Wang, L.H.; Zhao, J.; Yuan, H.; Tumaneng, K.; Li, H.; et al. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell 2012, 150, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Filardo, E.J.; Quinn, J.A.; Bland, K.I.; Frackelton, A.R., Jr. Estrogen-induced activation of ERK-1 and ERK-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol. Endocrinol. 2000, 14, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.C.; You, B.; Yang, Y.L.; Zhang, W.Q.; Wang, Y.C.; Xu, Z.; Dai, Y.; Liu, S.; Yang, C.T.; Li, H.; et al. YAP promotes erlotinib resistance in human non-small cell lung cancer cells. Oncotarget 2016, 7, 51922–51933. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.; Adamovich, Y.; Reuven, N.; Shaul, Y. YAP1 phosphorylation by c-Abl is a critical step in selective activation of proapoptotic genes in response to DNA damage. Mol. Cell 2008, 29, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Dobbelstein, M.; Strano, S.; Roth, J.; Blandino, G. P73-induced apoptosis: A question of compartments and cooperation. Biochem. Biophys. Res. Commun. 2005, 331, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.G.; Hwang, K.L.; Brown, K.K.; Evason, K.; Beltz, S.; Tsomides, A.; O’Connor, K.; Galli, G.G.; Yimlamai, D.; Chhangawala, S.; et al. YAP reprograms glutamine metabolism to increase nucleotide biosynthesis and enable liver growth. Nat. Cell Biol. 2016, 18, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Willers, H.; Azzoli, C.G.; Santivasi, W.L.; Xia, F. Basic mechanisms of therapeutic resistance to radiation and chemotherapy in lung cancer. Cancer J. 2013, 19, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Rosano, L.; Bagnato, A. Disrupting the endothelin and WNT relationship to overcome chemoresistance. Mol. Cell Oncol. 2015, 2, e995025. [Google Scholar] [CrossRef] [PubMed]
- Cianfrocca, R.; Rosano, L.; Tocci, P.; Sestito, R.; Caprara, V.; Di Castro, V.; De Maria, R.; Bagnato, A. Blocking endothelin-1-receptor/β-catenin circuit sensitizes to chemotherapy in colorectal cancer. Cell Death Differ. 2017, 24, 1811–1820. [Google Scholar] [CrossRef] [PubMed]
- Krause, M.; Dubrovska, A.; Linge, A.; Baumann, M. Cancer stem cells: Radioresistance, prediction of radiotherapy outcome and specific targets for combined treatments. Adv. Drug Deliv. Rev. 2017, 109, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Azzolin, L.; Panciera, T.; Soligo, S.; Enzo, E.; Bicciato, S.; Dupont, S.; Bresolin, S.; Frasson, C.; Basso, G.; Guzzardo, V.; et al. YAP/TAZ incorporation in the β-catenin destruction complex orchestrates the WNT response. Cell 2014, 158, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Pan, P.; Wang, Z.; Zhang, Y.; Xie, P.; Geng, D.; Jiang, Y.; Yu, R.; Zhou, X. β-catenin-mediated YAP signaling promotes human glioma growth. J. Exp. Clin. Cancer Res. 2017, 36, 136. [Google Scholar] [CrossRef] [PubMed]
- Deng, F.; Peng, L.; Li, Z.; Tan, G.; Liang, E.; Chen, S.; Zhao, X.; Zhi, F. YAP triggers the WNT/β-catenin signalling pathway and promotes enterocyte self-renewal, regeneration and tumorigenesis after DSS-induced injury. Cell Death Dis. 2018, 9, 153. [Google Scholar] [CrossRef] [PubMed]
- Ghiso, E.; Migliore, C.; Ciciriello, V.; Morando, E.; Petrelli, A.; Corso, S.; De Luca, E.; Gatti, G.; Volante, M.; Giordano, S. YAP-dependent AXL overexpression mediates resistance to EGFR inhibitors in NSCLC. Neoplasia 2017, 19, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Tricker, E.M.; Xu, C.; Uddin, S.; Capelletti, M.; Ercan, D.; Ogino, A.; Pratilas, C.A.; Rosen, N.; Gray, N.S.; Wong, K.K.; et al. Combined EGFR/MEK inhibition prevents the emergence of resistance in EGFR-mutant lung cancer. Cancer Discov. 2015, 5, 960–971. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. Met amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Zhang, Z.; Miao, L.; Yang, Z.; Yang, J.; Wang, Y.; Qian, D.; Cai, H.; Wang, Y. Anexelekto (AXL) increases resistance to EGFR-TKI and activation of AKT and ERK1/2 in non-small cell lung cancer cells. Oncol. Res. 2016, 24, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Wakelee, H.A.; Aragaki, A.K.; Tang, J.Y.; Kurian, A.W.; Manson, J.E.; Stefanick, M.L. Protective effects of statins in cancer: Should they be prescribed for high-risk patients? Curr. Atheroscler. Rep. 2016, 18, 72. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Fu, J.; Yuan, X.; Hu, C. Simvastatin inhibits the proliferation of A549 lung cancer cells through oxidative stress and up-regulation of SOD2. Pharmazie 2014, 69, 610–614. [Google Scholar] [PubMed]
- Yu, X.; Pan, Y.; Ma, H.; Li, W. Simvastatin inhibits proliferation and induces apoptosis in human lung cancer cells. Oncol. Res. 2013, 20, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, Z.; Li, Y.; Li, W.; Chen, Y. Simvastatin prevents proliferation and bone metastases of lung adenocarcinoma in vitro and in vivo. Neoplasma 2013, 60, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Cardwell, C.R.; Mc Menamin, U.; Hughes, C.M.; Murray, L.J. Statin use and survival from lung cancer: A population-based cohort study. Cancer Epidemiol. Biomarkers Prev. 2015, 24, 833–841. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.J.; Ezer, N.; Sigel, K.; Mhango, G.; Wisnivesky, J.P. The effect of statins on survival in patients with stage IV lung cancer. Lung Cancer 2016, 99, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Hung, M.S.; Chen, I.C.; Lee, C.P.; Huang, R.J.; Chen, P.C.; Tsai, Y.H.; Yang, Y.H. Statin improves survival in patients with EGFR-TKI lung cancer: A nationwide population-based study. PLoS ONE 2017, 12, e0171137. [Google Scholar] [CrossRef] [PubMed]
- Fiala, O.; Pesek, M.; Finek, J.; Minarik, M.; Benesova, L.; Bortlicek, Z.; Topolcan, O. Statins augment efficacy of EGFR-TKIs in patients with advanced-stage non-small cell lung cancer harbouring KRAS mutation. Tumour Biol. 2015, 36, 5801–5805. [Google Scholar] [CrossRef] [PubMed]
- Mantha, A.J.; McFee, K.E.; Niknejad, N.; Goss, G.; Lorimer, I.A.; Dimitroulakos, J. Epidermal growth factor receptor-targeted therapy potentiates lovastatin-induced apoptosis in head and neck squamous cell carcinoma cells. J. Cancer Res. Clin. Oncol. 2003, 129, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Hwang, K.E.; Kwon, S.J.; Kim, Y.S.; Park, D.S.; Kim, B.R.; Yoon, K.H.; Jeong, E.T.; Kim, H.R. Effect of simvastatin on the resistance to EGFR tyrosine kinase inhibitors in a non-small cell lung cancer with the T790M mutation of EGFR. Exp. Cell Res. 2014, 323, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Giroux Leprieur, E.; Dumenil, C.; Julie, C.; Giraud, V.; Dumoulin, J.; Labrune, S.; Chinet, T. Immunotherapy revolutionises non-small-cell lung cancer therapy: Results, perspectives and new challenges. Eur. J. Cancer 2017, 78, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Lu, X.; Dey, P.; Deng, P.; Wu, C.C.; Jiang, S.; Fang, Z.; Zhao, K.; Konaparthi, R.; Hua, S.; et al. Targeting YAP-dependent MDSC infiltration impairs tumor progression. Cancer Discov. 2016, 6, 80–95. [Google Scholar] [CrossRef] [PubMed]
- Solito, S.; Marigo, I.; Pinton, L.; Damuzzo, V.; Mandruzzato, S.; Bronte, V. Myeloid-derived suppressor cell heterogeneity in human cancers. Ann. N. Y. Acad. Sci. 2014, 1319, 47–65. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, M.L.; Lu, L.; Ramachandran, I.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the development of lung cancer. Cancer Immunol. Res. 2014, 2, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Huang, H.; Zhu, Y.; Yu, G.; Gao, X.; Xu, Y.; Liu, C.; Hou, J.; Zhang, X. A novel subset of B7-H3+CD14+HLA-DR−/low myeloid-derived suppressor cells are associated with progression of human NSCLC. Oncoimmunology 2015, 4, e977164. [Google Scholar] [CrossRef] [PubMed]
- Adah, D.; Hussain, M.; Qin, L.; Qin, L.; Zhang, J.; Chen, X. Implications of MDSCs-targeting in lung cancer chemo-immunotherapeutics. Pharmacol. Res. 2016, 110, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.; Hsu, P.C.; Yang, Y.L.; Xu, Z.; Dai, Y.; Wang, Y.; Chan, G.; Huang, Z.; Hu, B.; Li, H.; et al. YAP regulates PD-L1 expression in human NSCLC cells. Oncotarget 2017, 8, 114576–114587. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.S.; Park, D.I.; Lee, D.H.; Lee, J.E.; Yeo, M.K.; Park, Y.H.; Lim, D.S.; Choi, W.; Lee, D.H.; Yoo, G.; et al. Hippo effector YAP directly regulates the expression of PD-L1 transcripts in EGFR-TKI-resistant lung adenocarcinoma. Biochem. Biophys. Res. Commun. 2017, 491, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016, 8, 328rv324. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.; Fang, W.; Yu, J.; Chen, N.; Zhan, J.; Ma, Y.; Yang, Y.; Huang, Y.; Zhao, H.; Zhang, L. Expression of programmed death ligand-1 on tumor cells varies pre and post chemotherapy in non-small cell lung cancer. Sci. Rep. 2016, 6, 20090. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Li, G.; Wang, Y.; Wang, Y.; Zhao, S.; Haihong, P.; Zhao, H.; Wang, Y. Pd-L1 expression in lung cancer and its correlation with driver mutations: A meta-analysis. Sci. Rep. 2017, 7, 10255. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic representation of the Hippo pathway core components and their upstream regulators in mammals. The extracellular matrix (ECM), the cytoplasm, and the nucleus of cells are represented. Arrows indicate activation, while blunt lines indicate inhibition. Activation indicates an increase in protein levels or activity, while inhibition indicates a decrease in protein levels or activity. Red indicates eventual activation of nuclear Yes Associated Protein (YAP)/Trascriptional Coactivator with PDZ-binding motif (TAZ), while dark blue indicates eventual inhibition of nuclear YAP/TAZ. Light blue arrows indicate phosphorylation of proteins by kinases. Orange or light blue balls indicate phosphorylation sites of target proteins. The Hippo core kinase cassette is represented inside a black rectangle. Cell polarity and cell junction proteins activate Hippo pathway core kinases and inhibit YAP/TAZ nuclear function. Conversely, G-protein-coupled receptor (GPCR) signaling, mitogenic signals, inflammation, and mechanical stress coming from the ECM inhibit the Hippo pathway core kinases through mechanisms either dependent on or independent of the Rho GTPase signaling which in turn stabilizes the actin cytoskeleton core kinases thereby activating YAP/TAZ nuclear activity. Lipid biosynthesis and glycolysis activate nuclear YAP and TAZ. In the nucleus, YAP and TAZ interact with different transcription factors and the resulting transcriptional outcome is context-specific, depending on the incoming signals to which cells are exposed and the cell type subjected to such signals.
Figure 1.
Schematic representation of the Hippo pathway core components and their upstream regulators in mammals. The extracellular matrix (ECM), the cytoplasm, and the nucleus of cells are represented. Arrows indicate activation, while blunt lines indicate inhibition. Activation indicates an increase in protein levels or activity, while inhibition indicates a decrease in protein levels or activity. Red indicates eventual activation of nuclear Yes Associated Protein (YAP)/Trascriptional Coactivator with PDZ-binding motif (TAZ), while dark blue indicates eventual inhibition of nuclear YAP/TAZ. Light blue arrows indicate phosphorylation of proteins by kinases. Orange or light blue balls indicate phosphorylation sites of target proteins. The Hippo core kinase cassette is represented inside a black rectangle. Cell polarity and cell junction proteins activate Hippo pathway core kinases and inhibit YAP/TAZ nuclear function. Conversely, G-protein-coupled receptor (GPCR) signaling, mitogenic signals, inflammation, and mechanical stress coming from the ECM inhibit the Hippo pathway core kinases through mechanisms either dependent on or independent of the Rho GTPase signaling which in turn stabilizes the actin cytoskeleton core kinases thereby activating YAP/TAZ nuclear activity. Lipid biosynthesis and glycolysis activate nuclear YAP and TAZ. In the nucleus, YAP and TAZ interact with different transcription factors and the resulting transcriptional outcome is context-specific, depending on the incoming signals to which cells are exposed and the cell type subjected to such signals.

Figure 2.
Main proteins and pathways that influence YAP and TAZ in lung development and tumorigenesis. During lung development, the Crb3 polarity protein sequesters YAP and TAZ at the apical plasma membrane preventing their oncogenic function in the nucleus. Amot, a component of the tight junction complexes, exerts a similar function. GPCR receptors, EGFR receptor, and other RTKs, when activated by ligands, induce the RAS/RAF/MEK/ERK and the PI3K/AKT/mTOR pathways (inside a red rectangle), which, in turn, activate oncogenic YAP and TAZ through mechanisms either dependent on or independent of Hippo pathway core kinases (inside black rectangle). Upon DNA damage, cell contact, or other stress stimuli, RASSF1A activates the MST1/2-LATS1/2 kinase cassete, inhibiting YAP and TAZ oncogenic function. In the nucleus, Vgll4 inhibits the oncogenic role of YAP and TAZ through impairing YAP and TAZ binding with TEAD transcription factors. Arrows indicate activation of the indicated proteins, while blunt lines indicate repression of targeted proteins. Lines or arrows in red indicate signals which eventually activate nuclear YAP/TAZ, while lines or arrows in blue indicate signals or proteins which eventually inhibit nuclear YAP/TAZ, through mechanisms either dependent on or independent of Hippo pathway core kinases. Dashed arrows indicate reciprocal crosstalk. Abbreviations: MAPK, mitogen-activated protein kinase; RTK: receptor tyrosine kinase; EGF: epidermal growth factor; EGFR: EGF receptor; GPCR: G-protein-coupled receptor.
Figure 2.
Main proteins and pathways that influence YAP and TAZ in lung development and tumorigenesis. During lung development, the Crb3 polarity protein sequesters YAP and TAZ at the apical plasma membrane preventing their oncogenic function in the nucleus. Amot, a component of the tight junction complexes, exerts a similar function. GPCR receptors, EGFR receptor, and other RTKs, when activated by ligands, induce the RAS/RAF/MEK/ERK and the PI3K/AKT/mTOR pathways (inside a red rectangle), which, in turn, activate oncogenic YAP and TAZ through mechanisms either dependent on or independent of Hippo pathway core kinases (inside black rectangle). Upon DNA damage, cell contact, or other stress stimuli, RASSF1A activates the MST1/2-LATS1/2 kinase cassete, inhibiting YAP and TAZ oncogenic function. In the nucleus, Vgll4 inhibits the oncogenic role of YAP and TAZ through impairing YAP and TAZ binding with TEAD transcription factors. Arrows indicate activation of the indicated proteins, while blunt lines indicate repression of targeted proteins. Lines or arrows in red indicate signals which eventually activate nuclear YAP/TAZ, while lines or arrows in blue indicate signals or proteins which eventually inhibit nuclear YAP/TAZ, through mechanisms either dependent on or independent of Hippo pathway core kinases. Dashed arrows indicate reciprocal crosstalk. Abbreviations: MAPK, mitogen-activated protein kinase; RTK: receptor tyrosine kinase; EGF: epidermal growth factor; EGFR: EGF receptor; GPCR: G-protein-coupled receptor.

Figure 3.
Distribution of the genetic alterations of the TP53 gene in human cancer. Data obtained from the cBioPortal for Cancer Genomics website [149] after querying for TP53 alterations in 54,483 samples from 215 studies of different cancer types. Each column represents the alteration frequency of the TP53 gene in each cancer type indicated. Sometimes the same cancer type is indicated more than once because it has been analyzed in different independent studies. Red boxes indicate lung cancer.
Figure 3.
Distribution of the genetic alterations of the TP53 gene in human cancer. Data obtained from the cBioPortal for Cancer Genomics website [149] after querying for TP53 alterations in 54,483 samples from 215 studies of different cancer types. Each column represents the alteration frequency of the TP53 gene in each cancer type indicated. Sometimes the same cancer type is indicated more than once because it has been analyzed in different independent studies. Red boxes indicate lung cancer.

Figure 4.
(A) Co-occurrence of TP53, YAP1, and LATS2 mutations in lung cancer after querying 1144 lung cancers collected in the cBioportal website database [158]. (B) Putative driver mutations of TP53 co-occur with YAP and TAZ amplification or with LATS2 deletion in lung cancer (from the cBioportal website).
Figure 4.
(A) Co-occurrence of TP53, YAP1, and LATS2 mutations in lung cancer after querying 1144 lung cancers collected in the cBioportal website database [158]. (B) Putative driver mutations of TP53 co-occur with YAP and TAZ amplification or with LATS2 deletion in lung cancer (from the cBioportal website).

Figure 5.
Schematic representation of the pharmacological compounds (black boxes) that inhibit YAP and TAZ oncogenic function through targeting YAP and TAZ or the main proteins and pathways that influence YAP and TAZ in lung cancer. Honokiol reactivates LKB1 and inhibits the YAP/TAZ/β-catenin oncogenic pathway. Statins and norcantharidin (NCTD) inhibit the mevalonate pathway and the production of monounsaturated fatty acids—two biosynthetic pathways of lipids that are increased in tumorigenesis. Through the inhibition of these two pathways, statins and norcantharidin inhibit oncogenic YAP, TAZ, and β-catenin signaling. Statins also inhibit EGFR autophosphorylation and β-catenin activation and nuclear translocation. Rottlerin is a natural polyphenolic compound which inhibits oncogenic TAZ in lung. EGFR inhibitors are too many to be listed here, but they inhibit the EGFR receptors and their downstream effectors and their inhibition has a synergistic effect when combined with YAP inhibition mediated by Verteporfin. This latter impairs the binding of YAP with the oncogenic transcription factors TEADs. Black arrows indicate pharmacological activation of the targeted proteins or pathways. Black blunt lines indicate pharmacological inhibition of the targeted proteins or pathways. As in previous figures, lines or arrows in red indicate signals which eventually activate nuclear YAP/TAZ, while lines or arrows in blue indicate signals or proteins which eventually inhibit nuclear YAP/TAZ, through mechanisms either dependent on or independent of Hippo pathway core kinases. Dashed arrows indicate reciprocal crosstalk between receptors and their downstream transduction.
Figure 5.
Schematic representation of the pharmacological compounds (black boxes) that inhibit YAP and TAZ oncogenic function through targeting YAP and TAZ or the main proteins and pathways that influence YAP and TAZ in lung cancer. Honokiol reactivates LKB1 and inhibits the YAP/TAZ/β-catenin oncogenic pathway. Statins and norcantharidin (NCTD) inhibit the mevalonate pathway and the production of monounsaturated fatty acids—two biosynthetic pathways of lipids that are increased in tumorigenesis. Through the inhibition of these two pathways, statins and norcantharidin inhibit oncogenic YAP, TAZ, and β-catenin signaling. Statins also inhibit EGFR autophosphorylation and β-catenin activation and nuclear translocation. Rottlerin is a natural polyphenolic compound which inhibits oncogenic TAZ in lung. EGFR inhibitors are too many to be listed here, but they inhibit the EGFR receptors and their downstream effectors and their inhibition has a synergistic effect when combined with YAP inhibition mediated by Verteporfin. This latter impairs the binding of YAP with the oncogenic transcription factors TEADs. Black arrows indicate pharmacological activation of the targeted proteins or pathways. Black blunt lines indicate pharmacological inhibition of the targeted proteins or pathways. As in previous figures, lines or arrows in red indicate signals which eventually activate nuclear YAP/TAZ, while lines or arrows in blue indicate signals or proteins which eventually inhibit nuclear YAP/TAZ, through mechanisms either dependent on or independent of Hippo pathway core kinases. Dashed arrows indicate reciprocal crosstalk between receptors and their downstream transduction.


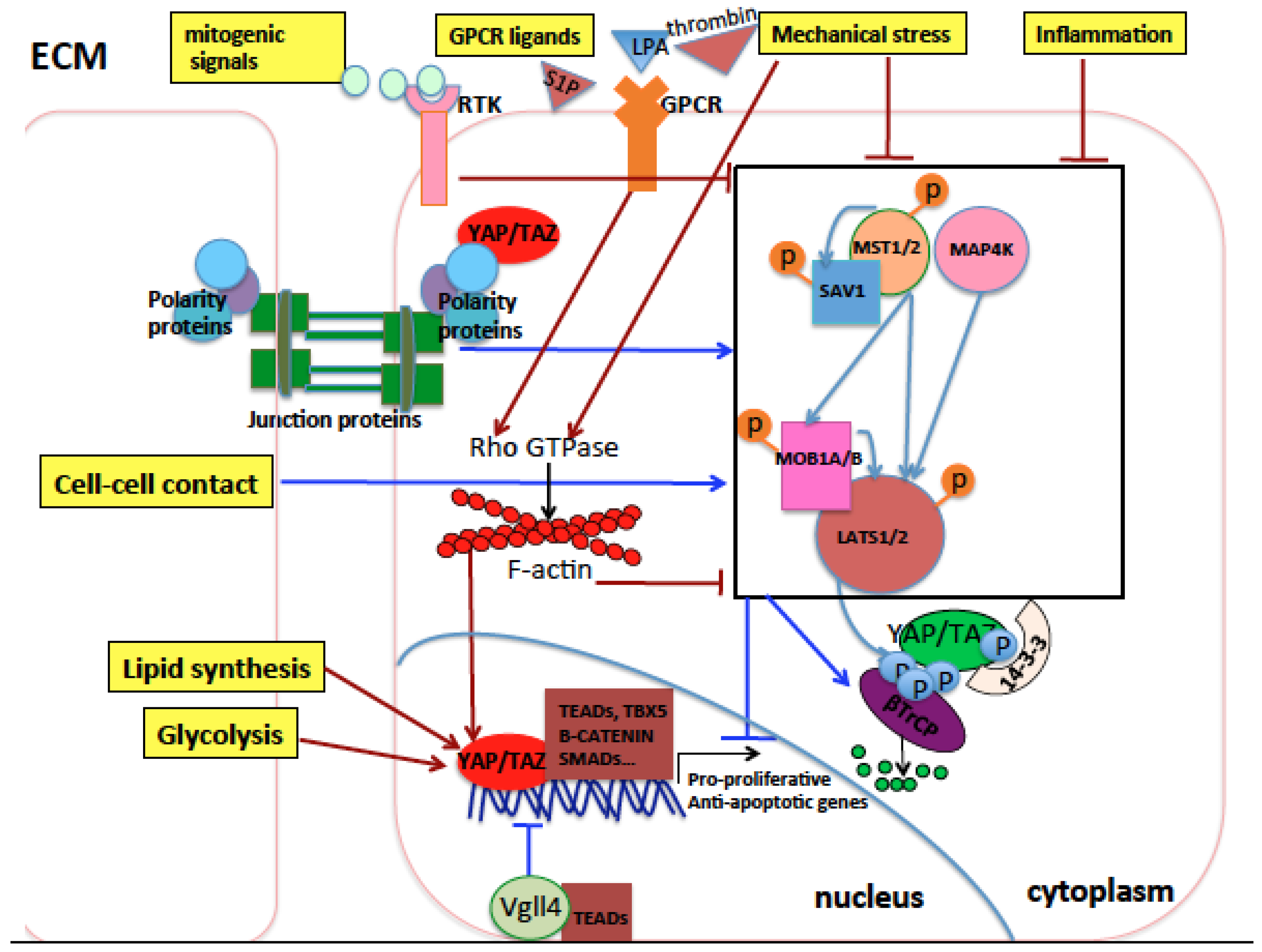{kind=link}
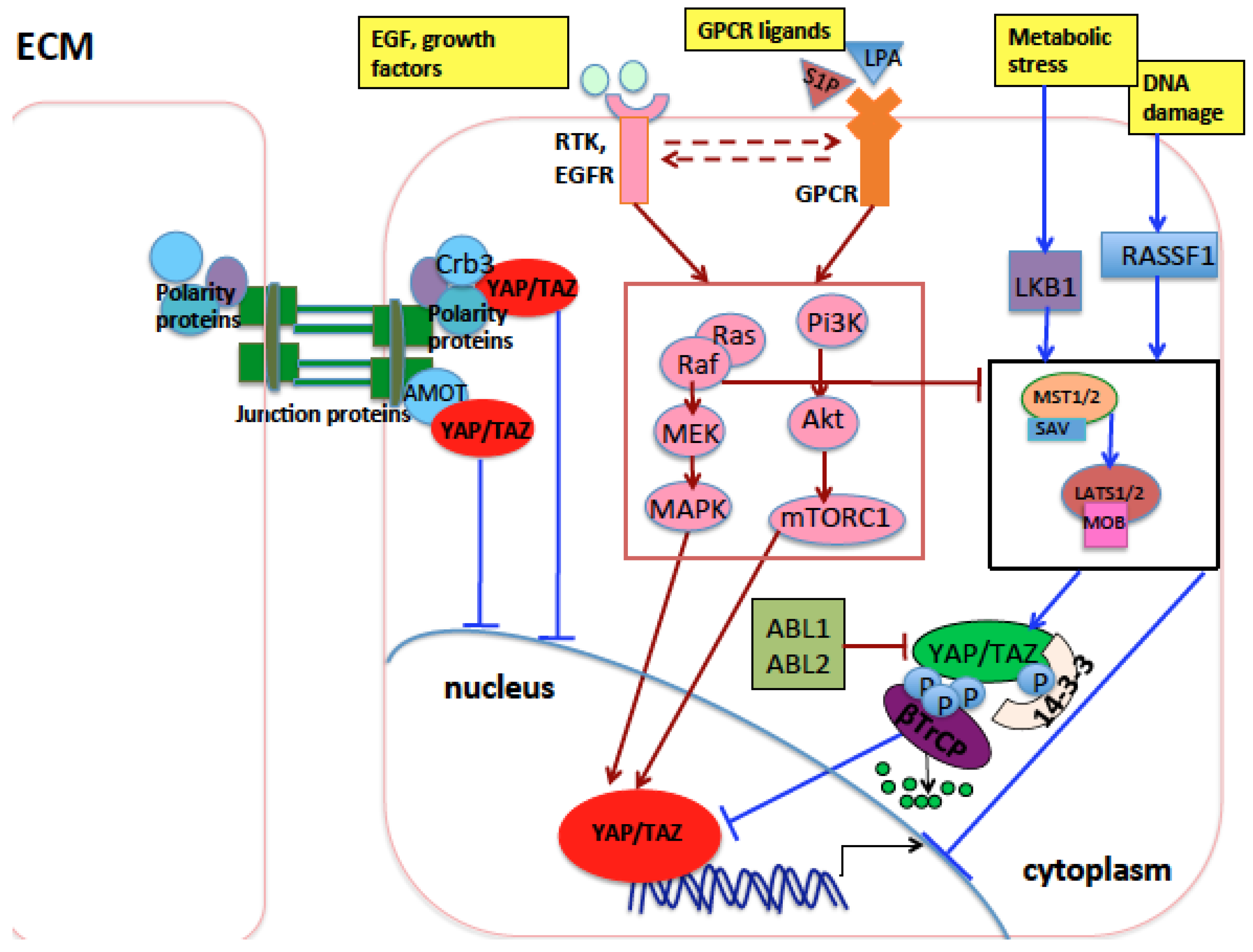{kind=link}
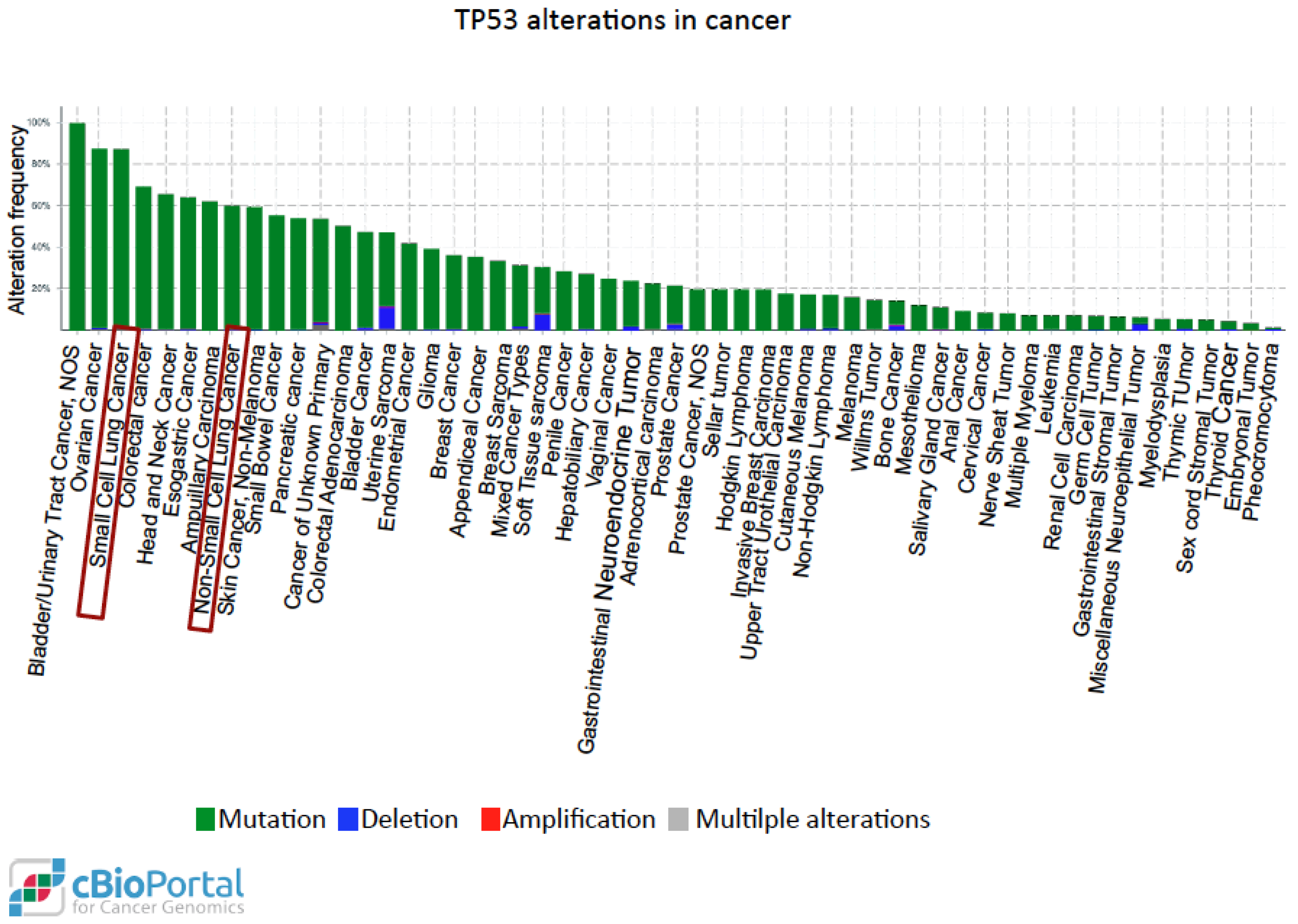{kind=link}
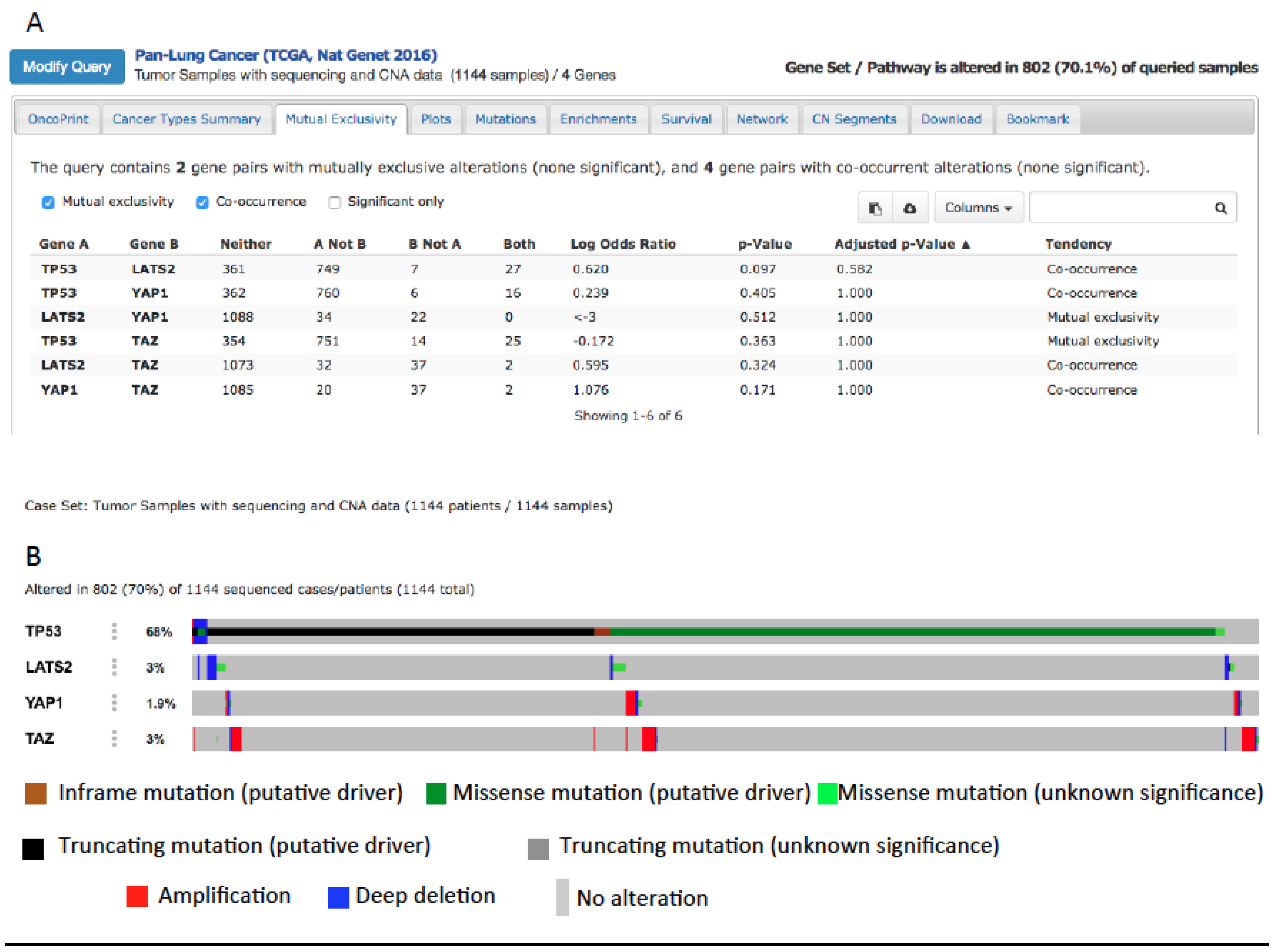{kind=link}
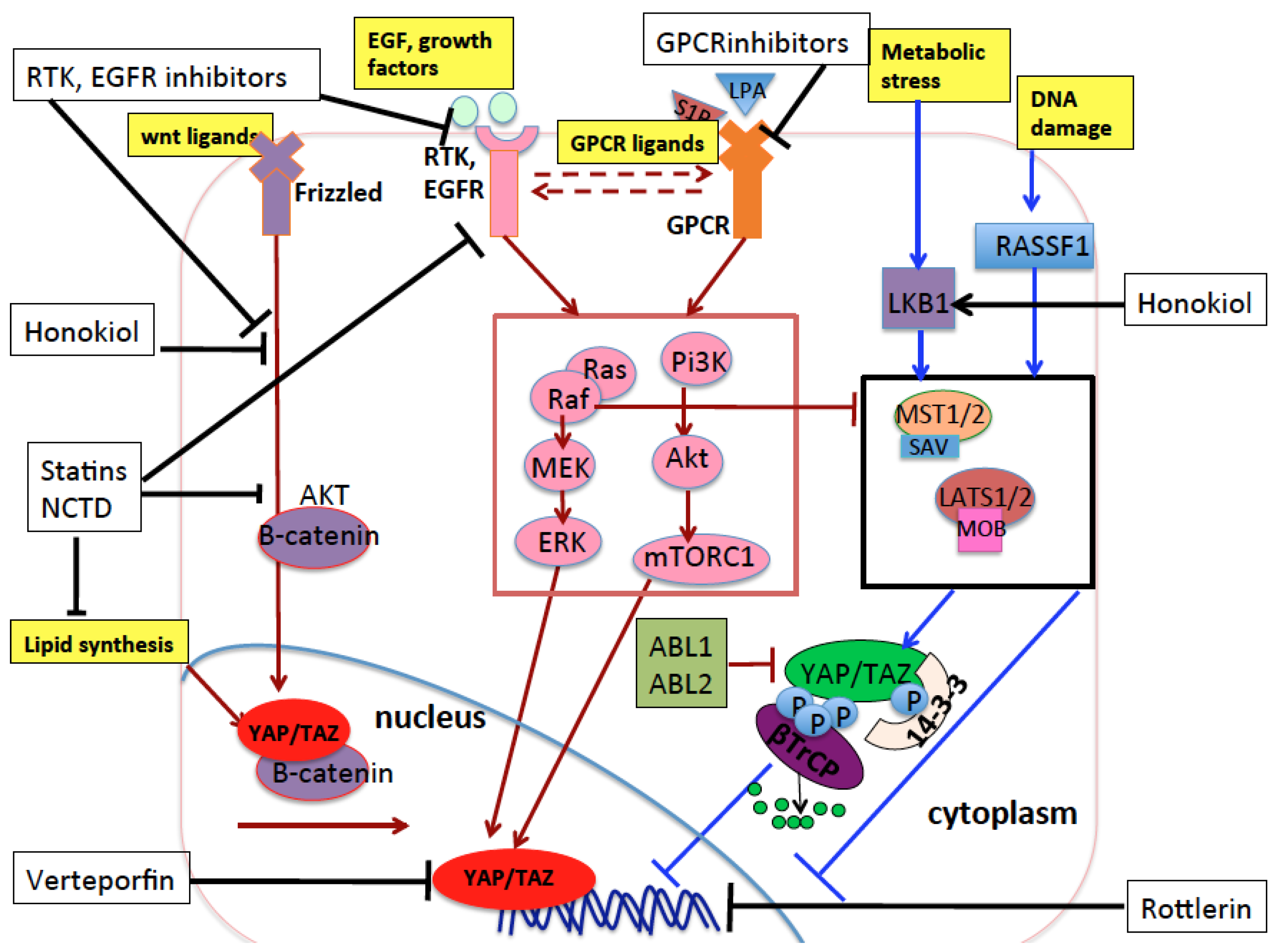{kind=link}
Table 1.
Hippo pathway components and upstream regulators found to be dysregulated in non-small cell lung cancer (NSCLC) in previous experimental reports.
Table 1.
Hippo pathway components and upstream regulators found to be dysregulated in non-small cell lung cancer (NSCLC) in previous experimental reports.
| Hippo Pathway Component/ Regulator | Increase/Decrease/Prognosis | Reference | N |
|---|---|---|---|
| YAP | Protein level increases in 70% NSCLC tissues. Bad prognosis. Correlation with late T stage, TNM stage and lymph node metastasis. | Su, L.L., et al.,2012 [72] | 40 |
| Protein level increased in 66.3% NSCLC tissues and predominantly nuclear. Bad prognosis. Correlation with TNM stage, lymph node metastasis, shorter O.S. | Wang, Y., et al.,2010 [73] | 92 | |
| YAP gene amplified in NSCLC cell lines and in 23% NSCLC tissues. | Lorenzetto E., et al, 2014 [74] | 77 | |
| Protein Level and Nuclear localization Increased in LAC. Bad prognosis. correlation with TNM stage, cyclinA overexpression, increased EGFR copy number. | Kim, J.M., et al., 2011 [75] | 168 | |
| Increased Nuclear localization. Increased nuclear and reduced cytoplasmic expression in NSCLC cell lines and tissues. | Guo, J., et al., 2017 [76] | 4 | |
| Protein level increased in 87.8% LAC. Bad prognosis. | Cui, Z.L., et al., 2012 [77] | 49 | |
| YAP1 R331W Missense germline mutation in 1.1% patients with LAC with respect to 0.18% in healthy control. Predictive of LAC predisposition. | Chen, H.Y., et al.,2015 [81] | 1312 LAC 1135 healthy controls | |
| Bad prognosis. Shorter survival when overexpressed with TAZ, SCD1 and β-catenin. | Noto, A., et al., 2017 [64] | 10 | |
| TAZ | Bad prognosis. Shorter survival when overexpressed with YAP, SCD1 and β-catenin. | Noto, A., et al., 2017 [64] | 10 |
| Overexpressed in NSCLC cells. | Zhou, Z., et al., 2011 [78] | cells | |
| Overexpressed in 66.8% NSCLC. Bad prognosis. Correlated with TNM stage and lymph node metastasis, invasion, shorter O.S and D.F.S. | Xie, M., et al., 2012 [79] | 181 | |
| Bad prognosis. Shorter survival. High TAZ expression at the genomic, mRNA, and protein levels in Squamous cell Carcinoma patients with respect to controls. | Noguchi, S., et al., 2014 [80] | 345 NSCLC patients 18 healthy controls | |
| LATS2 | Transcript Downregulated in NSCLC. | Strazisar, M., et al., 2009 [103] | 129 |
| Promoter hypermethylated in 71% NSCLC. | Malik, S.A., et al., 2017 [105] | 79 | |
| Lower protein and trasnscript expression in LAC. Good prognosis. Increased O.S and D.F.S. | Luo, S., et al., 2014 [108] | 79 | |
| LATS1 | Promoter hypermethylated in 66.66% NSCLC. | Malik, S.A., et al., 2017 [105] | 79 |
| Good prognosis. correlated with TNM stage, lymph node metastasis. | Lin, X.Y., et al., 2014 [109] | 136 | |
| AMOT | Downregulated in 45.8% NSCLC patients | Hsu, Y.L., et al., 2014 [111] | 24 |
| ABL1, ABL2 | ABL1 and 2 are somatically mutated in 1.5% and 4% NSCLC patients, ABL2 is amplified in 8% NSCLC. | Testoni, E., et al., 2016 [88] | N.A |
| LKB1 | Gene mutated in 18% LAC patients. | Ding, L., et al., 2008 [124] | 188 |
| Gene inactivated in 54% of LAC cell lines. | Carretero, J., et al., 2004 [125] | 11 | |
| Gene inactivated in 33% NSCLC patients. | Sanchez-Cespedes, M., et al., 2002 [127] | 20 | |
| Gene mutated in 8% NSCLC patients (7/91) and in 39% (20/51) NSCLC cell lines. Good prognosis. | Matsumoto, S., et al., 2007 [128] | 91 patients 51 cell lines | |
| RASSF1A | Promoter hypermethylated in 40% of primary lung tumors, missense mutation in 10% of primary lung tumors. | Dammann, R., et al., 2000 [117] | 60 |
| Promoter hypermethylated in 72% of SCLC, 34% of NSCLC. Loss of heterozigosity (LOH) in 86% SCLC (31/36), 91% (10/11) Squamous cell carcinoma, 71% (15/21) LAC. | Agathanggelou, A., et al., 2001 [118] | 29 SCLC, 41 NSCLC (hypermet study) 36 SCLC, 48 NSCLC (LOH study) | |
| Transcript Not expressed in 100% SCLC, 64% NSCLC. Promoter hypermethylated in 100% SCLC. Good prognosis. Hypermethylation was associated with impaired patient survival. | Burbee, D.G., et al., 2001 [119] | 47 SCLC, 107 NSCLC | |
| Promoter hypermethylated in 21.8% NSCLC patients. Good prognosis in patients expressing higher levels of RASSF1A. | de Fraipont, F., et al., 2012 [120] | 202 | |
| VGLL4 | Transcript downregulated in LAC tumor respect to non tumoral tissues (29/30) Lower levels of nuclear protein in LAC compared to normal lungs: 92.6% of patients (25/27) exhibited high nuclear VGLL4 expression in their normal lungs, whereas only 22.1% of patients (17 out of 77) had high nuclear expression of VGLL4 in their lung ADCs | Zhang, W., et al., 2014 [113] | 30 |
| Ski | Promoter hypermethylated in 56% of NSCLC tumor samples and in NSCLC cell lines Good prognosis. | Xie, M., et al., 2017 [130] | 168 |
Red indicates proteins that activate YAP or TAZ oncogenic function, are upregulated or hyperactivated in NSCLC, and whose upregulation is associated with poorer prognosis in NSCLC. Blue indicates proteins that inhibit YAP and TAZ oncogenic function, are downregulated in NSCLC and whose upregulation is associated with better prognosis. When control patients are not indicated, number of patients indicates only those who bear NSCLC. When the number of control patients is indicated, the study compares NSCLC patients with healthy controls. LAC = lung adenocarcinoma; TNM = tumor node metastasis stage; D.F.S. = disease-free survival; O.S. = overall survival; N = number of patients or cell lines.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lo Sardo, F.; Strano, S.; Blandino, G. YAP and TAZ in Lung Cancer: Oncogenic Role and Clinical Targeting. Cancers 2018, 10, 137. https://doi.org/10.3390/cancers10050137
AMA Style
Lo Sardo F, Strano S, Blandino G. YAP and TAZ in Lung Cancer: Oncogenic Role and Clinical Targeting. Cancers. 2018; 10(5):137. https://doi.org/10.3390/cancers10050137
Chicago/Turabian StyleLo Sardo, Federica, Sabrina Strano, and Giovanni Blandino. 2018. "YAP and TAZ in Lung Cancer: Oncogenic Role and Clinical Targeting" Cancers 10, no. 5: 137. https://doi.org/10.3390/cancers10050137
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.