The Roles of p53 in Mitochondrial Dynamics and Cancer Metabolism: The Pendulum between Survival and Death in Breast Cancer?


Abstract
:1. Introduction
1.1. Overview of Breast Cancer
1.2. Breast Cancer Treatment Resistance and Recurrence
2. The Multifaceted Tumor Suppressor Roles of p53
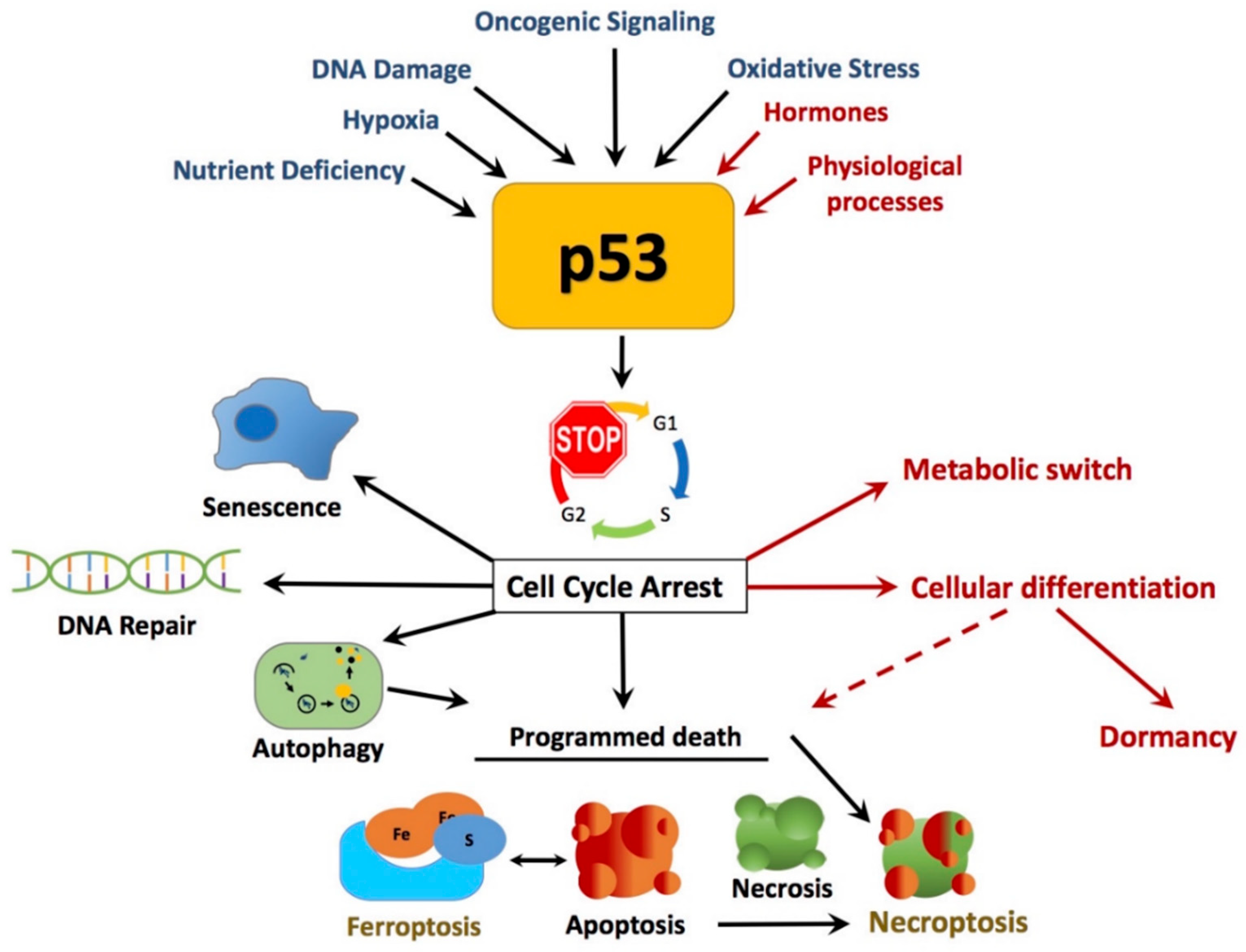
2.1. Canonical and Non-Canonical Functions of p53 in Cancer Protection
2.1.1. DNA Damage
2.1.2. Apoptosis
2.1.3. Necrosis
2.1.4. Autophagy
2.1.5. Dormancy
2.1.6. Cellular Senescence or Quiescence or Dormancy
2.1.7. Ferroptosis
3. Emerging Roles of p53 in Cancer Metabolism
3.1. Metabolic Reprogramming for Cellular Proliferation
3.2. The Warburg/Weinhouse Debate
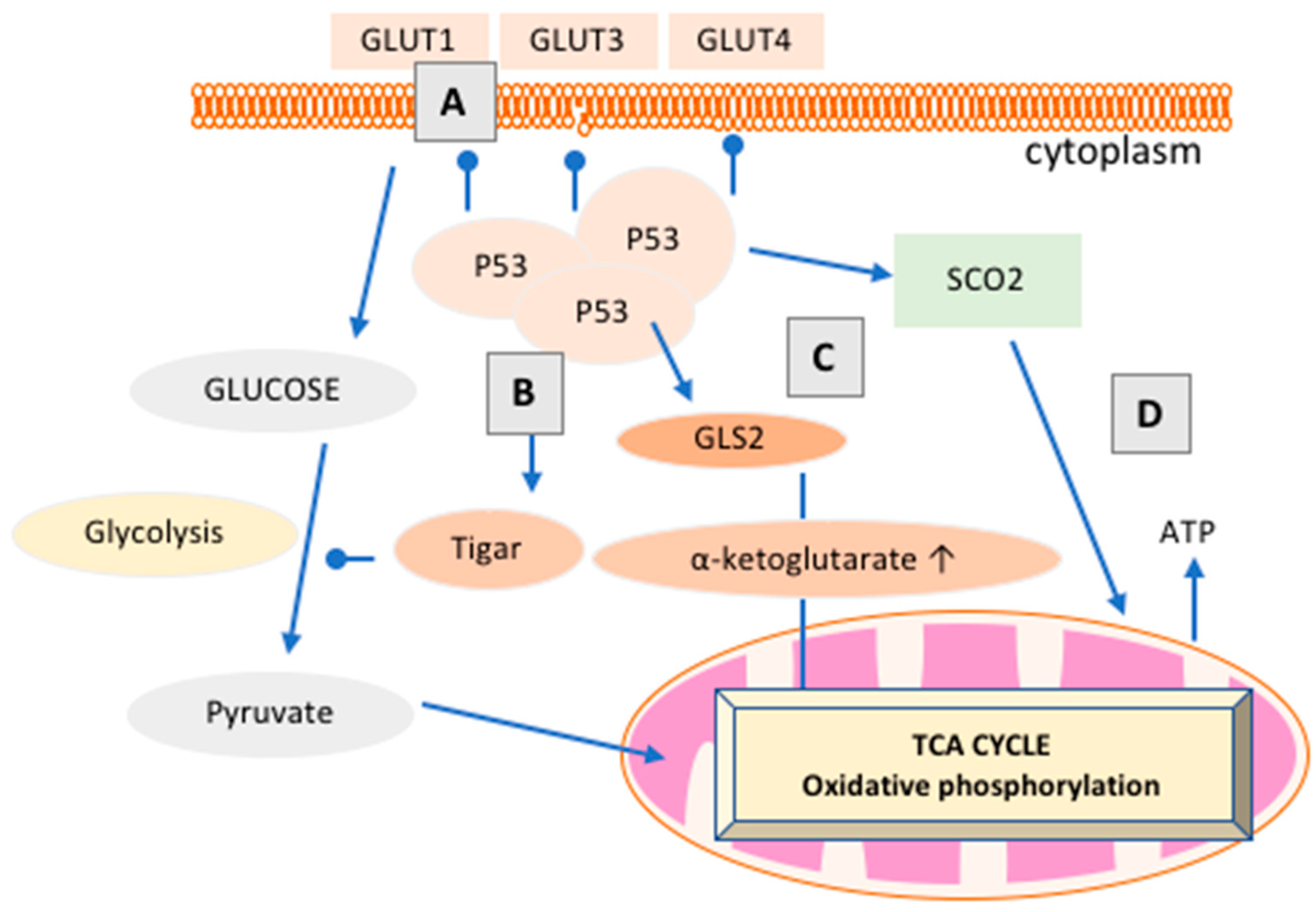
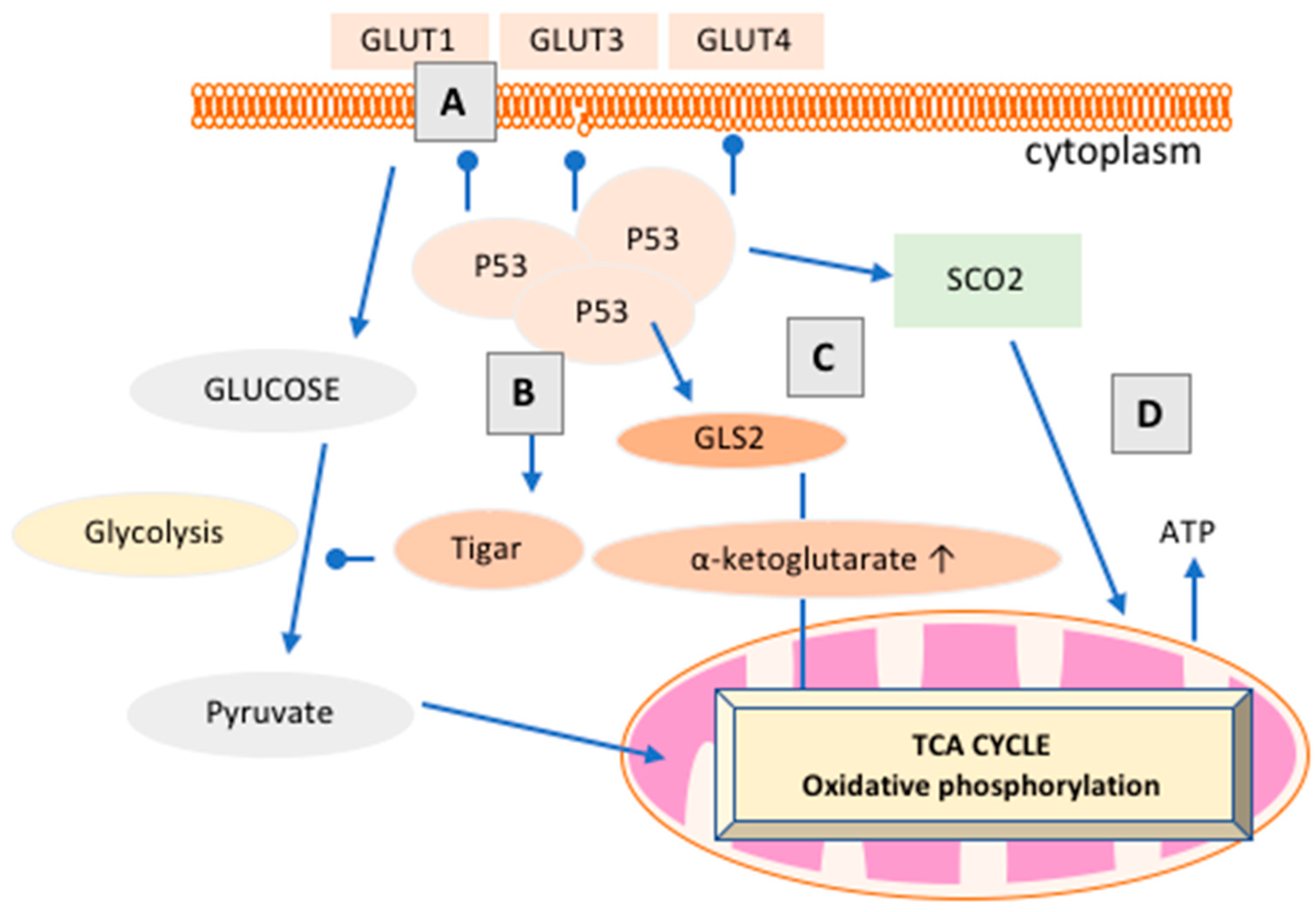
3.3. How Does p53 Regulate Cancer Metabolism?
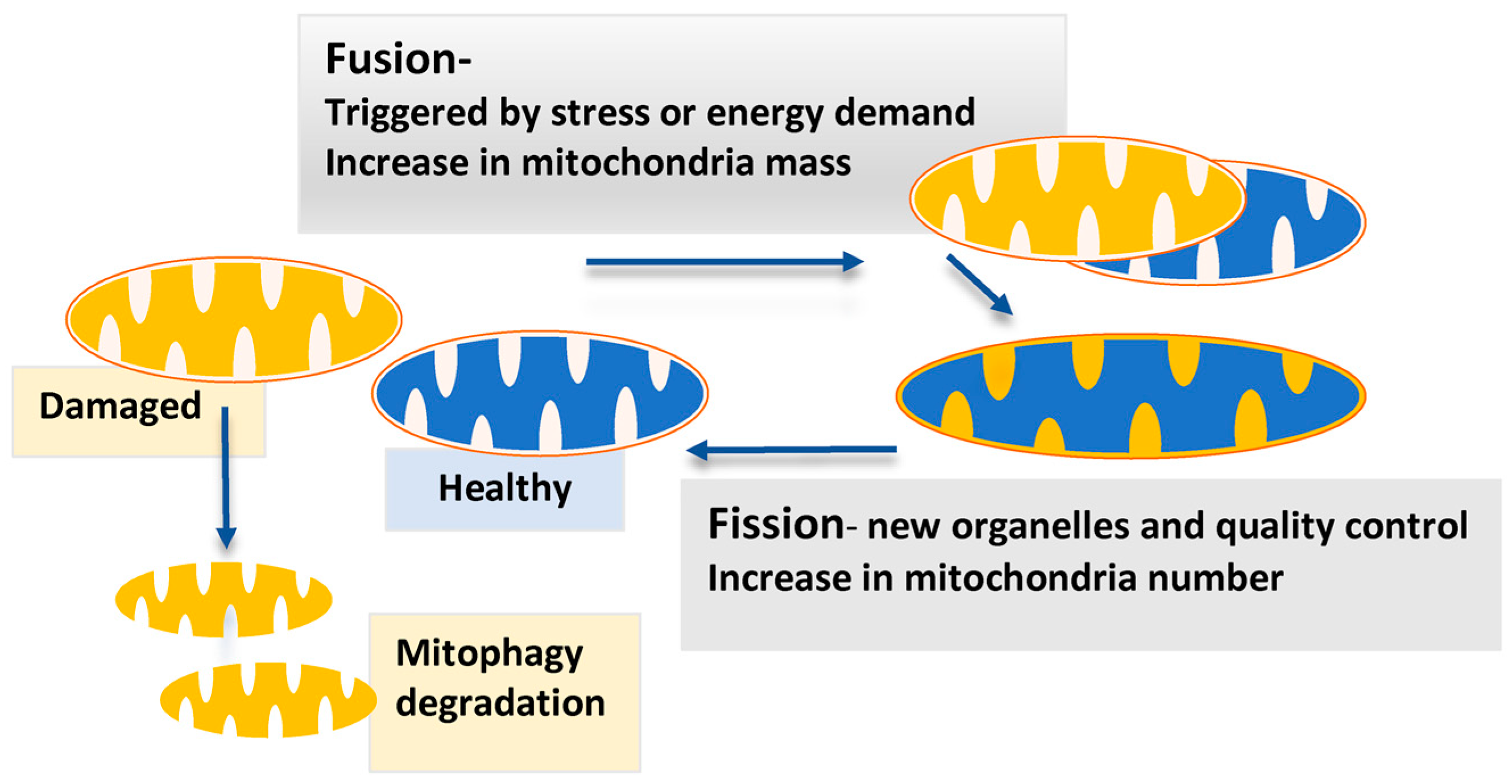
3.4. Mitochondrial Dynamics and p53
3.5. p53 Family Members p63 and p73 Play Significant Roles in the Cancer Metabolic Switch
4. p53 and Breast Cancer Metabolism
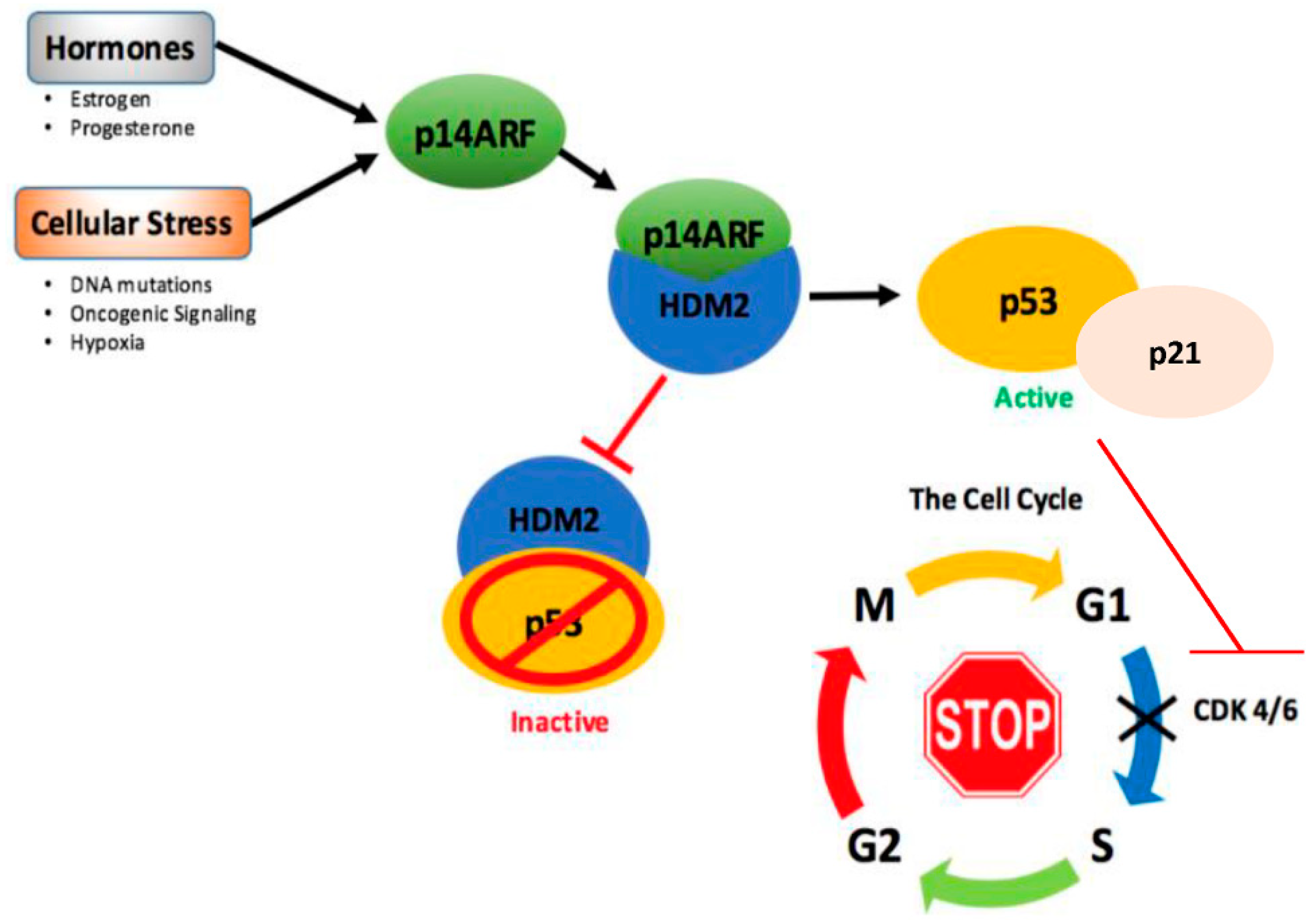
4.1. Upstream Regulation and Reactivation of p53 in Breast Cancer
4.2. p53 Regulates Mitochondria Dynamics in Breast Cancer
4.3. Novel Avenues for Targeting p53 in Drug Resistance, Recurrence, and Metastasis in Breast Cancer
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Vousden, K.H.; Ryan, K.M. P53 and metabolism. Nat. Rev. Cancer 2009, 9, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Prives, C. Blinded by the light: The growing complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Itahana, Y.; Itahana, K. Emerging roles of p53 family members in glucose metabolism. Int. J. Mol. Sci. 2018, 19, 776. [Google Scholar] [CrossRef] [PubMed]
- Gnanapradeepan, K.; Basu, S.; Barnoud, T.; Budina-Kolomets, A.; Kung, C.P.; Murphy, M.E. The p53 tumor suppressor in the control of metabolism and ferroptosis. Front. Endocrinol. 2018, 9, 124. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H. Functions of p53 in metabolism and invasion. Biochem. Soc. Trans. 2009, 37, 511–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Santis, M.C.; Porporato, P.E.; Martini, M.; Morandi, A. Signaling pathways regulating redox balance in cancer metabolism. Front. Oncol. 2018, 8, 126. [Google Scholar] [CrossRef] [PubMed]
- Olovnikov, I.A.; Kravchenko, J.E.; Chumakov, P.M. Homeostatic functions of the p53 tumor suppressor: Regulation of energy metabolism and antioxidant defense. Semin. Cancer Biol. 2009, 19, 32–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGowan, E.M.; Lin, Y.; Hatoum, D. Good guy or bad guy? The duality of wild-type p53 in breast cancer origin, treatment, and recurrence. Cancers 2018, 10, 172. [Google Scholar] [CrossRef] [PubMed]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. p53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Balint, E.E.; Vousden, K.H. Activation and activities of the p53 tumour suppressor protein. Br. J. Cancer 2001, 85, 1813–1823. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Mak, T.W. The current state of cancer metabolism. Nat. Rev. Cancer 2016, 16, 613–614. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Hu, W.; Feng, Z. Tumor suppressor p53 and its mutants in cancer metabolism. Cancer Lett. 2015, 356, 197–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berkers, C.R.; Maddocks, O.D.K.; Cheung, E.C.; Mor, I.; Vousden, K.H. Metabolic regulation by p53 family members. Cell Metab. 2013, 18, 617–633. [Google Scholar] [CrossRef] [PubMed]
- Matoba, S.; Kang, J.G.; Patino, W.D.; Wragg, A.; Boehm, M.; Gavrilova, O.; Hurley, P.J.; Bunz, F.; Hwang, P.M. p53 regulates mitochondrial respiration. Science 2006, 312, 1650–1653. [Google Scholar] [CrossRef] [PubMed]
- Stewart, B.; Wild, C.P. World Cancer Report 2014; WHO: Geneva, Switzerland, 2017. [Google Scholar]
- Jemal, A.; Ward, E.M.; Johnson, C.J.; Cronin, K.A.; Ma, J.; Ryerson, B.; Mariotto, A.; Lake, A.J.; Wilson, R.; Sherman, R.L.; et al. Annual report to the nation on the status of cancer, 1975–2014, featuring survival. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [PubMed]
- Ghoncheh, M.; Pournamdar, Z.; Salehiniya, H. Incidence and mortality and epidemiology of breast cancer in the world. Asian Pac. J. Cancer Prev. 2016, 17, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.; Tyson, J.J.; Dixon, J.M. Endocrine resistance in breast cancer—An overview and update. Mol. Cell. Endocrinol. 2015, 418, 220–234. [Google Scholar] [CrossRef] [PubMed]
- Althuis, M.D.; Fergenbaum, J.H.; Garcia-Closas, M.; Brinton, L.A.; Madigan, M.P.; Sherman, M.E. Etiology of hormone receptor-defined breast cancer: A systematic review of the literature. Cancer Epidemiol. Biomark. Prev. 2004, 13, 1558–1568. [Google Scholar]
- Allred, D.C. Issues and updates: Evaluating estrogen receptor-α, progesterone receptor, and HER2 in breast cancer. Mod. Pathol. 2010, 23, S52–S59. [Google Scholar] [CrossRef] [PubMed]
- Coates, A.S.; Winer, E.P.; Goldhirsch, A.; Gelber, R.D.; Gnant, M.; Piccart-Gebhart, M.; Thürlimann, B.; Senn, H.-J. Tailoring therapies—Improving the management of early breast cancer: St galleninternational expert consensus on the primary therapy of early breast cancer 2015. Ann. Oncol. 2015, 26, 1533–1546. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Konorev, E.A.; Kotamraju, S.; Joseph, J.; Kalivendi, S.; Kalyanaraman, B. Doxorubicin induces apoptosis in normal and tumor cells via distinctly different mechanisms: Intermediacy of H2O2- and p53-dependent pathways. J. Biol. Chem. 2004, 279, 25535–25543. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.; Watson, M.B.; O’Kane, S.L.; Drew, P.J.; Lind, M.J.; Cawkwell, L. The analysis of doxorubicin resistance in human breast cancer cells using antibody microarrays. Mol. Cancer Ther. 2006, 5, 2115–2120. [Google Scholar] [CrossRef] [PubMed]
- Balmer, M.T.; Katz, R.D.; Liao, S.; Goodwine, J.S.; Gal, S. Doxorubicin and 5-fluorouracil induced accumulation and transcriptional activity of p53 are independent of the phosphorylation at serine 15 in MCF-7 breast cancer cells. Cancer Biol. Ther. 2014, 15, 1000–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gudkov, A.V.; Komarova, E.A. The role of p53 in determining sensitivity to radiotherapy. Nat. Rev. Cancer 2003, 3, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Pinto, A.C.; Ades, F.; de Azambuja, E.; Piccart-Gebhart, M. Trastuzumab for patients with HER2 positive breast cancer: Delivery, duration and combination therapies. Breast 2013, 22, S152–S155. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.R.D.; Dowsett, M. Aromatase inhibitors for breast cancer: Lessons from the laboratory. Nat. Rev. Cancer 2003, 3, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Chen, A.; Bai, Z. Integrative investigation on breast cancer in ER, PR and HER2-defined subgroups using mrna and mirna expression profiling. Sci. Rep. 2014, 4, 6566. [Google Scholar] [CrossRef] [PubMed]
- Hudis, C.A.; Gianni, L. Triple-negative breast cancer: An unmet medical need. Oncologist 2011, 16, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Linden, H.M.; Anderson, B.O.; Li, C.I. Trends in 5-year survival rates among breast cancer patients by hormone receptor status and stage. Breast Cancer Res. Treat. 2014, 147, 609–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.Y.; Wu, C.Y.; Petrossian, K.; Huang, T.T.; Tseng, L.M.; Chen, S. Treatment for the endocrine resistant breast cancer: Current options and future perspectives. J. Steroid Biochem. Mol. Biol. 2017, 172, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Tryfonidis, K.; Zardavas, D.; Katzenellenbogen, B.S.; Piccart, M. Endocrine treatment in breast cancer: Cure, resistance and beyond. Cancer Treat. Rev. 2016, 50, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Early Breast Cancer Trialists’ Collaborative Group. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: An overview of the randomised trials. Lancet 2005, 365, 1687–1717. [Google Scholar]
- Ma, C.X.; Sanchez, C.G.; Ellis, M.J. Predicting endocrine therapy responsiveness in breast cancer. Oncology 2009, 23, 133–142. [Google Scholar] [PubMed]
- Roy, R.; Chun, J.; Powell, S.N. Brca1 and brca2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2012, 12, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.J.; Vousden, K.H. p53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Du, C.W.; Kwan, M.; Liang, S.X.; Zhang, G.J. The impact of p53 in predicting clinical outcome of breast cancer patients with visceral metastasis. Sci. Rep. 2013, 3, 2246. [Google Scholar] [CrossRef] [PubMed]
- Alsner, J.; Yilmaz, M.; Guldberg, P.; Hansen, L.L.; Overgaard, J. Heterogeneity in the clinical phenotype of TP53 mutations in breast cancer patients. Am. Assoc. Cancer Res. 2000, 6, 3923–3931. [Google Scholar] [CrossRef]
- Fresno Vara, J.A.; Casado, E.; de Castro, J.; Cejas, P.; Belda-Iniesta, C.; Gonzalez-Baron, M. PI3K/AKT signalling pathway and cancer. Cancer Treat. Rev. 2004, 30, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Bertheau, P.; Espié, M.; Turpin, E.; Lehmann, J.; Plassa, L.F.; Varna, M.; Janin, A.; de Thé, H. TP53 status and response to chemotherapy in breast cancer. Pathobiology 2008, 75, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Bertheau, P.; Lehmann-Che, J.; Varna, M.; Dumay, A.; Poirot, B.; Porcher, R.; Turpin, E.; Plassa, L.F.; de Roquancourt, A.; Bourstyn, E.; et al. p53 in breast cancer subtypes and new insights into response to chemotherapy. Breast 2013, 22, S27–S29. [Google Scholar] [CrossRef] [PubMed]
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More than just a powerhouse. Curr. Biol. 2006, 16, R551–R560. [Google Scholar] [CrossRef] [PubMed]
- Bosc, C.; Selak, M.A.; Sarry, J.E. Resistance is futile: Targeting mitochondrial energetics and metabolism to overcome drug resistance in cancer treatment. Cell Metab. 2017, 26, 705–707. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Lu, X. Live or let die: The cell’s response to p53. Nat. Rev. Cancer 2002, 2, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 2014, 14, 359–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khoo, K.H.; Verma, C.S.; Lane, D.P. Drugging the p53 pathway: Understanding the route to clinical efficacy. Nat. Rev. Drug Discov. 2014, 13, 217–236. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Kroemer, G. Ferroptosis in p53-dependent oncosuppression and organismal homeostasis. Cell Death Differ. 2015, 22, 1237–1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumann, K. Cell death: Multitasking p53 promotes necrosis. Nat. Rev. Mol. Cell Biol. 2012, 13, 480–481. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Kroemer, G. Cytoplasmic functions of the tumour suppressor p53. Nature 2009, 458, 1127–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Liu, P.; Li, J. Necroptosis: An emerging form of programmed cell death. Crit. Rev. Oncol. Hematol. 2012, 82, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Wang, B.; Zhao, X.; Wu, X.; Zhang, Q.; Wei, C.; Shi, M.; Li, Y.; Tang, W.; Zhang, J.; et al. Gain of function in the mouse model of a recurrent mutation p53(n236s) promotes the formation of double minute chromosomes and the oncogenic potential of p19(ARF). Mol. Carcinog. 2018, 57, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Escoll, M.; Gargini, R.; Cuadrado, A.; Anton, I.M.; Wandosell, F. Mutant p53 oncogenic functions in cancer stem cells are regulated by WIP through YAP/TAZ. Oncogene 2017, 36, 3515–3527. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Cheng, X.; Lu, J.; Wei, J.; Fu, G.; Zhu, F.; Jia, C.; Zhou, L.; Xie, H.; Zheng, S. Mitofusin-2 is a novel direct target of p53. Biochem. Biophys. Res. Commun. 2010, 400, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Saleem, A.; Iqbal, S.; Zhang, Y.; Hood, D.A. Effect of p53 on mitochondrial morphology, import, and assembly in skeletal muscle. Am. J. Physiol. Cell Physiol. 2015, 308, C319–C329. [Google Scholar] [CrossRef] [PubMed]
- McGowan, E.M.; Alling, N.; Jackson, E.A.; Yagoub, D.; Haass, N.K.; Allen, J.D.; Martinello-Wilks, R. Evaluation of cell cycle arrest in estrogen responsive MCF-7 breast cancer cells: Pitfalls of the mts assay. PLoS ONE 2011, 6, e20623. [Google Scholar] [CrossRef] [PubMed]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.N.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Riley, T.; Sontag, E.; Chen, P.; Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008, 9, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Menendez, D.; Inga, A.; Resnick, M.A. The expanding universe of p53 targets. Nat. Rev. Cancer 2009, 9, 724–737. [Google Scholar] [CrossRef] [PubMed]
- Proskuryakov, S.Y.; Konoplyannikov, A.G.; Gabai, V.L. Necrosis: A specific form of programmed cell death? Exp. Cell Res. 2003, 283, 1–16. [Google Scholar] [CrossRef]
- Lakin, N.D.; Jackson, S.P. Regulation of p53 in response to DNA damage. Oncogene 1999, 18, 7644–7655. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Siddik, Z.H.; Huang, Z.; Wang, R.; Koomen, J.; Kobayashi, R.; Khokhar, A.R.; Kuang, J. Induction of p21 by p53 following DNA damage inhibits both CDK4 and CDK2 activities. Oncogene 2005, 24, 2929–2943. [Google Scholar] [CrossRef] [PubMed]
- Brooks, C.L.; Gu, W. New insights into p53 activation. Cell Res. 2010, 20, 614–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musgrove, E.A.; Caldon, C.E.; Barraclough, J.; Stone, A.; Sutherland, R.L. Cyclin D as a therapeutic target in cancer. Nat. Rev. Cancer 2011, 11, 558–572. [Google Scholar] [CrossRef] [PubMed]
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef] [PubMed]
- Poletto, M.; Legrand, A.J.; Fletcher, S.C.; Dianov, G.L. p53 coordinates base excision repair to prevent genomic instability. Nucleic Acids Res. 2016, 44, 3165–3175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.L.; Seo, Y.R. p53 regulation of DNA excision repair pathways. Mutagenesis 2002, 17, 149–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, K.; Vousden, K.H. Puma, a novel proapoptotic gene, is induced by p53. Mol. Cell 2001, 7, 683–694. [Google Scholar] [CrossRef]
- Jeffers, J.R.; Parganas, E.; Lee, Y.; Yang, C.; Wang, J.; Brennan, J.; MacLean, K.H.; Han, J.; Chittenden, T.; Ihle, J.N.; et al. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell 2003, 4, 321–328. [Google Scholar] [CrossRef] [Green Version]
- Oda, E.; Ohki, R.; Murasawa, H.; Nemoto, J.; Shibue, T.; Yamashita, T.; Tokino, T.; Taniguchi, T.; Tanaka, N. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 2000, 288, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, J.E. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004, 303, 1010–1014. [Google Scholar] [CrossRef] [PubMed]
- Fridman, J.S.; Lowe, S.W. Control of apoptosis by p53. Oncogene 2003, 22, 9030–9040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Kepp, O.; Krautwald, S.; Kroemer, G.; Linkermann, A. Molecular mechanisms of regulated necrosis. Semin. Cell Dev. Biol. 2014, 35, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Yang, Z.; Xie, L.; DeWitt, J.P.; Chen, Y. Cancer therapy in the necroptosis era. Cell Death Differ. 2016, 23, 748–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, F.K.; Luz, N.F.; Moriwaki, K. Programmed necrosis in the cross talk of cell death and inflammation. Annu. Rev. Immunol. 2015, 33, 79–106. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, J.D.; White, E. Autophagy and metabolism. Science 2010, 330, 1344. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiuri, M.C.; Galluzzi, L.; Morselli, E.; Kepp, O.; Malik, S.A.; Kroemer, G. Autophagy regulation by p53. Curr. Opin. Cell Biol. 2010, 22, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Vitale, I.; Djavaheri-Mergny, M.; D’Amelio, M.; Criollo, A.; Morselli, E.; Zhu, C.; Harper, F.; et al. Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. 2008, 10, 676–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, B.; Abrams, J. P53: The janus of autophagy? Nat. Cell Biol. 2008, 10, 637–639. [Google Scholar] [CrossRef] [PubMed]
- Tasdemir, E.; Chiara Maiuri, M.; Morselli, E.; Criollo, A.; D’Amelio, M.; Djavaheri-Mergny, M.; Cecconi, F.; Tavernarakis, N.; Kroemer, G. A dual role of p53 in the control of autophagy. Autophagy 2008, 4, 810–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, K.; Kondo-Okamoto, N. Mitochondria and autophagy: Critical interplay between the two homeostats. Biochim. Biophys. Acta 2012, 1820, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef] [PubMed]
- Manjili, M.H. Tumor dormancy and relapse: From a natural byproduct of evolution to a disease state. Cancer Res. 2017, 77, 2564–2569. [Google Scholar] [CrossRef] [PubMed]
- Babot, M.; Galkin, A. Molecular mechanism and physiological role of active-deactive transition of mitochondrial complex i. Biochem. Soc. Trans. 2013, 41, 1325–1330. [Google Scholar] [CrossRef] [PubMed]
- Karrison, T.G.; Ferguson, D.J.; Meier, P. Dormancy of mammary carcinoma after mastectomy. J. Natl. Cancer Inst. 1999, 91, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Aguirre-Ghiso, J.A. Models, mechanisms and clinical evidence for cancer dormancy. Nat. Rev. Cancer 2007, 7, 834–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef] [PubMed]
- McGowan, E.M.; Tran, N.; Alling, N.; Yagoub, D.; Sedger, L.M.; Martiniello-Wilks, R. P14arf post-transcriptional regulation of nuclear cyclin d1 in MCF-7 breast cancer cells: Discrimination between a good and bad prognosis? PLoS ONE 2012, 7, e42246. [Google Scholar] [CrossRef] [PubMed]
- Hatoum, D.; Yagoub, D.; Ahadi, A.; Nassif, N.T.; McGowan, E.M. Annexin/s100a protein family regulation through p14arf-p53 activation: A role in cell survival and predicting treatment outcomes in breast cancer. PLoS ONE 2017, 12, e0169925. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.-K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.; et al. Crucial role of p53-dependent cellular senescence in suppression of PTEN-deficient tumorigenesis. Nature 2005, 436, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Rufini, A.; Tucci, P.; Celardo, I.; Melino, G. Senescence and aging: The critical roles of p53. Oncogene 2013, 32, 5129–5143. [Google Scholar] [CrossRef] [PubMed]
- Xue, W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, Y.; Chen, X. Senescence regulation by the p53 protein family. Methods Mol. Biol. 2013, 965, 37–61. [Google Scholar] [PubMed]
- Itahana, K.; Dimri, G.P.; Hara, E.; Itahana, Y.; Zou, Y.; Desprez, P.-Y.; Campisi, J. A role for p53 in maintaining and establishing the quiescence growth arrest in human cells. J. Biol. Chem. 2002, 277, 18206–18214. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Elf, S.E.; Miyata, Y.; Sashida, G.; Liu, Y.; Huang, G.; Di Giandomenico, S.; Lee, J.M.; Deblasio, A.; Menendez, S.; et al. p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell 2009, 4, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Zhao, W.; Yu, J.; Li, S.; Lin, L.; Chen, X. Induction of ferroptosis and mitochondrial dysfunction by oxidative stress in PC12 cells. Sci. Rep. 2018, 8, 574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Kon, N.; Jiang, L.; Tan, M.; Ludwig, T.; Zhao, Y.; Baer, R.; Gu, W. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 2012, 149, 1269–1283. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Peiris-Pages, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer metabolism: A therapeutic perspective. Nat. Rev. Clin. Oncol. 2016, 14, 11. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.P.; Sabatini, D.M. Cancer cell metabolism: Warburg and beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Salazar-Roa, M.; Malumbres, M. Fueling the cell division cycle. Trends Cell Biol. 2017, 27, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.H.; Stoeber, K. The cell cycle and cancer. J. Pathol. 2012, 226, 352–364. [Google Scholar] [CrossRef] [PubMed]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Hay, N. Reprogramming glucose metabolism in cancer: Can it be exploited for cancer therapy? Nat. Rev. Cancer 2016, 16, 635–649. [Google Scholar] [CrossRef] [PubMed]
- Szablewski, L. Expression of glucose transporters in cancers. Biochim. Biophys. Acta Rev. Cancer 2013, 1835, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef] [PubMed]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef] [PubMed]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V., Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. The metabolism of carcinoma cells. J. Cancer Res. 1925, 9, 148–163. [Google Scholar] [CrossRef]
- Petros, J.A.; Baumann, A.K.; Ruiz-Pesini, E.; Amin, M.B.; Sun, C.Q.; Hall, J.; Lim, S.; Issa, M.M.; Flanders, W.D.; Hosseini, S.H.; et al. Mtdna mutations increase tumorigenicity in prostate cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Kachhap, S.; Sun, W.; Wu, G.; Chuang, A.; Poeta, L.; Grumbine, L.; Mithani, S.K.; Chatterjee, A.; Koch, W.; et al. Frequency and phenotypic implications of mitochondrial DNA mutations in human squamous cell cancers of the head and neck. Proc. Natl. Acad. Sci. USA 2007, 104, 7540–7545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Bayley, J.P.; Devilee, P. Warburg tumours and the mechanisms of mitochondrial tumour suppressor genes. Barking up the right tree? Curr. Opin. Genet. Dev. 2010, 20, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Burk, D.; Schade, A.L. On respiratory impairment in cancer cells. Science 1956, 124, 270–272. [Google Scholar] [PubMed]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [PubMed]
- Weinhouse, S. On respiratory impairment in cancer cells. Science 1956, 124, 267–269. [Google Scholar] [CrossRef] [PubMed]
- Weinhouse, S.; Millington, R.H.; Wenner, C.E. Metabolism of neoplastic tissue. I. The oxidation of carbohydrate and fatty acids in transplanted tumors. Cancer Res. 1951, 11, 845–850. [Google Scholar] [PubMed]
- Senyilmaz, D.; Teleman, A.A. Chicken or the egg: Warburg effect and mitochondrial dysfunction. F1000Prime Rep. 2015, 7, 41. [Google Scholar] [CrossRef] [PubMed]
- Vega-Naredo, I.; Loureiro, R.; Mesquita, K.A.; Barbosa, I.A.; Tavares, L.C.; Branco, A.F.; Erickson, J.R.; Holy, J.; Perkins, E.L.; Carvalho, R.A.; et al. Mitochondrial metabolism directs stemness and differentiation in p19 embryonal carcinoma stem cells. Cell Death Differ. 2014, 21, 1560–1574. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penkert, J.; Ripperger, T.; Schieck, M.; Schlegelberger, B.; Steinemann, D.; Illig, T. On metabolic reprogramming and tumor biology: A comprehensive survey of metabolism in breast cancer. Oncotarget 2016, 7, 67626–67649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boroughs, L.K.; DeBerardinis, R.J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 2015, 17, 351–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, A.J.; Puzio-Kuter, A.M. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 2010, 330, 1340. [Google Scholar] [CrossRef] [PubMed]
- Iurlaro, R.; Leon-Annicchiarico, C.L.; Munoz-Pinedo, C. Regulation of cancer metabolism by oncogenes and tumor suppressors. Methods Enzymol. 2014, 542, 59–80. [Google Scholar] [PubMed]
- Jones, R.G.; Thompson, C.B. Tumor suppressors and cell metabolism: A recipe for cancer growth. Genes Dev. 2009, 23, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Madan, E.; Gogna, R.; Bhatt, M.; Pati, U.; Kuppusamy, P.; Mahdi, A.A. Regulation of glucose metabolism by p53: Emerging new roles for the tumor suppressor. Oncotarget 2011, 2, 948–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, W.; Zhang, C.; Wu, R.; Sun, Y.; Levine, A.; Feng, Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc. Natl. Acad. Sci. USA 2010, 107, 7455–7460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathupala, S.P.; Heese, C.; Pedersen, P.L. Glucose catabolism in cancer cells. The type ii hexokinase promoter contains functionally active response elements for the tumor suppressor p53. J. Biol. Chem. 1997, 272, 22776–22780. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Lozano, P.; Hixon, M.L.; Wagner, M.W.; Flores, A.I.; Ikawa, S.; Baldwin, A.S., Jr.; Chien, K.R.; Gualberto, A. P53 is a transcriptional activator of the muscle-specific phosphoglycerate mutase gene and contributes in vivo to the control of its cardiac expression. Cell Growth Differ. 1999, 10, 295–306. [Google Scholar] [PubMed]
- Maddocks, O.D.; Berkers, C.R.; Mason, S.M.; Zheng, L.; Blyth, K.; Gottlieb, E.; Vousden, K.H. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 2013, 493, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Westermann, B. Bioenergetic role of mitochondrial fusion and fission. Biochim. Biophys. Acta Bioenerg. 2012, 1817, 1833–1838. [Google Scholar] [CrossRef] [PubMed]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Wai, T.; Langer, T. Mitochondrial dynamics and metabolic regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Saleem, A.; Adhihetty, P.J.; Hood, D.A. Role of p53 in mitochondrial biogenesis and apoptosis in skeletal muscle. Physiol. Genom. 2009, 37, 58–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamponi, N.; Zamponi, E.; Cannas, S.A.; Billoni, O.V.; Helguera, P.R.; Chialvo, D.R. Mitochondrial network complexity emerges from fission/fusion dynamics. Sci. Rep. 2018, 8, 363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, A.Y.; Joseph, A.M.; Dutta, D.; Hwang, J.C.; Aris, J.P.; Leeuwenburgh, C. New insights into the role of mitochondria in aging: Mitochondrial dynamics and more. J. Cell Sci. 2010, 123, 2533–2542. [Google Scholar] [CrossRef] [PubMed]
- Bourdon, J.C.; Fernandes, K.; Murray-Zmijewski, F.; Liu, G.; Diot, A.; Xirodimas, D.P.; Saville, M.K.; Lane, D.P. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005, 19, 2122–2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avery-Kiejda, K.A.; Morten, B.; Wong-Brown, M.W.; Mathe, A.; Scott, R.J. The relative mrna expression of p53 isoforms in breast cancer is associated with clinical features and outcome. Carcinogenesis 2014, 35, 586–596. [Google Scholar] [CrossRef] [PubMed]
- Romano, R.A.; Smalley, K.; Magraw, C.; Serna, V.A.; Kurita, T.; Raghavan, S.; Sinha, S. Deltanp63 knockout mice reveal its indispensable role as a master regulator of epithelial development and differentiation. Development 2012, 139, 772–782. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.L.; Sengupta, S.; Gurdziel, K.; Bell, G.W.; Jacks, T.; Flores, E.R. P63 and p73 transcriptionally regulate genes involved in DNA repair. PLoS Genet. 2009, 5, e1000680. [Google Scholar] [CrossRef] [PubMed]
- Moll, U.M.; Slade, N. p63 and p73: Roles in development and tumor formation. Mol. Cancer Res. 2004, 2, 371–386. [Google Scholar] [PubMed]
- Napoli, M.; Flores, E.R. The p53 family orchestrates the regulation of metabolism: Physiological regulation and implications for cancer therapy. Br. J. Cancer 2017, 116, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Tomasini, R.; McKeon, F.D.; Mak, T.W.; Melino, G. The p53 family: Guardians of maternal reproduction. Nat. Rev. Mol. Cell Biol. 2011, 12, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Jiang, P.; Mancuso, A.; Stonestrom, A.; Brewer, M.D.; Minn, A.J.; Mak, T.W.; Wu, M.; Yang, X. Tap73 enhances the pentose phosphate pathway and supports cell proliferation. Nat. Cell Biol. 2013, 15, 991–1000. [Google Scholar] [CrossRef] [PubMed]
- Venkatanarayan, A.; Raulji, P.; Norton, W.; Chakravarti, D.; Coarfa, C.; Su, X.; Sandur, S.K.; Ramirez, M.S.; Lee, J.; Kingsley, C.V.; et al. IAPP-driven metabolic reprogramming induces regression of p53-deficient tumours in vivo. Nature 2015, 517, 626–630. [Google Scholar] [CrossRef] [PubMed]
- Giacobbe, A.; Bongiorno-Borbone, L.; Bernassola, F.; Terrinoni, A.; Markert, E.K.; Levine, A.J.; Feng, Z.; Agostini, M.; Zolla, L.; Agro, A.F.; et al. p63 regulates glutaminase 2 expression. Cell Cycle 2013, 12, 1395–1405. [Google Scholar] [PubMed] [Green Version]
- Rufini, A.; Niklison-Chirou, M.V.; Inoue, S.; Tomasini, R.; Harris, I.S.; Marino, A.; Federici, M.; Dinsdale, D.; Knight, R.A.; Melino, G.; et al. Tap73 depletion accelerates aging through metabolic dysregulation. Genes Dev. 2012, 26, 2009–2014. [Google Scholar] [CrossRef] [PubMed]
- Harrelson, J.P.; Lee, M.W. Expanding the view of breast cancer metabolism: Promising molecular targets and therapeutic opportunities. Pharmacol. Ther. 2016, 167, 60–73. [Google Scholar] [CrossRef] [PubMed]
- Ogrodzinski, M.P.; Bernard, J.J.; Lunt, S.Y. Deciphering metabolic rewiring in breast cancer subtypes. Transl. Res. 2017, 189, 105–122. [Google Scholar] [CrossRef] [PubMed]
- Won, K.Y.; Lim, S.J.; Kim, G.Y.; Kim, Y.W.; Han, S.A.; Song, J.Y.; Lee, D.K. Regulatory role of p53 in cancer metabolism via SCO2 and tigar in human breast cancer. Hum. Pathol. 2012, 43, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Hatoum, D.; McGowan, E.M. Recent advances in the use of metformin: Can treating diabetes prevent breast cancer? BioMed Res. Int. 2015, 2015, 548436. [Google Scholar] [CrossRef] [PubMed]
- Wade, M.; Li, Y.-C.; Wahl, G.M. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer 2013, 13, 83–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Ludwig, R.L.; Jensen, J.P.; Pierre, S.A.; Medaglia, M.V.; Davydov, I.V.; Safiran, Y.J.; Oberoi, P.; Kenten, J.H.; Phillips, A.C.; et al. Small molecule inhibitors of HDM2 ubiquitin ligase activity stabilize and activate p53 in cells. Cancer Cell 2005, 7, 547–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgess, A.; Chia, K.M.; Haupt, S.; Thomas, D.; Haupt, Y.; Lim, E. Clinical overview of MDM2/X-targeted therapies. Front. Oncol. 2016, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Crane, E.K.; Kwan, S.Y.; Izaguirre, D.I.; Tsang, Y.T.; Mullany, L.K.; Zu, Z.; Richards, J.S.; Gershenson, D.M.; Wong, K.K. Nutlin-3a: A potential therapeutic opportunity for TP53 wild-type ovarian carcinomas. PLoS ONE 2015, 10, e0135101. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Dilnawaz, F.; Sahoo, S.K. Targeted nutlin-3a loaded nanoparticles inhibiting p53-MDM2 interaction: Novel strategy for breast cancer therapy. Nanomedicine 2011, 6, 489–507. [Google Scholar] [CrossRef] [PubMed]
- Chene, P. Inhibiting the p53-MDM2 interaction: An important target for cancer therapy. Nat. Rev. Cancer 2003, 3, 102–109. [Google Scholar] [CrossRef] [PubMed]
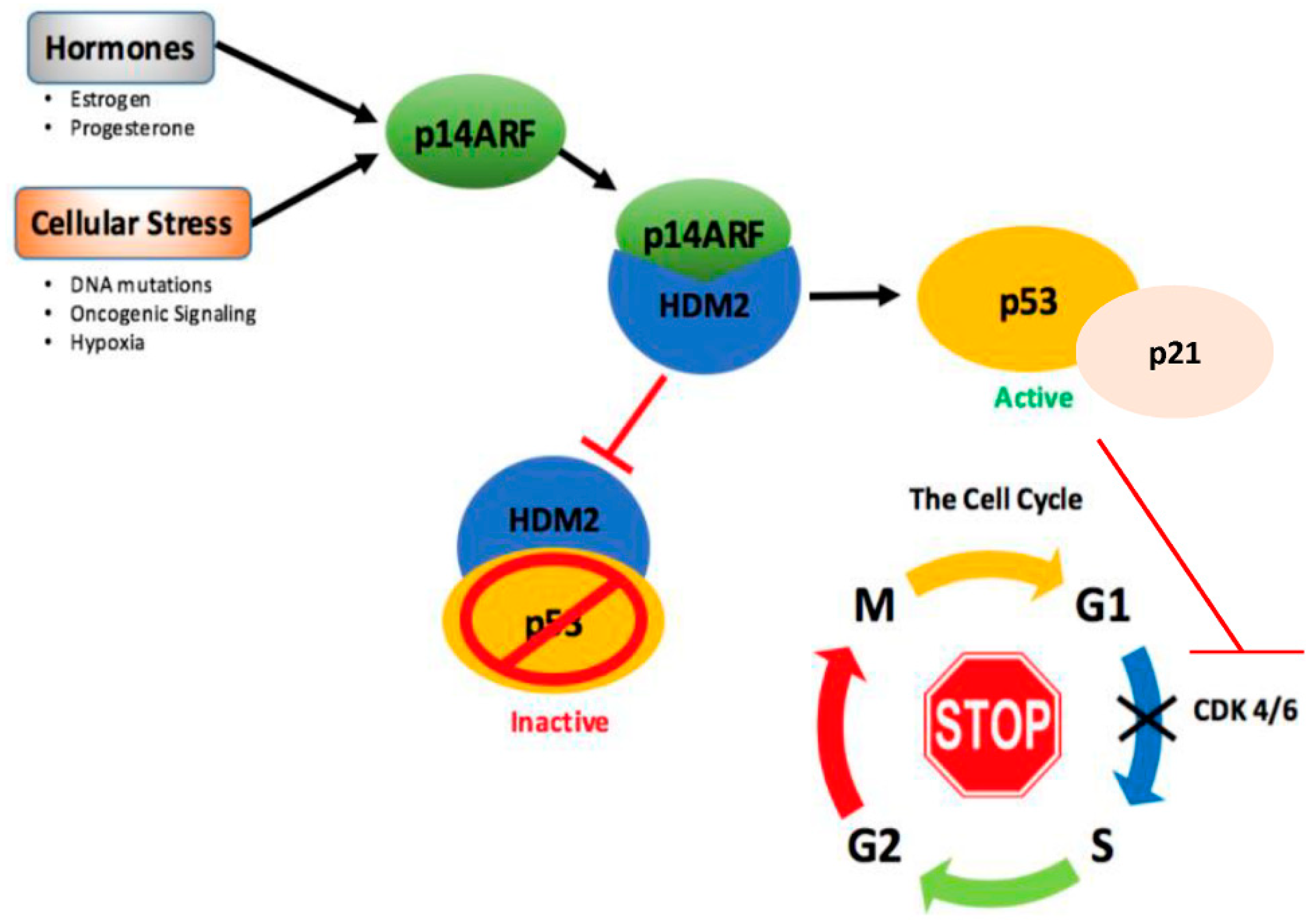
- Agrawal, A.; Yang, J.; Murphy, R.F.; Agrawal, D.K. Regulation of the p14arf-MDM2-p53 pathway: An overview in breast cancer. Exp. Mol. Pathol. 2006, 81, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Llanos, S.; Clark, P.A.; Rowe, J.; Peters, G. Stabilization of p53 by p14arf without relocation of MDM2 to the nucleolus. Nat. Cell Biol. 2001, 3, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Bates, S.; Phillips, A.C.; Clark, P.A.; Stott, F.; Peters, G.; Ludwig, R.L.; Vousden, K.H. P14arf links the tumour suppressors RB and p53. Nature 1998, 395, 124–125. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; Shepard, A.; Kittrell, F.; Mulac-Jericevic, B.; Medina, D.; Said, T.K. P19arf determines the balance between normal cell proliferation rate and apoptosis during mammary gland development. Mol. Biol. Cell 2004, 15, 2302–2311. [Google Scholar] [CrossRef] [PubMed]
- Kuperwasser, C.; Pinkas, J.; Hurlbut, G.D.; Naber, S.P.; Jerry, D.J. Cytoplasmic sequestration and functional repression of p53 in the mammary epithelium is reversed by hormonal treatment. Cancer Res. 2000, 60, 2723–2729. [Google Scholar] [PubMed]
- Tsubura, A.; Uehara, N.; Matsuoka, Y.; Yoshizawa, K.; Yuri, T. Estrogen and progesterone treatment mimicking pregnancy for protection from breast cancer. In Vivo 2008, 22, 191–201. [Google Scholar] [PubMed]
- Harland, M.; Taylor, C.F.; Chambers, P.A.; Kukalizch, K.; Randerson-Moor, J.A.; Gruis, N.A.; de Snoo, F.A.; ter Huurne, J.A.; Goldstein, A.M.; Tucker, M.A.; et al. A mutation hotspot at the p14arf splice site. Oncogene 2005, 24, 4604–4608. [Google Scholar] [CrossRef] [PubMed]
- Burri, N.; Shaw, P.; Bouzourene, H.; Sordat, I.; Sordat, B.; Gillet, M.; Schorderet, D.; Bosman, F.T.; Chaubert, P. Methylation silencing and mutations of the p14arf and p16ink4a genes in colon cancer. Lab. Investig. 2001, 81, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Tyler, T.; Saadatmandi, N.; Lee, C.; Borgstrom, P.; Gjerset, R.A. Enhanced tumor suppression by a p14arf/p53 bicistronic adenovirus through increased p53 protein translation and stability. Cancer Res. 2003, 63, 3646–3653. [Google Scholar] [PubMed]
- Bandorowicz-Pikula, J.; Wos, M.; Sekrecka-Belniak, A.; Pikula, S. Annexins in mitochondria. Postepy Biochem. 2016, 62, 216–223. [Google Scholar] [PubMed]
- Chlystun, M.; Campanella, M.; Law, A.L.; Duchen, M.R.; Fatimathas, L.; Levine, T.P.; Gerke, V.; Moss, S.E. Regulation of mitochondrial morphogenesis by annexin a6. PLoS ONE 2013, 8, e53774. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.T.; Shin, H.; Westerling, T.; Liu, X.S.; Brown, M. Estrogen receptor prevents p53-dependent apoptosis in breast cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 18060–18065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Konduri, S.D.; Bansal, S.; Nayak, B.K.; Rajasekaran, S.A.; Karuppayil, S.M.; Rajasekaran, A.K.; Das, G.M. Estrogen receptor-α binds p53 tumor suppressor protein directly and represses its function. J. Biol. Chem. 2006, 281, 9837–9840. [Google Scholar] [CrossRef] [PubMed]
- Konduri, S.D.; Medisetty, R.; Liu, W.; Kaipparettu, B.A.; Srivastava, P.; Brauch, H.; Fritz, P.; Swetzig, W.M.; Gardner, A.E.; Khan, S.A.; et al. Mechanisms of estrogen receptor antagonism toward p53 and its implications in breast cancer therapeutic response and stem cell regulation. Proc. Natl. Acad. Sci. USA 2010, 107, 15081–15086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osborne, C.K. Steroid hormone receptors in breast cancer management. Breast Cancer Res. Treat. 1998, 51, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Salvatori, L.; Pallante, P.; Ravenna, L.; Chinzari, P.; Frati, L.; Russo, M.A.; Petrangeli, E. Oestrogens and selective oestrogen receptor (ER) modulators regulate EGF receptor gene expression through human er [α] and [β] subtypes via a sp1 site. Oncogene 0000, 22, 4875–4881. [Google Scholar] [CrossRef] [PubMed]
- Jordan, V.C. Chemoprevention of breast cancer with selective oestrogen-receptor modulators. Nat. Rev. Cancer 2007, 7, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Islam, R.A.; Hossain, S.; Chowdhury, E.H. Potential therapeutic targets in energy metabolism pathways of breast cancer. Curr. Cancer Drug Targets 2017, 17, 707–721. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.M. Physical activity and cancer prevention--data from epidemiologic studies. Med. Sci. Sports Exerc. 2003, 35, 1823–1827. [Google Scholar] [CrossRef] [PubMed]
- Holloszy, J.O. Biochemical adaptations in muscle. Effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle. J. Biol. Chem. 1967, 242, 2278–2282. [Google Scholar] [PubMed]
- San-Millan, I.; Brooks, G.A. Reexamining cancer metabolism: Lactate production for carcinogenesis could be the purpose and explanation of the warburg effect. Carcinogenesis 2017, 38, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Turcotte, L.P.; Richter, E.A.; Kiens, B. Increased plasma FFA uptake and oxidation during prolonged exercise in trained vs. Untrained humans. Am. J. Physiol. 1992, 262, E791–E799. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, J.D.; Close, G.L.; Drust, B.; Morton, J.P. The emerging role of p53 in exercise metabolism. Sports Med. 2014, 44, 303–309. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Receptor Status [21,31] | Prevalence (%) | Endogenous Ligand | Standard Therapy Type |
|---|---|---|---|
| ERα+/PR+ | 70–80% | Estradiol/Progesterone | Endocrine Therapy |
| HER2+ | 15–20% | EGF, TGF-α | HER2-targeted Therapy |
| TNBC | 15% | - | Chemotherapy/Radiotherapy |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moulder, D.E.; Hatoum, D.; Tay, E.; Lin, Y.; McGowan, E.M. The Roles of p53 in Mitochondrial Dynamics and Cancer Metabolism: The Pendulum between Survival and Death in Breast Cancer? Cancers 2018, 10, 189. https://doi.org/10.3390/cancers10060189
Moulder DE, Hatoum D, Tay E, Lin Y, McGowan EM. The Roles of p53 in Mitochondrial Dynamics and Cancer Metabolism: The Pendulum between Survival and Death in Breast Cancer? Cancers. 2018; 10(6):189. https://doi.org/10.3390/cancers10060189
Chicago/Turabian StyleMoulder, David E., Diana Hatoum, Enoch Tay, Yiguang Lin, and Eileen M. McGowan. 2018. "The Roles of p53 in Mitochondrial Dynamics and Cancer Metabolism: The Pendulum between Survival and Death in Breast Cancer?" Cancers 10, no. 6: 189. https://doi.org/10.3390/cancers10060189