Oncolytic Reovirus and Immune Checkpoint Inhibition as a Novel Immunotherapeutic Strategy for Breast Cancer

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
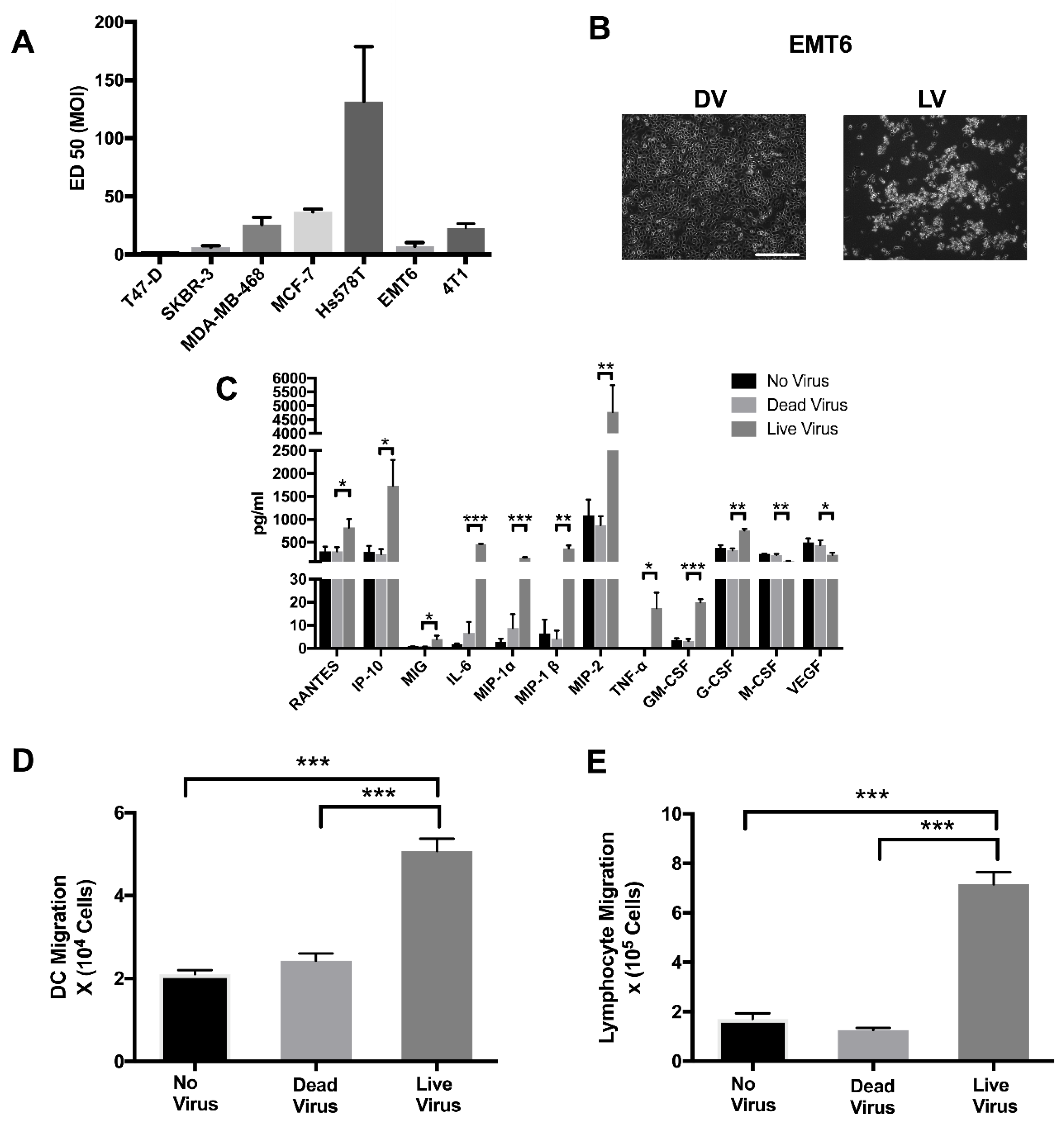
2.1. Reovirus Infection of Human and Murine Breast Cancer Cell Lines Result in Oncolysis, Cytokine Production and Immune Cell Migration
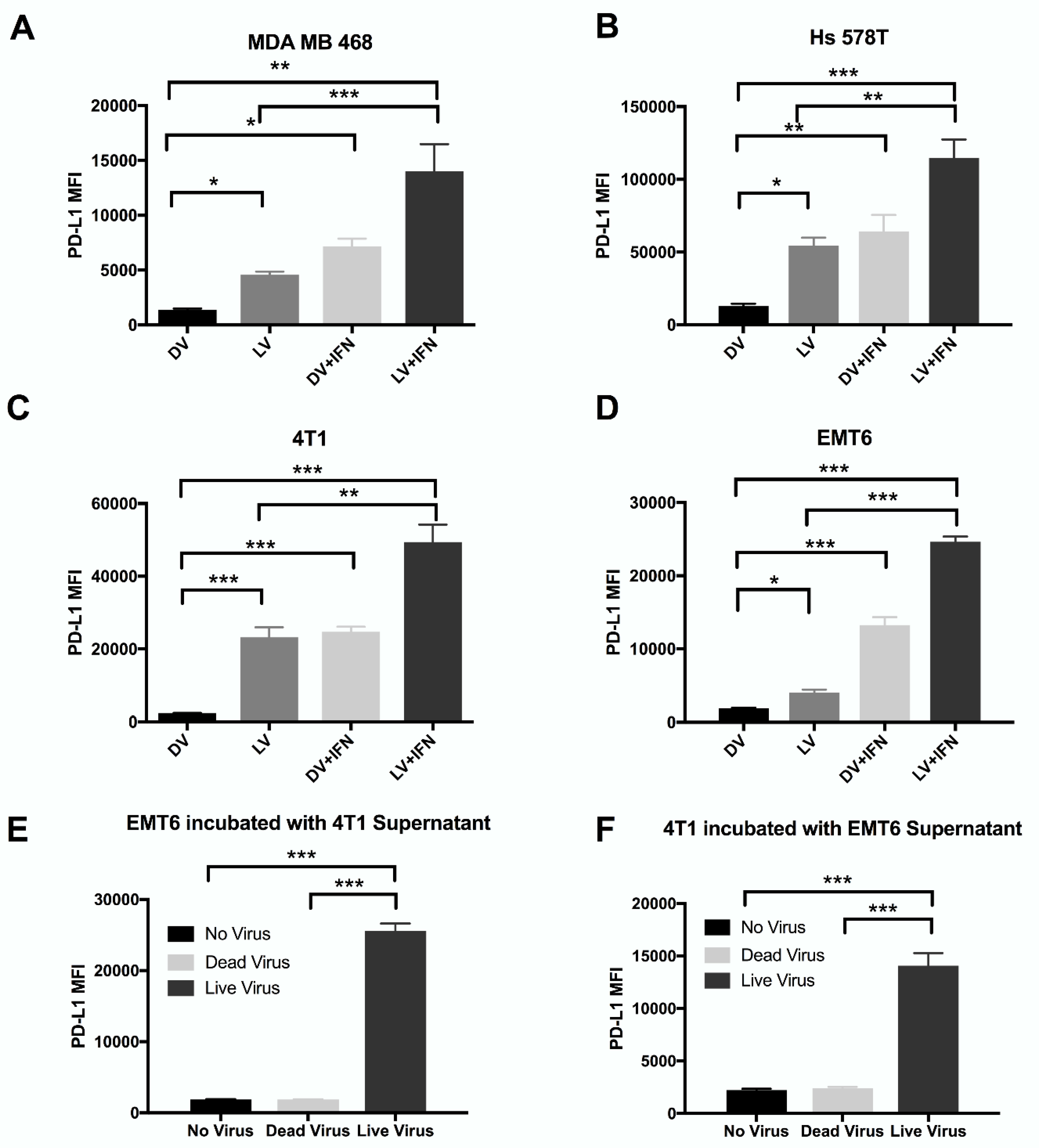
2.2. Reovirus Induces Expression of PD-L1 on the Surface of Human and Murine Breast Cancer Cell Lines Independent of the Presence of Virus
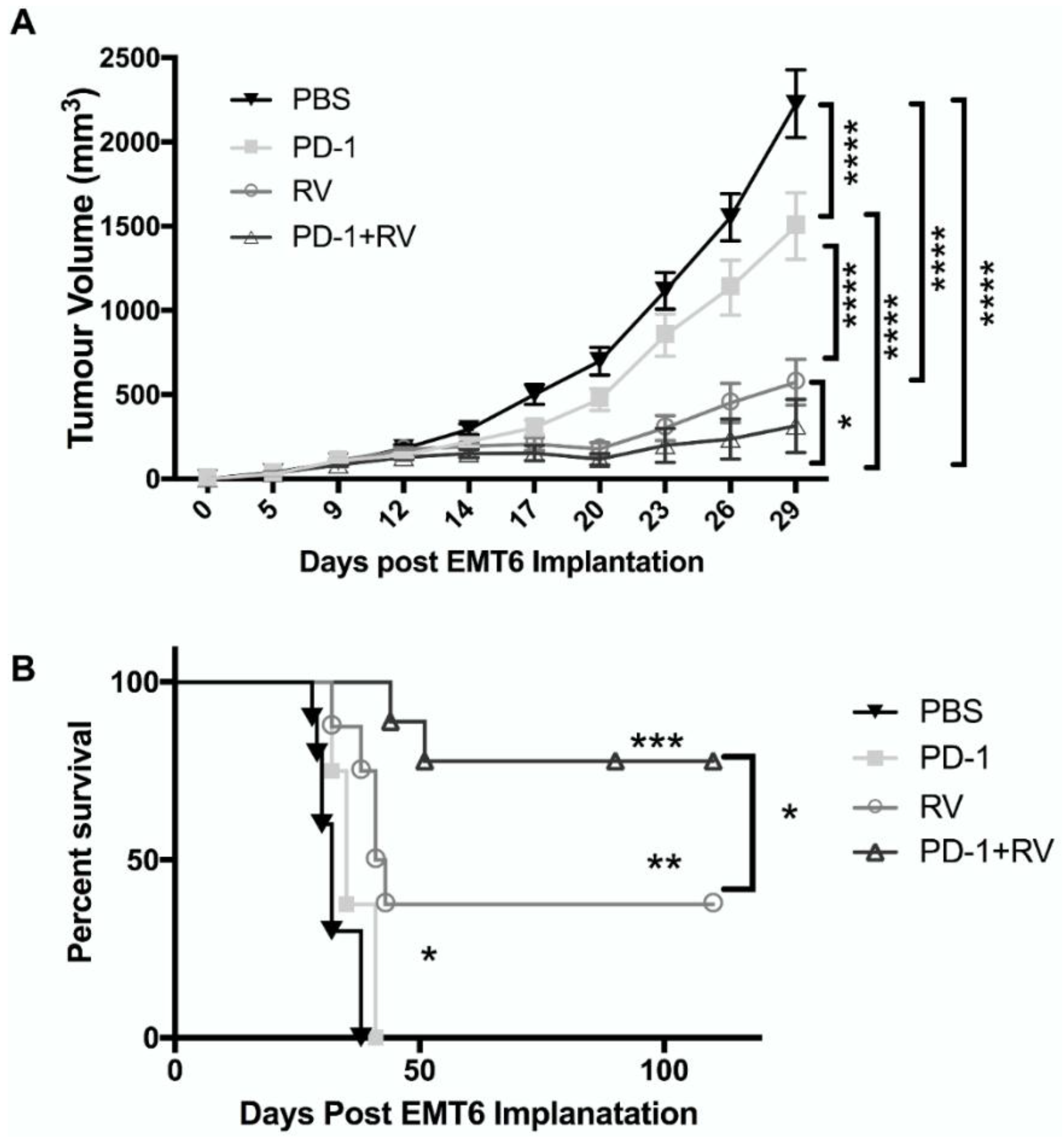
2.3. Reovirus Demonstrates In Vivo Therapeutic Efficacy as a Monotherapy and Is Significantly Enhanced by Combination with PD-1 Inhibition
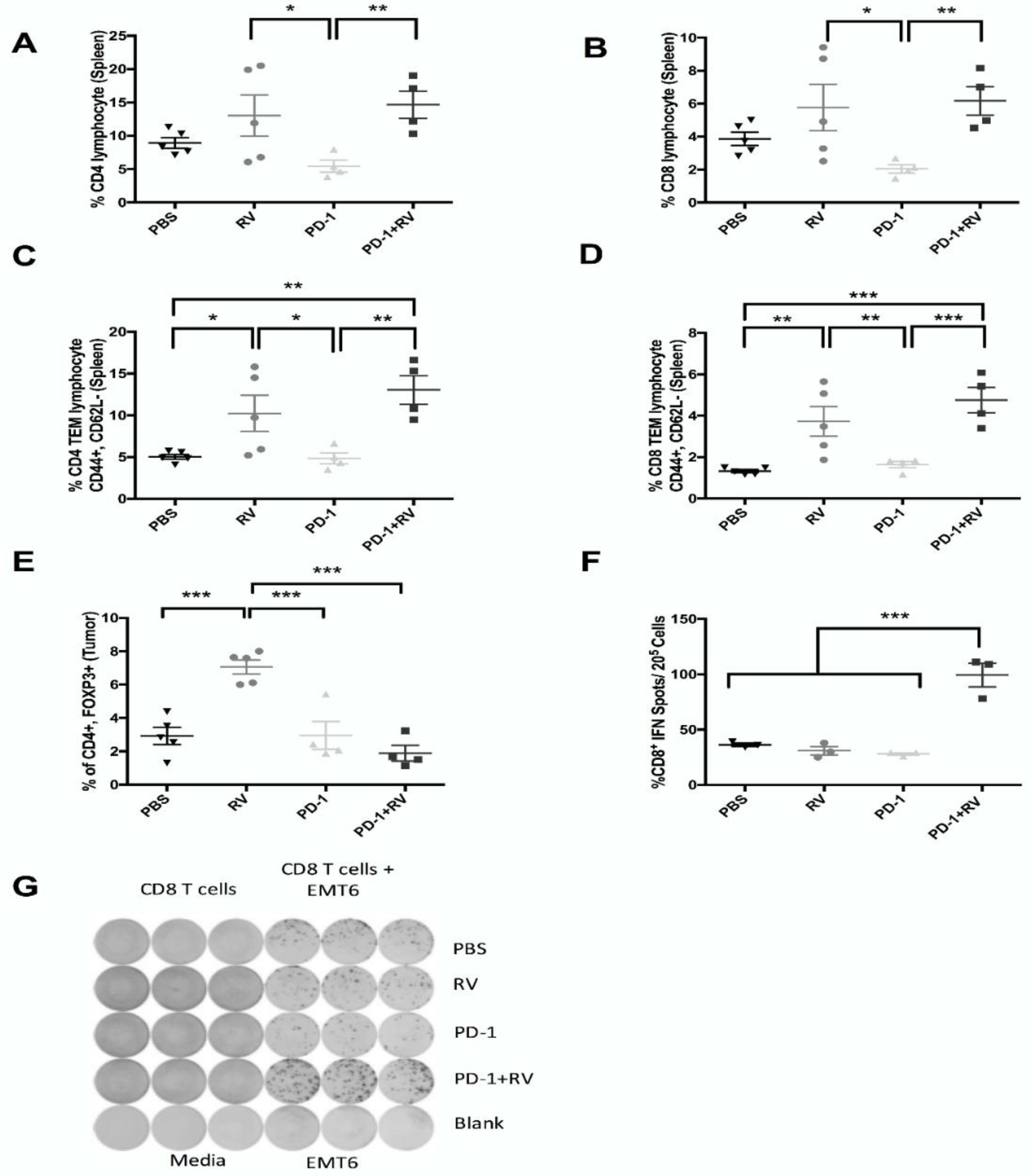
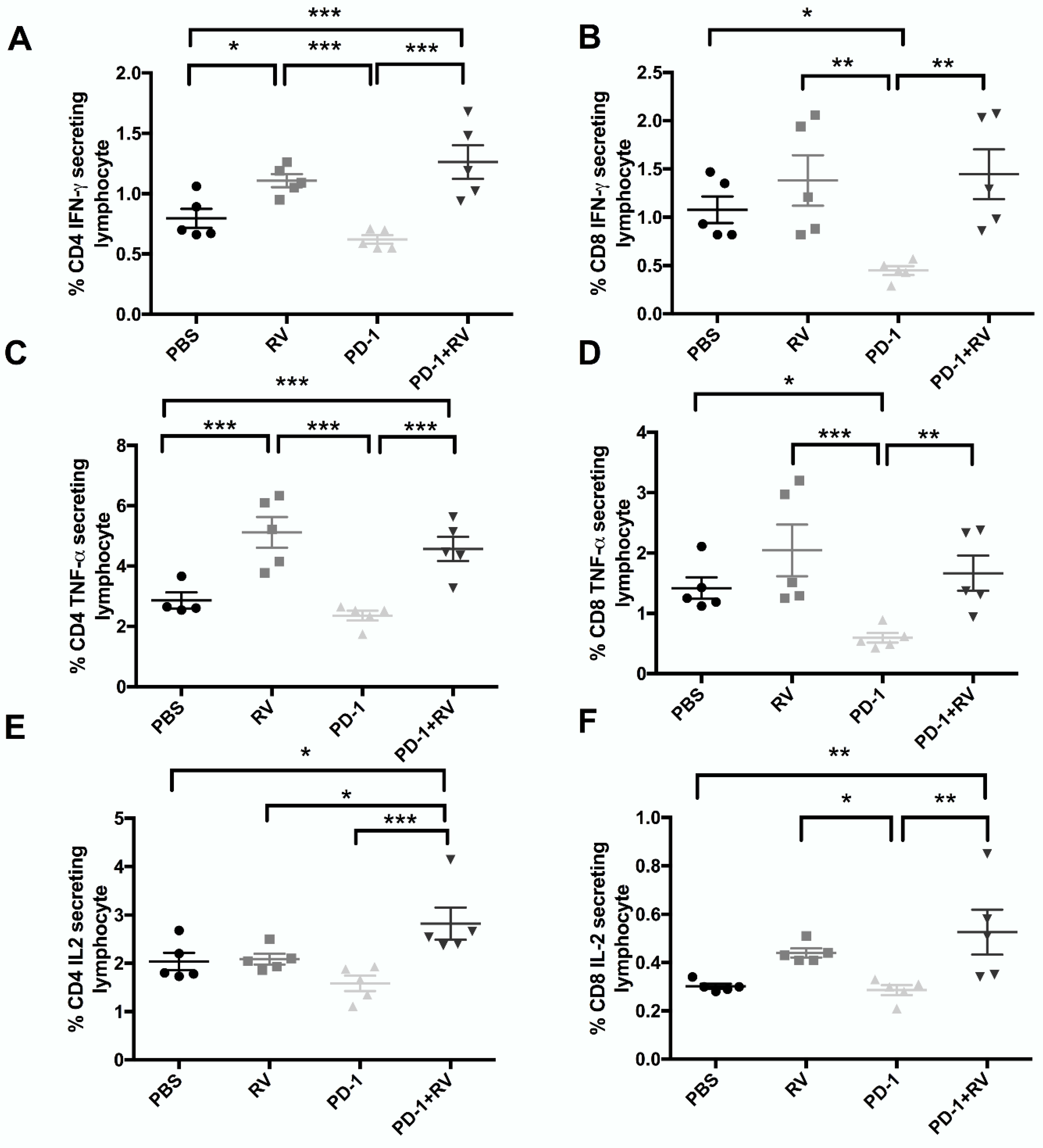
2.4. PD-1 Inhibition Augments Reovirus-Mediated Antitumor Immune Response through Recruitment of Memory T-Cell Populations and Enhanced Inflammatory Cytokine Production
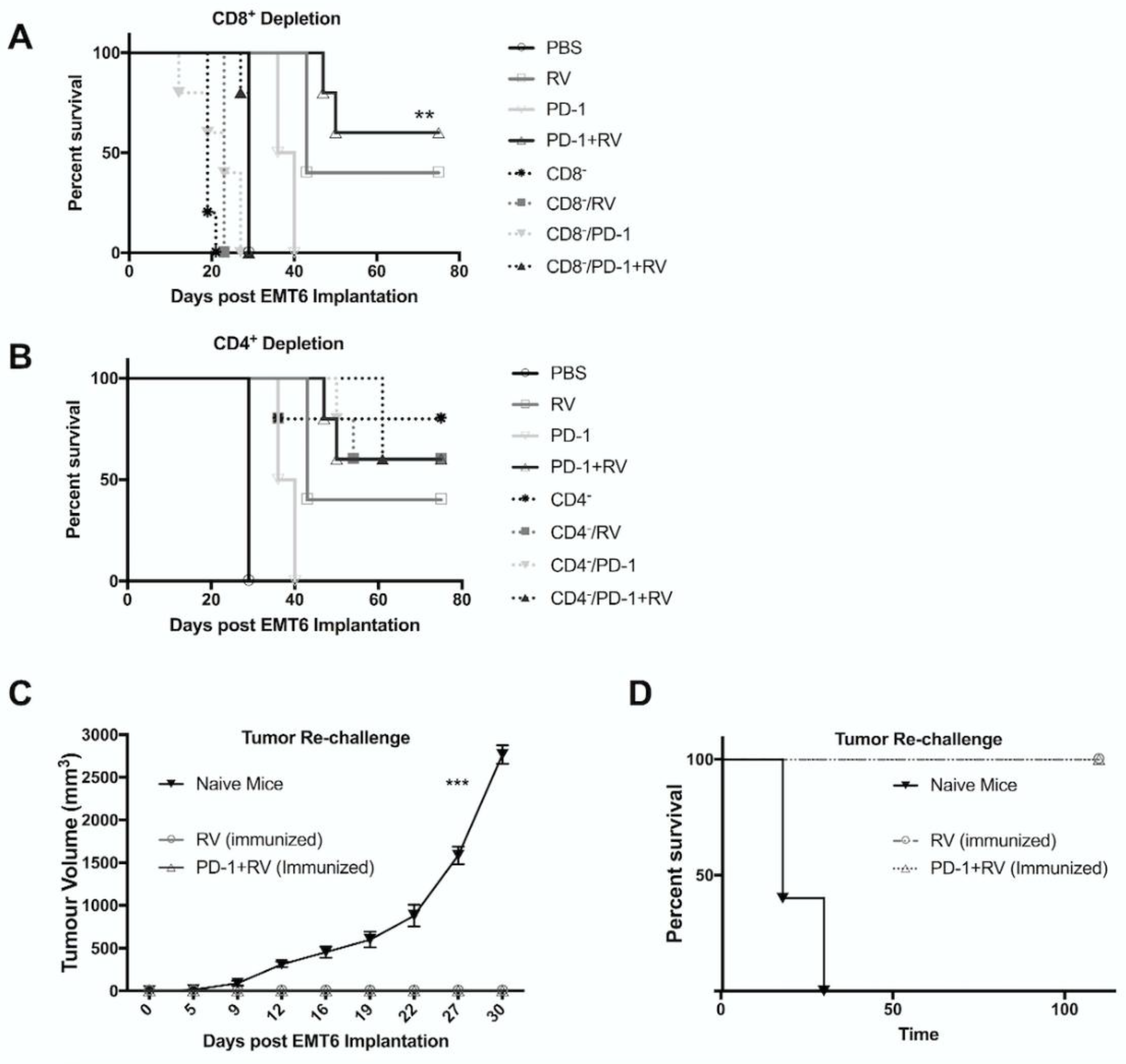
2.5. Treatment with Reovirus and PD-1 Induces Protective Immunity against Tumor Re-Challenge, and Treatment Benefits Depend on the Presence of CD8+ T Cells
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Reagents
4.2. Cell Viability Assay and Determination of RV ED50
4.3. Transwell Chemotaxis Assay
4.4. Cytokine Expression Assay
4.5. Assessment of PD-L1 Expression via Flow Cytometry
4.6. 4T1 and EMT6 Supernatant Cross-Incubation and PDL-1 Expression Analysis
4.7. Immunocompetent Syngeneic Murine Breast Cancer Model
4.8. CD8+ Enrichment
4.9. Immunostaining and Flow Cytometry of In Vivo Samples
4.10. ELISPOT Assay
4.11. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Toss, A.; Cristofanilli, M. Molecular characterization and targeted therapeutic approaches in breast cancer. Breast Cancer Res. 2015, 17, 60. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA-Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Dieci, M.V.; Criscitiello, C.; Goubar, A.; Viale, G.; Conte, P.; Guarneri, V.; Ficarra, G.; Mathieu, M.C.; Delaloge, S.; Curigliano, G.; et al. Prognostic value of tumor-infiltrating lymphocytes on residual disease after primary chemotherapy for triple-negative breast cancer: A retrospective multicenter study. Ann. Oncol. 2014, 25, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Loi, S.; Michiels, S.; Salgado, R.; Sirtaine, N.; Jose, V.; Fumagalli, D.; Kellokumpu-Lehtinen, P.-L.; Bono, P.; Kataja, V.; Desmedt, C.; et al. Tumor infiltrating lymphocytes are prognostic in triple negative breast cancer and predictive for trastuzumab benefit in early breast cancer: Results from the FinHER trial. Ann. Oncol. 2014, 25, 1544–1550. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, S.M.A.; Paish, E.C.; Powe, D.G.; Macmillan, R.D.; Grainge, M.J.; Lee, A.H.S.; Ellis, I.O.; Green, A.R. Tumor-infiltrating CD8+ lymphocytes predict clinical outcome in breast cancer. J. Clin. Oncol. 2011, 29, 1949–1955. [Google Scholar] [CrossRef] [PubMed]
- Comins, C.; Heinemann, L.; Harrington, K.; Melcher, A.; De Bono, J.; Pandha, H. Reovirus: Viral Therapy for Cancer “as Nature Intended”. Clin. Oncol. 2008, 20, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Marcato, P.; Shmulevitz, M.; Pan, D.; Stoltz, D.; Lee, P.W. Ras Transformation Mediates Reovirus Oncolysis by Enhancing Virus Uncoating, Particle Infectivity, and Apoptosis-dependent Release. Mol. Ther. 2007, 15, 1522–1530. [Google Scholar] [CrossRef] [PubMed]
- Coffey, M.C.; Strong, J.E.; Forsyth, P.A.; Lee, P.W. Reovirus therapy of tumors with activated Ras pathway. Science 1998, 282, 1332–1334. [Google Scholar] [CrossRef] [PubMed]
- Kelly, K.R.; Espitia, C.M.; Zhao, W.; Wendlandt, E.; Tricot, G.; Zhan, F.; Carew, J.S.; Nawrocki, S.T. Junctional adhesion molecule-A is overexpressed in advanced multiple myeloma and determines response to oncolytic reovirus. Oncotarget 2015, 6, 41275–41289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hata, Y.; Etoh, T.; Inomata, M.; Shiraishi, N.; Nishizono, A.; Kitano, S. Efficacy of oncolytic reovirus against human breast cancer cells. Oncol. Rep. 2008, 19, 1395–1398. [Google Scholar] [PubMed]
- Norman, K.L.; Coffey, M.C.; Hirasawa, K.; Demetrick, D.J.; Nishikawa, S.G.; DiFrancesco, L.M.; Strong, J.E.; Lee, P.W.K. Reovirus oncolysis of human breast cancer. Hum. Gene Ther. 2002, 13, 641–652. [Google Scholar] [CrossRef] [PubMed]
- Kelly, K.R.; Wu, K.; Tsao-Wei, D.; Groshen, S.; Triche, T.J.; Mohrbacher, A.; Chang, G.; Fernando, D.; Siddiqi, I.N.; Coffey, M.; et al. Oncolytic Reovirus Immune Priming: A Phase 1b Study of Reolysin with Bortezomib and Dexamethasone in Patients with Relapsed/Refractory Multiple Myeloma. Blood 2016, 128, 4507. [Google Scholar]
- Lawson, K.A.; Mostafa, A.A.; Shi, Z.Q.; Spurrell, J.; Chen, W.; Kawakami, J.; Gratton, K.; Thakur, S.; Morris, D.G. Repurposing Sunitinib with Oncolytic Reovirus as a Novel Immunotherapeutic Strategy for Renal Cell Carcinoma. Clin. Cancer Res. 2016, 22, 5839–5850. [Google Scholar] [CrossRef] [PubMed]
- Thirukkumaran, C.; Morris, D.G. Oncolytic Viral Therapy Using Reovirus. Methods Mol. Biol. 2015, 1317, 187–223. [Google Scholar] [PubMed]
- Thirukkumaran, C.M.; Shi, Z.Q.; Luider, J.; Kopciuk, K.; Gao, H.; Bahlis, N.; Neri, P.; Pho, M.; Stewart, D.; Mansoor, A.; et al. Reovirus as a viable therapeutic option for the treatment of multiple myeloma. Clin. Cancer Res. 2012, 18, 4962–4972. [Google Scholar] [CrossRef] [PubMed]
- Campion, C.A.; Soden, D.; Forde, P.F. Antitumour responses induced by a cell-based Reovirus vaccine in murine lung and melanoma models. BMC Cancer 2016, 16, 462. [Google Scholar] [CrossRef] [PubMed]
- Villalona-Calero, M.A.; Lam, E.; Otterson, G.A.; Zhao, W.; Timmons, M.; Subramaniam, D.; Hade, E.M.; Gill, G.M.; Coffey, M.; Selvaggi, G.; et al. Oncolytic reovirus in combination with chemotherapy in metastatic or recurrent non-small cell lung cancer patients with KRAS-activated tumors. Cancer 2016, 122, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, V.; Ellard, S.L.; Dent, S.F.; Tu, D.; Mates, M.; Dhesy-Thind, S.K.; Panasci, L.; Gelmon, K.A.; Salim, M.; Song, X.; et al. A randomized phase II study of weekly paclitaxel with or without pelareorep in patients with metastatic breast cancer: Final analysis of Canadian Cancer Trials Group IND.213. Breast Cancer Res. Treat. 2017, 33, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crinò, L.; Eberhardt, W.E.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. CheckMate 025 Investigators Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, J.E.; Hoffman-Censits, J.; Powles, T.; van der Heijden, M.S.; Balar, A.V.; Necchi, A.; Dawson, N.; O’Donnell, P.H.; Balmanoukian, A.; Loriot, Y.; et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet 2016, 387, 1909–1920. [Google Scholar] [CrossRef]
- Muenst, S.; Schaerli, A.R.; Gao, F.; Däster, S.; Trella, E.; Droeser, R.A.; Muraro, M.G.; Zajac, P.; Zanetti, R.; Gillanders, W.E.; et al. Expression of programmed death ligand 1 (PD-L1) is associated with poor prognosis in human breast cancer. Breast Cancer Res. Treat. 2014, 146, 15–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittendorf, E.A.; Philips, A.V.; Meric-Bernstam, F.; Qiao, N.; Wu, Y.; Harrington, S.; Su, X.; Wang, Y.; Gonzalez-Angulo, A.M.; Akcakanat, A.; et al. PD-L1 Expression in Triple-Negative Breast Cancer. Cancer Immunol. Res. 2014, 2, 361–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Sun, H.; Zhao, S.; Wang, Y.; Pu, H.; Zhang, Q. Expression of PD-L1 and prognosis in breast cancer: A meta-analysis. Oncotarget 2017, 8, 31347–31354. [Google Scholar] [CrossRef] [PubMed]
- Rajani, K.; Parrish, C.; Kottke, T.; Thompson, J.; Zaidi, S.; Ilett, L.; Shim, K.G.; Diaz, R.M.; Pandha, H.; Harrington, K.; et al. Combination Therapy With Reovirus and Anti-PD-1 Blockade Controls Tumor Growth Through Innate and Adaptive Immune Responses. Mol. Ther. 2016, 24, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Cockle, J.V.; Rajani, K.; Zaidi, S.; Kottke, T.; Thompson, J.; Diaz, R.M.; Shim, K.; Peterson, T.; Parney, I.F.; Short, S.; et al. Combination viroimmunotherapy with checkpoint inhibition to treat glioma, based on location-specific tumor profiling. Neuro Oncol. 2016, 18, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Zamarin, D.; Holmgaard, R.B.; Subudhi, S.K.; Park, J.S.; Mansour, M.; Palese, P.; Merghoub, T.; Wolchok, J.D.; Allison, J.P. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci. Transl. Med. 2014, 6, 226ra32. [Google Scholar] [CrossRef] [PubMed]
- Ilett, E.; Kottke, T.; Thompson, J.; Rajani, K.; Zaidi, S.; Evgin, L.; Coffey, M.; Ralph, C.; Diaz, R.; Pandha, H.; et al. Prime-boost using separate oncolytic viruses in combination with checkpoint blockade improves anti-tumour therapy. Gene Ther. 2017, 24, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Lawler, S.E.; Speranza, M.-C.; Cho, C.-F.; Chiocca, E.A. Oncolytic Viruses in Cancer Treatment: A Review. JAMA Oncol. 2017, 3, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Meyers, D.E.; Wang, A.A.; Thirukkumaran, C.M.; Morris, D.G. Current Immunotherapeutic Strategies to Enhance Oncolytic Virotherapy. Front. Oncol. 2017, 7, 114. [Google Scholar] [CrossRef] [PubMed]
- Thirukkumaran, C.M.; Nodwell, M.J.; Hirasawa, K.; Shi, Z.Q.; Diaz, R.; Luider, J.; Johnston, R.N.; Forsyth, P.A.; Magliocco, A.M.; Lee, P.; et al. Oncolytic viral therapy for prostate cancer: Efficacy of reovirus as a biological therapeutic. Cancer Res. 2010, 70, 2435–2444. [Google Scholar] [CrossRef] [PubMed]
- Gorczynski, R.M.; Chen, Z.; Erin, N.; Khatri, I.; Podnos, A. Comparison of immunity in mice cured of primary/metastatic growth of EMT6 or 4THM breast cancer by chemotherapy or immunotherapy. PLoS ONE 2014, 9, e113597. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Ouzounova, M.; Lee, E.; Piranlioglu, R.; El Andaloussi, A.; Kolhe, R.; Demirci, M.F.; Marasco, D.; Asm, I.; Chadli, A.; Hassan, K.A.; et al. Monocytic and granulocytic myeloid derived suppressor cells differentially regulate spatiotemporal tumour plasticity during metastatic cascade. Nat. Commun. 2017, 8, 14979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, P.; Lee, Y.; Liu, W.; Krausz, T.; Chong, A.; Schreiber, H.; Fu, Y.-X. Intratumor depletion of CD4+ cells unmasks tumor immunogenicity leading to the rejection of late-stage tumors. J. Exp. Med. 2005, 201, 779–791. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Nanda, R.; Chow, L.Q.M.; Dees, E.C.; Berger, R.; Gupta, S.; Geva, R.; Pusztai, L.; Pathiraja, K.; Aktan, G.; Cheng, J.D.; et al. Pembrolizumab in Patients With Advanced Triple-Negative Breast Cancer: Phase Ib KEYNOTE-012 Study. J. Clin. Oncol. 2016, 34, 2460–2467. [Google Scholar] [CrossRef] [PubMed]
- Rugo, H.; Delord, J.P.; Im, S.A.; Ott, P.A.; Piha-Paul, S.A. Preliminary Efficacy And Safety Of Pembrolizumab In Patients With Pd-L1 Positive, Estrogen Receptor-Positive/HER2-Negative Advanced Breast Cancer. In Proceedings of the 38th Annual San Antonio Breast Cancer Symposium, San Antonio, TX, USA, 8–12 December 2015. [Google Scholar]
- McSherry, E.A.; McGee, S.F.; Jirstrom, K.; Doyle, E.M.; Brennan, D.J.; Landberg, G.; Dervan, P.A.; Hopkins, A.M.; Gallagher, W.M. JAM-A expression positively correlates with poor prognosis in breast cancer patients. Int. J. Cancer 2009, 125, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Naik, M.U.; Naik, T.U.; Suckow, A.T.; Duncan, M.K.; Naik, U.P. Attenuation of junctional adhesion molecule-A is a contributing factor for breast cancer cell invasion. Cancer Res. 2008, 68, 2194–2203. [Google Scholar] [CrossRef] [PubMed]
- Clements, D.R.; Sterea, A.M.; Kim, Y.; Helson, E.; Dean, C.A.; Nunokawa, A.; Coyle, K.M.; Sharif, T.; Marcato, P.; Gujar, S.A.; et al. Newly recruited CD11b+, GR-1+, Ly6C(high) myeloid cells augment tumor-associated immunosuppression immediately following the therapeutic administration of oncolytic reovirus. J. Immunol. 2015, 194, 4397–4412. [Google Scholar] [CrossRef] [PubMed]
- Steele, L.; Errington, F.; Prestwich, R.; Ilett, E.; Harrington, K.; Pandha, H.; Coffey, M.; Selby, P.; Vile, R.; Melcher, A. Pro-inflammatory cytokine/chemokine production by reovirus treated melanoma cells is PKR/NF-κB mediated and supports innate and adaptive anti-tumour immune priming. Mol. Cancer 2011, 10, 20. [Google Scholar] [CrossRef] [PubMed]
- Abiko, K.; Matsumura, N.; Hamanishi, J.; Horikawa, N.; Murakami, R.; Yamaguchi, K.; Yoshioka, Y.; Baba, T.; Konishi, I.; Mandai, M. IFN-γ from lymphocytes induces PD-L1 expression and promotes progression of ovarian cancer. Br. J. Cancer 2015, 112, 1501–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fend, L.; Yamazaki, T.; Remy, C.; Fahrner, C.; Gantzer, M.; Nourtier, V.; Preville, X.; Quéméneur, E.; Kepp, O.; Adam, J.; et al. Immune checkpoint blockade, immunogenic chemotherapy or IFN-α blockade boost the local and abscopal effects of oncolytic virotherapy. Cancer Res. 2017, 77, 4146. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Wang, H.; Kottke, T.; White, C.; Twigger, K.; Diaz, R.M.; Thompson, J.; Selby, P.; de Bono, J.; Melcher, A.; et al. Cyclophosphamide facilitates antitumor efficacy against subcutaneous tumors following intravenous delivery of reovirus. Clin. Cancer Res. 2008, 14, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Stanton, S.E.; Disis, M.L. Clinical significance of tumor-infiltrating lymphocytes in breast cancer. J. Immunother. Cancer 2016, 4, 59. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lu, P.; Li, B.; Zhang, W.; Yang, R.; Chu, Y.; Luo, K. Interleukin 2 and interleukin 10 function synergistically to promote CD8(+) T cell cytotoxicity, which is suppressed by regulatory T cells in breast cancer. Int. J. Biochem. Cell Biol. 2017, 87, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Pulaski, B.A.; Ostrand-Rosenberg, S. Mouse 4T1 breast tumor model. Curr. Protoc. Immunol. 2001, 39, 20–22. [Google Scholar]
- Bourgeois-Daigneault, M.-C.; Roy, D.G.; Aitken, A.S.; El Sayes, N.; Martin, N.T.; Varette, O.; Falls, T.; St-Germain, L.E.; Pelin, A.; Lichty, B.D.; et al. Neoadjuvant oncolytic virotherapy before surgery sensitizes triple-negative breast cancer to immune checkpoint therapy. Sci. Transl. Med. 2018, 10, eaao1641. [Google Scholar] [CrossRef] [PubMed]
- DeBiasi, R.L.; Edelstein, C.L.; Sherry, B.; Tyler, K.L. Calpain inhibition protects against virus-induced apoptotic myocardial injury. J. Virol. 2001, 75, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Duraiswamy, J.; Freeman, G.J.; Coukos, G. Therapeutic PD-1 Pathway Blockade Augments with Other Modalities of Immunotherapy T-Cell Function to Prevent Immune Decline in Ovarian Cancer. Cancer Res. 2013, 73, 6900–6912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mostafa, A.A.; Codner, D.; Hirasawa, K.; Komatsu, Y.; Young, M.N.; Steimle, V.; Drover, S. Activation of ERα signaling differentially modulates IFN-γ induced HLA-class II expression in breast cancer cells. PLoS ONE 2014, 9, e87377. [Google Scholar] [CrossRef] [PubMed] [Green Version]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mostafa, A.A.; Meyers, D.E.; Thirukkumaran, C.M.; Liu, P.J.; Gratton, K.; Spurrell, J.; Shi, Q.; Thakur, S.; Morris, D.G. Oncolytic Reovirus and Immune Checkpoint Inhibition as a Novel Immunotherapeutic Strategy for Breast Cancer. Cancers 2018, 10, 205. https://doi.org/10.3390/cancers10060205
Mostafa AA, Meyers DE, Thirukkumaran CM, Liu PJ, Gratton K, Spurrell J, Shi Q, Thakur S, Morris DG. Oncolytic Reovirus and Immune Checkpoint Inhibition as a Novel Immunotherapeutic Strategy for Breast Cancer. Cancers. 2018; 10(6):205. https://doi.org/10.3390/cancers10060205
Chicago/Turabian StyleMostafa, Ahmed A., Daniel E. Meyers, Chandini M. Thirukkumaran, Peter J. Liu, Kathy Gratton, Jason Spurrell, Qiao Shi, Satbir Thakur, and Don G. Morris. 2018. "Oncolytic Reovirus and Immune Checkpoint Inhibition as a Novel Immunotherapeutic Strategy for Breast Cancer" Cancers 10, no. 6: 205. https://doi.org/10.3390/cancers10060205