Human Oncoviruses and p53 Tumor Suppressor Pathway Deregulation at the Origin of Human Cancers

 ,
,  , and
, and
Abstract
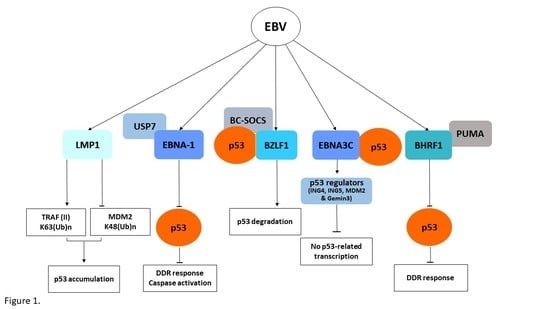
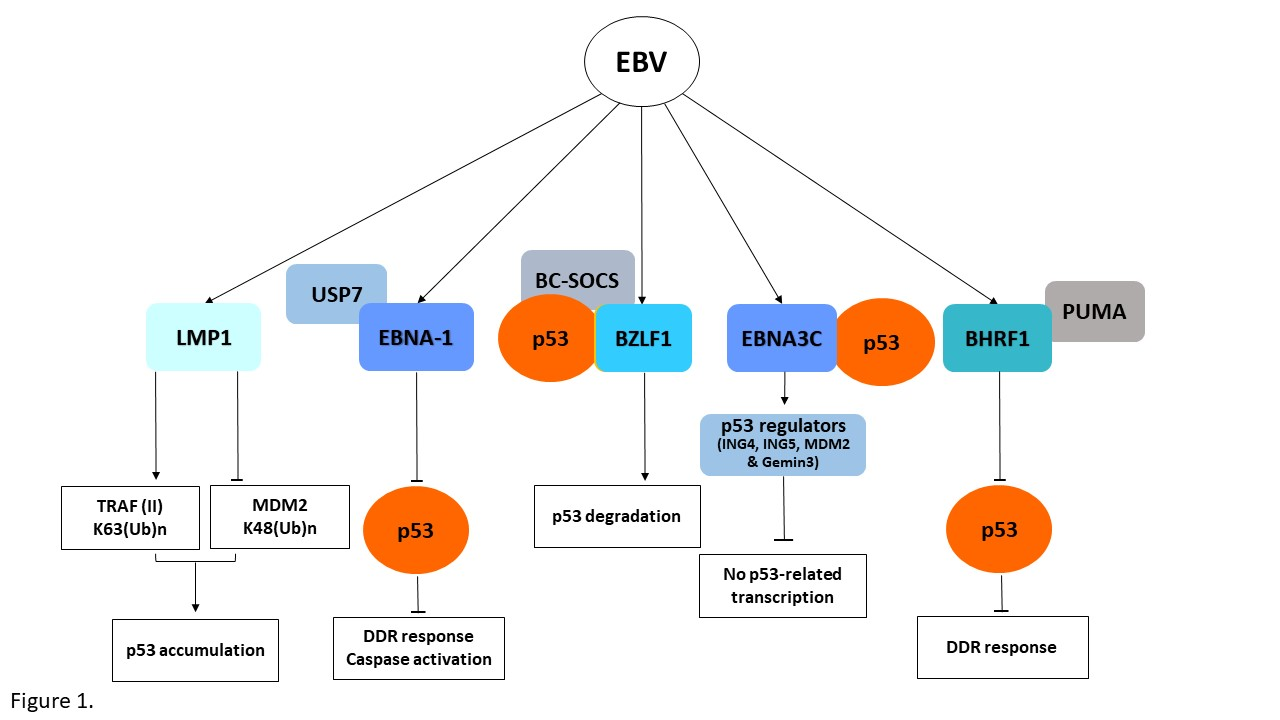
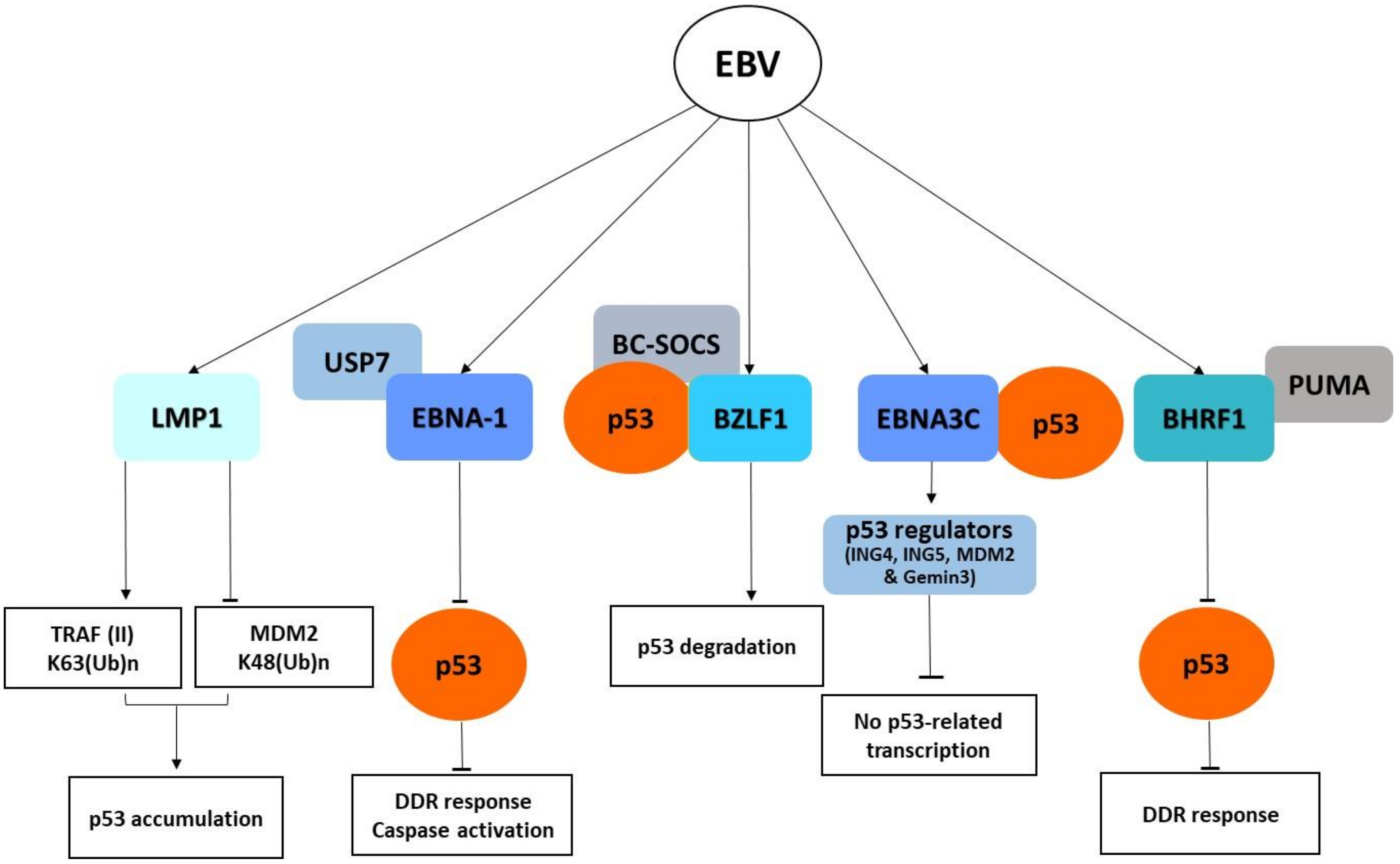
:
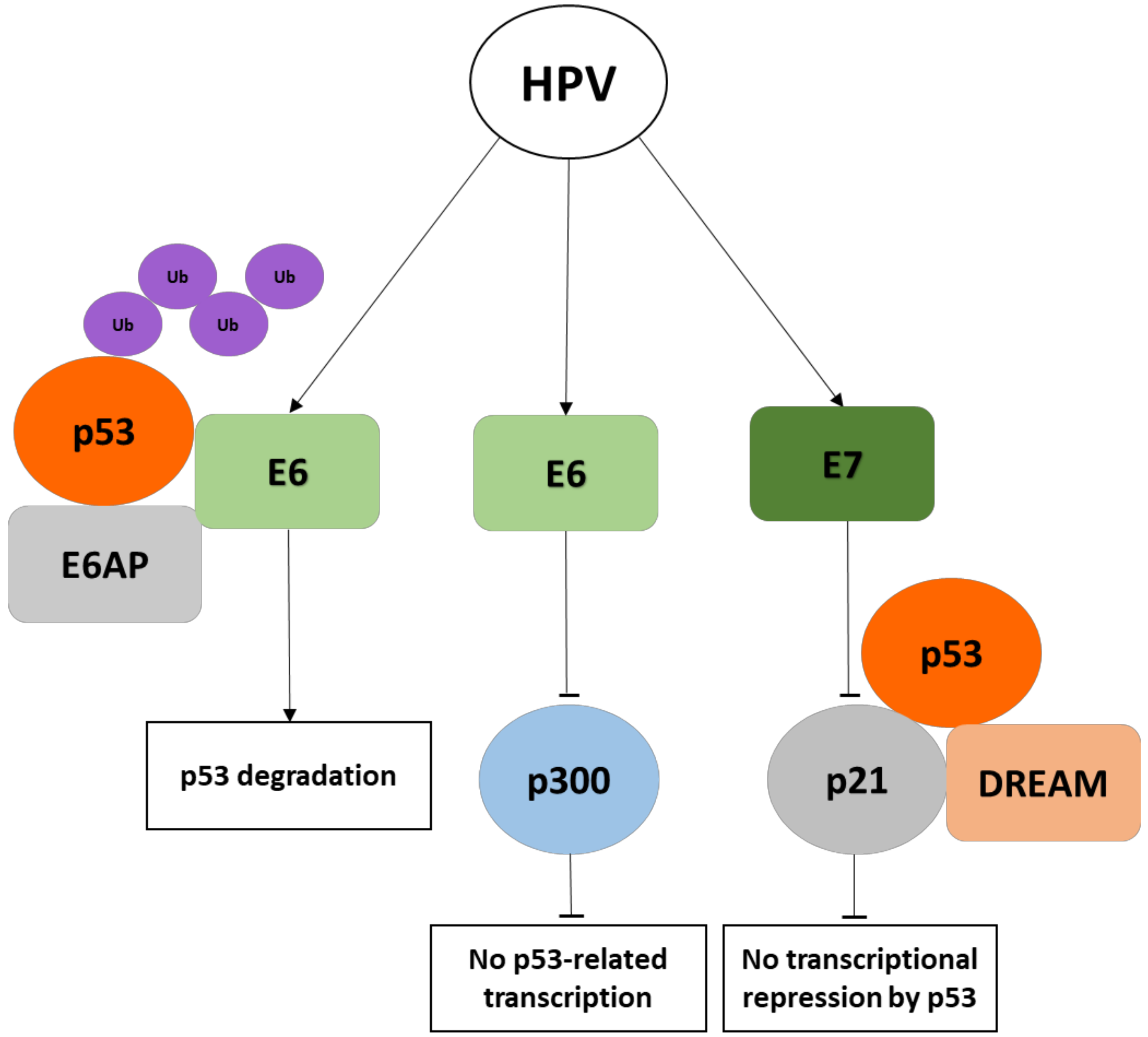
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. The Tumor Suppressor p53
3. The Epstein–Barr Virus (EBV)
4. The Hepatitis B Virus
5. The Human T Cell Lymphotropic Virus-1 (HTLV-1)
6. The Human Papillomavirus (HPV)
7. The Hepatitis C Virus
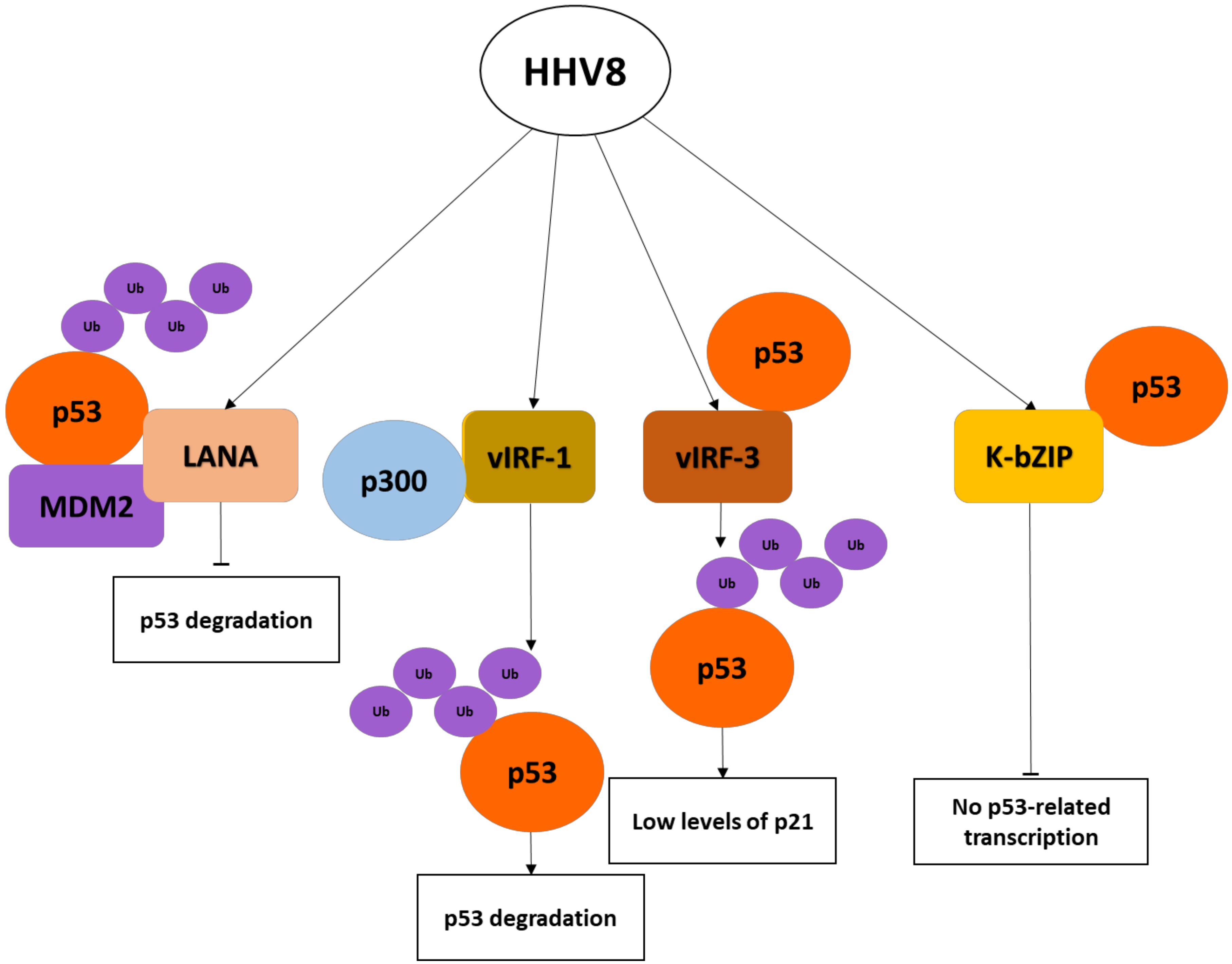
8. The Kaposi’s Sarcoma Herpesvirus (HHV8)
9. The Merkel Cell Polyomavirus (MCPyV)
10. Targeting the p53 Proteasome in Virus-Related Human Cancers
11. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Plummer, M.; de Martel, C.; Vignat, J.; Ferlay, J.; Bray, F.; Franceschi, S. Global burden of cancers attributable to infections in 2012: A synthetic analysis. Lancet Glob. Health 2016, 4, e609–e616. [Google Scholar] [CrossRef]
- Bouvard, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A review of human carcinogens—Part B: Biological agents. Lancet Oncol. 2009, 10, 321–322. [Google Scholar] [CrossRef]
- Mesri, E.A.; Feitelson, M.A.; Munger, K. Human viral oncogenesis: A cancer hallmarks analysis. Cell Host. Microbe 2014, 15, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Tornesello, M.L.; Buonaguro, L.; Buonaguro, F.M. An overview of new biomolecular pathways in pathogen-related cancers. Futur. Oncol. 2015, 11, 1625–1639. [Google Scholar] [CrossRef] [PubMed]
- Saenz-Robles, M.T.; Sullivan, C.S.; Pipas, J.M. Transforming functions of Simian Virus 40. Oncogene 2001, 20, 7899–7907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazo, P.A.; Santos, C.R. Interference with p53 functions in human viral infections, a target for novel antiviral strategies? Rev. Med. Virol. 2011, 21, 285–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Zapien, D.; Ruiz, F.X.; Poirson, J.; Mitschler, A.; Ramirez, J.; Forster, A.; Cousido-Siah, A.; Masson, M.; Pol, S.V.; Podjarny, A.; et al. Structure of the E6/E6AP/p53 complex required for HPV-mediated degradation of p53. Nature 2016, 529, 541–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aloni-Grinstein, R.; Charni-Natan, M.; Solomon, H.; Rotter, V. p53 and the Viral Connection: Back into the Future (double dagger). Cancers 2018, 10, 178. [Google Scholar] [CrossRef] [PubMed]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 2014, 14, 359–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meek, D.W.; Anderson, C.W. Posttranslational modification of p53: Cooperative integrators of function. Cold Spring Harb. Perspect. Biol. 2009, 1, a000950. [Google Scholar] [CrossRef] [PubMed]
- Teufel, D.P.; Bycroft, M.; Fersht, A.R. Regulation by phosphorylation of the relative affinities of the N-terminal transactivation domains of p53 for p300 domains and Mdm2. Oncogene 2009, 28, 2112–2118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.W.; Ferreon, J.C.; Ferreon, A.C.; Arai, M.; Wright, P.E. Graded enhancement of p53 binding to CREB-binding protein (CBP) by multisite phosphorylation. Proc. Natl. Acad. Sci. USA 2010, 107, 19290–19295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakaguchi, K.; Herrera, J.E.; Saito, S.; Miki, T.; Bustin, M.; Vassilev, A.; Anderson, C.W.; Appella, E. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev. 1998, 12, 2831–2841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, S.M.; Quelle, D.E. p53 Acetylation: Regulation and Consequences. Cancers 2014, 7, 30–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, C.L.; Gu, W. p53 ubiquitination: Mdm2 and beyond. Mol. Cell 2006, 21, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Iwakuma, T.; Lozano, G. MDM2, an introduction. Mol. Cancer Res. 2003, 1, 993–1000. [Google Scholar] [PubMed]
- Perry, M.E. The regulation of the p53-mediated stress response by MDM2 and MDM4. Cold Spring Harb. Perspect. Biol. 2010, 2, a000968. [Google Scholar] [CrossRef] [PubMed]
- Kussie, P.H.; Gorina, S.; Marechal, V.; Elenbaas, B.; Moreau, J.; Levine, A.J.; Pavletich, N.P. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 1996, 274, 948–953. [Google Scholar] [CrossRef] [PubMed]
- Soussi, T. TP53 mutations in human cancer: Database reassessment and prospects for the next decade. Adv. Cancer Res. 2011, 110, 107–139. [Google Scholar] [PubMed]
- Rivlin, N.; Brosh, R.; Oren, M.; Rotter, V. Mutations in the p53 Tumor Suppressor Gene: Important Milestones at the Various Steps of Tumorigenesis. Genes Cancer 2011, 2, 466–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Coillie, S.V.; Fang, J.Y.; Xu, J. Gain of function of mutant p53: R282W on the peak? Oncogenesis 2016, 5, e196. [Google Scholar] [CrossRef] [PubMed]
- Klein, G. Tumor Associations of EBV—Historical Perspectives. Curr. Top. Microbiol. Immunol. 2015, 390, 17–22. [Google Scholar] [PubMed]
- Fitzsimmons, L.; Kelly, G.L. EBV and Apoptosis: The Viral Master Regulator of Cell Fate? Viruses 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Chevallier-Greco, A.; Manet, E.; Chavrier, P.; Mosnier, C.; Daillie, J.; Sergeant, A. Both Epstein-Barr virus (EBV)-encoded trans-acting factors, EB1 and EB2, are required to activate transcription from an EBV early promoter. EMBO J. 1986, 5, 3243–3249. [Google Scholar] [PubMed]
- Sato, Y.; Kamura, T.; Shirata, N.; Murata, T.; Kudoh, A.; Iwahori, S.; Nakayama, S.; Isomura, H.; Nishiyama, Y.; Tsurumi, T.; et al. Degradation of phosphorylated p53 by viral protein-ECS E3 ligase complex. PLoS Pathog. 2009, 5, e1000530. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Shirata, N.; Kudoh, A.; Iwahori, S.; Nakayama, S.; Murata, T.; Isomura, H.; Nishiyama, Y.; Tsurumi, T. Expression of Epstein-Barr virus BZLF1 immediate-early protein induces p53 degradation independent of MDM2, leading to repression of p53-mediated transcription. Virology 2009, 388, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Guo, Y.; Xiao, B.; Banerjee, S.; Saha, A.; Lu, J.; Glisovic, T.; Robertson, E.S. Epstein-Barr virus nuclear antigen 3C stabilizes Gemin3 to block p53-mediated apoptosis. PLoS. Pathog. 2011, 7, e1002418. [Google Scholar] [CrossRef] [PubMed]
- Yi, F.; Saha, A.; Murakami, M.; Kumar, P.; Knight, J.S.; Cai, Q.; Choudhuri, T.; Robertson, E.S. Epstein-Barr virus nuclear antigen 3C targets p53 and modulates its transcriptional and apoptotic activities. Virology 2009, 388, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Saridakis, V.; Sheng, Y.; Sarkari, F.; Holowaty, M.N.; Shire, K.; Nguyen, T.; Zhang, R.G.; Liao, J.; Lee, W.; Edwards, A.M.; et al. Structure of the p53 binding domain of HAUSP/USP7 bound to Epstein-Barr nuclear antigen 1 implications for EBV-mediated immortalization. Mol. Cell 2005, 18, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Murakami, M.; Verma, S.C.; Cai, Q.; Haldar, S.; Kaul, R.; Wasik, M.A.; Middeldorp, J.; Robertson, E.S. Epstein-Barr Virus nuclear antigen 1 (EBNA1) confers resistance to apoptosis in EBV-positive B-lymphoma cells through up-regulation of survivin. Virology 2011, 410, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Kvansakul, M.; Wei, A.H.; Fletcher, J.I.; Willis, S.N.; Chen, L.; Roberts, A.W.; Huang, D.C.S.; Colman, P.M. Structural basis for apoptosis inhibition by Epstein-Barr virus BHRF1. PLoS. Pathog. 2010, 6, e1001236. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, W.; Xiao, L.; Xu, J.; Chen, X.; Tang, M.; Dong, Z.; Tao, Q.; Cao, Y. Viral oncoprotein LMP1 disrupts p53-induced cell cycle arrest and apoptosis through modulating K63-linked ubiquitination of p53. Cell Cycle 2012, 11, 2327–2336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClain, S.L.; Clippinger, A.J.; Lizzano, R.; Bouchard, M.J. Hepatitis B virus replication is associated with an HBx-dependent mitochondrion-regulated increase in cytosolic calcium levels. J. Virol. 2007, 81, 12061–12065. [Google Scholar] [CrossRef] [PubMed]
- Soria, C.; Estermann, F.E.; Espantman, K.C.; O'Shea, C.C. Heterochromatin silencing of p53 target genes by a small viral protein. Nature 2010, 466, 1076–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xian, L.; Zhao, J.; Wang, J.; Fang, Z.; Peng, B.; Wang, W.; Ji, X.; Yu, L. p53 Promotes proteasome-dependent degradation of oncogenic protein HBx by transcription of MDM2. Mol. Biol. Rep. 2010, 37, 2935–2940. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.; Groopman, J.D. Interaction of mutant hepatitis B X protein with p53 tumor suppressor protein affects both transcription and cell survival. Mol. Carcinog. 2011, 50, 972–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, N.; Liu, Q.; Yang, X.; Zhang, F.; Li, X.; Ma, Y.; Guan, F.; Zhao, X.; Li, Z.; Zhang, L.; et al. Hepatitis B virus-upregulated lnc-HUR1 promotes cell proliferation and tumorigenesis by blocking p53 activity. Hepatology 2018. [Google Scholar] [CrossRef] [PubMed]
- Tornesello, M.L.; Buonaguro, L.; Tatangelo, F.; Botti, G.; Izzo, F.; Buonaguro, F.M. Mutations in TP53, CTNNB1 and PIK3CA genes in hepatocellular carcinoma associated with hepatitis B and hepatitis C virus infections. Genomics 2013, 102, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Gouas, D.A.; Shi, H.; Hautefeuille, A.H.; Ortiz-Cuaran, S.L.; Legros, P.C.; Szymanska, K.J.; Galy, O.; Egevad, L.A.; Abedi-Ardekani, B.; Wiman, K.G.; et al. Effects of the TP53 p.R249S mutant on proliferation and clonogenic properties in human hepatocellular carcinoma cell lines: Interaction with hepatitis B virus X protein. Carcinogenesis 2010, 31, 1475–1482. [Google Scholar] [CrossRef] [PubMed]
- Younis, I.; Yamamoto, B.; Phipps, A.; Green, P.L. Human T-cell leukemia virus type 1 expressing nonoverlapping tax and rex genes replicates and immortalizes primary human T lymphocytes but fails to replicate and persist in vivo. J. Virol. 2005, 79, 14473–14481. [Google Scholar] [CrossRef] [PubMed]
- Kannian, P.; Green, P.L. Human T Lymphotropic Virus Type 1 (HTLV-1): Molecular Biology and Oncogenesis. Viruses 2010, 2, 2037–2077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derse, D.; Hill, S.A.; Lloyd, P.A.; Chung, H.; Morse, B.A. Examining human T-lymphotropic virus type 1 infection and replication by cell-free infection with recombinant virus vectors. J. Virol. 2001, 75, 8461–8468. [Google Scholar] [CrossRef] [PubMed]
- Zane, L.; Yasunaga, J.; Mitagami, Y.; Yedavalli, V.; Tang, S.-W.; Chen, C.-Y.; Ratner, L.; Lu, X.; Jeang, K.-T. Wip1 and p53 contribute to HTLV-1 Tax-induced tumorigenesis. Retrovirology 2012, 9, 114. [Google Scholar] [CrossRef] [PubMed]
- Ohsugi, T.; Ishida, T.; Shimasaki, T.; Okada, S.; Umezawa, K. p53 dysfunction precedes the activation of nuclear factor-kappaB during disease progression in mice expressing Tax, a human T-cell leukemia virus type 1 oncoprotein. Carcinogenesis 2013, 34, 2129–2136. [Google Scholar] [CrossRef] [PubMed]
- De Villiers, E.M.; Fauquet, C.; Broker, T.R.; Bernard, H.U.; zur Hausen, H. Classification of papillomaviruses. Virology 2004, 324, 17–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Martel, C.; Plummer, M.; Vignat, J.; Franceschi, S. Worldwide burden of cancer attributable to HPV by site, country and HPV type. Int. J. Cancer 2017, 141, 664–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moody, C. Mechanisms by which HPV Induces a Replication Competent Environment in Differentiating Keratinocytes. Viruses 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Tommasino, M. The human papillomavirus family and its role in carcinogenesis. Semin. Cancer Biol. 2014, 26, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Zanier, K.; Charbonnier, S.; Sidi, A.O.; McEwen, A.G.; Ferrario, M.G.; Poussin-Courmontagne, P.; Cura, V.; Brimer, N.; Babah, K.O.; Ansari, T.; et al. Structural basis for hijacking of cellular LxxLL motifs by papillomavirus E6 oncoproteins. Science 2013, 339, 694–698. [Google Scholar] [CrossRef] [PubMed]
- Ganti, K.; Broniarczyk, J.; Manoubi, W.; Massimi, P.; Mittal, S.; Pim, D.; Szalmas, A.; Thatte, J.; Thomas, M.; Tomaić, V.; et al. The Human Papillomavirus E6 PDZ Binding Motif: From Life Cycle to Malignancy. Viruses 2015, 7, 3530–3551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phelps, W.C.; Yee, C.L.; Munger, K.; Howley, P.M. The human papillomavirus type 16 E7 gene encodes transactivation and transformation functions similar to those of adenovirus E1A. Cell 1988, 53, 539–547. [Google Scholar] [CrossRef]
- Hoppe-Seyler, K.; Bossler, F.; Braun, J.A.; Herrmann, A.L.; Hoppe-Seyler, F. The HPV E6/E7 Oncogenes: Key Factors for Viral Carcinogenesis and Therapeutic Targets. Trends Microbiol. 2018, 26, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Katzenellenbogen, R.A.; Vliet-Gregg, P.; Xu, M.; Galloway, D.A. NFX1-123 increases hTERT expression and telomerase activity posttranscriptionally in human papillomavirus type 16 E6 keratinocytes. J. Virol. 2009, 83, 6446–6456. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Piao, L.; Bullock, B.N.; Smith, A.; Su, T.; Zhang, M.; Teknos, T.N.; Arora, P.S.; Pan, Q. Targeting HPV16 E6-p300 interaction reactivates p53 and inhibits the tumorigenicity of HPV-positive head and neck squamous cell carcinoma. Oncogene 2014, 33, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Chand, V.; John, R.; Jaiswal, N.; Johar, S.S.; Nag, A. High-risk HPV16E6 stimulates hADA3 degradation by enhancing its SUMOylation. Carcinogenesis 2014, 35, 1830–1839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, N.A.; Galloway, D.A. Novel Functions of the Human Papillomavirus E6 Oncoproteins. Annu. Rev. Virol. 2015, 2, 403–423. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, F.; Banks, L. The human papillomavirus E6 protein and its contribution to malignant progression. Oncogene 2001, 20, 7874–7887. [Google Scholar] [CrossRef] [PubMed]
- Tommasino, M. The biology of beta human papillomaviruses. Virus Res. 2017, 231, 128–138. [Google Scholar] [CrossRef] [PubMed]
- White, E.A.; Kramer, R.E.; Tan, M.J.; Hayes, S.D.; Harper, J.W.; Howley, P.M. Comprehensive analysis of host cellular interactions with human papillomavirus E6 proteins identifies new E6 binding partners and reflects viral diversity. J. Virol. 2012, 86, 13174–13186. [Google Scholar] [CrossRef] [PubMed]
- Cornet, I.; Bouvard, V.; Campo, M.S.; Thomas, M.; Banks, L.; Gissmann, L.; Lamartine, J.; Sylla, B.S.; Accardi, R.; Tommasino, M.; et al. Comparative analysis of transforming properties of E6 and E7 from different beta human papillomavirus types. J. Virol. 2012, 86, 2366–2370. [Google Scholar] [CrossRef] [PubMed]
- Howie, H.L.; Koop, J.I.; Weese, J.; Robinson, K.; Wipf, G.; Kim, L.; Galloway, D.A. Beta-HPV 5 and 8 E6 promote p300 degradation by blocking AKT/p300 association. PLoS. Pathog. 2011, 7, e1002211. [Google Scholar] [CrossRef] [PubMed]
- Muench, P.; Probst, S.; Schuetz, J.; Leiprecht, N.; Busch, M.; Wesselborg, S.; Stubenrauch, F.; Iftner, T. Cutaneous papillomavirus E6 proteins must interact with p300 and block p53-mediated apoptosis for cellular immortalization and tumorigenesis. Cancer Res. 2010, 70, 6913–6924. [Google Scholar] [CrossRef] [PubMed]
- Songock, W.K.; Kim, S.M.; Bodily, J.M. The human papillomavirus E7 oncoprotein as a regulator of transcription. Virus Res. 2017, 231, 56–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demers, G.W.; Foster, S.A.; Halbert, C.L.; Galloway, D.A. Growth arrest by induction of p53 in DNA damaged keratinocytes is bypassed by human papillomavirus 16 E7. Proc. Natl. Acad. Sci. USA 1994, 91, 4382–4386. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.R.; Toth, Z.; Shin, Y.C.; Lee, J.-S.; Chang, H.; Gu, W.; Oh, T.-K.; Kim, M.H.; Jung, J.U. Kaposi’s sarcoma-associated herpesvirus viral interferon regulatory factor 4 targets MDM2 to deregulate the p53 tumor suppressor pathway. J. Virol. 2009, 83, 6739–6747. [Google Scholar] [CrossRef] [PubMed]
- Nor Rashid, N.; Yusof, R.; Watson, R.J. Disruption of repressive p130-DREAM complexes by human papillomavirus 16 E6/E7 oncoproteins is required for cell-cycle progression in cervical cancer cells. J. Gen. Virol. 2011, 92, 2620–2627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadasivam, S.; DeCaprio, J.A. The DREAM complex: Master coordinator of cell cycle-dependent gene expression. Nat. Rev. Cancer 2013, 13, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Uxa, S.; Stanko, C.; Magin, T.M.; Engeland, K. Human papilloma virus E7 oncoprotein abrogates the p53-p21-DREAM pathway. Sci. Rep. 2017, 7, 2603. [Google Scholar] [CrossRef] [PubMed]
- Engeland, K. Cell cycle arrest through indirect transcriptional repression by p53: I. have a DREAM. Cell Death. Differ. 2018, 25, 114–132. [Google Scholar] [CrossRef] [PubMed]
- Chevaliez, S.; Pawlotsky, J.-M. HCV Genome and Life Cycle. In Hepatitis C Viruses: Genomes and Molecular Biology; Tan, S.L., Ed.; Horizon Bioscience: Norfolk, UK, 2006; Available online: https://books.google.com.hk/books?hl=zh-TW&lr=&id=zf4C0V_7Lu4C&oi=fnd&pg=PA5&dq=HCV+Genome+and+Life+Cycle&ots=7tpNy0hlDm&sig=ANJB-KC6QM_Uan4RQfVcer5HIFc&redir_esc=y#v=onepage&q=HCV%20Genome%20and%20Life%20Cycle&f=false (accessed on 22 June 2018).
- Otsuka, M.; Kato, N.; Lan, K.; Yoshida, H.; Kato, J.; Goto, T.; Shiratori, Y.; Omata, M. Hepatitis C virus core protein enhances p53 function through augmentation of DNA binding affinity and transcriptional ability. J. Biol. Chem. 2000, 275, 34122–34130. [Google Scholar] [CrossRef] [PubMed]
- Bittar, C.; Shrivastava, S.; Bhanja, C.J.; Rahal, P.; Ray, R.B. Hepatitis C virus NS2 protein inhibits DNA damage pathway by sequestering p53 to the cytoplasm. PLoS ONE 2013, 8, e62581. [Google Scholar] [CrossRef] [PubMed]
- Lan, K.H.; Sheu, M.L.; Hwang, S.J.; Yen, S.-H.; Chen, S.-Y.; Wu, J.-C.; Wang, Y.-J.; Kato, N.; Omata, M.; Chang, F.-Y.; et al. HCV NS5A interacts with p53 and inhibits p53-mediated apoptosis. Oncogene 2002, 21, 4801–4811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, D.; Zhao, L.; Zhang, L.; Jiang, Y.; Tian, Y.; Xiao, X.; Gong, G. p53 controls hepatitis C virus non-structural protein 5A-mediated downregulation of GADD45α expression via the NF-κB and PI3K-Akt pathways. J. Gen. Virol. 2013, 94, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Russo, J.J.; Bohenzky, R.A.; Chien, M.C.; Chen, J.; Yan, M.; Maddalena, D.; Parry, J.P.; Peruzzi, D.; Edelman, I.S.; Chang, Y.; et al. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. USA 1996, 93, 14862–14867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.; Cesarman, E.; Pessin, M.S.; Lee, F.; Culpepper, J.; Knowles, D.M.; Moore, P.S. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 1994, 266, 1865–1869. [Google Scholar] [CrossRef] [PubMed]
- Cesarman, E.; Chang, Y.; Moore, P.S.; Said, J.W.; Knowles, D.M. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 1995, 332, 1186–1191. [Google Scholar] [CrossRef] [PubMed]
- Neipel, F.; Fleckenstein, B. The role of HHV-8 in Kaposi's sarcoma. Semin. Cancer Biol. 1999, 9, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Isobe, T.; Kitagawa, M.; Ueda, K. Kaposi's sarcoma-associated herpesvirus-encoded LANA positively affects on ubiquitylation of p53. Biochem. Biophys. Res. Commun. 2010, 403, 194–197. [Google Scholar] [CrossRef] [PubMed]
- Radkov, S.A.; Kellam, P.; Boshoff, C. The latent nuclear antigen of Kaposi sarcoma-associated herpesvirus targets the retinoblastoma-E2F pathway and with the oncogene Hras transforms primary rat cells. Nat. Med. 2000, 6, 1121–1127. [Google Scholar] [CrossRef] [PubMed]
- Si, H.; Robertson, E.S. Kaposi's sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen induces chromosomal instability through inhibition of p53 function. J. Virol. 2006, 80, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Li, M.; Zarycki, J.; Jung, J.U. Inhibition of p53 tumor suppressor by viral interferon regulatory factor. J. Virol. 2001, 75, 7572–7582. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.B.; Nicholas, J. Bim nuclear translocation and inactivation by viral interferon regulatory factor. PLoS Pathog. 2010, 6, e1001031. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.C.; Nakamura, H.; Liang, X.; Feng, P.; Chang, H.; Kowalik, T.F.; Jung, J.U. Inhibition of the ATM/p53 signal transduction pathway by Kaposi’s sarcoma-associated herpesvirus interferon regulatory factor 1. J. Virol. 2006, 80, 2257–2266. [Google Scholar] [CrossRef] [PubMed]
- Baresova, P.; Musilova, J.; Pitha, P.M.; Lubyova, B. p53 tumor suppressor protein stability and transcriptional activity are targeted by Kaposi's sarcoma-associated herpesvirus-encoded viral interferon regulatory factor 3. Mol. Cell Biol. 2014, 34, 386–399. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Seo, T.; Hwang, S.; Lee, D.; Gwack, Y.; Choe, J. The K-bZIP protein from Kaposi's sarcoma-associated herpesvirus interacts with p53 and represses its transcriptional activity. J. Virol. 2000, 74, 11977–11982. [Google Scholar] [CrossRef] [PubMed]
- Shuda, M.; Feng, H.; Kwun, H.J.; Rosen, S.T.; Gjoerup, O.; Moore, P.S.; Chang, Y. T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proc. Natl. Acad. Sci. USA 2008, 105, 16272–16277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef] [PubMed]
- Kwun, H.J.; Guastafierro, A.; Shuda, M.; Meinke, G.; Bohm, A.; Moore, P.S.; Chang, Y. The minimum replication origin of merkel cell polyomavirus has a unique large T-antigen loading architecture and requires small T-antigen expression for optimal replication. J. Virol. 2009, 83, 12118–12128. [Google Scholar] [CrossRef] [PubMed]
- Houben, R.; Shuda, M.; Weinkam, R.; Schrama, D.; Feng, H.; Chang, Y.; Moore, P.S.; Becker, J.C. Merkel cell polyomavirus-infected Merkel cell carcinoma cells require expression of viral T. antigens. J. Virol. 2010, 84, 7064–7072. [Google Scholar] [CrossRef] [PubMed]
- Borchert, S.; Czech-Sioli, M.; Neumann, F.; Schmidt, C.; Wimmer, P.; Dobner, T.; Grundhoff, A.; Fischer, N. High-affinity Rb binding, p53 inhibition, subcellular localization, and transformation by wild-type or tumor-derived shortened Merkel cell polyomavirus large T. antigens. J. Virol. 2014, 88, 3144–3160. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T. MDM2 inhibitors for cancer therapy. Trends Mol. Med. 2007, 13, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Kallen, J.; Goepfert, A.; Blechschmidt, A.; Izaac, A.; Geiser, M.; Tavares, G.; Ramage, P.; Furet, P.; Masuya, K.; Lisztwan, J.; et al. Crystal Structures of Human MdmX (HdmX) in Complex with p53 Peptide Analogues Reveal Surprising Conformational Changes. J. Biol. Chem. 2009, 284, 8812–8821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yee-Lin, V.; Pooi-Fong, W.; Soo-Beng, A.K. Nutlin-3, A p53-Mdm2 Antagonist for Nasopharyngeal Carcinoma Treatment. Mini Rev. Med. Chem. 2018, 18, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [PubMed]
- Apontes, P.; Leontieva, O.V.; Demidenko, Z.N.; Li, F.; Blagosklonny, M.V. Exploring long-term protection of normal human fibroblasts and epithelial cells from chemotherapy in cell culture. Oncotarget 2011, 2, 222–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voon, Y.L.; Ahmad, M.; Wong, P.F.; Husaini, R.; Ng, W.T.-W.; Leong, C.-O.; Lane, D.P.; Khoo, A.S.-B. Nutlin-3 sensitizes nasopharyngeal carcinoma cells to cisplatin-induced cytotoxicity. Oncol. Rep. 2015, 34, 1692–1700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renouf, B.; Hollville, E.; Pujals, A.; Tetaud, C.; Garibal, J.; Wiels, J. Activation of p53 by MDM2 antagonists has differential apoptotic effects on Epstein-Barr virus (EBV)-positive and EBV-negative Burkitt's lymphoma cells. Leukemia 2009, 23, 1557–1563. [Google Scholar] [CrossRef] [PubMed]
- Forte, E.; Luftig, M.A. MDM2-dependent inhibition of p53 is required for Epstein-Barr virus B-cell growth transformation and infected-cell survival. J. Virol. 2009, 83, 2491–2499. [Google Scholar] [CrossRef] [PubMed]
- Sarek, G.; Kurki, S.; Enback, J.; Iotzova, G.; Haas, J.; Laakkonen, P.; Laiho, M.; Ojala, P.M. Reactivation of the p53 pathway as a treatment modality for KSHV-induced lymphomas. J. Clin. Investig. 2007, 117, 1019–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarek, G.; Ojala, P.M. p53 reactivation kills KSHV lymphomas efficiently in vitro and in vivo: New hope for treating aggressive viral lymphomas. Cell Cycle 2007, 6, 2205–2209. [Google Scholar] [CrossRef] [PubMed]
- Petre, C.E.; Sin, S.H.; Dittmer, D.P. Functional p53 signaling in Kaposi's sarcoma-associated herpesvirus lymphomas: Implications for therapy. J. Virol. 2007, 81, 1912–1922. [Google Scholar] [CrossRef] [PubMed]
- Burgess, A.; Chia, K.M.; Haupt, S.; Thomas, D.; Haupt, Y.; Lim, E. Clinical Overview of MDM2/X.-Targeted Therapies. Front Oncol. 2016, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Carry, J.C.; Garcia-Echeverria, C. Inhibitors of the p53/hdm2 protein-protein interaction-path to the clinic. Bioorg. Med. Chem. Lett. 2013, 23, 2480–2485. [Google Scholar] [CrossRef] [PubMed]
- Lemos, A.; Leao, M.; Soares, J.; Palmeira, A.; Pinto, M.; Saraiva, L.; Sousa, M.E. Medicinal Chemistry Strategies to Disrupt the p53-MDM2/MDMX Interaction. Med. Res. Rev. 2016, 36, 789–844. [Google Scholar] [CrossRef] [PubMed]
- Bernal, F.; Wade, M.; Godes, M.; Davis, T.N.; Whitehead, D.G.; Kung, A.L.; Wahl, G.M.; Walensky, L.D. A stapled p53 helix overcomes HDMX-mediated suppression of p53. Cancer Cell 2010, 18, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, L.A.; Neriah, D.B.; Senecal, A.; Benard, L.; Thiruthuvanathan, V.; Yatsenko, T.; Narayanagari, S.-R.; Wheat, J.C.; Todorova, T.I.; Mitchell, K.; et al. Dual inhibition of MDMX and MDM2 as a therapeutic strategy in leukemia. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.Y.; Yoshida, N.; Christie, A.L.; Ghandi, M.; Dharia, N.V.; Dempster, J.; Murakami, M.; Shigemori, K.; Morrow, S.N.; Scoyk, A.; et al. Targetable vulnerabilities in T- and NK-cell lymphomas identified through preclinical models. Nat. Commun. 2018, 9, 2024. [Google Scholar] [CrossRef] [PubMed]
- Schiffman, M.; Doorbar, J.; Wentzensen, N.; De Sanjosé, S.; Fakhry, C.; Monk, B.J.; Stanley, M.A.; Franceschi, S. Carcinogenic human papillomavirus infection. Nat. Rev. Dis. Primers 2016, 2, 16086. [Google Scholar] [CrossRef] [PubMed]
- Vande Pol, S.B.; Klingelhutz, A.J. Papillomavirus E6 oncoproteins. Virology 2013, 445, 115–137. [Google Scholar] [CrossRef] [PubMed]
- Zanier, K.; Stutz, C.; Kintscher, S.; Reinz, E.; Sehr, P.; Bulkescher, J.; Hoppe-Seyler, K.; Trave, G.; Hoppe-Seyler, F. The E6AP binding pocket of the HPV16 E6 oncoprotein provides a docking site for a small inhibitory peptide unrelated to E6AP, indicating druggability of E6. PLoS ONE 2014, 9, e112514. [Google Scholar] [CrossRef] [PubMed]
- Stutz, C.; Reinz, E.; Honegger, A.; Bulkescher, J.; Schweizer, J.; Zanier, K.; Travé, G.; Lohrey, C.; Hoppe-Seyler, K.; Hoppe-Seyler, F.; et al. Intracellular Analysis of the Interaction between the Human Papillomavirus Type 16 E6 Oncoprotein and Inhibitory Peptides. PLoS ONE 2015, 10, e0132339. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tornesello, M.L.; Annunziata, C.; Tornesello, A.L.; Buonaguro, L.; Buonaguro, F.M. Human Oncoviruses and p53 Tumor Suppressor Pathway Deregulation at the Origin of Human Cancers. Cancers 2018, 10, 213. https://doi.org/10.3390/cancers10070213
Tornesello ML, Annunziata C, Tornesello AL, Buonaguro L, Buonaguro FM. Human Oncoviruses and p53 Tumor Suppressor Pathway Deregulation at the Origin of Human Cancers. Cancers. 2018; 10(7):213. https://doi.org/10.3390/cancers10070213
Chicago/Turabian StyleTornesello, Maria Lina, Clorinda Annunziata, Anna Lucia Tornesello, Luigi Buonaguro, and Franco Maria Buonaguro. 2018. "Human Oncoviruses and p53 Tumor Suppressor Pathway Deregulation at the Origin of Human Cancers" Cancers 10, no. 7: 213. https://doi.org/10.3390/cancers10070213