Independent Mechanisms Lead to Genomic Instability in Hodgkin Lymphoma: Microsatellite or Chromosomal Instability †

 , ,
, ,  ,
,  , ,
, , 
Abstract
:1. Introduction
2. Results
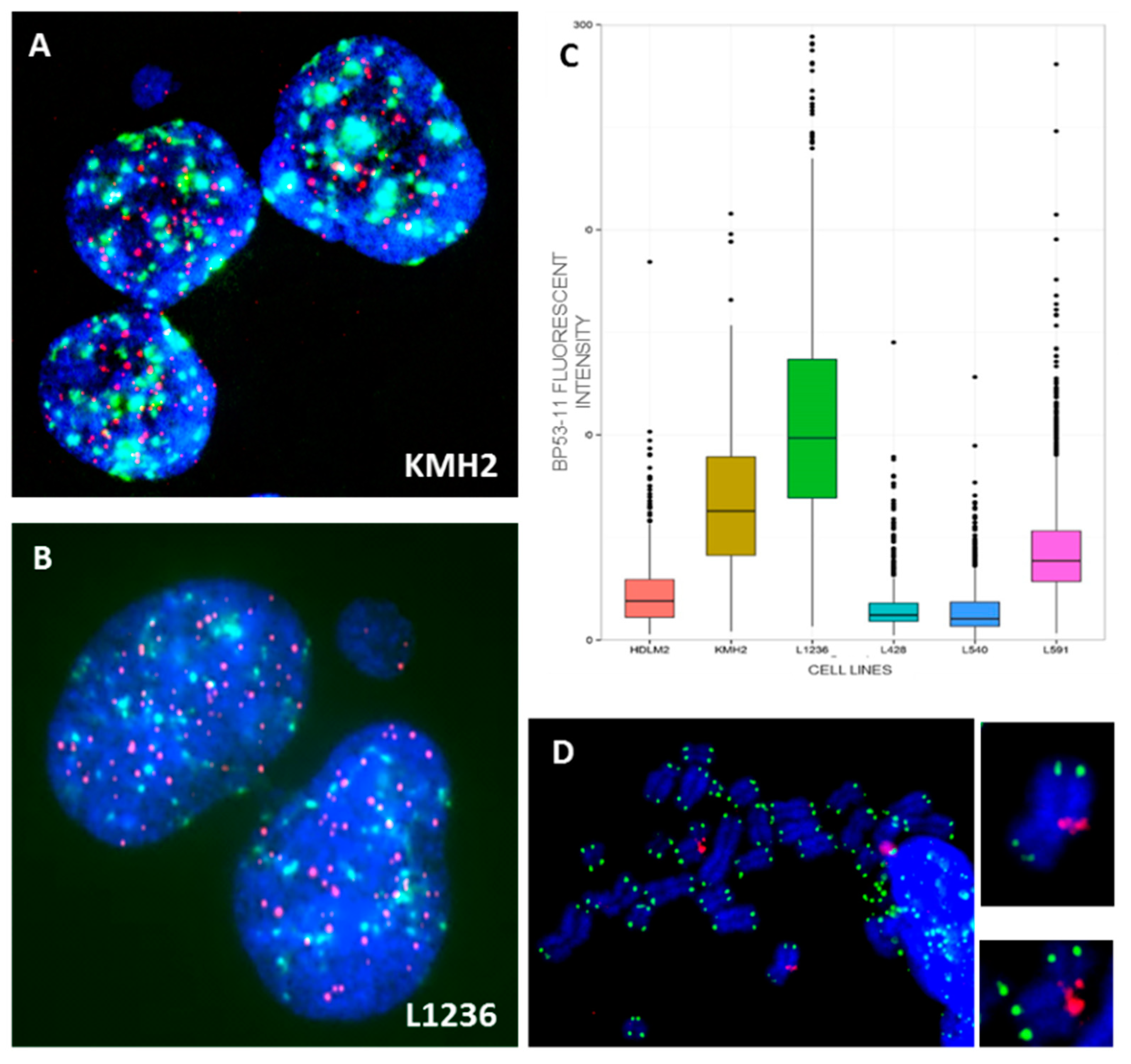
2.1. Genomic Instability in HL Cell Lines via Microsatellite Instability and P53 Status
2.2. Genomic Instability via Chromosomal Instability in HL Cell Lines
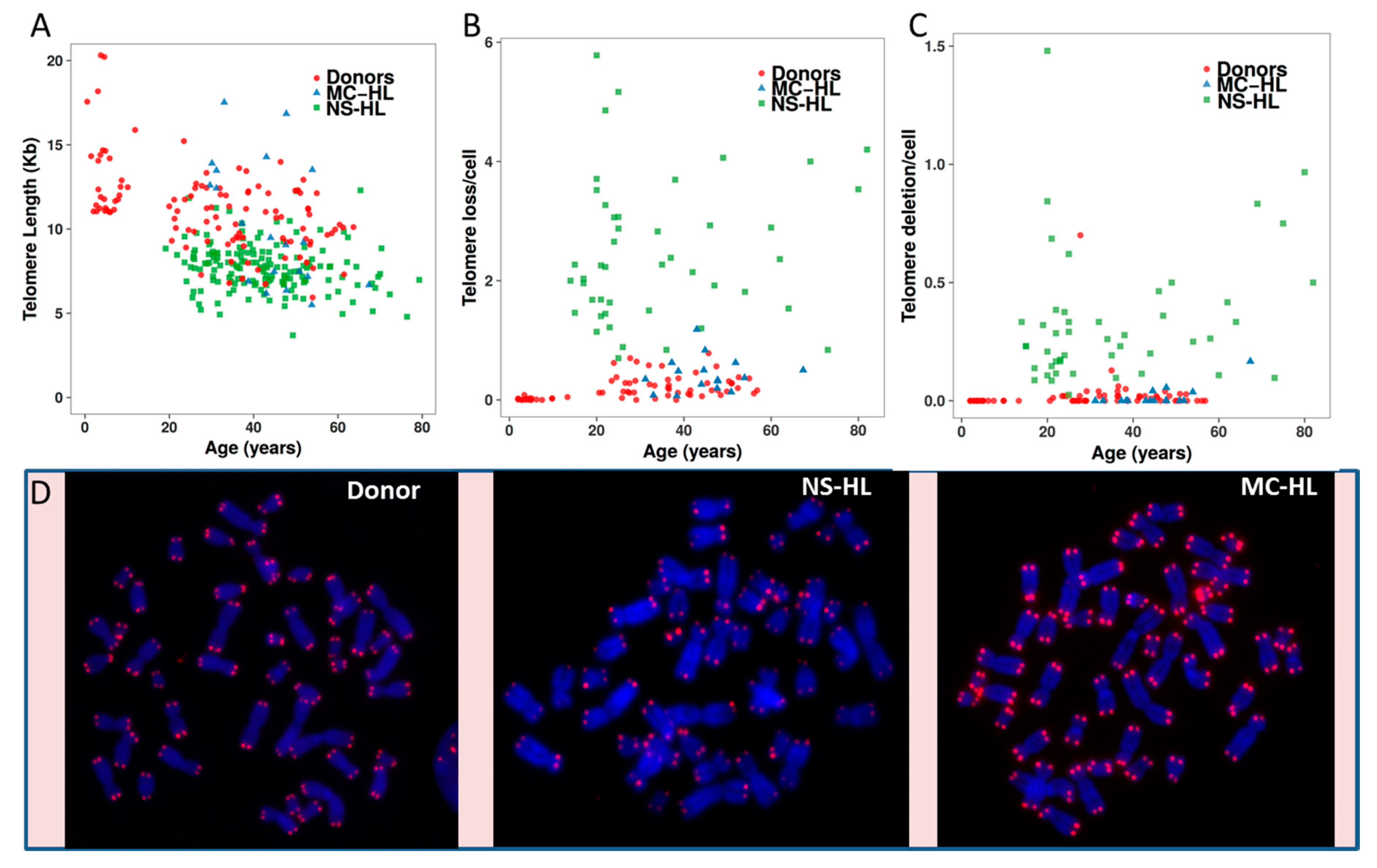
2.2.1. Telomere Dysfunction in HL Cell Lines
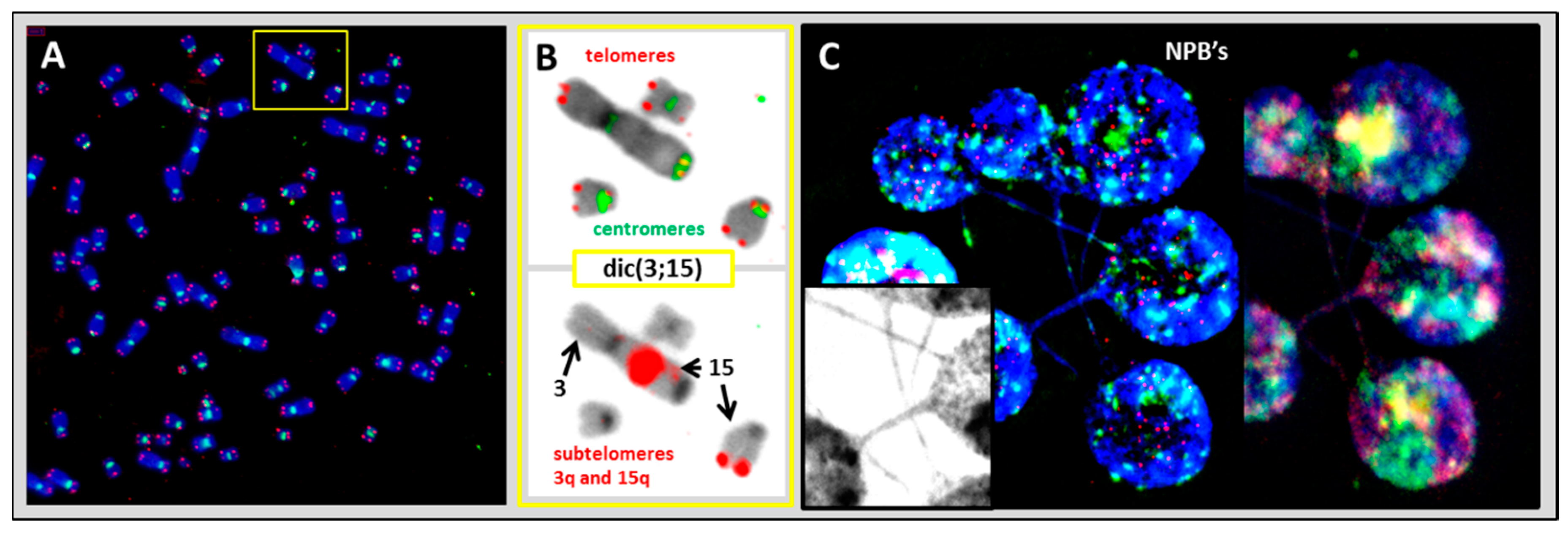
2.2.2. Aneuploidy, Dicentric Chromosomes, and Micronucleus Formation Lead to Chromosomal Instability in HL Cell Lines
2.2.3. Structural Chromosome Aberrations
3. Validation of Telomere Dysfunction Findings in Peripheral Blood Lymphocytes of HL Patients
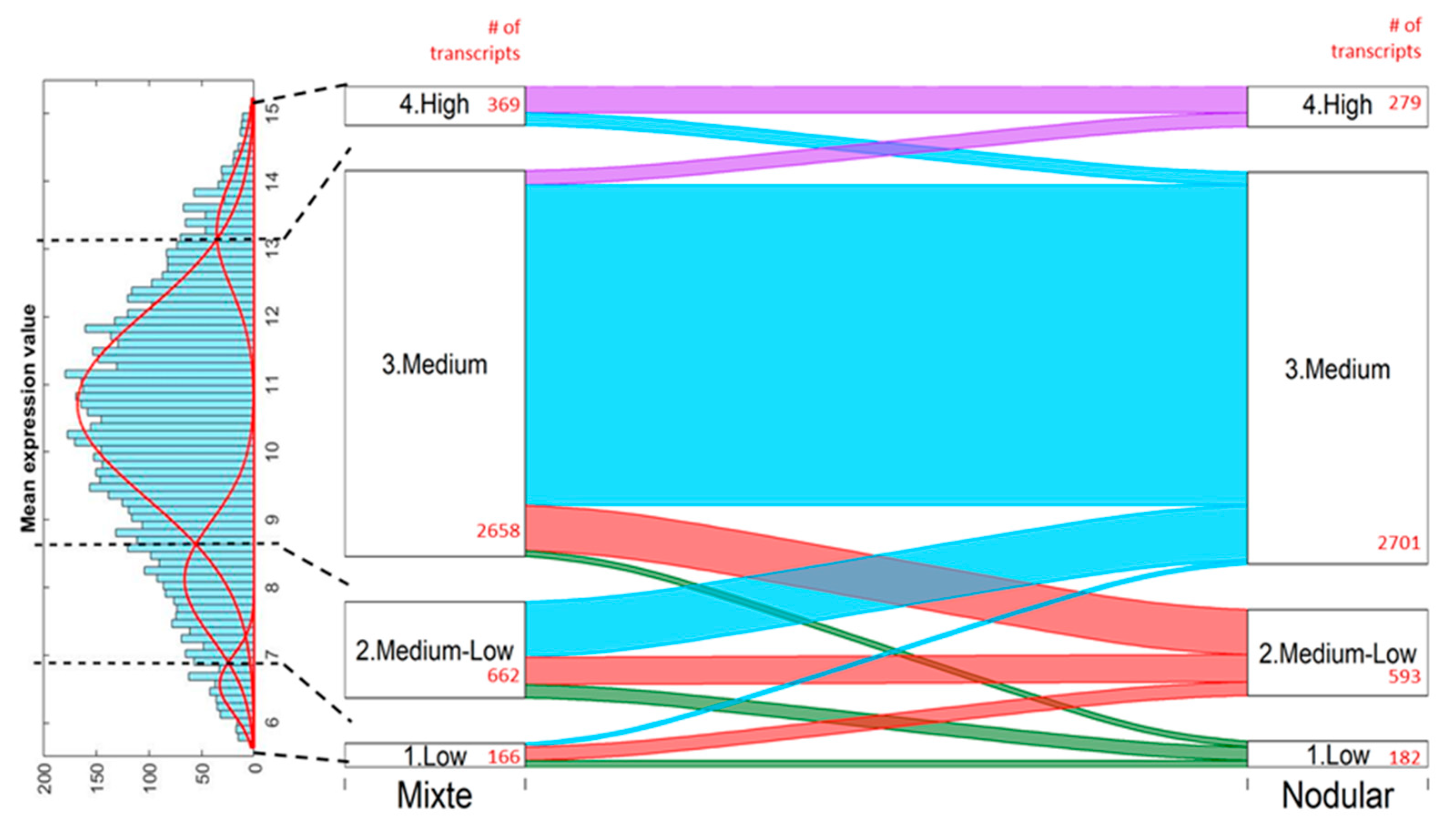
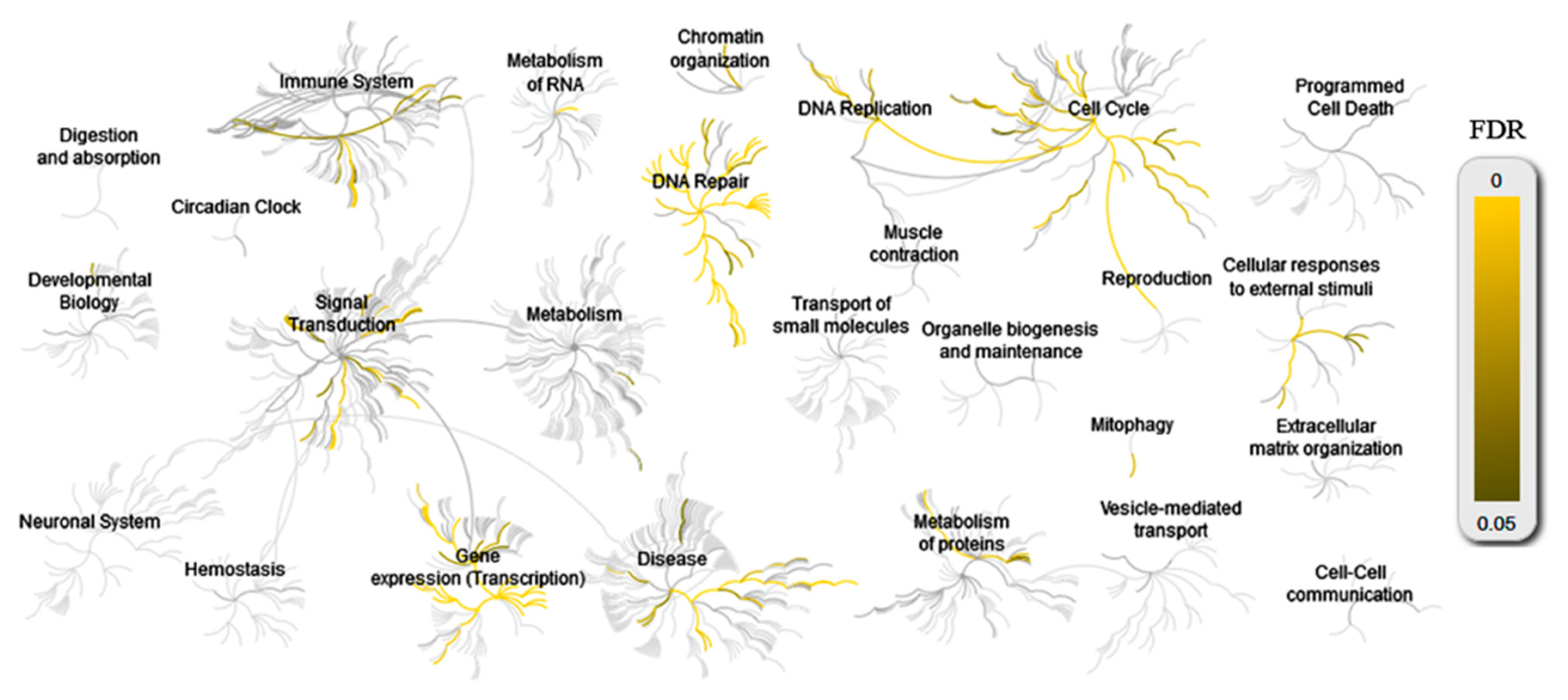
4. Transcriptome Analysis
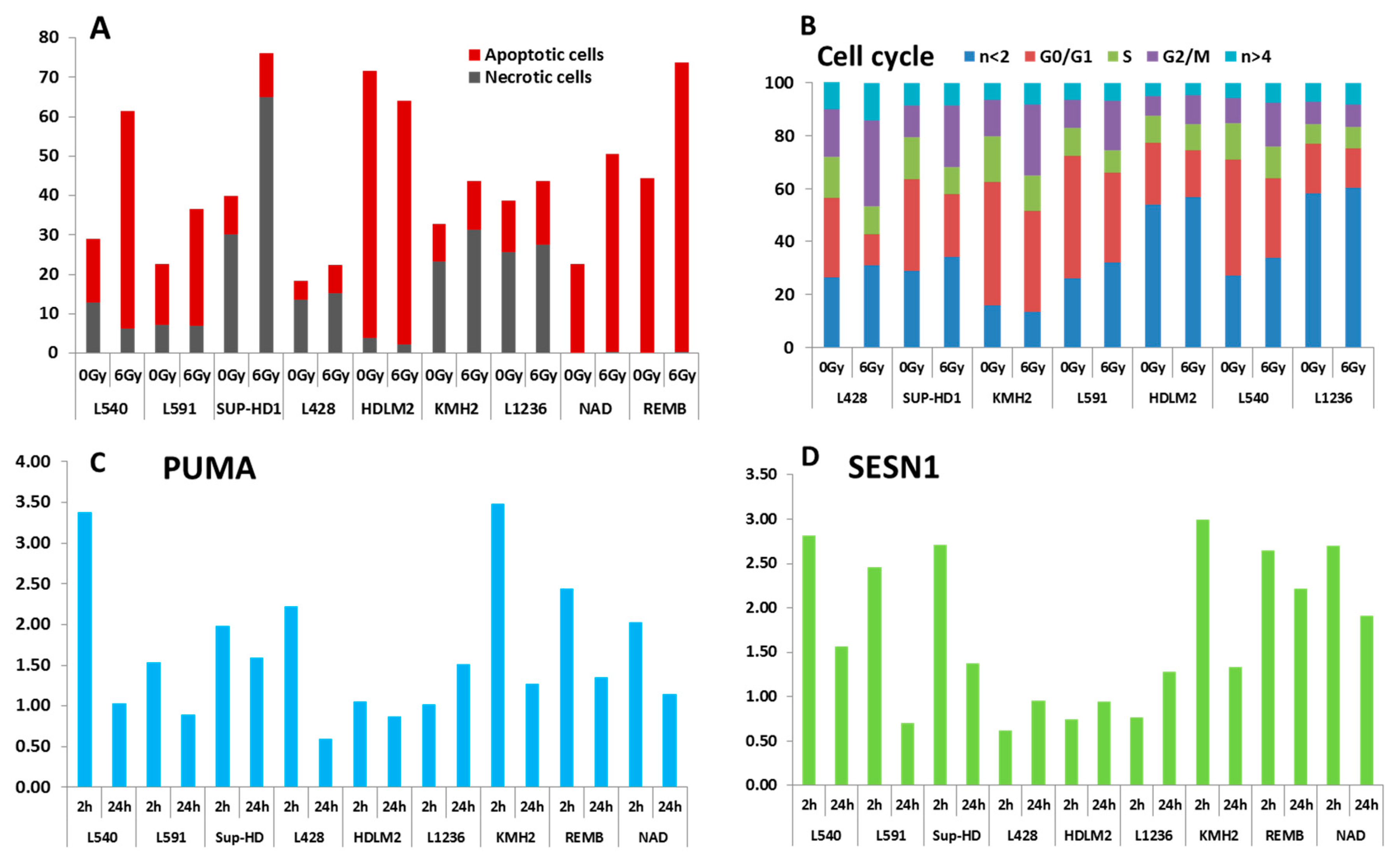
5. Altered Double-Strand Break Response in HL Cell Lines
6. Discussion
7. Materials and Methods
7.1. Cell Lines Used, Patients and Culture Conditions
7.2. Cytogenetic Slides (Preparation of Metaphase Spreads)
7.3. CD30 and CD15 Detection and Cell Cycle Analysis by Flow Cytometry
7.4. TP53 Functional Assay for Screening of the Cell Lines
7.5. Evaluation of Microsatellite Instability (MSI) in HL Cell Lines
7.6. Immunofluorescence and Immunofluorescent-FISH (IF-FISH)
7.7. Telomere Quantification
7.8. Telomere-Centromere Staining and M-FISH Staining
7.9. Micronucleus Assay
7.10. Western Blot Analysis
7.11. Apoptosis, Cell Cycles and Transcriptional Response after In Vitro Irradiation of HL Cell Lines
7.12. RNA Extraction and Transcriptome Analysis
7.13. Micronucleus and Chromosomal Aberration Scoring
7.14. Statistical Analysis
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stein, H.; Delsol, G.; Pileri, S.; Weiss, L.; Poppema, S.; Jaffe, E. Classical hodgkin lymphoma. In Who Classification of Tumours of Haematopoietic and Lymphoid Tissues; IARC: Lyon, France, 2008; p. 326. [Google Scholar]
- Matsuki, E.; Younes, A. Lymphomagenesis in Hodgkin lymphoma. Semin. Cancer Biol. 2015, 34, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Atkin, N.B. Cytogenetics of Hodgkin’s disease. Cytogenet. Cell Genet. 1998, 80, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Falzetti, D.; Crescenzi, B.; Matteuci, C.; Falini, B.; Martelli, M.F.; Van Den Berghe, H.; Mecucci, C. Genomic instability and recurrent breakpoints are main cytogenetic findings in Hodgkin’s disease. Haematologica 1999, 84, 298–305. [Google Scholar] [PubMed]
- Janz, M.; Stuhmer, T.; Vassilev, L.T.; Bargou, R.C. Pharmacologic activation of p53-dependent and p53-independent apoptotic pathways in Hodgkin/Reed-Sternberg cells. Leukemia 2007, 21, 772–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, S.; Crescenzi, B.; Schneider, M.; Ascani, S.; Hartmann, S.; Hansmann, M.L.; Falini, B.; Mecucci, C.; Tiacci, E.; Kuppers, R. Subclonal evolution of a classical Hodgkin lymphoma from a germinal center b-cell-derived mantle cell lymphoma. Int. J. Cancer 2014, 134, 832–843. [Google Scholar] [CrossRef] [PubMed]
- Re, D.; Wickenhauser, C.; Ahmadi, T.; Buchdunger, E.; Kochanek, M.; Diehl, V.; Wolf, J. Preclinical evaluation of the antiproliferative potential of STI571 in Hodgkin’s disease. Br. J. Cancer 2002, 86, 1333–1335. [Google Scholar] [CrossRef] [PubMed]
- Knecht, H.; Righolt, C.; Mai, S. Genomic instability: The driving force behind refractory/relapsing hodgkin’s lymphoma. Cancers 2013, 5, 714–725. [Google Scholar] [CrossRef] [PubMed]
- Cuceu, C.; Hempel, W.M.; Sabatier, L.; Bosq, J.; Carde, P.; M’Kacher, R. Chromosomal instability in hodgkin lymphoma: An in-depth review and perspectives. Cancers 2018, 10, 91. [Google Scholar] [CrossRef] [PubMed]
- Goel, A.; Nagasaka, T.; Arnold, C.N.; Inoue, T.; Hamilton, C.; Niedzwiecki, D.; Compton, C.; Mayer, R.J.; Goldberg, R.; Bertagnolli, M.M.; et al. The CPG island methylator phenotype and chromosomal instability are inversely correlated in sporadic colorectal cancer. Gastroenterology 2007, 132, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Buecher, B.; Cacheux, W.; Rouleau, E.; Dieumegard, B.; Mitry, E.; Lievre, A. Role of microsatellite instability in the management of colorectal cancers. Dig. Liver Dis. 2013, 45, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Re, D.; Benenson, L.; Wickenhauser, C.; Starostik, P.; Staratschek-Jox, A.; Muller-Hermelink, H.K.; Diehl, V.; Wolf, J. Proficient mismatch repair protein expression in Hodgkin and Reed Sternberg cells. Int. J. Cancer. 2002, 97, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Knecht, H.; Kongruttanachok, N.; Sawan, B.; Brossard, J.; Prevost, S.; Turcotte, E.; Lichtensztejn, Z.; Lichtensztejn, D.; Mai, S. Three-dimensional telomere signatures of Hodgkin- and Reed-Sternberg cells at diagnosis identify patients with poor response to conventional chemotherapy. Transl. Oncol. 2012, 5, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Guffei, A.; Sarkar, R.; Klewes, L.; Righolt, C.; Knecht, H.; Mai, S. Dynamic chromosomal rearrangements in Hodgkin’s lymphoma are due to ongoing three-dimensional nuclear remodeling and breakage-bridge-fusion cycles. Haematologica 2010, 95, 2038–2046. [Google Scholar] [CrossRef] [PubMed]
- M’Kacher, R.; Bennaceur-Griscelli, A.; Girinsky, T.; Koscielny, S.; Delhommeau, F.; Dossou, J.; Violot, D.; Leclercq, E.; Courtier, M.H.; Beron-Gaillard, N.; et al. Telomere shortening and associated chromosomal instability in peripheral blood lymphocytes of patients with Hodgkin’s lymphoma prior to any treatment are predictive of second cancers. Int. J. Radiat. Oncol. Biol. Phys. 2007, 68, 465–471. [Google Scholar] [CrossRef] [PubMed]
- M’Kacher, R.; Girinsky, T.; Colicchio, B.; Ricoul, M.; Dieterlen, A.; Jeandidier, E.; Heidingsfelder, L.; Cuceu, C.; Shim, G.; Frenzel, M.; et al. Telomere shortening: A new prognostic factor for cardiovascular disease post-radiation exposure. Radiat. Prot. Dosim. 2015, 164, 134–137. [Google Scholar] [CrossRef] [PubMed]
- Weber-Matthiesen, K.; Deerberg-Wittram, J.; Rosenwald, A.; Poetsch, M.; Grote, W.; Schlegelberger, B. Translocation t(2;5) is not a primary event in Hodgkin’s disease. Simultaneous immunophenotyping and interphase cytogenetics. Am. J. Pathol. 1996, 149, 463–468. [Google Scholar] [PubMed]
- Nagel, S.; Meyer, C.; Quentmeier, H.; Kaufmann, M.; Drexler, H.G.; MacLeod, R.A. Chromothripsis in hodgkin lymphoma. Genes Chromosom. Cancer 2013, 52, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sattarzadeh, A.; Diepstra, A.; Visser, L.; van den Berg, A. The microenvironment in classical hodgkin lymphoma: An actively shaped and essential tumor component. Semin. Cancer Biol. 2014, 24, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Jansen, M.P.; Hopman, A.H.; Haesevoets, A.M.; Gennotte, I.A.; Bot, F.J.; Arends, J.W.; Ramaekers, F.C.; Schouten, H.C. Chromosomal abnormalities in Hodgkin’s disease are not restricted to Hodgkin/Reed-Sternberg cells. J. Pathol. 1998, 185, 145–152. [Google Scholar] [CrossRef]
- Feuerborn, A.; Moritz, C.; Von Bonin, F.; Dobbelstein, M.; Trumper, L.; Sturzenhofecker, B.; Kube, D. Dysfunctional p53 deletion mutants in cell lines derived from Hodgkin’s lymphoma. Leuk. Lymphoma 2006, 47, 1932–1940. [Google Scholar] [CrossRef] [PubMed]
- M’Kacher, R.; Andreoletti, L.; Flamant, S.; Milliat, F.; Girinsky, T.; Dossou, J.; Violot, D.; Assaf, E.; Clausse, B.; Koscielny, S.; et al. JC human polyomavirus is associated to chromosomal instability in peripheral blood lymphocytes of Hodgkin’s lymphoma patients and poor clinical outcome. Ann. Oncol. 2010, 21, 826–832. [Google Scholar] [CrossRef] [PubMed]
- Marczyk, M.; Jaksik, R.; Polanski, A.; Polanska, J. Adaptive filtering of microarray gene expression data based on gaussian mixture decomposition. BMC Bioinform. 2013, 14, 101. [Google Scholar] [CrossRef] [PubMed]
- M’Kacher, R.; Cuceu, C.; Al Jawhari, M.; Morat, L.; Frenzel, M.; Shim, G.; Lenain, A.; Hempel, W.M.; Junker, S.; Girinsky, T.; et al. The transition between telomerase and alt mechanisms in Hodgkin lymphoma and its predictive value in clinical outcomes. Cancers 2018, 10, 169. [Google Scholar] [CrossRef] [PubMed]
- Badie, C.; Dziwura, S.; Raffy, C.; Tsigani, T.; Alsbeih, G.; Moody, J.; Finnon, P.; Levine, E.; Scott, D.; Bouffler, S. Aberrant CDKN1a transcriptional response associates with abnormal sensitivity to radiation treatment. Br. J. Cancer 2008, 98, 1845–1851. [Google Scholar] [CrossRef] [PubMed]
- Howitt, B.E.; Shukla, S.A.; Sholl, L.M.; Ritterhouse, L.L.; Watkins, J.C.; Rodig, S.; Stover, E.; Strickland, K.C.; D’Andrea, A.D.; Wu, C.J.; et al. Association of polymerase e-mutated and microsatellite-instable endometrial cancers with neoantigen load, number of tumor-infiltrating lymphocytes, and expression of PD-1 and PD-L1. JAMA Oncol. 2015, 1, 1319–1323. [Google Scholar] [CrossRef] [PubMed]
- Vanderwalde, A.; Spetzler, D.; Xiao, N.; Gatalica, Z.; Marshall, J. Microsatellite instability status determined by next-generation sequencing and compared with PD-L1 and tumor mutational burden in 11,348 patients. Cancer Med. 2018, 7, 746–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fridman, J.S.; Lowe, S.W. Control of apoptosis by p53. Oncogene 2003, 22, 9030–9040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robles, A.I.; Harris, C.C. Clinical outcomes and correlates of TP53 mutations and cancer. Cold Spring Harb. Perspect. Biol. 2010, 2, a001016. [Google Scholar] [CrossRef] [PubMed]
- Schulz, E.; Kashofer, K.; Heitzer, E.; Mhatre, K.N.; Speicher, M.R.; Hoefler, G.; Sill, H. Preexisting TP53 mutation in therapy-related acute myeloid leukemia. Ann. Hematol. 2015, 94, 527–529. [Google Scholar] [CrossRef] [PubMed]
- M’Kacher, R.; Bennaceur, A.; Farace, F.; Lauge, A.; Plassa, L.F.; Wittmer, E.; Dossou, J.; Violot, D.; Deutsch, E.; Bourhis, J.; et al. Multiple molecular mechanisms contribute to radiation sensitivity in mantle cell lymphoma. Oncogene 2003, 22, 7905–7912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Beato, M.; Sanchez-Aguilera, A.; Piris, M.A. Cell cycle deregulation in B-cell lymphomas. Blood 2003, 101, 1220–1235. [Google Scholar] [CrossRef] [PubMed]
- Barascu, A.; Le Chalony, C.; Pennarun, G.; Genet, D.; Zaarour, N.; Bertrand, P. Oxydative stress alters nuclear shape through lamins dysregulation: A route to senescence. Nucleus 2012, 3, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Jansen, M.P.; Machiels, B.M.; Hopman, A.H.; Broers, J.L.; Bot, F.J.; Arends, J.W.; Ramaekers, F.C.; Schouten, H.C. Comparison of a and B-type lamin expression in reactive lymph nodes and nodular sclerosing Hodgkin’s disease. Histopathology 1997, 31, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Gascoigne, K.E.; Cheeseman, I.M. Induced dicentric chromosome formation promotes genomic rearrangements and tumorigenesis. Chromosome Res. 2013, 21, 407–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maciejowski, J.; Li, Y.; Bosco, N.; Campbell, P.J.; de Lange, T. Chromothripsis and kataegis induced by telomere crisis. Cell 2015, 163, 1641–1654. [Google Scholar] [CrossRef] [PubMed]
- Fenech, M.; Kirsch-Volders, M.; Natarajan, A.T.; Surralles, J.; Crott, J.W.; Parry, J.; Norppa, H.; Eastmond, D.A.; Tucker, J.D.; Thomas, P. Molecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis 2011, 26, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.J.; de Lange, T. Significant role for P16ink4a in P53-independent telomere-directed senescence. Curr. Biol. 2004, 14, 2302–2308. [Google Scholar] [CrossRef] [PubMed]
- Toufektchan, E.; Toledo, F. The guardian of the genome revisited: P53 downregulates genes required for telomere maintenance, DNA repair, and centromere structure. Cancers 2018, 10, 135. [Google Scholar] [CrossRef] [PubMed]
- Berger, R.; Busson-Le Coniat, M. Centric and pericentric chromosome rearrangements in hematopoietic malignancies. Leukemia 1999, 13, 671–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gisselsson, D.; Pettersson, L.; Höglund, M.; Heidenblad, M.; Gorunova, L.; Wiegant, J.; Mertens, F.; Dal Cin, P.; Mitelman, F.; Mandahl, N. Chromosomal breakage-fusion-bridge events cause genetic intratumor heterogeneity. Proc. Natl. Acad. Sci. USA 2000, 97, 5357–5362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacKinnon, R.N.; Campbell, L.J. The role of dicentric chromosome formation and secondary centromere deletion in the evolution of myeloid malignancy. Genet. Res. Int. 2011, 2011, 643628. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Matsuoka, H.; Funakoshi, Y.; Yakushijin, K.; Okamura, A.; Itoh, T.; Minami, H. A novel dicentric chromosome, dic(9;9)(p12;q34), leading to trisomy 9 in follicular lymphoma without t(14;18). Leuk. Res. 2011, 35, e100–e103. [Google Scholar] [CrossRef] [PubMed]
- M’Kacher, R.; Maalouf, E.E.; Ricoul, M.; Heidingsfelder, L.; Laplagne, E.; Cuceu, C.; Hempel, W.M.; Colicchio, B.; Dieterlen, A.; Sabatier, L. New tool for biological dosimetry: Reevaluation and automation of the gold standard method following telomere and centromere staining. Mutat. Res. 2014, 770, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Lejeune, J.; Dutrillaux, B.; Rethore, M.O.; Prieur, M. Comparison of the structure of chromatids of homo sapiens and pan troglodytes (author’s transl). Chromosoma 1973, 43, 423–444. [Google Scholar] [CrossRef] [PubMed]
- Avarello, R.; Pedicini, A.; Caiulo, A.; Zuffardi, O.; Fraccaro, M. Evidence for an ancestral alphoid domain on the long arm of human chromosome 2. Hum. Genet. 1992, 89, 247–249. [Google Scholar] [CrossRef] [PubMed]
- Kramer, K.M.; Brock, J.A.; Bloom, K.; Moore, J.K.; Haber, J.E. Two different types of double-strand breaks in saccharomyces cerevisiae are repaired by similar RAD52-independent, nonhomologous recombination events. Mol. Cell. Biol. 1994, 14, 1293–1301. [Google Scholar] [CrossRef] [PubMed]
- Pennaneach, V.; Kolodner, R.D. Stabilization of dicentric translocations through secondary rearrangements mediated by multiple mechanisms in S. Cerevisiae. PLoS ONE 2009, 4, e6389. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Gawel, M.; Dominska, M.; Greenwell, P.W.; Hazkani-Covo, E.; Bloom, K.; Petes, T.D. Nonrandom distribution of interhomolog recombination events induced by breakage of a dicentric chromosome in saccharomyces cerevisiae. Genetics 2013, 194, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Stimpson, K.M.; Song, I.Y.; Jauch, A.; Holtgreve-Grez, H.; Hayden, K.E.; Bridger, J.M.; Sullivan, B.A. Telomere disruption results in non-random formation of de novo dicentric chromosomes involving acrocentric human chromosomes. PLoS Genet. 2010, 6. [Google Scholar] [CrossRef] [PubMed]
- Murnane, J.P.; Sabatier, L. Chromosome rearrangements resulting from telomere dysfunction and their role in cancer. BioEssays 2004, 26, 1164–1174. [Google Scholar] [CrossRef] [PubMed]
- Kaddour, A.; Colicchio, B.; Buron, D.; El Maalouf, E.; Laplagne, E.; Borie, C.; Ricoul, M.; Lenain, A.; Hempel, W.M.; Morat, L.; et al. Transmission of induced chromosomal aberrations through successive mitotic divisions in human lymphocytes after in vitro and in vivo radiation. Sci. Rep. 2017, 7, 3291. [Google Scholar] [CrossRef] [PubMed]
- Lopez, V.; Barinova, N.; Onishi, M.; Pobiega, S.; Pringle, J.R.; Dubrana, K.; Marcand, S. Cytokinesis breaks dicentric chromosomes preferentially at pericentromeric regions and telomere fusions. Genes Dev. 2015, 29, 322–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, J.; Ferme, C.; Noordijk, E.M.; Morschhauser, F.; Girinsky, T.; Gaillard, I.; Lugtenburg, P.J.; Andre, M.; Lybeert, M.L.M.; Stamatoullas, A.; et al. Comparison of 36 Gy, 20 Gy, or no radiation therapy after 6 cycles of EBVP chemotherapy and complete remission in early-stage Hodgkin lymphoma without risk factors: Results of the EORT-GELA H9-F intergroup randomized trial. Int. J. Radiat. Oncol. Biol. Phys. 2018, 100, 1133–1145. [Google Scholar] [CrossRef] [PubMed]
- Hasenclever, D.; Diehl, V. A prognostic score for advanced Hodgkin’s disease. International prognostic factors project on advanced Hodgkin’s disease. N. Engl. J. Med. 1998, 339, 1506–1514. [Google Scholar] [CrossRef] [PubMed]
- Schaadt, M.; Diehl, V.; Stein, H.; Fonatsch, C.; Kirchner, H.H. Two neoplastic cell lines with unique features derived from Hodgkin’s disease. Int. J. Cancer 1980, 26, 723–731. [Google Scholar] [CrossRef] [PubMed]
- Naumovski, L.; Utz, P.J.; Bergstrom, S.K.; Morgan, R.; Molina, A.; Toole, J.J.; Glader, B.E.; McFall, P.; Weiss, L.M.; Warnke, R.; et al. SUP-HD1: A new Hodgkin’s disease-derived cell line with lymphoid features produces interferon-gamma. Blood 1989, 74, 2733–2742. [Google Scholar] [PubMed]
- Diehl, V.; Pfreundschuh, M.; Fonatsch, C.; Stein, H.; Falk, M.; Burrichter, H.; Schaadt, M. Phenotypic and genotypic analysis of Hodgkin’s disease derived cell lines: Histopathological and clinical implications. Cancer Surv. 1985, 4, 399–419. [Google Scholar] [PubMed]
- Wolf, J.; Kapp, U.; Bohlen, H.; Kornacker, M.; Schoch, C.; Stahl, B.; Mucke, S.; von Kalle, C.; Fonatsch, C.; Schaefer, H.E.; et al. Peripheral blood mononuclear cells of a patient with advanced Hodgkin’s lymphoma give rise to permanently growing Hodgkin-Reed Sternberg cells. Blood 1996, 87, 3418–3428. [Google Scholar] [PubMed]
- Kamesaki, H.; Fukuhara, S.; Tatsumi, E.; Uchino, H.; Yamabe, H.; Miwa, H.; Shirakawa, S.; Hatanaka, M.; Honjo, T. Cytochemical, immunologic, chromosomal, and molecular genetic analysis of a novel cell line derived from Hodgkin’s disease. Blood 1986, 68, 285–292. [Google Scholar] [PubMed]
- Flaman, J.M.; Frebourg, T.; Moreau, V.; Charbonnier, F.; Martin, C.; Chappuis, P.; Sappino, A.P.; Limacher, I.M.; Bron, L.; Benhattar, J.; et al. A simple p53 functional assay for screening cell lines, blood, and tumors. Proc. Natl. Acad. Sci. USA 1995, 92, 3963–3967. [Google Scholar] [CrossRef] [PubMed]
- Suraweera, N.; Duval, A.; Reperant, M.; Vaury, C.; Furlan, D.; Leroy, K.; Seruca, R.; Iacopetta, B.; Hamelin, R. Evaluation of tumor microsatellite instability using five quasimonomorphic mononucleotide repeats and pentaplex PCR. Gastroenterology 2002, 123, 1804–1811. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Lenain, C.; Bauwens, S.; Rizzo, A.; Saint-Leger, A.; Poulet, A.; Benarroch, D.; Magdinier, F.; Morere, J.; Amiard, S.; et al. TFR2 and apollo cooperate with topoisomerase 2alpha to protect human telomeres from replicative damage. Cell 2010, 142, 230–242. [Google Scholar] [CrossRef] [PubMed]
- Cesare, A.J.; Kaul, Z.; Cohen, S.B.; Napier, C.E.; Pickett, H.A.; Neumann, A.A.; Reddel, R.R. Spontaneous occurrence of telomeric DNA damage response in the absence of chromosome fusions. Nat. Struct. Mol. Biol. 2009, 16, 1244–1251. [Google Scholar] [CrossRef] [PubMed]
- Finot, F.; Kaddour, A.; Morat, L.; Mouche, I.; Zaguia, N.; Cuceu, C.; Souverville, D.; Negrault, S.; Cariou, O.; Essahli, A.; et al. Genotoxic risk of ethyl-paraben could be related to telomere shortening. J. Appl. Toxicol. 2017, 37, 758–771. [Google Scholar] [CrossRef] [PubMed]
- Marotta, M.; Chen, X.; Watanabe, T.; Faber, P.W.; Diede, S.J.; Tapscott, S.; Tubbs, R.; Kondratova, A.; Stephens, R.; Tanaka, H. Homology-mediated end-capping as a primary step of sister chromatid fusion in the breakage-fusion-bridge cycles. Nucleic Acids Res. 2013, 41, 9732–9740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wendt, H.W. Dealing with a common problem in social science: A simplified rank-biserial coefficient of correlation based on the U statistic. Eur. J. Soc. 1972, 2, 463–465. [Google Scholar] [CrossRef]
- Cohen, J. Statistical Power Analysis for the Behavioral Sciences, 2nd ed.; Erlbaum: Hillsdale, NJ, USA, 1988. [Google Scholar]
- Ansell, S.M. Hodgkin lymphoma: 2016 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2016, 91, 434–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Microsatellite (MSI) * | ||||||
|---|---|---|---|---|---|---|
| Cell Line | PE83 NR21 | PE31 BAT26 | PE47 BAT25 | PE85 NR24 | PE84 NR2 | RATIO * |
| L428 | MSI | MSI | MSI | AIA | MSI | 4/5 |
| L591 | AIA | AIA | AIA | AIA | MSI | 1/5 |
| SUP-HD1 | AIA | AIA | AIA | AIA | MSI | 1/5 |
| L540 | MSI | AIA | AIA | AIA | AIA | 1/5 |
| HDLM-2 | MSI | MSI | MSI | AIA | AIA | 3/5 |
| L1236 | AIA | AIA | AIA | AIA | AIA | 0/5 |
| KMH2 | AIA | MSI | MSI | AIA | MSI | 3/5 |
| Cell Line | Origin | Histology Type | EBV Status | Gender (Age) |
|---|---|---|---|---|
| L428 | B-cell | Nodular Sclerosis (young) | − | Female (37) |
| L591 | B-cell | Nodular Sclerosis (young) | + | Female (31) |
| SUP-HD1 | B-cell | Nodular Sclerosis (young) | − | Male (33) |
| L540 | T-cell | Nodular Sclerosis (young) | − | Female (20) |
| HDLM2 | T-cell | Nodular Sclerosis (old) | − | Male (74) |
| L1236 | B-cell | Mixed Cellularity | − | Male (34) |
| KMH2 | B-cell | Mixed Cellularity | − | Male (37) |
| Characteristics | No. of Patients |
|---|---|
| Nodular Sclerosis (NS) | 100 |
| Male | 53 |
| Age | 33 (17–81) |
| Treatment | |
| Combined modality | 100% |
| Mixed Cellularity (MC) | 23 |
| Male | 15 |
| Age | 39 (19–69) |
| Treatment | |
| Combined modality | 100% |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuceu, C.; Colicchio, B.; Jeandidier, E.; Junker, S.; Plassa, F.; Shim, G.; Mika, J.; Frenzel, M.; AL Jawhari, M.; Hempel, W.M.; et al. Independent Mechanisms Lead to Genomic Instability in Hodgkin Lymphoma: Microsatellite or Chromosomal Instability. Cancers 2018, 10, 233. https://doi.org/10.3390/cancers10070233
Cuceu C, Colicchio B, Jeandidier E, Junker S, Plassa F, Shim G, Mika J, Frenzel M, AL Jawhari M, Hempel WM, et al. Independent Mechanisms Lead to Genomic Instability in Hodgkin Lymphoma: Microsatellite or Chromosomal Instability. Cancers. 2018; 10(7):233. https://doi.org/10.3390/cancers10070233
Chicago/Turabian StyleCuceu, Corina, Bruno Colicchio, Eric Jeandidier, Steffen Junker, François Plassa, Grace Shim, Justyna Mika, Monika Frenzel, Mustafa AL Jawhari, William M. Hempel, and et al. 2018. "Independent Mechanisms Lead to Genomic Instability in Hodgkin Lymphoma: Microsatellite or Chromosomal Instability" Cancers 10, no. 7: 233. https://doi.org/10.3390/cancers10070233