Embigin Promotes Prostate Cancer Progression by S100A4-Dependent and-Independent Mechanisms

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
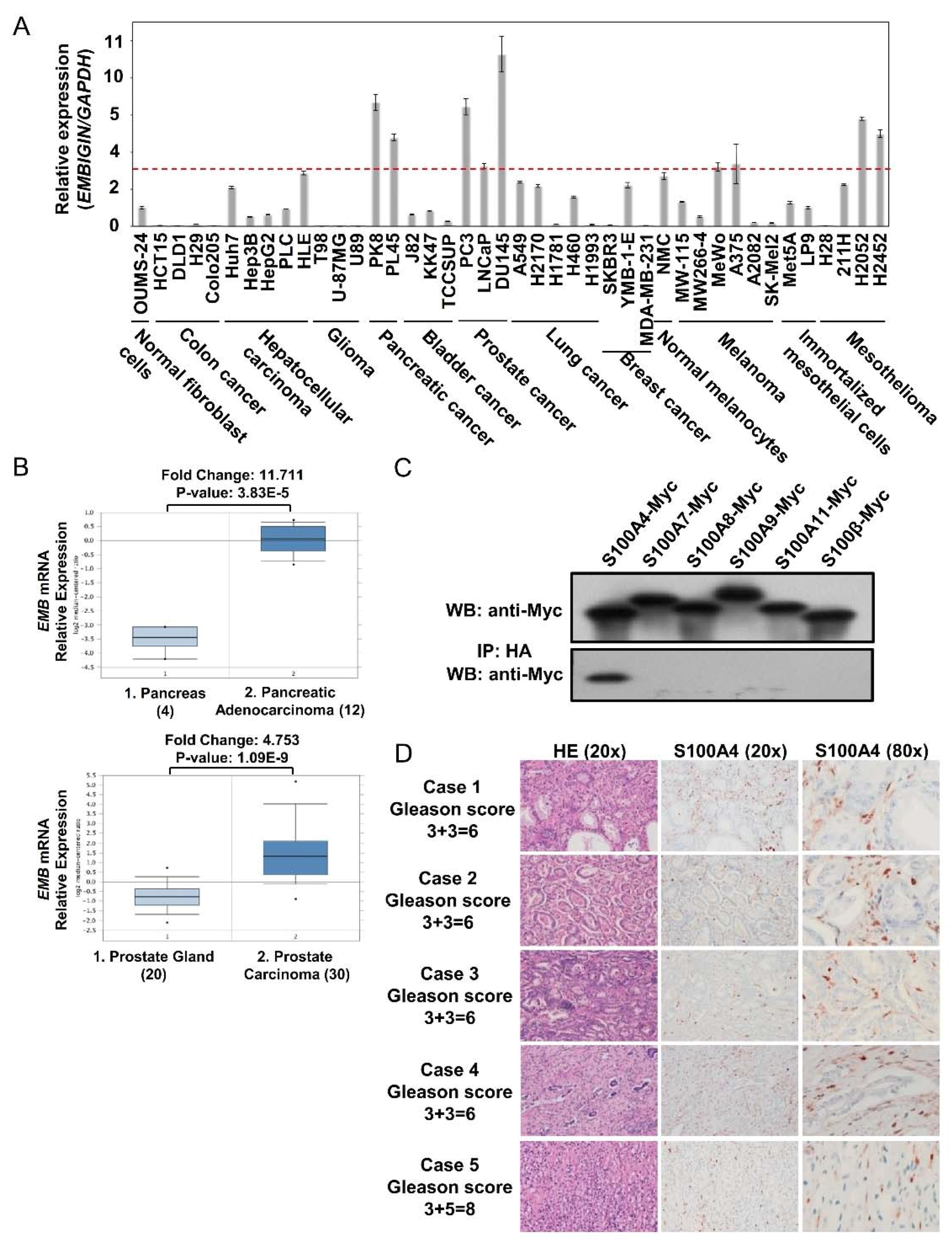
2.1. Embigin Expression Level Is High in Prostate Cancer and Embigin Binds to S100A4
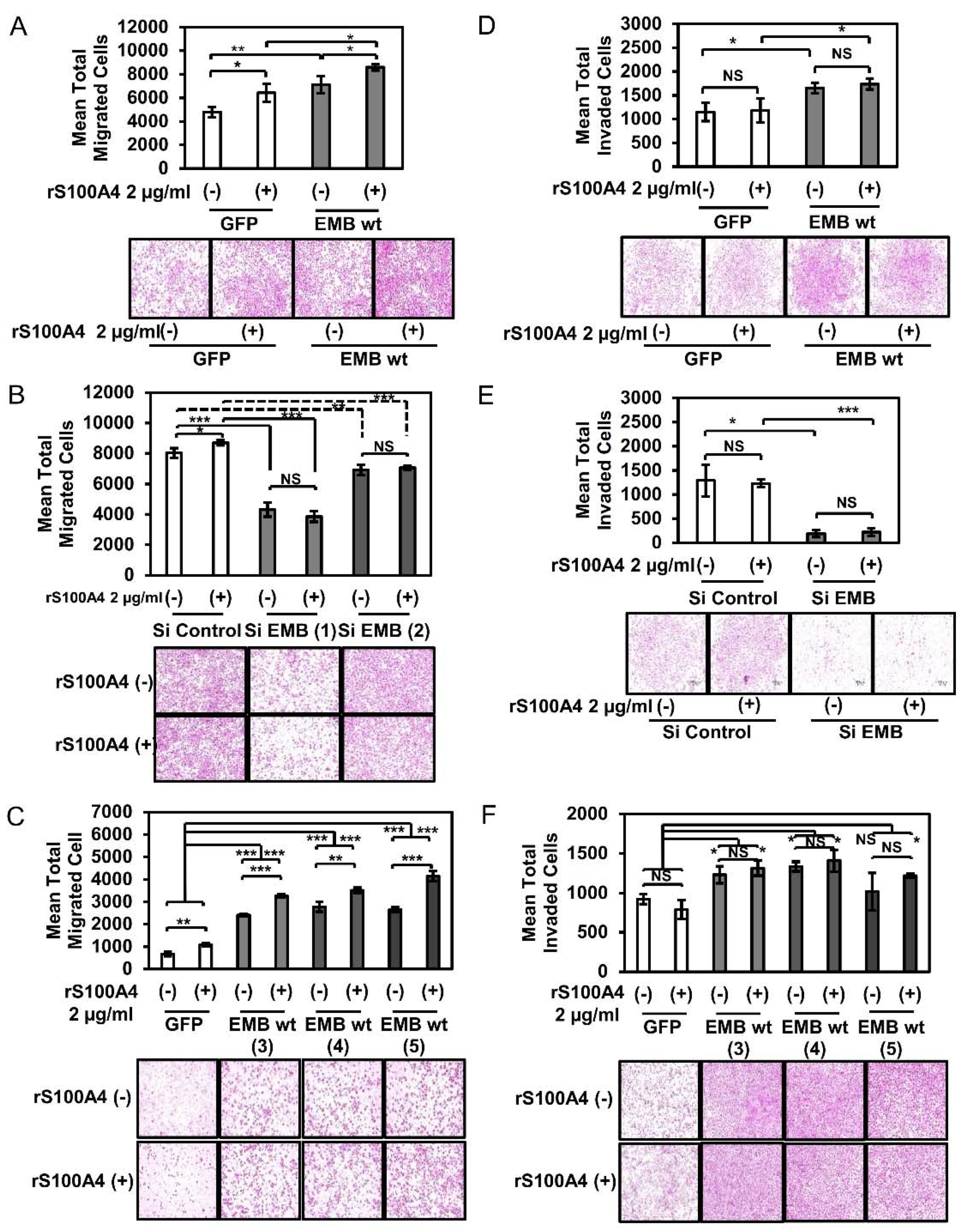
2.2. S100A4 Binding to Embigin Augments Migration Ability of Prostate Cancer Cells
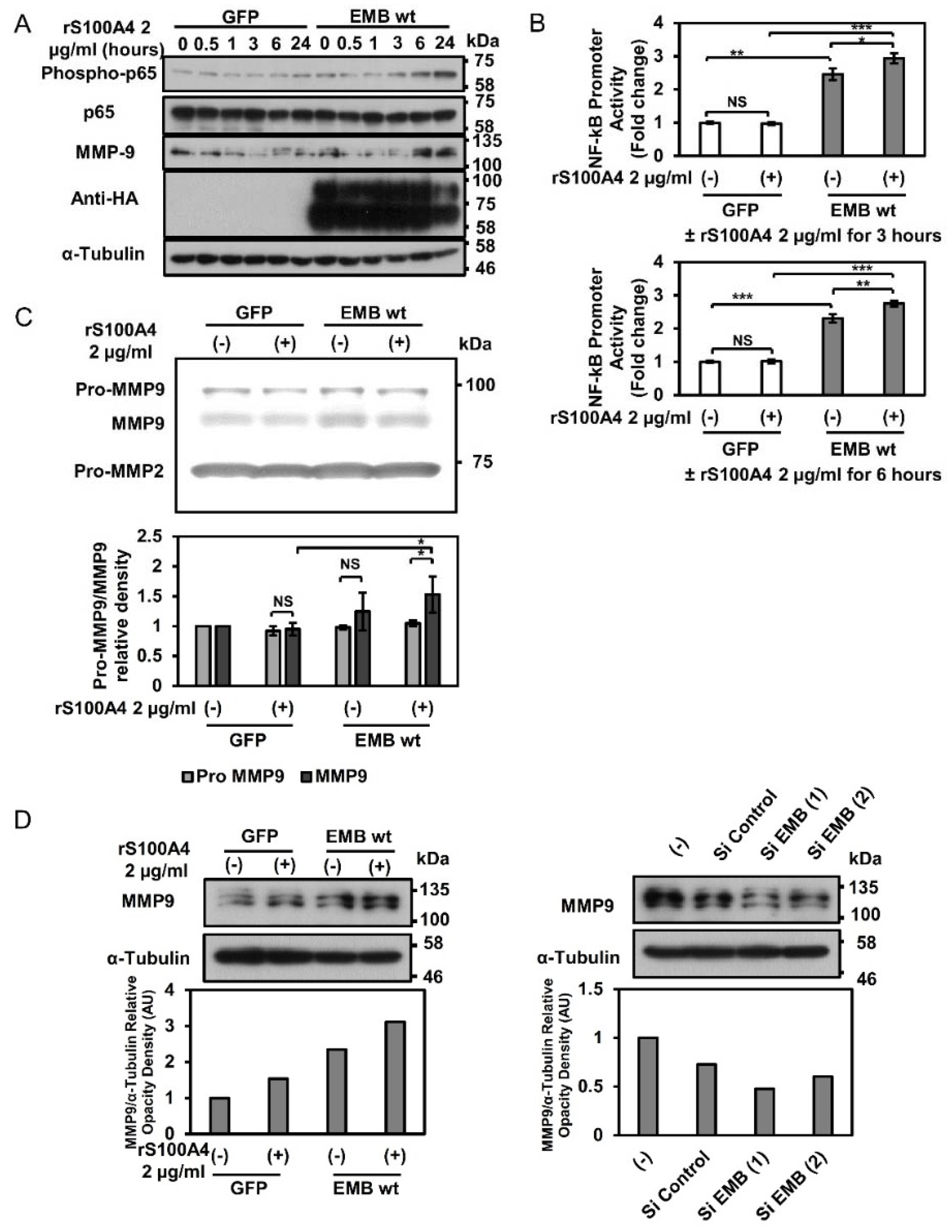
2.3. Embigin Induces NF-κB and MMP9 Activation
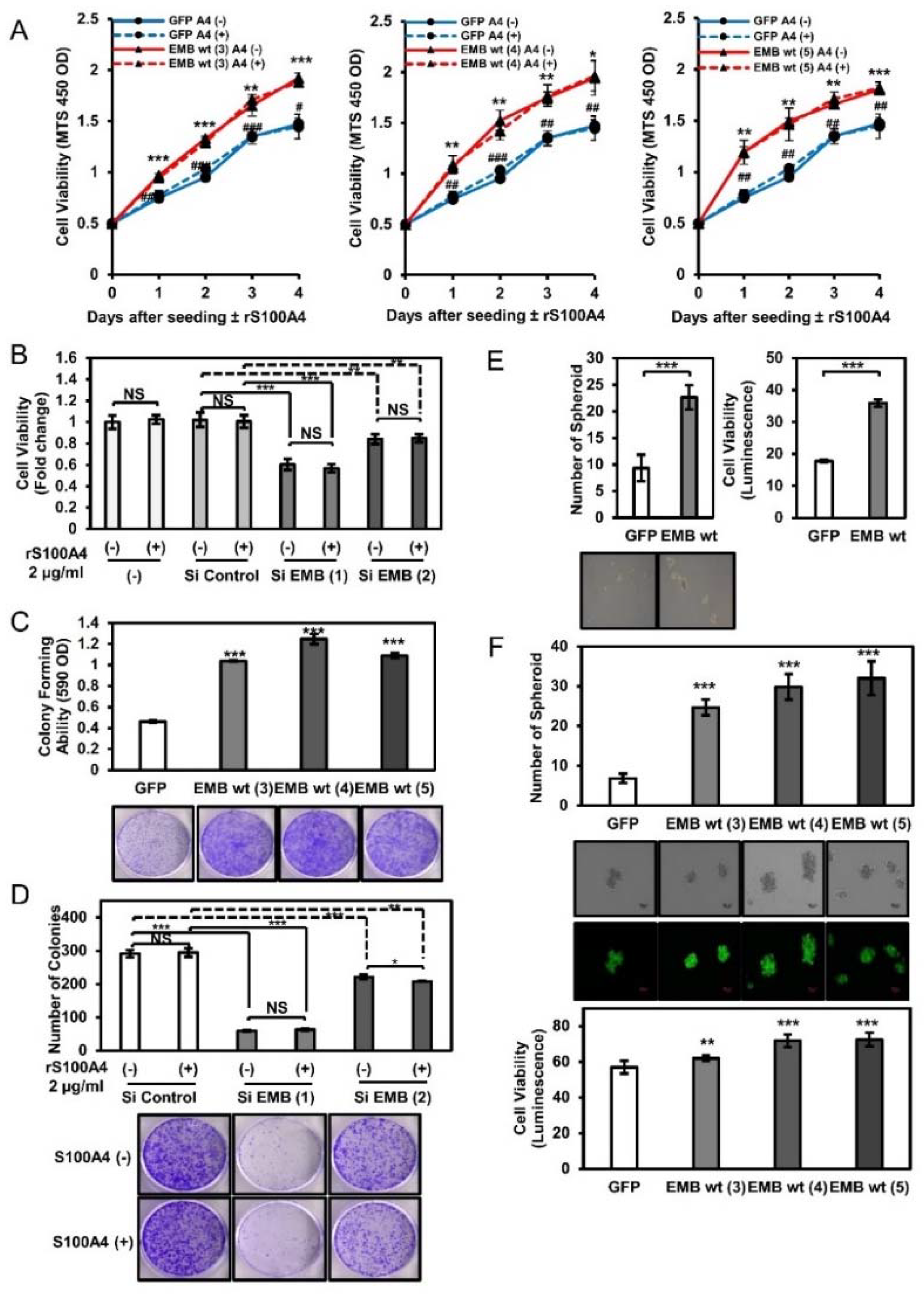
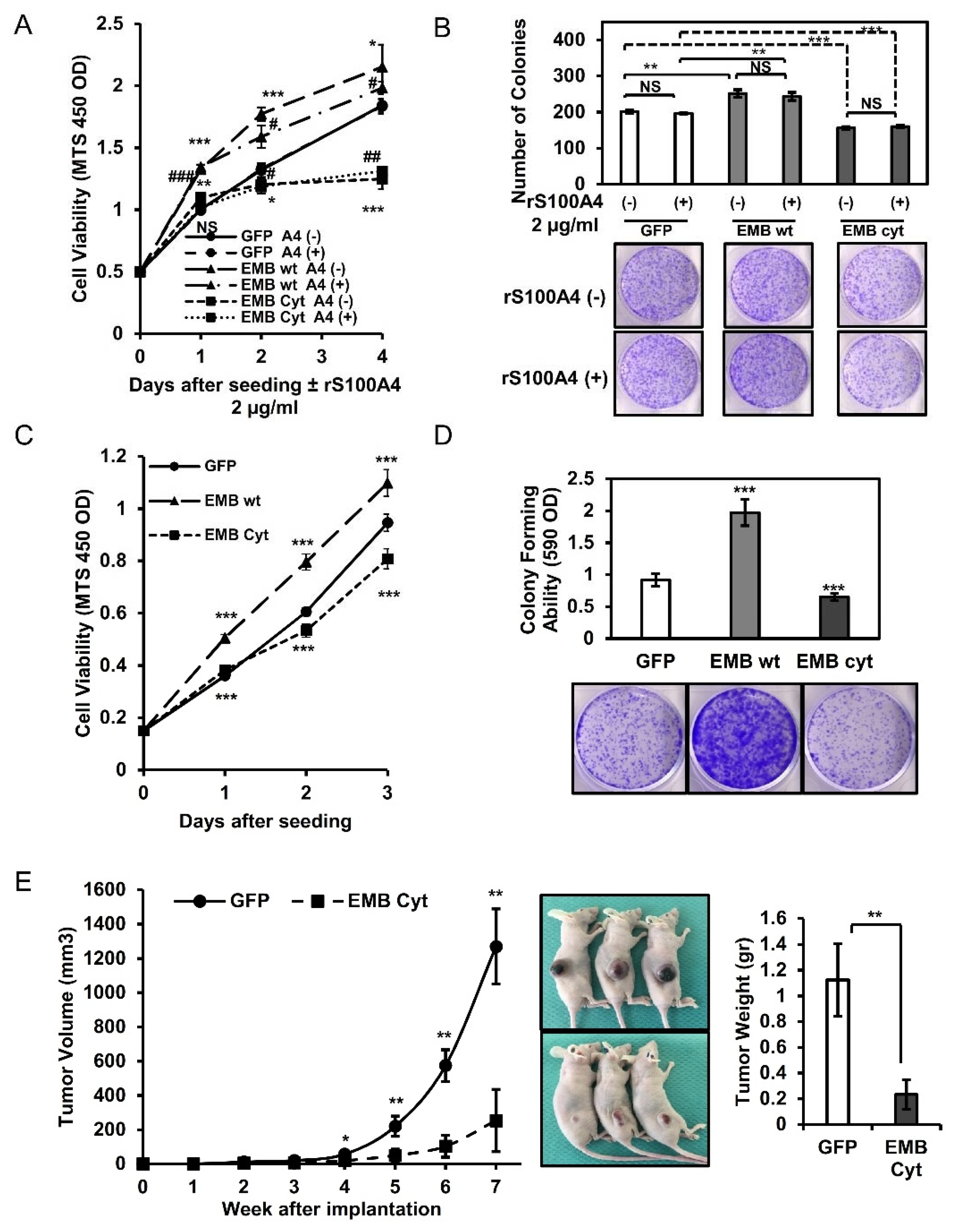
2.4. Embigin Induces Tumor Growth and Colony- and Spheroid-Forming Ability Independently of S100A4
2.5. Intracellular Cytoplasmic Tail of Embigin Is Vital for Prostate Cancer Growth
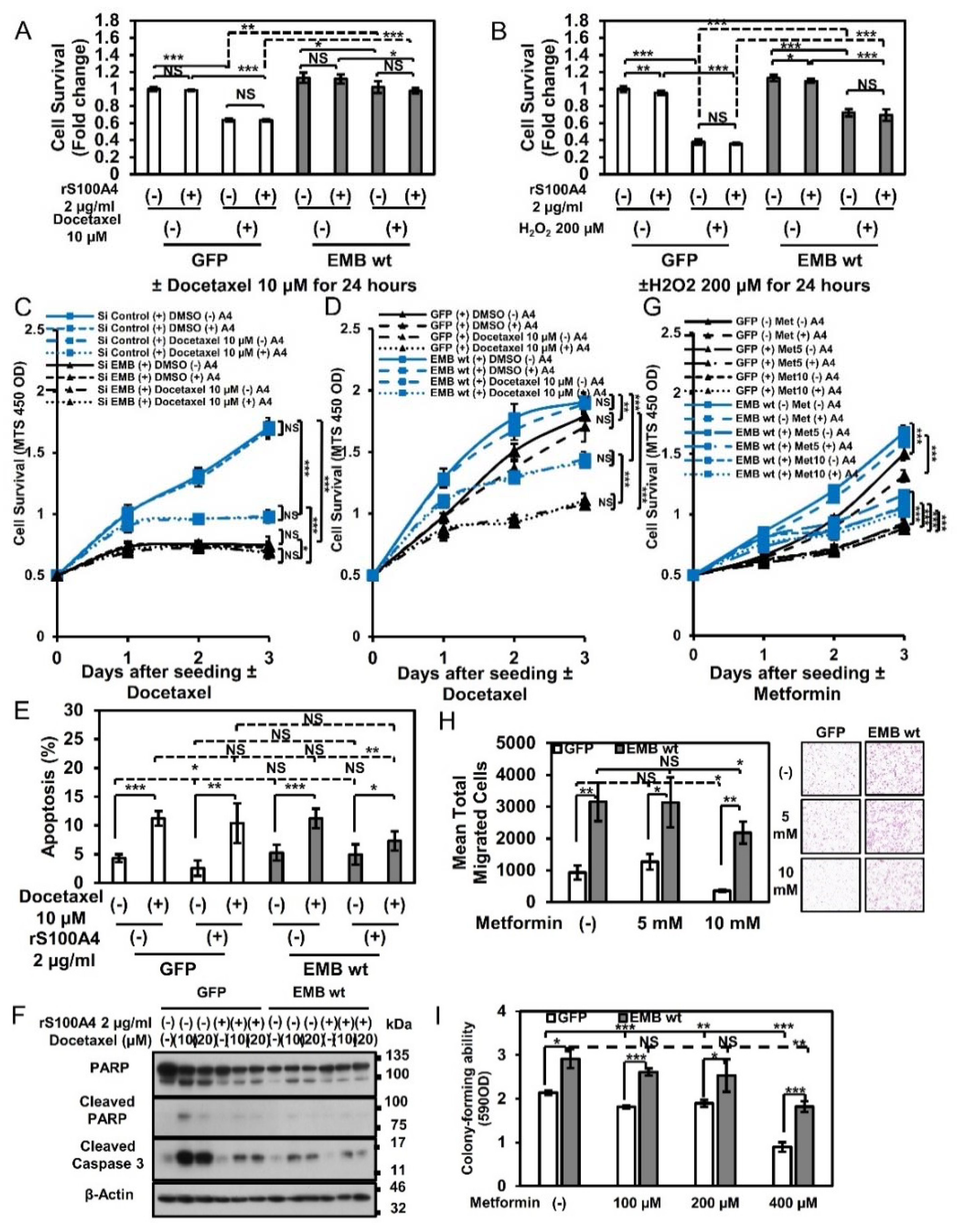
2.6. Embigin Mediates Prostate Cancer Cells Survival upon Chemotherapy and Oxidative Stress
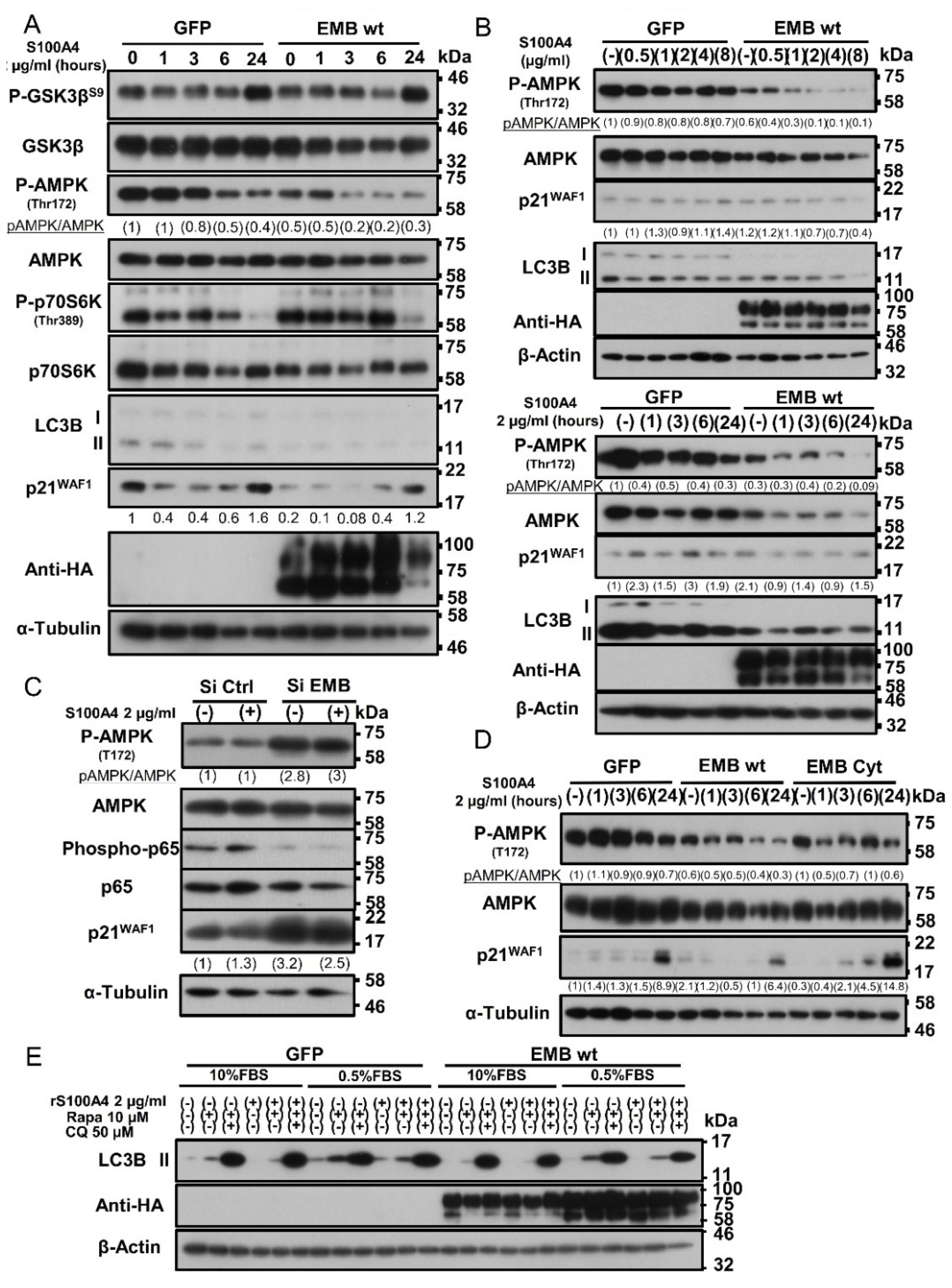
2.7. Binding of S100A4 to Embigin Mediates its Biological Functions through AMPK/mTOR/p21 Signaling and Inhibits Autophagy
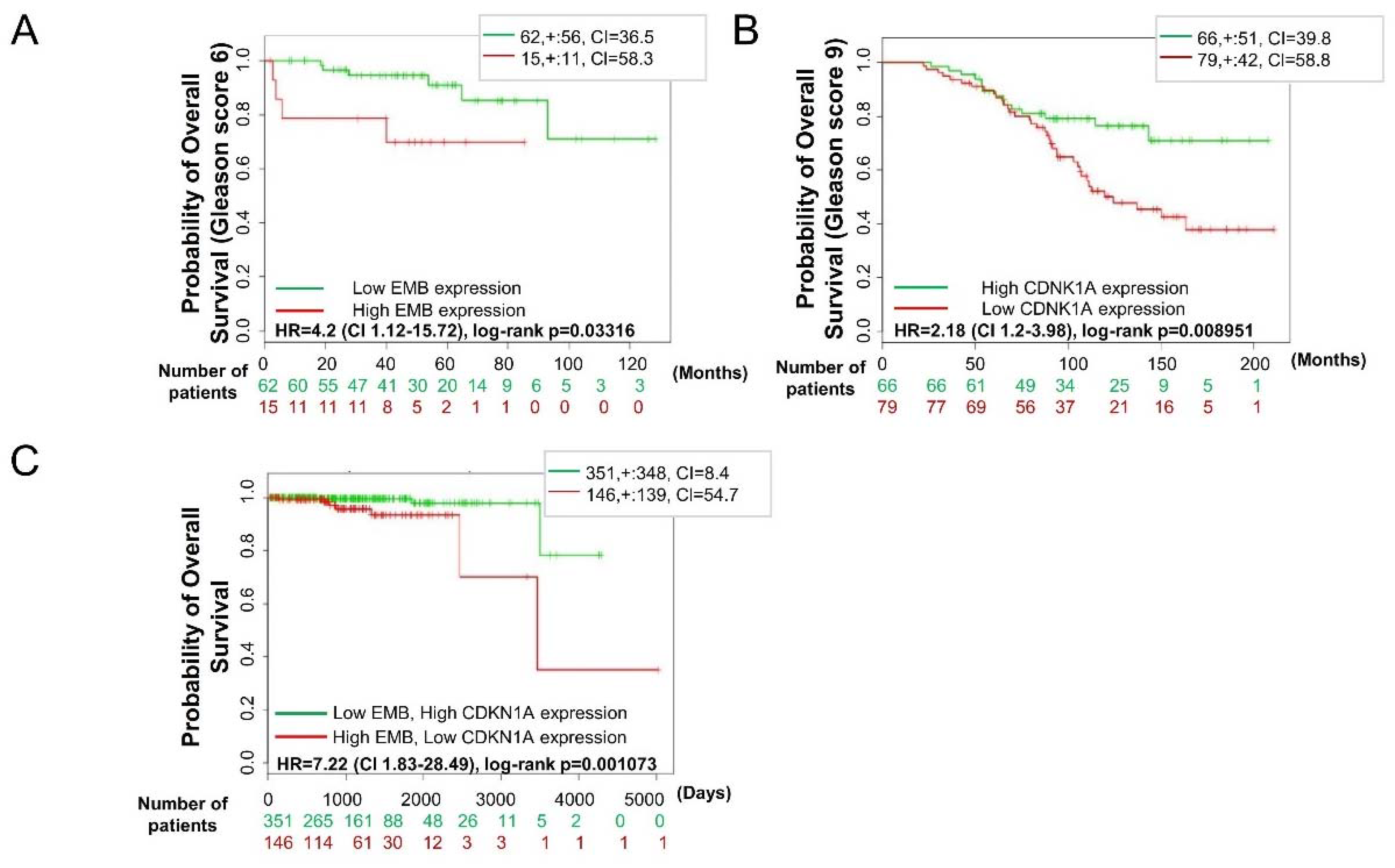
2.8. Embigin and p21WAF1 Expression in Prostate Cancer Tissue can Predict Survival
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Chemicals
4.2. Preparation of S100A4 Protein
4.3. Preparation of a Plasmid Vector
4.4. Embigin Knockdown Experiment with Short Interference RNA (siRNA)
4.5. Embigin-S100 Proteins Binding Assays
4.6. Cell Migration and Invasion Assays
4.7. Luciferase Reporter Assay
4.8. Zymography
4.9. RNA Extraction and Real-Time Quantitative Reverse Transcription (qRT)-PCR Analysis
4.10. Cell Viability Assay (MTS Assay)
4.11. Colony Formation Assay
4.12. Spheroid Formation Assay
4.13. Cell Apoptosis Assay
4.14. Western Blotting
4.15. Immunohistochemistry
4.16. In vivo Growth Assay
4.17. Western Blot Analysis of Mouse Tumors
4.18. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AMPK | Adenosine monophosphate-activated protein kinase |
| mTORC1 | Mammalian target of rapamycin complex 1 |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| MMP9 | Matrix Metalloproteinase 9 |
References
- Guenette, R.S.; Sridhar, S.; Herley, M.; Mooibroek, M.; Wong, P.; Tenniswood, M. Embigin, a developmentally expressed member of the immunoglobulin super family, is also expressed during regression of prostate and mammary gland. Dev. Genet. 1997, 21, 268–278. [Google Scholar] [CrossRef]
- Tachikui, H.; Kurosawa, N.; Kadomatsu, K.; Muramatsu, T. Genomic organization and promoter activity of embigin, a member of the immunoglobulin superfamily. Gene 1999, 240, 325–332. [Google Scholar] [CrossRef]
- Pridans, C.; Holmes, M.L.; Polli, M.; Wettenhall, J.M.; Dakic, A.; Corcoran, L.M.; Smyth, G.K.; Nutt, S.L. Identification of Pax5 target genes in early B cell differentiation. J. Immunol. 2008, 180, 1719–1728. [Google Scholar] [CrossRef] [PubMed]
- Silberstein, L.; Goncalves, K.A.; Kharchenko, P.V.; Turcotte, R.; Kfoury, Y.; Mercier, F.; Baryawno, N.; Severe, N.; Bachand, J.; Spencer, J.A.; et al. Proximity-Based Differential Single-Cell Analysis of the Niche to Identify Stem/Progenitor Cell Regulators. Cell Stem Cell 2016, 19, 530–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, F.; Zhang, J.; Zhang, Y.; Liu, H.; Yang, C.; Wang, J.; Guo, Y.; Wen, X.H.; Zhang, K.; Huang, B.; et al. Embigin, regulated by HOXC8, plays a suppressive role in breast tumorigenesis. Oncotarget 2015, 6, 23496–23509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, D.E.; Kim, J.M.; Kim, C.; Song, S.Y. Embigin is overexpressed in pancreatic ductal adenocarcinoma and regulates cell motility through epithelial to mesenchymal transition via the TGF-β pathway. Mol. Carcinog. 2016, 55, 633–645. [Google Scholar] [CrossRef] [PubMed]
- Haase-Kohn, C.; Wolf, S.; Herwig, N.; Mosch, B.; Pietzsch, J. Metastatic potential of B16-F10 melanoma cells is enhanced by extracellular S100A4 derived from RAW264.7 macrophages. Biochem. Biophys. Res. Commun. 2014, 446, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Hansen, B.; Ornas, D.; Grigorian, M.; Klingelhofer, J.; Tulchinsky, E.; Lukanidin, E.; Ambartsumian, N. Extracellular S100A4(mts1) stimulates invasive growth of mouse endothelial cells and modulates MMP-13 matrix metalloproteinase activity. Oncogene 2004, 23, 5487–5495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahriari, K.; Shen, F.; Worrede-Mahdi, A.; Liu, Q.; Gong, Y.; Garcia, F.U.; Fatatis, A. Cooperation among heterogeneous prostate cancer cells in the bone metastatic niche. Oncogene 2017, 36, 2846–2856. [Google Scholar] [CrossRef] [PubMed]
- Semov, A.; Moreno, M.J.; Onichtchenko, A.; Abulrob, A.; Ball, M.; Ekiel, I.; Pietrzynski, G.; Stanimirovic, D. Alakhov Valery. Metastasis-associated protein S100A4 induces angiogenesis through interaction with Annexin II and accelerated plasmin formation. J. Biol. Chem. 2005, 280, 20833–20841. [Google Scholar] [CrossRef] [PubMed]
- Kiryushko, D.; Novitskaya, V.; Soroka, V.; Klingelhofer, J.; Lukanidin, E.; Berezin, V.; Bock, E. Molecular mechanisms of Ca2+ signaling in neurons induced by the S100A4 protein. Mol. Cell. Biol. 2006, 26, 3625–3638. [Google Scholar] [CrossRef] [PubMed]
- Yammani, R.R.; Carlson, C.S.; Bresnick, A.R.; Loeser, R.F. Increase in production of matrix metalloproteinase 13 by human articular chondrocytes due to stimulation with S100A4: role of the receptor for advanced glycation end products. Arthritis Rheum. 2006, 54, 2901–2911. [Google Scholar] [CrossRef] [PubMed]
- Belot, N.; Pochet, R.; Heizmann, C.W.; Kiss, R.; Decaestecker, C. Extracellular S100A4 stimulates the migration rate of astrocytic tumor cells by modifying the organization of their actin cytoskeleton. Biochim. Biophys. Acta 2002, 1600, 74–83. [Google Scholar] [CrossRef]
- Iacobuzio-Donahue, C.A.; Maitra, A.; Olsen, M.; Lowe, A.W.; van Heek, N.T.; Rosty, C.; Walter, K.; Sato, N.; Parker, A.; Ashfaq, R.; et al. Exploration of global gene expression patterns in pancreatic adenocarcinoma using cDNA microarrays. Am. J. Pathol. 2003, 162, 1151–1162. [Google Scholar] [CrossRef]
- Tomlins, S.A.; Mehra, R.; Rhodes, D.R.; Cao, X.; Wang, L.; Dhanasekaran, S.M.; Kalyana-Sundaram, S.; Wei, J.T.; Rubin, M.A.; Pienta, K.J.; et al. Integrative molecular concept modeling of prostate cancer progression. Nat. Genet. 2007, 39, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Ruma, I.M.; Putranto, E.W.; Kondo, E.; Murata, H.; Watanabe, M.; Huang, P.; Kinoshita, R.; Futami, J.; Inoue, Y.; Yamauchi, A.; et al. MCAM, as a novel receptor for S100A8/A9, mediates progression of malignant melanoma through prominent activation of NF-κB and ROS formation upon ligand binding. Clin. Exp. Metastasis 2016, 33, 609–627. [Google Scholar] [CrossRef] [PubMed]
- Hibino, T.; Sakaguchi, M.; Miyamoto, S.; Yamamoto, M.; Motoyama, A.; Hosoi, J.; Shimokata, T.; Ito, T.; Tsuboi, R.; Huh, N.H. S100A9 is a novel ligand of EMMPRIN that promotes melanoma metastasis. Cancer Res. 2013, 73, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Helfman, D.M.; Kim, E.J.; Lukanidin, E.; Grigorian, M. The metastasis associated protein S100A4: role in tumour progression and metastasis. Br. J. Cancer 2005, 92, 1955–1958. [Google Scholar] [CrossRef] [PubMed]
- Medapati, M.R.; Dahlmann, M.; Ghavami, S.; Pathak, K.A.; Lucman, L.; Klonisch, T.; Hoang-Vu, C.; Stein, U.; Hombach-Klonisch, S. RAGE Mediates the Pro-Migratory Response of Extracellular S100A4 in Human Thyroid Cancer Cells. Thyroid 2015, 25, 514–527. [Google Scholar] [CrossRef] [PubMed]
- Saleem, M.; Kweon, M.H.; Johnson, J.J.; Adhami, V.M.; Elcheva, I.; Khan, N.; Hafeez, B.B.; Bhat, K.M.R.; Sarfaraz, S.; Reagan-Shaw, S.; et al. S100A4 accelerates tumorigenesis and invasion of human prostate cancer through the transcriptional regulation of matrix metalloproteinase 9. Proc. Natl. Acad. Sci. USA 2006, 103, 14825–14830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddique, H.R.; Adhami, V.M.; Parray, A.; Johnson, J.J.; Siddiqui, I.A.; Shekhani, M.T.; Murtaza, I.; Ambartsumian, N.; Konety, B.R.; Mukhtar, H.; et al. The S100A4 Oncoprotein Promotes Prostate Tumorigenesis in a Transgenic Mouse Model, Regulating NFκB through the RAGE Receptor. Genes Cancer 2013, 4, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Kramer, G.; Schwarz, S.; Hägg, M.; Havelka, A.M.; Linder, S. Docetaxel induces apoptosis in hormone refractory prostate carcinomas during multiple treatment cycles. Br. J. Cancer 2006, 94, 1592–1598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tannock, I.F.; de Wit, R.; Berry, W.R.; Horti, J.; Pluzanska, A.; Chi, K.N.; Oudard, S.; Théodore, C.; James, N.D.; Turesson, I.; et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N. Engl. J. Med. 2004, 351, 1502–1512. [Google Scholar] [CrossRef] [PubMed]
- Alexandre, J.; Batteux, F.; Nicco, C.; Chéreau, C.; Laurent, A.; Guillevin, L.; Weill, B.; Goldwasser, F. Accumulation of hydrogen peroxide is an early and crucial step for paclitaxel-induced cancer cell death both in vitro and in vivo. Int. J. Cancer 2006, 119, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Ben Sahra, I.; Laurent, K.; Loubat, A.; Giorgetti-Peraldi, S.; Colosetti, P.; Auberger, P.; Tanti, J.F.; Marchand-Brustel, Y.L.; Bost, F. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene 2008, 27, 3576–3586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardie, D.G.; Alessi, D.R. LKB1 and AMPK and the cancer-metabolism link-ten years after. BMC Biol. 2013, 11, 36. [Google Scholar] [CrossRef] [PubMed]
- Menon, S.; Manning, B.D. Common corruption of the mTOR signaling network in human tumors. Oncogene 2009, 27, S43–S51. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.J.; Thompson, C.B. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell. 2005, 18, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Shao, S.H.; Xu, Z.X.; Hennessy, B.; Ding, Z.; Larrea, M.; Kondo, S.; Dumont, D.J.; Gutterman, J.U.; Walker, C.L.; et al. The energy sensing LKB1–AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat. Cell. Biol. 2007, 9, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, S.; Ross, F.A.; Vara Ciruelos, D.; Gray, A.; Gowans, G.J.; Hardie, D.G. AMPK Causes Cell Cycle Arrest in LKB1-Deficient Cells via Activation of CAMKK2. Mol. Cancer Res. 2016, 8, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Levin, V.A.; Panchabhai, S.C.; Shen, L.; Kornblau, S.M.; Qiu, Y.; Baggerly, K.A. Different changes in protein and phosphoprotein levels result from serum starvation of high-grade glioma and adenocarcinoma cell lines. J. Proteome. Res. 2010, 9, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Aguirre-Gamboa, R.; Gomez-Rueda, H.; Martínez-Ledesma, E.; Martínez-Torteya, A.; Chacolla-Huaringa, R.; Rodriguez-Barrientos, A.; Tamez-Peña, J.G.; Treviño, V. SurvExpress, an online biomarker validation tool and database for cancer gene expression data using survival analysis. PLoS ONE 2013, 8, e74250. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Kollmeyer, T.M.; Morlan, B.W.; Anderson, S.K.; Bergstralh, E.J.; Davis, B.J.; Asmann, Y.W.; Klee, G.G.; Ballman, K.V.; Jenkins, R.B. A tissue biomarker panel predicting systemic progression after PSA recurrence post-definitive prostate cancer therapy. PLoS ONE 2008, 3, e2318. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.C.; Meredith, D.; Fox, J.E.; Manoharan, C.; Davies, A.J.; Halestrap, A.P. Basigin (CD147) is the target for organomercurial inhibition of monocarboxylate transporter isoforms 1 and 4: the ancillary protein for the insensitive MCT2 is EMBIGIN (gp70). J. Biol. Chem. 2005, 280, 27213–27221. [Google Scholar] [CrossRef] [PubMed]
- Pertega-Gomes, N.; Vizcaino, J.R.; Felisbino, S.; Warren, A.Y.; Shaw, G.; Kay, J.; Whitaker, H.; Lynch, A.G.; Fryer, L.; Neal, D.E.; Massie, C.E. Epigenetic and oncogenic regulation of SLC16A7 (MCT2) results in protein over-expression, impacting on signalling and cellular phenotypes in prostate cancer. Oncotarget 2015, 6, 21675–21684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valença, I.; Pértega-Gomes, N.; Vizcaino, J.R.; Henrique, R.M.; Lopes, C.; Baltazar, F.; Ribeiro, D. Localization of MCT2 at peroxisomes is associated with malignant transformation in prostate cancer. J. Cell. Mol. Med. 2015, 19, 723–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, F.R.; Chiche, J.; Marchiq, I.; Naiken, T.; Ilc, K.; Murray, C.M.; Critchlow, S.E.; Roux, D.; Simon, M.P.; Pouysségur, J. CD147 subunit of lactate/H+ symporters MCT1 and hypoxia-inducible MCT4 is critical for energetics and growth of glycolytic tumors. Proc. Natl. Acad. Sci. USA 2011, 108, 16663–16668. [Google Scholar] [Green Version]
- Marchiq, I.; Le, F.R.; Roux, D.; Simon, M.P.; Pouyssegur, J. Genetic disruption of lactate/H+ symporters (MCTs) and their subunit CD147/BASIGIN sensitizes glycolytic tumor cells to phenformin. Cancer Res. 2015, 75, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Carling, D.; Mayer, F.V.; Sanders, M.J.; Gamblin, S.J. AMP-activated protein kinase: nature’s energy sensor. Nat Chem. Biol. 2011, 7, 512–528. [Google Scholar] [CrossRef] [PubMed]
- Dange, M.C.; Bhonsle, H.S.; Godbole, R.K.; More, S.K.; Bane, S.M.; Kulkarni, M.J.; Kalraiyas, R.D. Mass spectrometry based identification of galectin-3 interacting proteins potentially involved in lung cancer metastasis. Mol. BioSyst. 2017, 13, 2303–2309. [Google Scholar] [CrossRef] [PubMed]
- Nakano, A.; Kato, H.; Watanabe, T.; Min, K.D.; Yamazaki, S.; Asano, Y.; Seguchi, O.; Higo, S.; Shintani, Y.; Asanuma, H.; et al. AMPK controls the speed of microtubule polymerization and directional cell migration through CLIP-170 phosphorylation. Nat. Cell. Biol. 2010, 12, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Bae, H.B.; Zmijewski, J.W.; Deshane, J.S.; Tadie, J.M.; Chaplin, D.D.; Takashima, S.; Abraham, E. AMP-activated protein kinase enhances the phagocytic ability of macrophages and neutrophils. FASEB J. 2011, 25, 4358–4368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, E.; Ata, R.; Thavarajah, T.; Medvedev, S.; Bowden, P.; Marshall, J.G.; Antonescu, C.N. AMP-Activated Protein Kinase Regulates the Cell Surface Proteome and Integrin Membrane Traffic. PLoS ONE 2015, 10, e0128013. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Tsukamoto, O.; Nakano, A.; Kato, H.; Kioka, H.; Ito, N.; Higo, S.; Yamazaki, S.; Shintani, Y.; Matsuoka, K.; et al. Augmented AMPK activity inhibits cell migration by phosphorylating the novel substrate Pdlim5. Nat. Commun. 2015, 6, 6137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, C.C.; Lee, K.H.; Lai, I.L.; Wang, D.; Mo, X.; Kulp, S.K.; Shapiro, C.L.; Chen, C.S. AMPK reverses the mesenchymal phenotype of cancer cells by targeting the Akt-MDM2-Foxo3a signaling axis. Cancer Res. 2014, 74, 4783–4795. [Google Scholar] [CrossRef] [PubMed]
- Bungard, D.; Fuerth, B.J.; Zeng, P.Y.; Faubert, B.; Maas, N.L.; Viollet, B.; Carling, D.; Thompson, C.B.; Jones, R.G.; Berger, S.L. Signaling kinase AMPK activates stress-promoted transcription via histone H2B phosphorylation. Science 2010, 329, 1201–1205. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, A.C.; Liu, Y.; Edlind, M.P.; Ingolia, N.T.; Janes, M.R.; Sher, A.; Shi, E.Y.; Stumpf, C.R.; Christensen, C.; Bonham, M.J.; et al. The translational landscape of mTOR signaling steers cancer initiation and metastasis. Nature 2012, 485, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Rosner, M.; Siegel, N.; Valli, A.; Fuchs, C.; Hengstschläger, M. mTOR phosphorylated at S2448 binds to raptor and rictor. Amino Acids 2010, 38, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Ehlers, A.; Burkhardt, L.; Sirma, H.; Steuber, T.; Graefen, M.; Sauter, G.; Minner, S.; Simon, R.; Schlomm, T.; et al. Loss of pSer2448-mTOR expression is linked to adverse prognosis and tumor progression in ERG-fusion-positive cancers. Int. J. Cancer 2013, 132, 1333–1340. [Google Scholar] [CrossRef] [PubMed]
- Mertz, K.D.; Setlur, S.R.; Dhanasekaran, S.M.; Demichelis, F.; Perner, S.; Tomlins, S.; Tchinda, J.; Laxman, B.; Vessella, R.L.; Beroukhimt, R.; et al. Molecular characterization of TMPRSS2-ERG gene fusion in the NCI-H660 prostate cancer cell line: a new perspective for an old model. Neoplasia 2007, 9, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.P.; Li, J.; Yadav, S.S.; Tewari, A.K. Recent insights into NF-kB signaling pathways and the link between inflammation and prostate cancer. BJU Int. 2014, 114, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Dan, H.C.; Cooper, M.J.; Cogswell, P.C.; Duncan, J.A.; Ting, J.P.; Baldwin, A.S. Akt dependent regulation of NF-κB is controlled by mTOR and Raptor in association with IKK. Genes Dev. 2008, 22, 1490–1500. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Gartenhaus, R.B.; Lapidus, R.G.; Hussain, A.; Zhang, Y.; Wang, X.; Wang, X.H. Differential IKK/NF-κB Activity Is Mediated by TSC2 through mTORC1 in PTEN-Null Prostate Cancer and Tuberous Sclerosis Complex Tumor Cells. Mol. Cancer Res. 2015, 13, 1602–1614. [Google Scholar] [CrossRef] [PubMed]
- Tomlins, S.A.; Laxman, B.; Dhanasekaran, S.M.; Helgeson, B.E.; Cao, X.; Morris, D.S.; Menon, A.; Jing, X.J.; Cao Q Han, B.; et al. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature 2007, 448, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.D.; Jin, F.; Shin, S.; Oh, S.; Lightfoot, S.A.; Grande, J.P.; Johnson, A.J.; van Deursen, J.M.; Wren, J.D.; Janknecht, R. Histone demethylase JMJD2A drives prostate tumorigenesis through transcription factor ETV1. J. Clin. Investig. 2016, 126, 706–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakaguchi, M.; Watanabe, M.; Kinoshita, R.; Kaku, H.; Ueki, H.; Futami, J.; Murata, H.; Inoue, Y.; Li, S.A.; Huang, P.; et al. Dramatic increase in expression of a transgene by insertion of promoters downstream of the cargo gene. Mol. Biotechnol. 2014, 56, 621–630. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruma, I.M.W.; Kinoshita, R.; Tomonobu, N.; Inoue, Y.; Kondo, E.; Yamauchi, A.; Sato, H.; Sumardika, I.W.; Chen, Y.; Yamamoto, K.-I.; et al. Embigin Promotes Prostate Cancer Progression by S100A4-Dependent and-Independent Mechanisms. Cancers 2018, 10, 239. https://doi.org/10.3390/cancers10070239
Ruma IMW, Kinoshita R, Tomonobu N, Inoue Y, Kondo E, Yamauchi A, Sato H, Sumardika IW, Chen Y, Yamamoto K-I, et al. Embigin Promotes Prostate Cancer Progression by S100A4-Dependent and-Independent Mechanisms. Cancers. 2018; 10(7):239. https://doi.org/10.3390/cancers10070239
Chicago/Turabian StyleRuma, I Made Winarsa, Rie Kinoshita, Nahoko Tomonobu, Yusuke Inoue, Eisaku Kondo, Akira Yamauchi, Hiroki Sato, I Wayan Sumardika, Youyi Chen, Ken-Ichi Yamamoto, and et al. 2018. "Embigin Promotes Prostate Cancer Progression by S100A4-Dependent and-Independent Mechanisms" Cancers 10, no. 7: 239. https://doi.org/10.3390/cancers10070239