Circulating Tumor Cells Develop Resistance to TRAIL-Induced Apoptosis Through Autophagic Removal of Death Receptor 5: Evidence from an In Vitro Model



Abstract
1. Introduction
2. Results
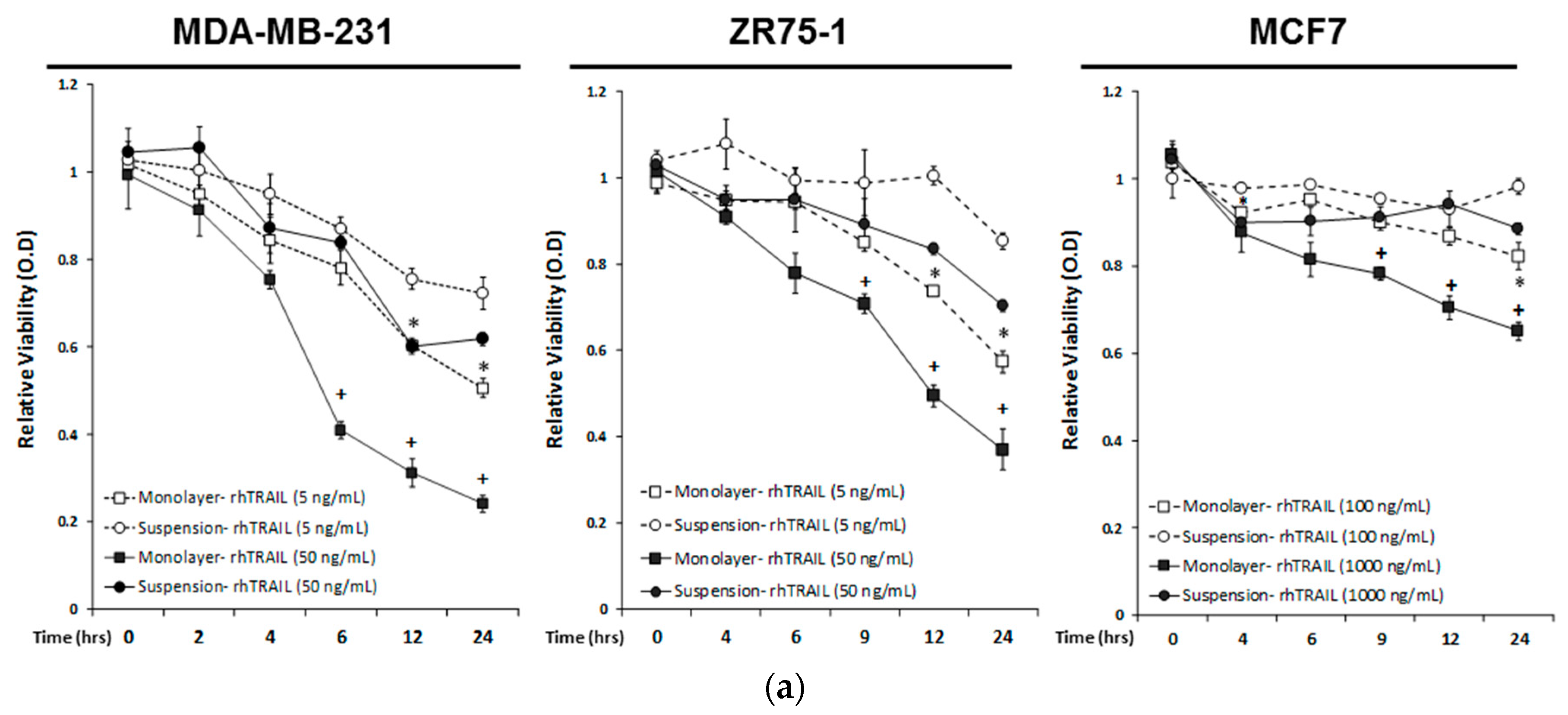
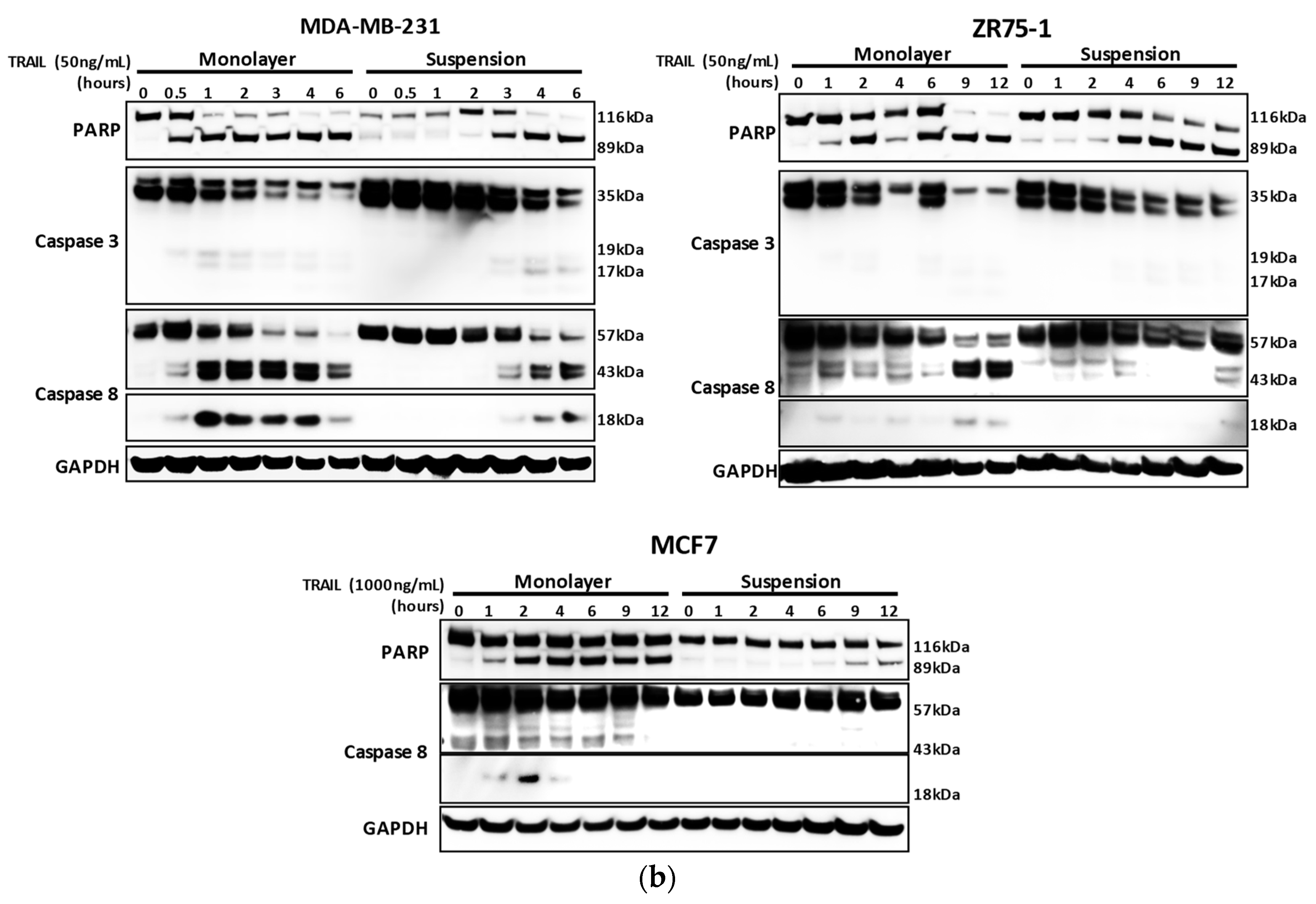
2.1. Breast Cancer Cells Develop Resistance to Recombinant Human (rh)TRAIL Induced Apoptosis under Non-Adherent Culture Conditions
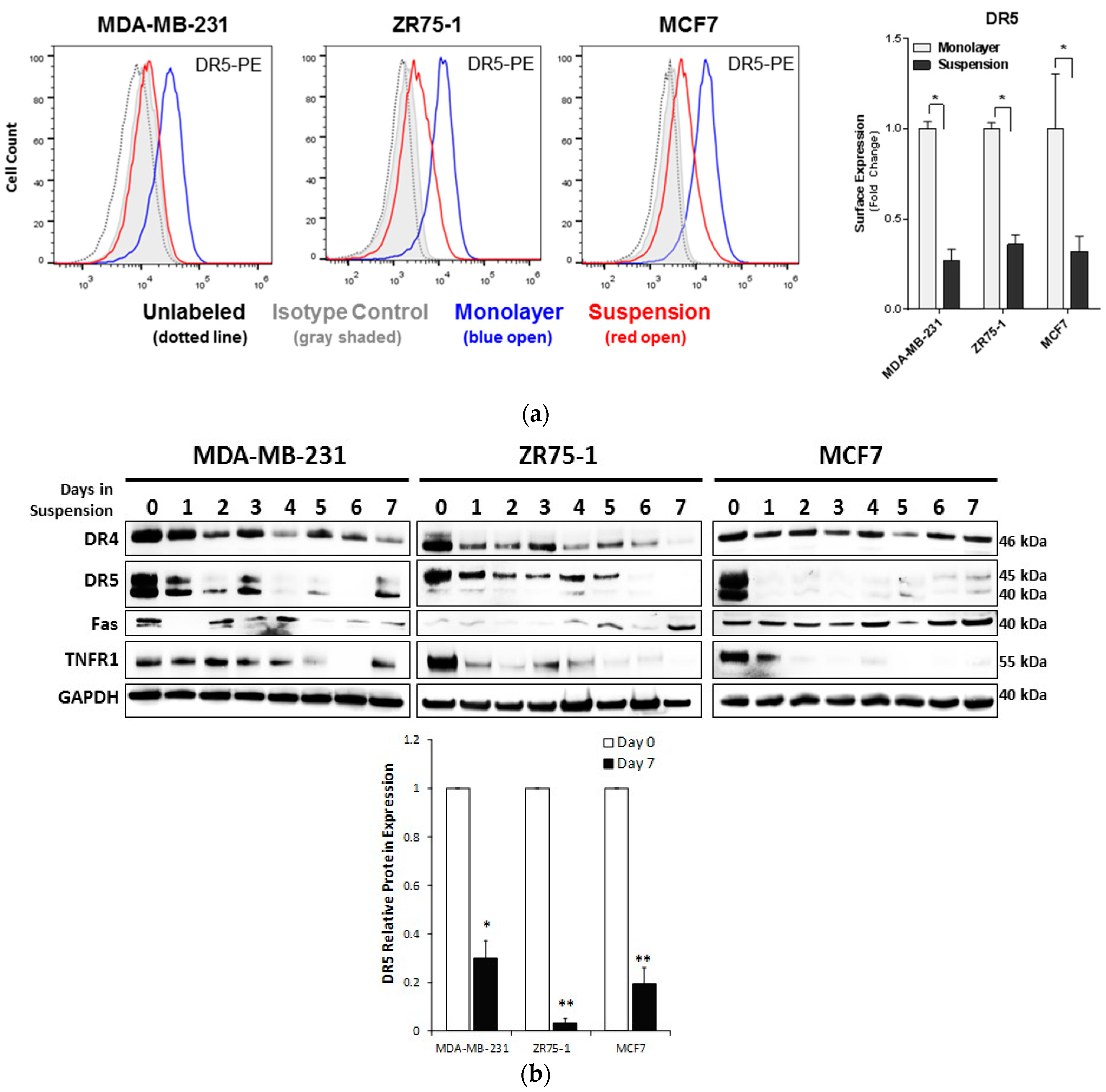
2.2. Non-Adherent Culture Decreases the DR5 Surface and Total Protein Expression
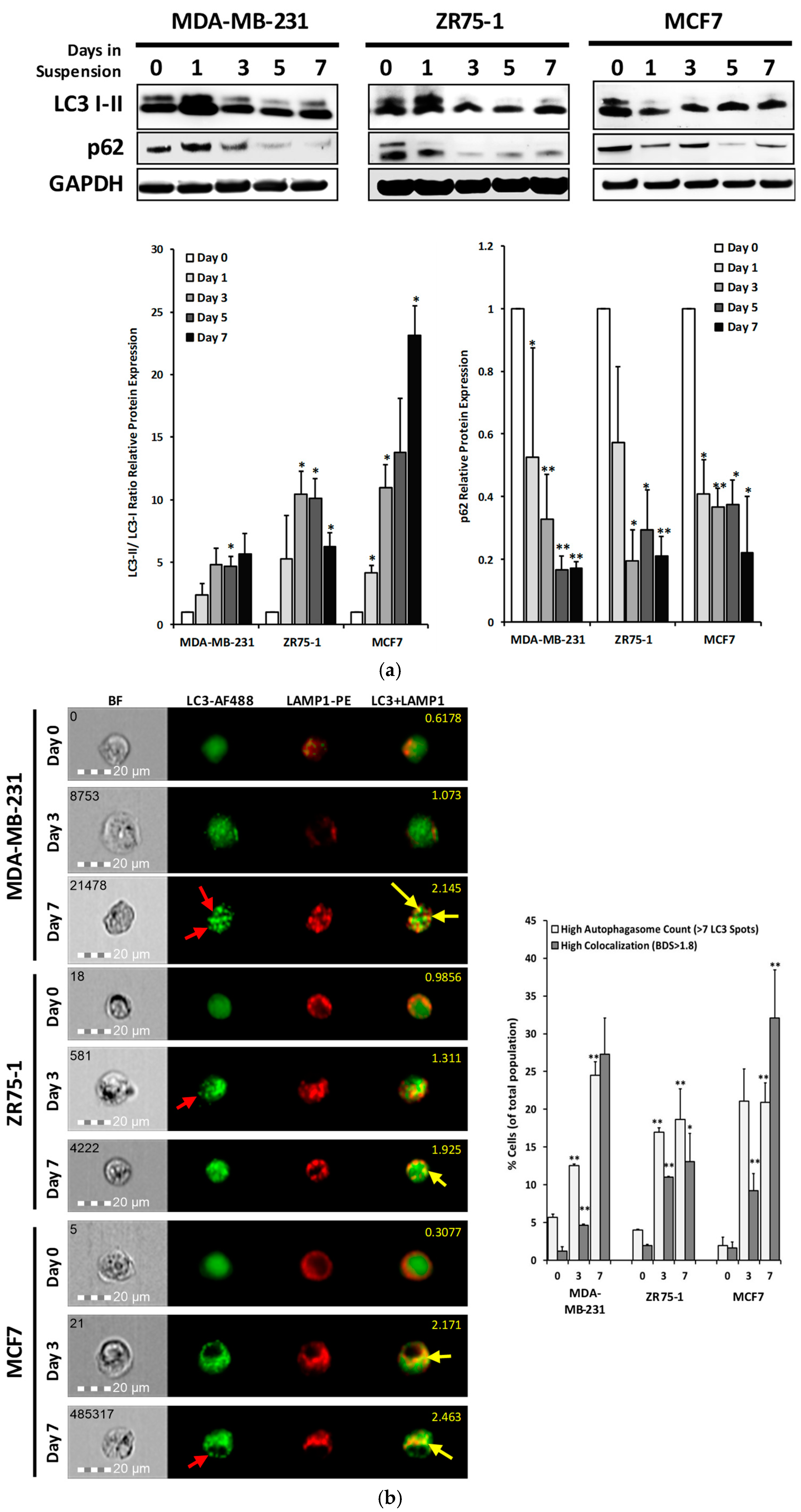
2.3. Autophagic Flux is Upregulated in Cells Cultured in Non-Adherent Conditions
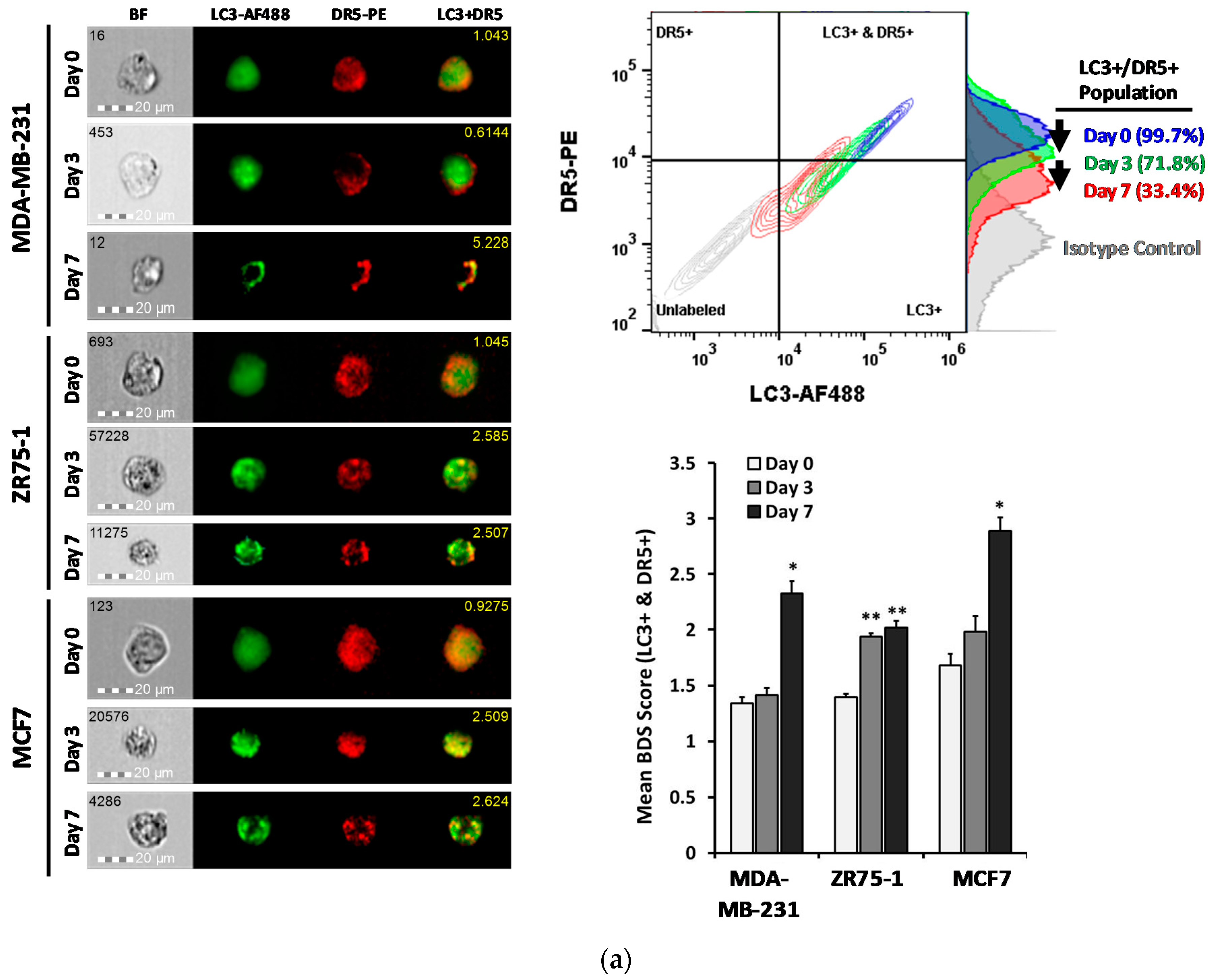
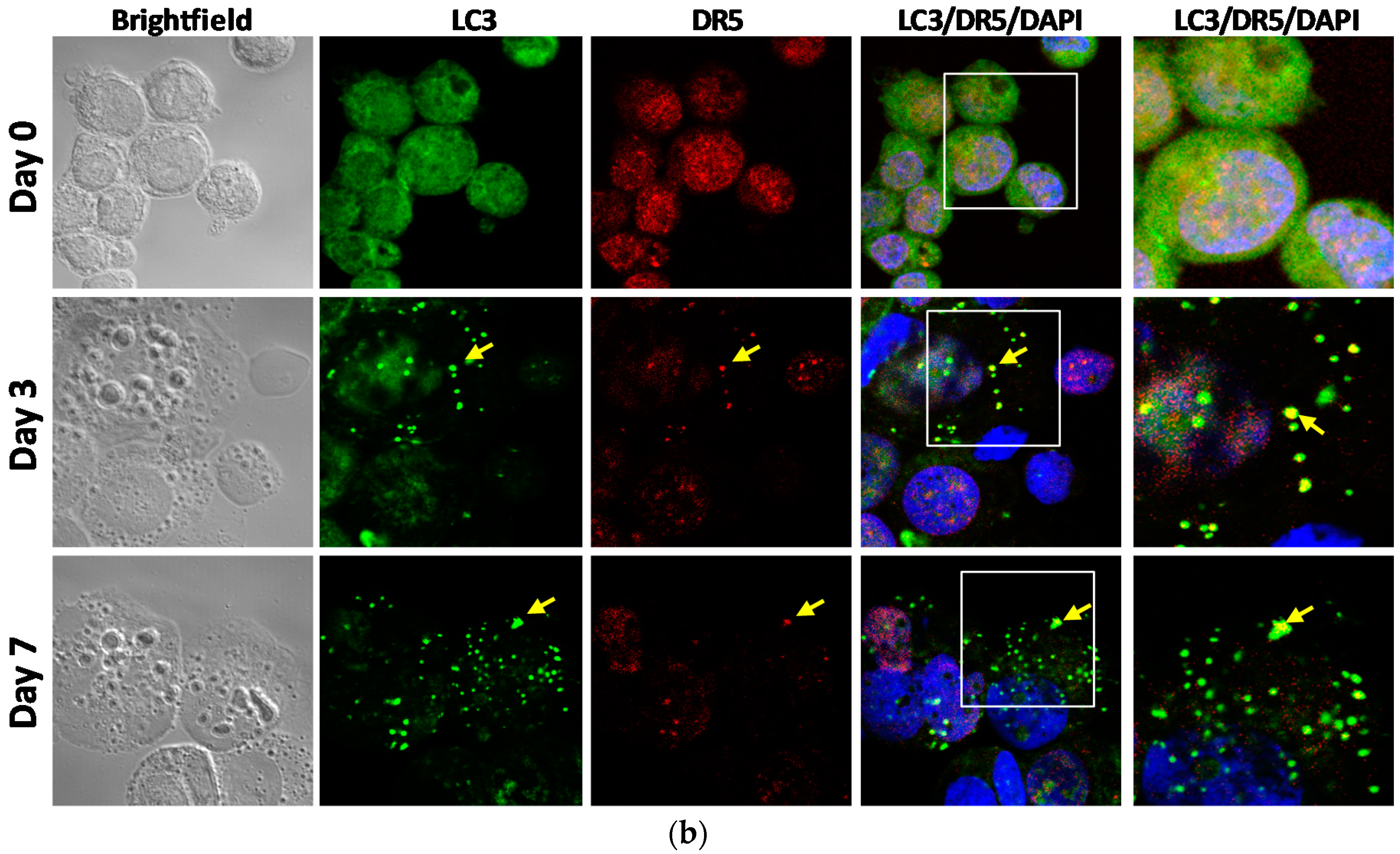
2.4. DR5 is Localized to Autophagosomes for Degradation
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture Conditions
4.2. TRAIL Cytotoxicity Assay
4.3. Cell Viability Assays
4.4. Flow Cytometry Detection of Surface Death Receptors
4.5. Western Blotting
4.6. Gene Expression Analysis
4.7. Imaging Flow Cytometry for Autophagic Flux Analysis
4.8. Immunocytochemistry
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- León-Mateos, L.; Casas, H.; Abalo, A.; Vieito, M.; Abreu, M.; Anido, U.; Gómez-Tato, A.; López, R.; Abal, M.; Muinelo-Romay, L. Improving circulating tumor cells enumeration and characterization to predict outcome in first line chemotherapy mCRPC patients. Oncotarget 2017, 8, 54708–54721. [Google Scholar] [CrossRef] [PubMed]
- Smerage, J.B.; Barlow, W.E.; Hortobagyi, G.N.; Winer, E.P.; Leyland-Jones, B.; Srkalovic, G.; Tejwani, S.; Schott, A.F.; O‘Rourke, M.A.; Lew, D.L.; et al. Circulating tumor cells and response to chemotherapy in metastatic breast cancer: SWOG S0500. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2014, 32, 3483–3489. [Google Scholar] [CrossRef] [PubMed]
- Cristofanilli, M. Circulating Tumor Cells, Disease Progression, and Survival in Metastatic Breast Cancer. Semin. Oncol. 2006, 33, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.P.; Bahig, H.; Wang, J.; Cardenas, C.E.; Lucci, A.; Hall, C.S.; Meas, S.; Sarli, V.N.; Yuan, Y.; Urbauer, D.L.; et al. Predicting treatment Response based on Dual assessment of magnetic resonance Imaging kinetics and Circulating Tumor cells in patients with Head and Neck cancer (PREDICT-HN): Matching ‘liquid biopsy‘ and quantitative tumor modeling. BMC Cancer 2018, 18, 903. [Google Scholar] [CrossRef]
- Keup, C.; Mach, P.; Aktas, B.; Tewes, M.; Kolberg, H.-C.; Hauch, S.; Sprenger-Haussels, M.; Kimmig, R.; Kasimir-Bauer, S. RNA Profiles of Circulating Tumor Cells and Extracellular Vesicles for Therapy Stratification of Metastatic Breast Cancer Patients. Clin. Chem. 2018, 64, 1054–1062. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.-T.; Cui, X.; Chen, Q.; Li, Y.-F.; Cui, Y.-H.; Wang, Y.; Jiang, J. Circulating tumor cell status monitors the treatment responses in breast cancer patients: A meta-analysis. Sci. Rep. 2017, 7, 43464. [Google Scholar] [CrossRef] [PubMed]
- Politaki, E.; Agelaki, S.; Apostolaki, S.; Hatzidaki, D.; Strati, A.; Koinis, F.; Perraki, M.; Saloustrou, G.; Stoupis, G.; Kallergi, G.; et al. A Comparison of Three Methods for the Detection of Circulating Tumor Cells in Patients with Early and Metastatic Breast Cancer. Cell. Physiol. Biochem. 2017, 44, 594–606. [Google Scholar] [CrossRef]
- Janni, W.J.; Rack, B.; Terstappen, L.W.M.M.; Pierga, J.-Y.; Taran, F.-A.; Fehm, T.; Hall, C.; de Groot, M.R.; Bidard, F.-C.; Friedl, T.W.P.; et al. Pooled Analysis of the Prognostic Relevance of Circulating Tumor Cells in Primary Breast Cancer. Clin. Cancer Res. 2016, 22, 2583. [Google Scholar] [CrossRef]
- Hartkopf, A.D.; Wagner, P.; Wallwiener, D.; Fehm, T.; Rothmund, R. Changing Levels of Circulating Tumor Cells in Monitoring Chemotherapy Response in Patients with Metastatic Breast Cancer. Anticancer Res. 2011, 31, 979–984. [Google Scholar]
- Kim, Y.-N.; Koo, K.H.; Sung, J.Y.; Yun, U.-J.; Kim, H. Anoikis Resistance: An Essential Prerequisite for Tumor Metastasis. Int. J. Cell Biol. 2012, 2012. [Google Scholar] [CrossRef]
- Mitchell, M.J.; King, M.R. Fluid Shear Stress Sensitizes Cancer Cells to Receptor-Mediated Apoptosis via Trimeric Death Receptors. New J. Phys. 2013, 15, 015008. [Google Scholar] [CrossRef] [PubMed]
- Rejniak, K.A. Circulating Tumor Cells: When a Solid Tumor Meets a Fluid Microenvironment. Adv. Exp. Med. Biol. 2016, 936, 93–106. [Google Scholar]
- Paoli, P.; Giannoni, E.; Chiarugi, P. Anoikis molecular pathways and its role in cancer progression. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2013, 1833, 3481–3498. [Google Scholar] [CrossRef] [PubMed]
- Laguinge, L.M.; Samara, R.N.; Wang, W.; El-Deiry, W.S.; Corner, G.; Augenlicht, L.; Mishra, L.; Jessup, J.M. TRAIL DR5 Receptor Mediates Anoikis in Human Colorectal Carcinoma Cell Lines. Cancer Res. 2008, 68, 909. [Google Scholar] [CrossRef] [PubMed]
- Weiss, L.; Mayhew, E.; Rapp, D.G.; Holmes, J.C. Metastatic inefficiency in mice bearing B16 melanomas. Br. J. Cancer 1982, 45, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Fung, C.; Lock, R.; Gao, S.; Salas, E.; Debnath, J. Induction of Autophagy during Extracellular Matrix Detachment Promotes Cell Survival. Mol. Biol. Cell 2008, 19, 797–806. [Google Scholar] [CrossRef]
- Avivar-Valderas, A.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Bardeesy, N.; Debnath, J.; Aguirre-Ghiso, J.A. Regulation of autophagy during ECM detachment is linked to a selective inhibition of mTORC1 by PERK. Oncogene 2013, 32, 4932–4940. [Google Scholar] [CrossRef]
- Lippai, M.; Low, P. The Role of the Selective Adaptor p62 and Ubiquitin-Like Proteins in Autophagy. BioMed Res. Int. 2014, 2014, 11. [Google Scholar] [CrossRef]
- Yang, X.; Yu, D.-D.; Yan, F.; Jing, Y.-Y.; Han, Z.-P.; Sun, K.; Liang, L.; Hou, J.; Wei, L.-X. The role of autophagy induced by tumor microenvironment in different cells and stages of cancer. Cell Biosci. 2015, 5, 14. [Google Scholar] [CrossRef]
- Ojha, R.; Jha, V.; Singh, S.K.; Bhattacharyya, S. Autophagy inhibition suppresses the tumorigenic potential of cancer stem cell enriched side population in bladder cancer. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2014, 1842, 2073–2086. [Google Scholar] [CrossRef]
- Apel, A.; Herr, I.; Schwarz, H.; Rodemann, H.P.; Mayer, A. Blocked Autophagy Sensitizes Resistant Carcinoma Cells to Radiation Therapy. Cancer Res. 2008, 68, 1485. [Google Scholar] [CrossRef] [PubMed]
- Eritja, N.; Chen, B.-J.; Rodríguez-Barrueco, R.; Santacana, M.; Gatius, S.; Vidal, A.; Martí, M.D.; Ponce, J.; Bergadà, L.; Yeramian, A.; et al. Autophagy orchestrates adaptive responses to targeted therapy in endometrial cancer. Autophagy 2017, 13, 608–624. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, B. TRAIL Resistance of Breast Cancer Cells Is Associated with Constitutive Endocytosis of Death Receptors 4 and 5. Mol. Cancer Res. 2008, 6, 1861–1871. [Google Scholar] [CrossRef]
- Di, X.; Zhang, G.; Zhang, Y.; Takeda, K.; Rivera Rosado, L.A.; Zhang, B. Accumulation of autophagosomes in breast cancer cells induces TRAIL resistance through downregulation of surface expression of death receptors 4 and 5. Oncotarget 2013, 4, 1349–1364. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yang, X.; Xu, T.; Kong, Q.; Zhang, Y.; Shen, Y.; Wei, Y.; Wang, G.; Chang, K.-J. Overcoming resistance to TRAIL-induced apoptosis in solid tumor cells by simultaneously targeting death receptors, c-FLIP and IAPs. Int. J. Oncol. 2016, 49, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhou, J.-Y.; Wei, W.-Z.; Wu, G.S. Activation of the Akt Survival Pathway Contributes to TRAIL Resistance in Cancer Cells. PLoS ONE 2010, 5, e10226. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.-F.; Yang, X.-R.; Zhou, J.; Qiu, S.-J.; Fan, J.; Xu, Y. Circulating tumor cells: Advances in detection methods, biological issues, and clinical relevance. J. Cancer Res. Clin. Oncol. 2011, 137, 1151–1173. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.M.; Ramani, V.C.; Jeffrey, S.S. Circulating tumor cell technologies. Mol. Oncol. 2016, 10, 374–394. [Google Scholar] [CrossRef]
- Kallergi, G.; Politaki, E.; Alkahtani, S.; Stournaras, C.; Georgoulias, V. Evaluation of Isolation Methods for Circulating Tumor Cells (CTCs). Cell. Physiol. Biochem. 2016, 40, 411–419. [Google Scholar] [CrossRef]
- Au, S.H.; Edd, J.; Stoddard, A.E.; Wong, K.H.K.; Fachin, F.; Maheswaran, S.; Haber, D.A.; Stott, S.L.; Kapur, R.; Toner, M. Microfluidic Isolation of Circulating Tumor Cell Clusters by Size and Asymmetry. Sci. Rep. 2017, 7, 2433. [Google Scholar] [CrossRef]
- Adams, D.L.; Zhu, P.; Makarova, O.V.; Martin, S.S.; Charpentier, M.; Chumsri, S.; Li, S.; Amstutz, P.; Tang, C.-M. The systematic study of circulating tumor cell isolation using lithographic microfilters. RSC Adv. 2014, 9, 4334–4342. [Google Scholar] [CrossRef] [PubMed]
- Calvet, C.Y.; André, F.M.; Mir, L.M. The Culture of Cancer Cell Lines as Tumorspheres Does Not Systematically Result in Cancer Stem Cell Enrichment. PLoS ONE 2014, 9, e89644. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, S.; Marshall, J.R.; Messing, J.A.; Hsu, J.-W.; King, M.R. TRAIL-Mediated Apoptosis in Breast Cancer Cells Cultured as 3D Spheroids. PLoS ONE 2014, 9, e111487. [Google Scholar] [CrossRef]
- Liu, G.-C.; Zhang, J.; Liu, S.-G.; Gao, R.; Long, Z.-F.; Tao, K.; Ma, Y.-F. Detachment of esophageal carcinoma cells from extracellular matrix causes relocalization of death receptor 5 and apoptosis. World J. Gastroenterol. WJG 2009, 15, 836–844. [Google Scholar] [CrossRef] [PubMed]
- Smit, L.; Berns, K.; Spence, K.; Ryder, W.D.; Zeps, N.; Madiredjo, M.; Beijersbergen, R.; Bernards, R.; Clarke, R.B. An integrated genomic approach identifies that the PI3K/AKT/FOXO pathway is involved in breast cancer tumor initiation. Oncotarget 2016, 7, 2596–2610. [Google Scholar] [CrossRef] [PubMed]
- Nanou, A.; Crespo, M.; Flohr, P.; De Bono, J.; Terstappen, L. Scanning Electron Microscopy of Circulating Tumor Cells and Tumor-Derived Extracellular Vesicles. Cancers 2018, 10, 416. [Google Scholar] [CrossRef]
- Chen, J.-J.; Jennifer Shen, H.C.; Rivera Rosado, L.A.; Zhang, Y.; Di, X.; Zhang, B. Mislocalization of death receptors correlates with cellular resistance to their cognate ligands in human breast cancer cells. Oncotarget 2012, 3, 833–842. [Google Scholar] [PubMed]
- Kagawa, S.; Gu, J.; Honda, T.; McDonnell, T.J.; Swisher, S.G.; Roth, J.A.; Fang, B. Deficiency of Caspase-3 in MCF7 Cells Blocks Bax-mediated Nuclear Fragmentation but not Cell Death. Clin. Cancer Res. 2001, 7, 1474–1480. [Google Scholar] [PubMed]
- Dufour, F.; Rattier, T.; Constantinescu, A.A.; Zischler, L.; Morlé, A.; Ben Mabrouk, H.; Humblin, E.; Jacquemin, G.; Szegezdi, E.; Delacote, F.; et al. TRAIL receptor gene editing unveils TRAIL-R1 as a master player of apoptosis induced by TRAIL and ER stress. Oncotarget 2016, 8, 9974–9985. [Google Scholar] [CrossRef]
- Zamai, L.; Ponti, C.; Mirandola, P.; Gobbi, G.; Papa, S.; Galeotti, L.; Cocco, L.; Vitale, M. NK Cells and Cancer. J. Immunol. 2007, 178, 4011–4016. [Google Scholar] [CrossRef]
- Park, J.Y.; Jeong, A.L.; Joo, H.J.; Han, S.; Kim, S.-H.; Kim, H.-Y.; Lim, J.-S.; Lee, M.-S.; Choi, H.-K.; Yang, Y. Development of suspension cell culture model to mimic circulating tumor cells. Oncotarget 2018, 9, 622–640. [Google Scholar] [CrossRef] [PubMed]
- Gencoglu, M.F.; Barney, L.E.; Hall, C.L.; Brooks, E.A.; Schwartz, A.D.; Corbett, D.C.; Stevens, K.R.; Peyton, S.R. Comparative Study of Multicellular Tumor Spheroid Formation Methods and Implications for Drug Screening. ACS Biomater. Sci. Eng. 2018, 4, 410–420. [Google Scholar] [CrossRef] [PubMed]
- Gravelle, P.; Jean, C.; Familiades, J.; Decaup, E.; Blanc, A.; Bezombes-Cagnac, C.; Laurent, C.; Savina, A.; Fournié, J.-J.; Laurent, G. Cell Growth in Aggregates Determines Gene Expression, Proliferation, Survival, Chemoresistance, and Sensitivity to Immune Effectors in Follicular Lymphoma. Am. J. Pathol. 2014, 184, 282–295. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-J.; Knudsen, S.; Mazin, W.; Dahlgaard, J.; Zhang, B. A 71-Gene Signature of TRAIL Sensitivity in Cancer Cells. Mol. Cancer Ther. 2012, 11, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Eckhardt, S.G.; Kurzrock, R.; Ebbinghaus, S.; O’Dwyer, P.J.; Gordon, M.S.; Novotny, W.; Goldwasser, M.A.; Tohnya, T.M.; Lum, B.L.; et al. Phase I Dose-Escalation Study of Recombinant Human Apo2L/TRAIL, a Dual Proapoptotic Receptor Agonist, in Patients With Advanced Cancer. J. Clin. Oncol. 2010, 28, 2839–2846. [Google Scholar] [CrossRef] [PubMed]
- Hotte, S.J.; Hirte, H.W.; Chen, E.X.; Siu, L.L.; Le, L.H.; Corey, A.; Iacobucci, A.; MacLean, M.; Lo, L.; Fox, N.L.; et al. A Phase 1 Study of Mapatumumab (Fully Human Monoclonal Antibody to TRAIL-R1) in Patients with Advanced Solid Malignancies. Clin. Cancer Res. 2008, 14, 3450–3455. [Google Scholar] [CrossRef] [PubMed]
- Forero-Torres, A.; Shah, J.; Wood, T.; Posey, J.; Carlisle, R.; Copigneaux, C.; Luo, F.R.; Wojtowicz-Praga, S.; Percent, I.; Saleh, M. Phase I trial of weekly tigatuzumab, an agonistic humanized monoclonal antibody targeting death receptor 5 (DR5). Cancer Biother. Radiopharm. 2010, 25, 13–19. [Google Scholar] [CrossRef]
- Camidge, D.R. Apomab: An agonist monoclonal antibody directed against Death Receptor 5/TRAIL-Receptor 2 for use in the treatment of solid tumors. Expert Opin. Biol. Ther. 2008, 8, 1167–1176. [Google Scholar] [CrossRef]
- Han, J.; Hou, W.; Goldstein, L.A.; Lu, C.; Stolz, D.B.; Yin, X.-M.; Rabinowich, H. Involvement of Protective Autophagy in TRAIL Resistance of Apoptosis-defective Tumor Cells. J. Biol. Chem. 2008, 283, 19665–19677. [Google Scholar] [CrossRef]
- Monma, H.; Iida, Y.; Moritani, T.; Okimoto, T.; Tanino, R.; Tajima, Y.; Harada, M. Chloroquine augments TRAIL-induced apoptosis and induces G2/M phase arrest in human pancreatic cancer cells. PLoS ONE 2018, 13, e0193990. [Google Scholar] [CrossRef]
- Reis, C.R.; van der Sloot, A.M.; Natoni, A.; Szegezdi, E.; Setroikromo, R.; Meijer, M.; Sjollema, K.; Stricher, F.; Cool, R.H.; Samali, A.; et al. Rapid and efficient cancer cell killing mediated by high-affinity death receptor homotrimerizing TRAIL variants. Cell Death Dis. 2010, 1, e83. [Google Scholar] [CrossRef] [PubMed]
- Rajan, R.; Karbowniczek, M.; Pugsley, H.R.; Sabnani, M.K.; Astrinidis, A.; La-Beck, N.M. Quantifying autophagosomes and autolysosomes in cells using imaging flow cytometry. Cytom. Part A 2015, 87, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Phadwal, K.; Alegre-Abarrategui, J.; Watson, A.S.; Pike, L.; Anbalagan, S.; Hammond, E.M.; Wade-Martins, R.; McMichael, A.; Klenerman, P.; Simon, A.K. A novel method for autophagy detection in primary cells. Autophagy 2012, 8, 677–689. [Google Scholar] [CrossRef] [PubMed]
- Pugsley, H.R. Quantifying autophagy: Measuring LC3 puncta and autolysosome formation in cells using multispectral imaging flow cytometry. Methods 2017, 112, 147–156. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Gene ID | MDA-MB-231 Day 7/Day 0 | MCF7 Day 7/Day 0 |
|---|---|---|---|
| DR4 | TNFRSF10a | −0.711 (0.034) | 0.066 (0.835) |
| DR5 | TNFRSF10b | 0.281 (0.260) | 0.302 (0.386) |
| Fas | FAS | 0.408 (0.293) | 0.029 (0.976) |
| TNFR1 | TNFRSF1A | 0.151 (0.530) | 0.220 (0.188) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Twomey, J.D.; Zhang, B. Circulating Tumor Cells Develop Resistance to TRAIL-Induced Apoptosis Through Autophagic Removal of Death Receptor 5: Evidence from an In Vitro Model. Cancers 2019, 11, 94. https://doi.org/10.3390/cancers11010094
Twomey JD, Zhang B. Circulating Tumor Cells Develop Resistance to TRAIL-Induced Apoptosis Through Autophagic Removal of Death Receptor 5: Evidence from an In Vitro Model. Cancers. 2019; 11(1):94. https://doi.org/10.3390/cancers11010094
Chicago/Turabian StyleTwomey, Julianne D., and Baolin Zhang. 2019. "Circulating Tumor Cells Develop Resistance to TRAIL-Induced Apoptosis Through Autophagic Removal of Death Receptor 5: Evidence from an In Vitro Model" Cancers 11, no. 1: 94. https://doi.org/10.3390/cancers11010094
APA StyleTwomey, J. D., & Zhang, B. (2019). Circulating Tumor Cells Develop Resistance to TRAIL-Induced Apoptosis Through Autophagic Removal of Death Receptor 5: Evidence from an In Vitro Model. Cancers, 11(1), 94. https://doi.org/10.3390/cancers11010094