NGF-Enhanced Vasculogenic Properties of Epithelial Ovarian Cancer Cells Is Reduced by Inhibition of the COX-2/PGE2 Signaling Axis

 , ,
, ,  , and
, and
Abstract
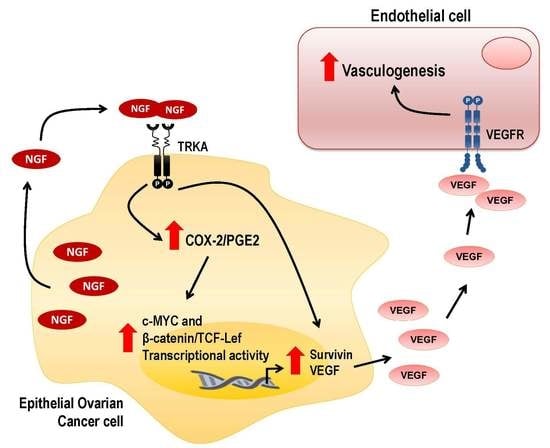
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
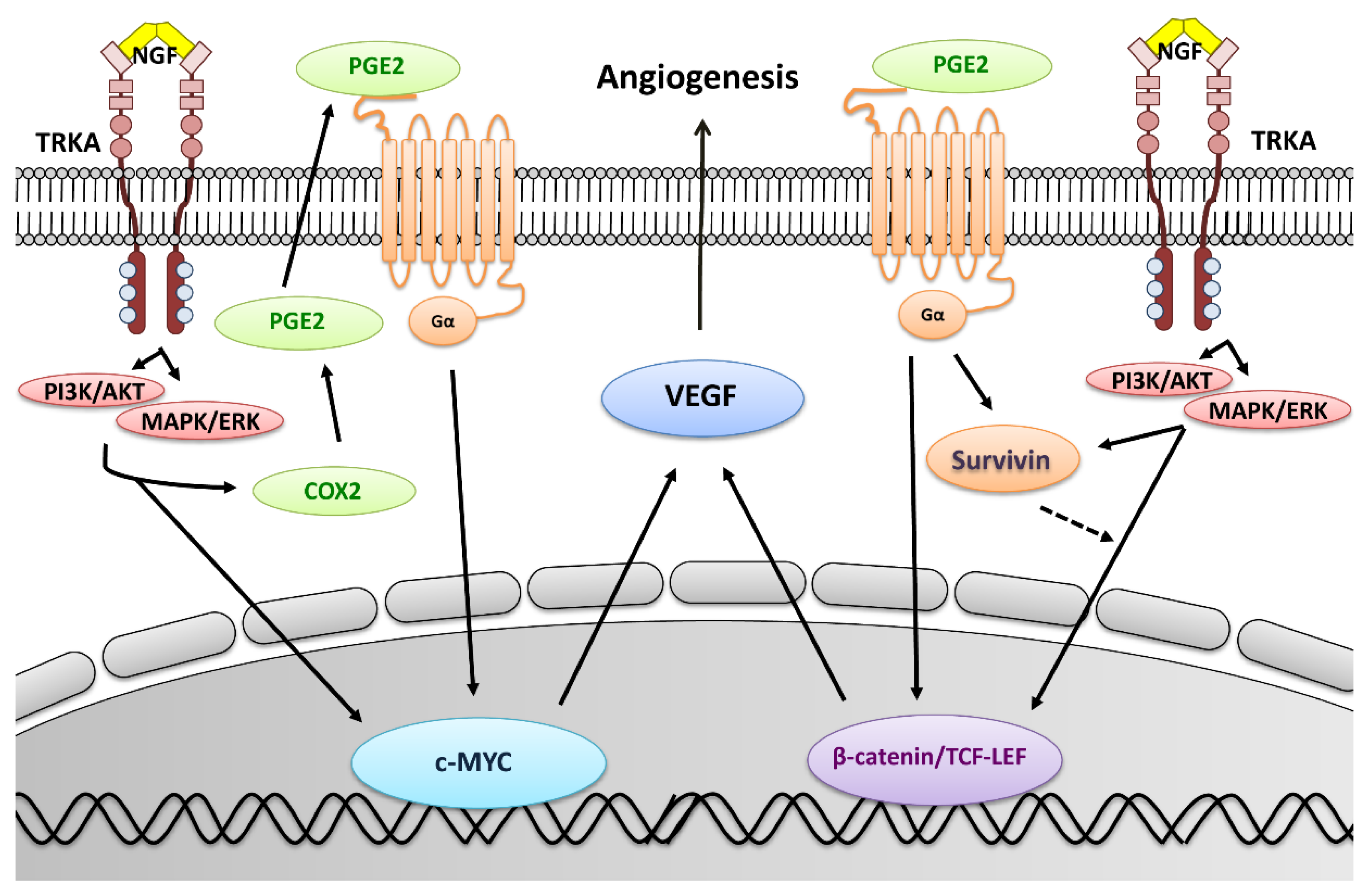
2.1. COX-2 Expression Increases During EOC Progression
2.2. NGF Increases COX-2 Expression in EOC Cells
2.3. NGF Increases PGE2 Secretion by EOC Cells
2.4. Role of COX/2-PGE2 in VEGF Secretion by Ovarian Cells
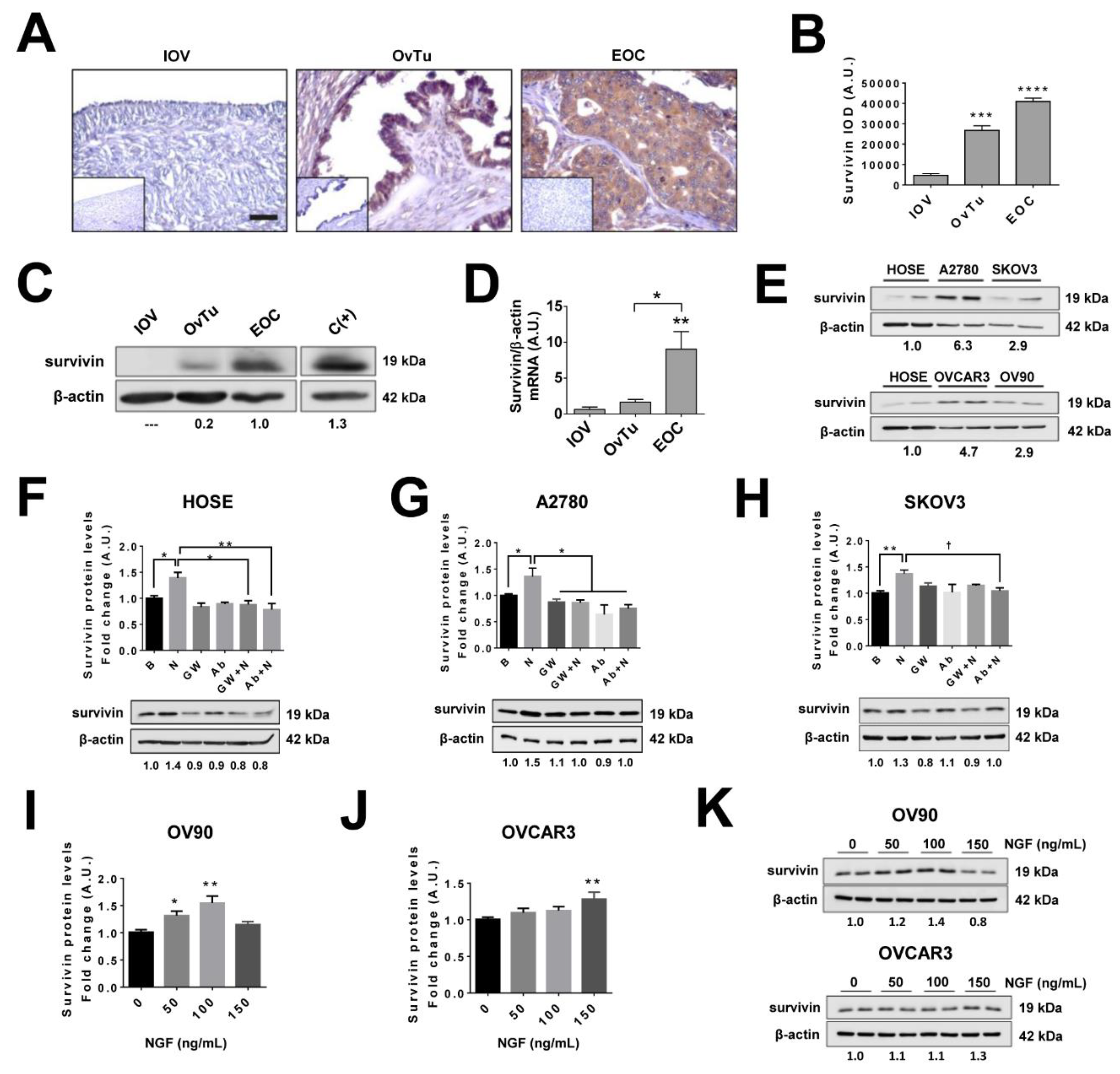
2.5. Survivin Levels Increase During EOC Progression
2.6. NGF Increases Survivin Levels in Epithelial Ovarian Cells
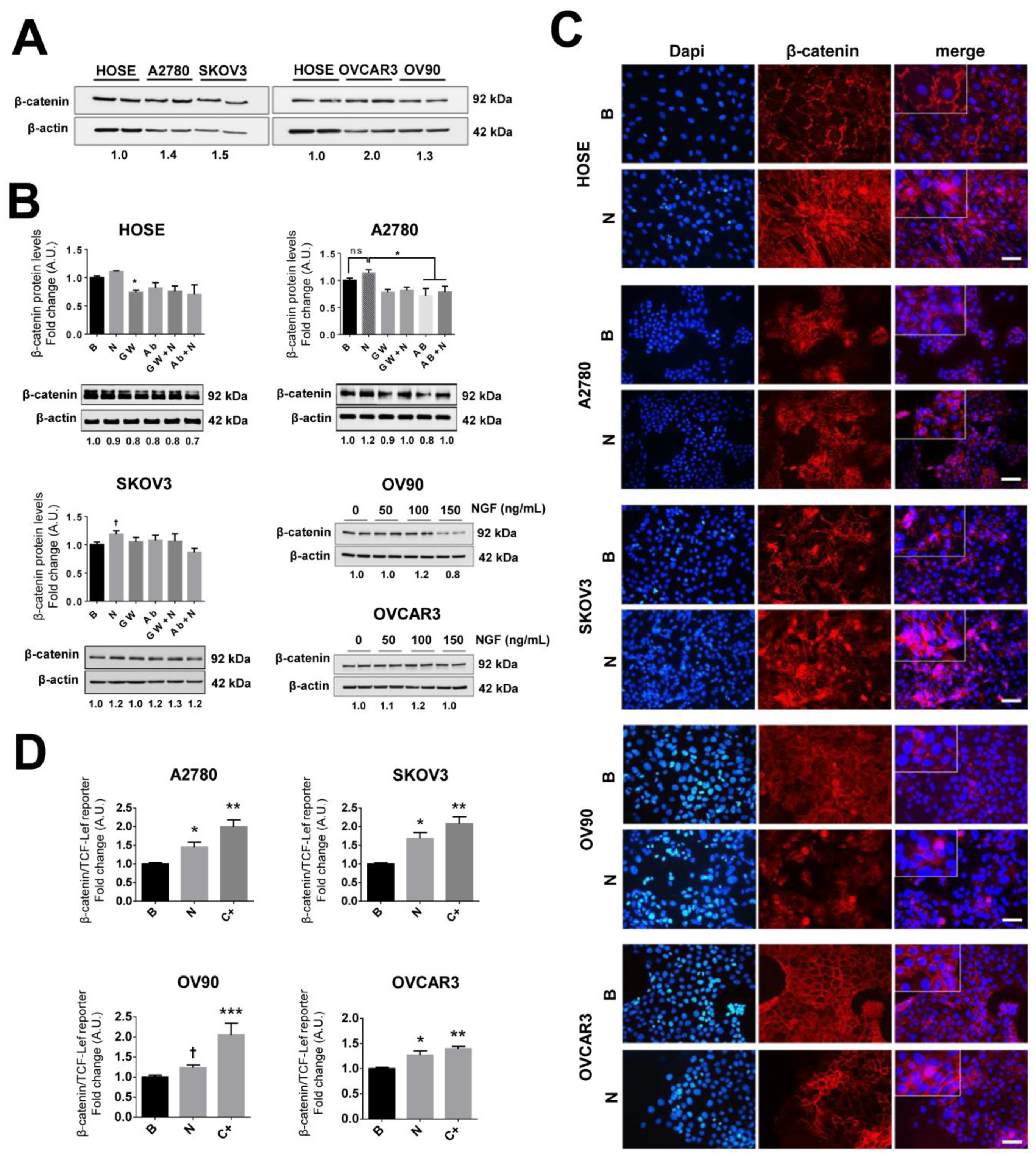
2.7. NGF Increases β-Catenin-TCF-Lef Transcriptional Activity in EOC Cells
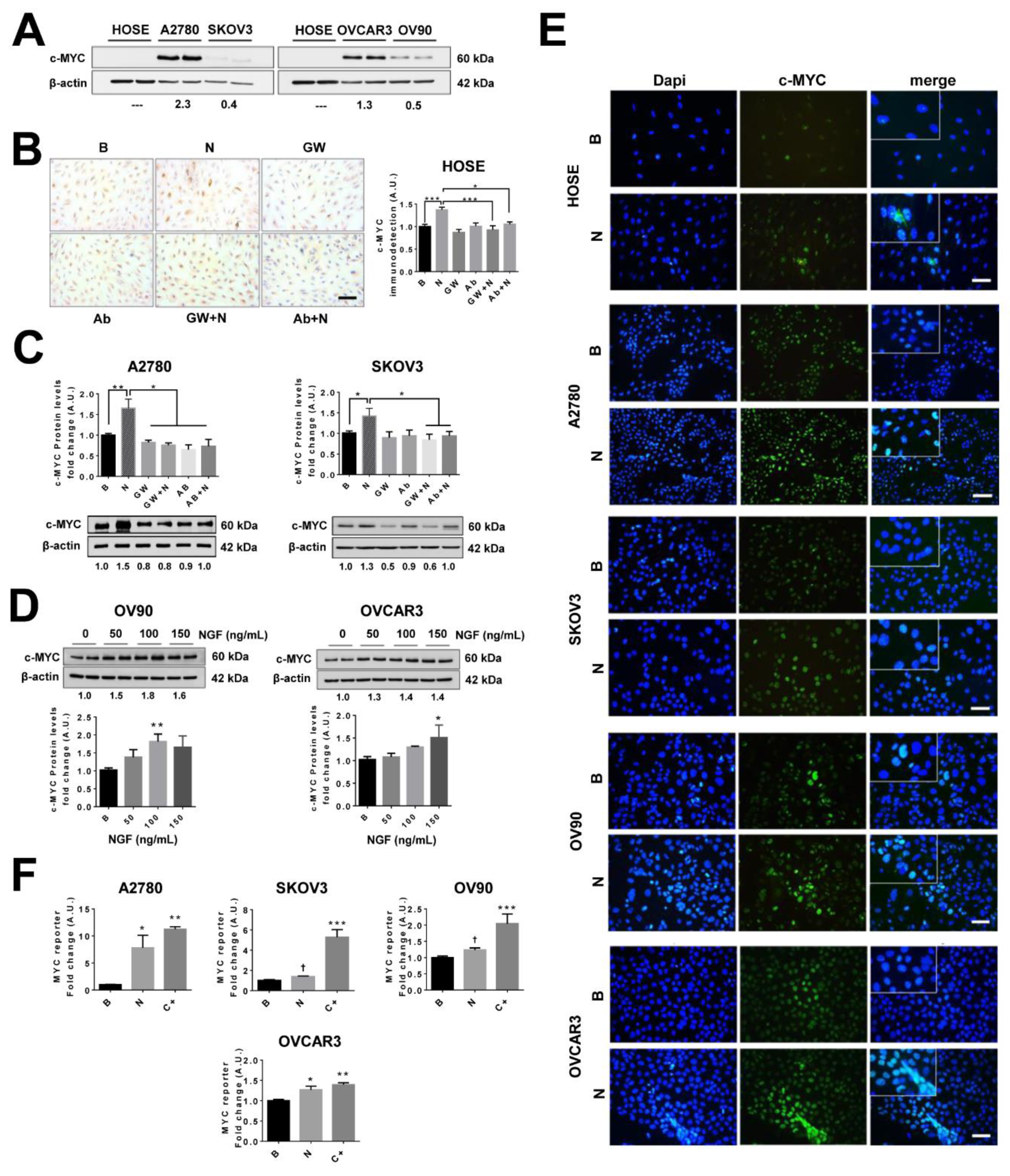
2.8. NGF Increases c-MYC Levels and Its Transcriptional Activity in EOC Cells
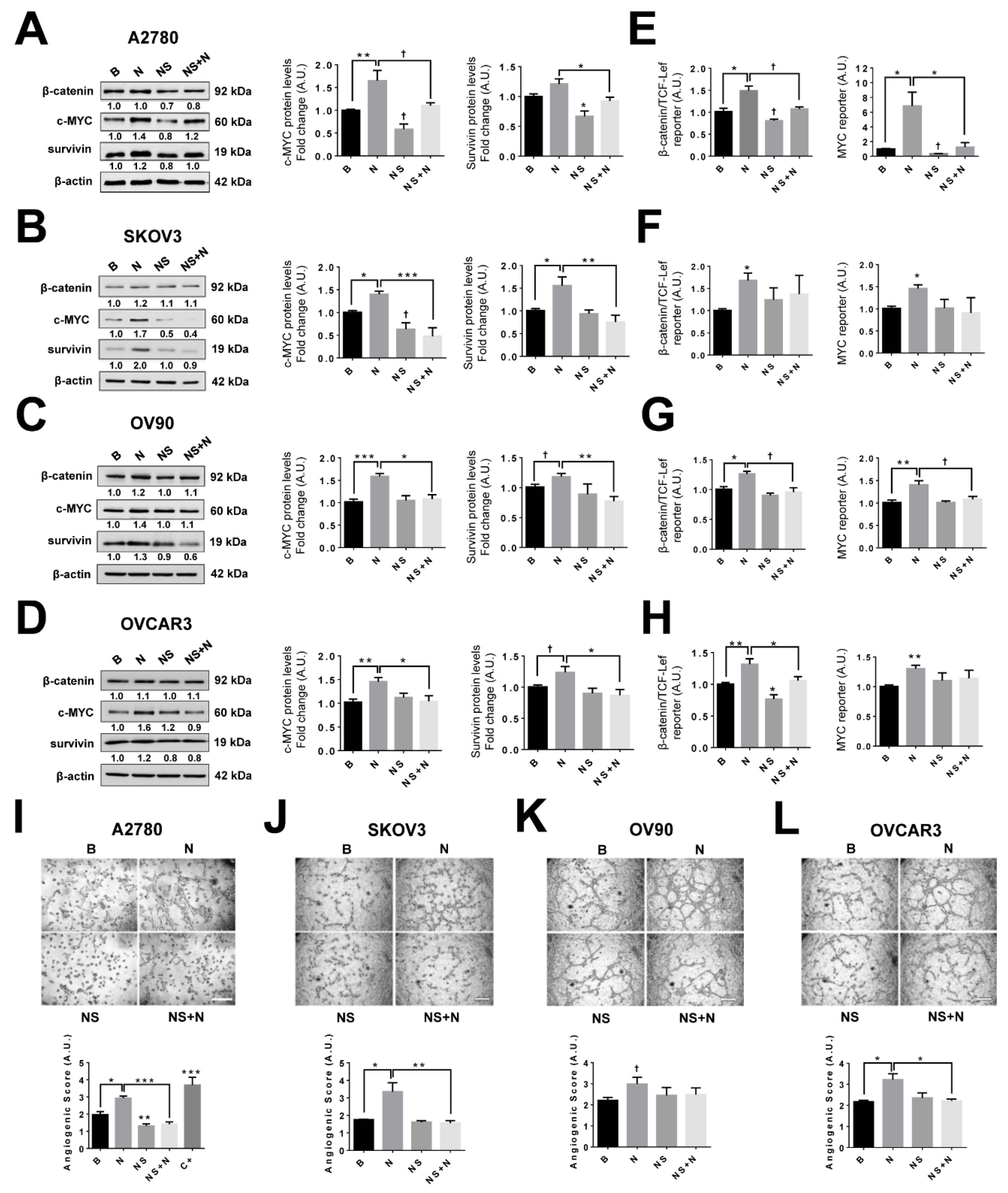
2.9. Inhibition of the COX-2/PGE2 Axis Prevents NGF Stimulated Increases in Pro-Angiogenic Proteins in EOC Cells
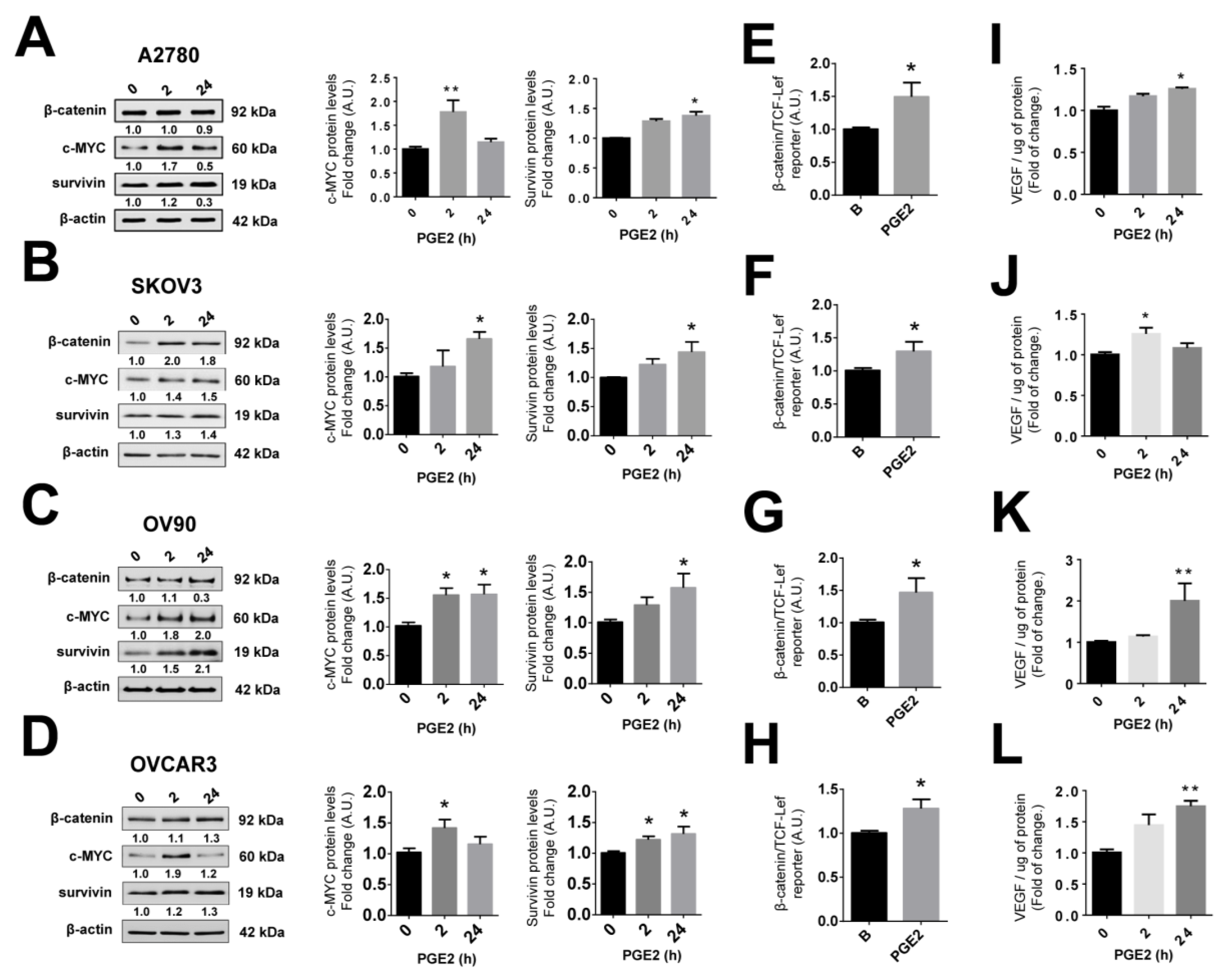
2.10. PGE2 Stimulation Increases the Expression of Pro-Angiogenic Proteins in EOC Cells
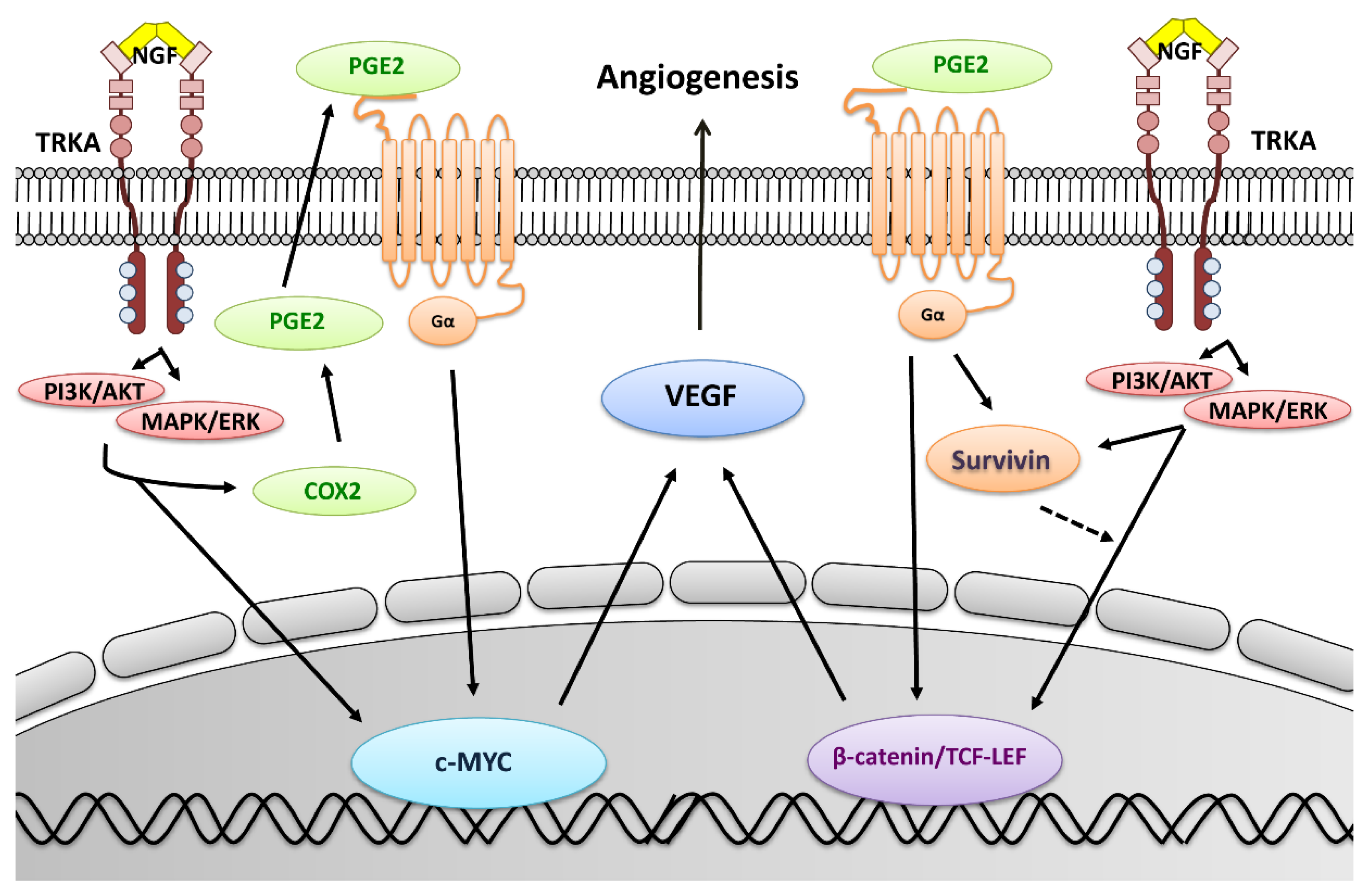
3. Discussion
4. Methods
4.1. Tissue Samples
4.2. Cell Culture and Treatments
4.3. Immunohistochemistry (IHQ)
4.4. Immunocytochemistry (ICQ)
4.5. Western Blotting
4.6. Total RNA Extraction and RT-PCR
4.7. PGE2 and VEGF ELISA
4.8. β-catenin/Tcf-Lef Reporter Assay
4.9. MYC Reporter Assay
4.10. Vasculogenesis Assay
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Reid, B.M.; Permuth, J.B.; Sellers, T.A. Epidemiology of ovarian cancer: A review. Cancer Biol. Med. 2017, 14, 9–32. [Google Scholar] [PubMed] [Green Version]
- World Ovarian Cancer Coalition. The World Ovarian Cancer Coalition Atlas. Global Trends in Incidence, Mortality and Survival. 2018. Available online: https://worldovariancancercoalition.org/wp-content/uploads/2018/10/THE-WORLD-OVARIAN-CANCER-COALITION-ATLAS-2018.pdf (accessed on 20 November 2019).
- Cancer Research UK. Ovarian Cancer. Available online: https://www.cancerresearchuk.org/about-cancer/ovarian-cancer/survival (accessed on 20 November 2019).
- American Cancer Society Ovarian Cancer, Early Detection, Diagnosis and Staging, Survival Rates for Ovarian Cancer, by Stage. Available online: https://www.cancer.org/cancer/ovarian-cancer/detection-diagnosis-staging/survival-rates.html (accessed on 20 November 2019).
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hennessy, B.T.; Coleman, R.L.; Markman, M. Ovarian cancer. Lancet 2009, 374, 1371–1382. [Google Scholar] [CrossRef]
- Doubeni, C.A.; Doubeni, A.R.; Myers, A.E. Diagnosis and Management of Ovarian Cancer. Am. Fam. Physician 2016, 93, 937–944. [Google Scholar]
- Duncan, T.J.; Al-Attar, A.; Rolland, P.; Scott, I.V.; Deen, S.; Liu, D.T.; Spendlove, I.; Durrant, L.G. Vascular endothelial growth factor expression in ovarian cancer: A model for targeted use of novel therapies? Clin. Cancer Res. 2008, 14, 3030–3035. [Google Scholar] [CrossRef] [Green Version]
- Tapia, V.; Gabler, F.; Munoz, M.; Yazigi, R.; Paredes, A.; Selman, A.; Vega, M.; Romero, C. Tyrosine kinase A receptor (trkA): A potential marker in epithelial ovarian cancer. Gynecol. Oncol. 2011, 121, 13–23. [Google Scholar] [CrossRef]
- Urzua, U.; Tapia, V.; Geraldo, M.P.; Selman, A.; Vega, M.; Romero, C. Nerve growth factor stimulates cellular proliferation of human epithelial ovarian cancer. Horm. Metab. Res. 2012, 44, 656–661. [Google Scholar] [CrossRef] [Green Version]
- Campos, X.; Munoz, Y.; Selman, A.; Yazigi, R.; Moyano, L.; Weinstein-Oppenheimer, C.; Lara, H.E.; Romero, C. Nerve growth factor and its high-affinity receptor trkA participate in the control of vascular endothelial growth factor expression in epithelial ovarian cancer. Gynecol. Oncol. 2007, 104, 168–175. [Google Scholar] [CrossRef]
- Garrido, M.P.; Vera, C.; Vega, M.; Quest, A.F.G.; Romero, C. Metformin prevents nerve growth factor-dependent proliferative and proangiogenic effects in epithelial ovarian cancer cells and endothelial cells. Ther. Adv. Med. Oncol. 2018, 10, 1758835918770984. [Google Scholar] [CrossRef]
- Eibl, G.; Bruemmer, D.; Okada, Y.; Duffy, J.P.; Law, R.E.; Reber, H.A.; Hines, O.J. PGE (2) is generated by specific COX-2 activity and increases VEGF production in COX-2—Expressing human pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2003, 306, 887–897. [Google Scholar] [CrossRef]
- Ding, Y.B.; Shi, R.H.; Tong, J.D.; Li, X.Y.; Zhang, G.X.; Xiao, W.M.; Yang, J.G.; Bao, Y.; Wu, J.; Yan, Z.G.; et al. PGE2 up-regulates vascular endothelial growth factor expression in MKN28 gastric cancer cells via epidermal growth factor receptor signaling system. Exp. Oncol. 2005, 27, 108–113. [Google Scholar] [PubMed]
- Wang, X.; Klein, R.D. Prostaglandin E2 induces vascular endothelial growth factor secretion in prostate cancer cells through EP2 receptor-mediated cAMP pathway. Mol. Carcinog. 2007, 46, 912–923. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Chakraborty, G.; Raja, R.; Kale, S.; Kundu, G.C. Prostaglandin E2 regulates tumor angiogenesis in prostate cancer. Cancer Res. 2008, 68, 7750–7759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, J.G.; Rodriguez, D.A.; Valenzuela, M.; Calderon, C.; Urzua, U.; Munroe, D.; Rosas, C.; Lemus, D.; Diaz, N.; Wright, M.C.; et al. Survivin expression promotes VEGF-induced tumor angiogenesis via PI3K/Akt enhanced beta-catenin/Tcf-Lef dependent transcription. Mol. Cancer 2014, 13, 209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun, K.S.; Langenbach, R. The prostaglandin E2 receptor, EP2, regulates survivin expression via an EGFR/STAT3 pathway in UVB-exposed mouse skin. Mol. Carcinog. 2011, 50, 439–448. [Google Scholar] [CrossRef]
- Olsen, J.J.; Pohl, S.O.; Deshmukh, A.; Visweswaran, M.; Ward, N.C.; Arfuso, F.; Agostino, M.; Dharmarajan, A. The Role of Wnt Signalling in Angiogenesis. Clin. Biochem. Rev. 2017, 38, 131–142. [Google Scholar]
- Shao, J.; Lee, S.B.; Guo, H.; Evers, B.M.; Sheng, H. Prostaglandin E2 stimulates the growth of colon cancer cells via induction of amphiregulin. Cancer Res. 2003, 63, 5218–5223. [Google Scholar]
- Bazzani, L.; Donnini, S.; Finetti, F.; Christofori, G.; Ziche, M. PGE2/EP3/SRC signaling induces EGFR nuclear translocation and growth through EGFR ligands release in lung adenocarcinoma cells. Oncotarget 2017, 8, 31270–31287. [Google Scholar] [CrossRef] [Green Version]
- Sobolewski, C.; Cerella, C.; Dicato, M.; Diederich, M. Cox-2 inhibitors induce early c-Myc downregulation and lead to expression of differentiation markers in leukemia cells. Cell Cycle 2011, 10, 2978–2993. [Google Scholar] [CrossRef]
- Morita, I. Distinct functions of COX-1 and COX-2. Prostaglandins Other Lipid Mediat. 2002, 68, 165–175. [Google Scholar] [CrossRef]
- Rouzer, C.A.; Marnett, L.J. Cyclooxygenases: Structural and functional insights. J. Lipid Res. 2009, 50, 29–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vane, J.R.; Bakhle, Y.S.; Botting, R.M. Cyclooxygenases 1 and 2. Annu. Rev. Pharm. Toxicol. 1998, 38, 97–120. [Google Scholar] [CrossRef] [PubMed]
- Ristimaki, A.; Honkanen, N.; Jankala, H.; Sipponen, P.; Harkonen, M. Expression of cyclooxygenase-2 in human gastric carcinoma. Cancer Res. 1997, 57, 1276–1280. [Google Scholar] [PubMed]
- Parrett, M.; Harris, R.; Joarder, F.; Ross, M.; Clausen, K.; Robertson, F. Cyclooxygenase-2 gene expression in human breast cancer. Int. J. Oncol. 1997, 10, 503–507. [Google Scholar] [CrossRef] [PubMed]
- Kern, M.A.; Haugg, A.M.; Koch, A.F.; Schilling, T.; Breuhahn, K.; Walczak, H.; Fleischer, B.; Trautwein, C.; Michalski, C.; Schulze-Bergkamen, H.; et al. Cyclooxygenase-2 inhibition induces apoptosis signaling via death receptors and mitochondria in hepatocellular carcinoma. Cancer Res. 2006, 66, 7059–7066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, E.; Tominaga, K.; Watanabe, T.; Fujiwara, Y.; Oshitani, N.; Matsumoto, T.; Higuchi, K.; Tarnawski, A.S.; Arakawa, T. COX-2 is essential for EGF induction of cell proliferation in gastric RGM1 cells. Dig. Dis. Sci. 2003, 48, 2257–2262. [Google Scholar] [CrossRef]
- Cheng, A.S.; Chan, H.L.; To, K.F.; Leung, W.K.; Chan, K.K.; Liew, C.T.; Sung, J.J. Cyclooxygenase-2 pathway correlates with vascular endothelial growth factor expression and tumor angiogenesis in hepatitis B virus-associated hepatocellular carcinoma. Int. J. Oncol. 2004, 24, 853–860. [Google Scholar] [CrossRef]
- Reichardt, L.F. Neurotrophin-regulated signalling pathways. Philos. Trans. R. Soc. 2006, 361, 1545–1564. [Google Scholar] [CrossRef] [Green Version]
- Pai, R.; Szabo, I.L.; Soreghan, B.A.; Atay, S.; Kawanaka, H.; Tarnawski, A.S. PGE(2) stimulates VEGF expression in endothelial cells via ERK2/JNK1 signaling pathways. Biochem. Biophys. Res. Commun. 2001, 286, 923–928. [Google Scholar] [CrossRef]
- Baker, V.V.; Borst, M.P.; Dixon, D.; Hatch, K.D.; Shingleton, H.M.; Miller, D. c-myc amplification in ovarian cancer. Gynecol. Oncol. 1990, 38, 340–342. [Google Scholar] [CrossRef]
- Gatcliffe, T.A.; Monk, B.J.; Planutis, K.; Holcombe, R.F. Wnt signaling in ovarian tumorigenesis. Int. J. Gynecol. Cancer 2008, 18, 954–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rask, K.; Nilsson, A.; Brannstrom, M.; Carlsson, P.; Hellberg, P.; Janson, P.O.; Hedin, L.; Sundfeldt, K. Wnt-signalling pathway in ovarian epithelial tumours: Increased expression of beta-catenin and GSK3beta. Br. J. Cancer 2003, 89, 1298–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosgrave, N.; Hill, A.D.; Young, L.S. Growth factor-dependent regulation of survivin by c-myc in human breast cancer. J. Mol. Endocrinol. 2006, 37, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Baudino, T.A.; McKay, C.; Pendeville-Samain, H.; Nilsson, J.A.; Maclean, K.H.; White, E.L.; Davis, A.C.; Ihle, J.N.; Cleveland, J.L. c-Myc is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Dev. 2002, 16, 2530–2543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Duan, N.; Zhang, C.; Zhang, W. Survivin and Tumorigenesis: Molecular Mechanisms and Therapeutic Strategies. J. Cancer 2016, 7, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Sanhueza, C.; Wehinger, S.; Castillo Bennett, J.; Valenzuela, M.; Owen, G.I.; Quest, A.F. The twisted survivin connection to angiogenesis. Mol. Cancer 2015, 14, 198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, L.; Chang, H.M.; Cheng, J.C.; Leung, P.C.; Sun, Y.P. TGF-beta1 induces COX-2 expression and PGE2 production in human granulosa cells through Smad signaling pathways. J. Clin. Endocrinol. Metab. 2014, 99, E1217–E1226. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Liang, L.; Yan, X.; Liu, N.; Gong, L.; Pan, S.; Lin, F.; Zhang, Q.; Zhao, H.; Zheng, F. Survivin status affects prognosis and chemosensitivity in epithelial ovarian cancer. Int. J. Gynecol. Cancer 2013, 23, 256–263. [Google Scholar] [CrossRef] [Green Version]
- Mita, A.C.; Mita, M.M.; Nawrocki, S.T.; Giles, F.J. Survivin: Key regulator of mitosis and apoptosis and novel target for cancer therapeutics. Clin. Cancer Res. 2008, 14, 5000–5005. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Tenev, T.; Martins, L.M.; Downward, J.; Lemoine, N.R. The ubiquitin-proteasome pathway regulates survivin degradation in a cell cycle-dependent manner. J. Cell. Sci. 2000, 113 Pt 23, 4363–4371. [Google Scholar]
- Ye, Q.; Cai, W.; Zheng, Y.; Evers, B.M.; She, Q.B. ERK and AKT signaling cooperate to translationally regulate survivin expression for metastatic progression of colorectal cancer. Oncogene 2014, 33, 1828–1839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kargman, S.L.; O’Neill, G.P.; Vickers, P.J.; Evans, J.F.; Mancini, J.A.; Jothy, S. Expression of prostaglandin G/H synthase-1 and -2 protein in human colon cancer. Cancer Res. 1995, 55, 2556–2559. [Google Scholar] [PubMed]
- Denkert, C.; Kobel, M.; Pest, S.; Koch, I.; Berger, S.; Schwabe, M.; Siegert, A.; Reles, A.; Klosterhalfen, B.; Hauptmann, S. Expression of cyclooxygenase 2 is an independent prognostic factor in human ovarian carcinoma. Am. J. Pathol. 2002, 160, 893–903. [Google Scholar] [CrossRef] [Green Version]
- Dore, M.; Cote, L.C.; Mitchell, A.; Sirois, J. Expression of prostaglandin G/H synthase type 1, but not type 2, in human ovarian adenocarcinomas. J. Histochem. Cytochem. 1998, 46, 77–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juuti, A.; Louhimo, J.; Nordling, S.; Ristimaki, A.; Haglund, C. Cyclooxygenase-2 expression correlates with poor prognosis in pancreatic cancer. J. Clin. Pathol. 2006, 59, 382–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nozoe, T.; Ezaki, T.; Kabashima, A.; Baba, H.; Maehara, Y. Significance of immunohistochemical expression of cyclooxygenase-2 in squamous cell carcinoma of the esophagus. Am. J. Surg. 2005, 189, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Hoier, B.; Prats, C.; Qvortrup, K.; Pilegaard, H.; Bangsbo, J.; Hellsten, Y. Subcellular localization and mechanism of secretion of vascular endothelial growth factor in human skeletal muscle. FASEB J. 2013, 27, 3496–3504. [Google Scholar] [CrossRef]
- Rask, K.; Zhu, Y.; Wang, W.; Hedin, L.; Sundfeldt, K. Ovarian epithelial cancer: A role for PGE2-synthesis and signalling in malignant transformation and progression. Mol. Cancer 2006, 5, 62. [Google Scholar] [CrossRef] [Green Version]
- Asanuma, H.; Torigoe, T.; Kamiguchi, K.; Hirohashi, Y.; Ohmura, T.; Hirata, K.; Sato, M.; Sato, N. Survivin expression is regulated by coexpression of human epidermal growth factor receptor 2 and epidermal growth factor receptor via phosphatidylinositol 3-kinase/AKT signaling pathway in breast cancer cells. Cancer Res. 2005, 65, 11018–11025. [Google Scholar] [CrossRef] [Green Version]
- Daikoku, T.; Wang, D.; Tranguch, S.; Morrow, J.D.; Orsulic, S.; DuBois, R.N.; Dey, S.K. Cyclooxygenase-1 is a potential target for prevention and treatment of ovarian epithelial cancer. Cancer Res. 2005, 65, 3735–3744. [Google Scholar] [CrossRef] [Green Version]
- Wilson, A.J.; Fadare, O.; Beeghly-Fadiel, A.; Son, D.S.; Liu, Q.; Zhao, S.; Saskowski, J.; Uddin, M.J.; Daniel, C.; Crews, B.; et al. Aberrant over-expression of COX-1 intersects multiple pro-tumorigenic pathways in high-grade serous ovarian cancer. Oncotarget 2015, 6, 21353–21368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahid, I.; AlMalki, W.; AlRabia, M.; Ahmed, M.; Imam, M.; Saifullah, M.; Hafeez, M. Chapter: Recent Advances in Angiogenesis Assessment Methods and Their Clinical Applications. In Physiologic and Pathologic Angiogenesis—Signaling Mechanisms and Targeted Therapy; InTechOpen, Ed.; 2017; Available online: https://www.intechopen.com/books/physiologic-and-pathologic-angiogenesis-signaling-mechanisms-and-targeted-therapy/recent-advances-in-angiogenesis-assessment-methods-and-their-clinical-applications (accessed on 20 November 2019).
- Sanz-Nogues, C.; O’Brien, T. In vitro models for assessing therapeutic angiogenesis. Drug Discov. Today 2016, 21, 1495–1503. [Google Scholar] [CrossRef] [PubMed]
- Kohan-Ivani, K.; Gabler, F.; Selman, A.; Vega, M.; Romero, C. Role of dihydrotestosterone (DHT) on TGF-beta1 signaling pathway in epithelial ovarian cancer cells. J. Cancer Res. Clin. Oncol. 2016, 142, 47–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, D.A.; Tapia, J.C.; Fernandez, J.G.; Torres, V.A.; Munoz, N.; Galleguillos, D.; Leyton, L.; Quest, A.F. Caveolin-1-mediated suppression of cyclooxygenase-2 via a beta-catenin-Tcf/Lef-dependent transcriptional mechanism reduced prostaglandin E2 production and survivin expression. Mol. Biol. Cell 2009, 20, 2297–2310. [Google Scholar] [CrossRef] [Green Version]
- Aranda, E.; Owen, G.I. A semi-quantitative assay to screen for angiogenic compounds and compounds with angiogenic potential using the EA. hy926 endothelial cell line. Biol. Res. 2009, 42, 377–389. [Google Scholar] [CrossRef] [Green Version]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garrido, M.P.; Hurtado, I.; Valenzuela-Valderrama, M.; Salvatierra, R.; Hernández, A.; Vega, M.; Selman, A.; Quest, A.F.G.; Romero, C. NGF-Enhanced Vasculogenic Properties of Epithelial Ovarian Cancer Cells Is Reduced by Inhibition of the COX-2/PGE2 Signaling Axis. Cancers 2019, 11, 1970. https://doi.org/10.3390/cancers11121970
Garrido MP, Hurtado I, Valenzuela-Valderrama M, Salvatierra R, Hernández A, Vega M, Selman A, Quest AFG, Romero C. NGF-Enhanced Vasculogenic Properties of Epithelial Ovarian Cancer Cells Is Reduced by Inhibition of the COX-2/PGE2 Signaling Axis. Cancers. 2019; 11(12):1970. https://doi.org/10.3390/cancers11121970
Chicago/Turabian StyleGarrido, Maritza P., Iván Hurtado, Manuel Valenzuela-Valderrama, Renato Salvatierra, Andrea Hernández, Margarita Vega, Alberto Selman, Andrew F. G. Quest, and Carmen Romero. 2019. "NGF-Enhanced Vasculogenic Properties of Epithelial Ovarian Cancer Cells Is Reduced by Inhibition of the COX-2/PGE2 Signaling Axis" Cancers 11, no. 12: 1970. https://doi.org/10.3390/cancers11121970