Papillary Renal Cell Carcinomas Rewire Glutathione Metabolism and Are Deficient in Both Anabolic Glucose Synthesis and Oxidative Phosphorylation

 , and
, and
Abstract
:1. Introduction
2. Results
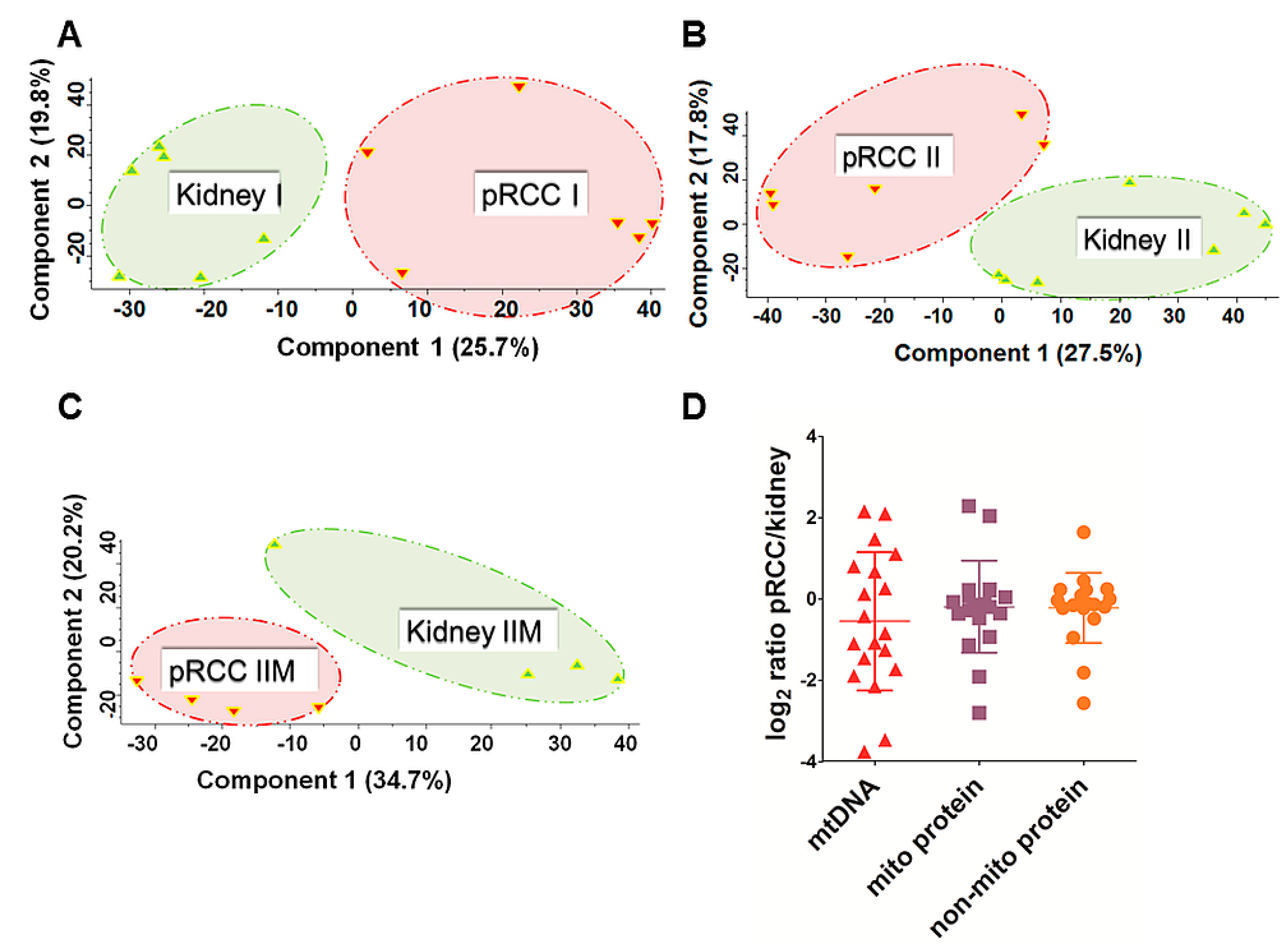
2.1. Proteome Profiling of pRCC
2.2. mtDNA Mutations in pRCC Did Not Reveal Any Major Impact on the Respiratory Chain
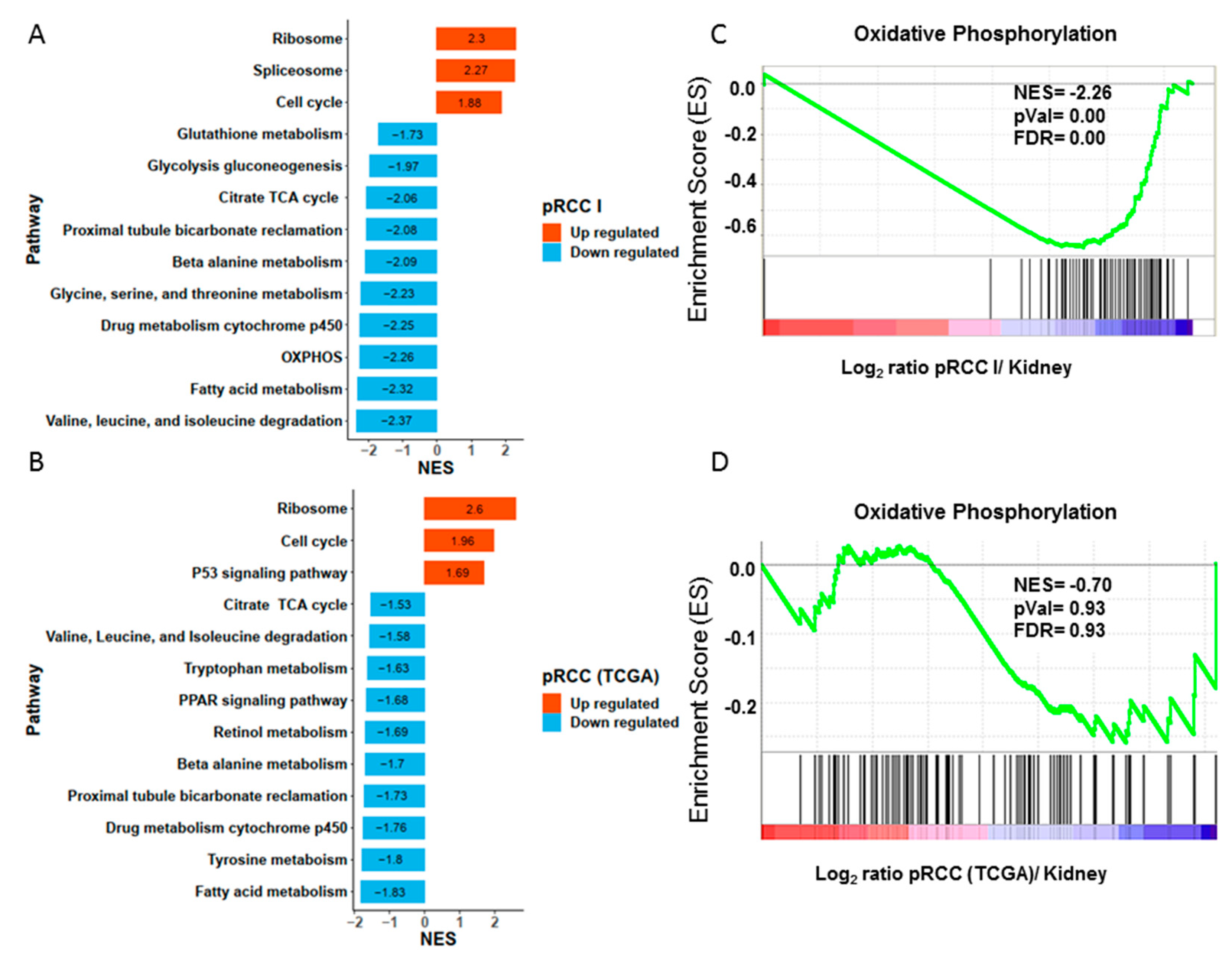
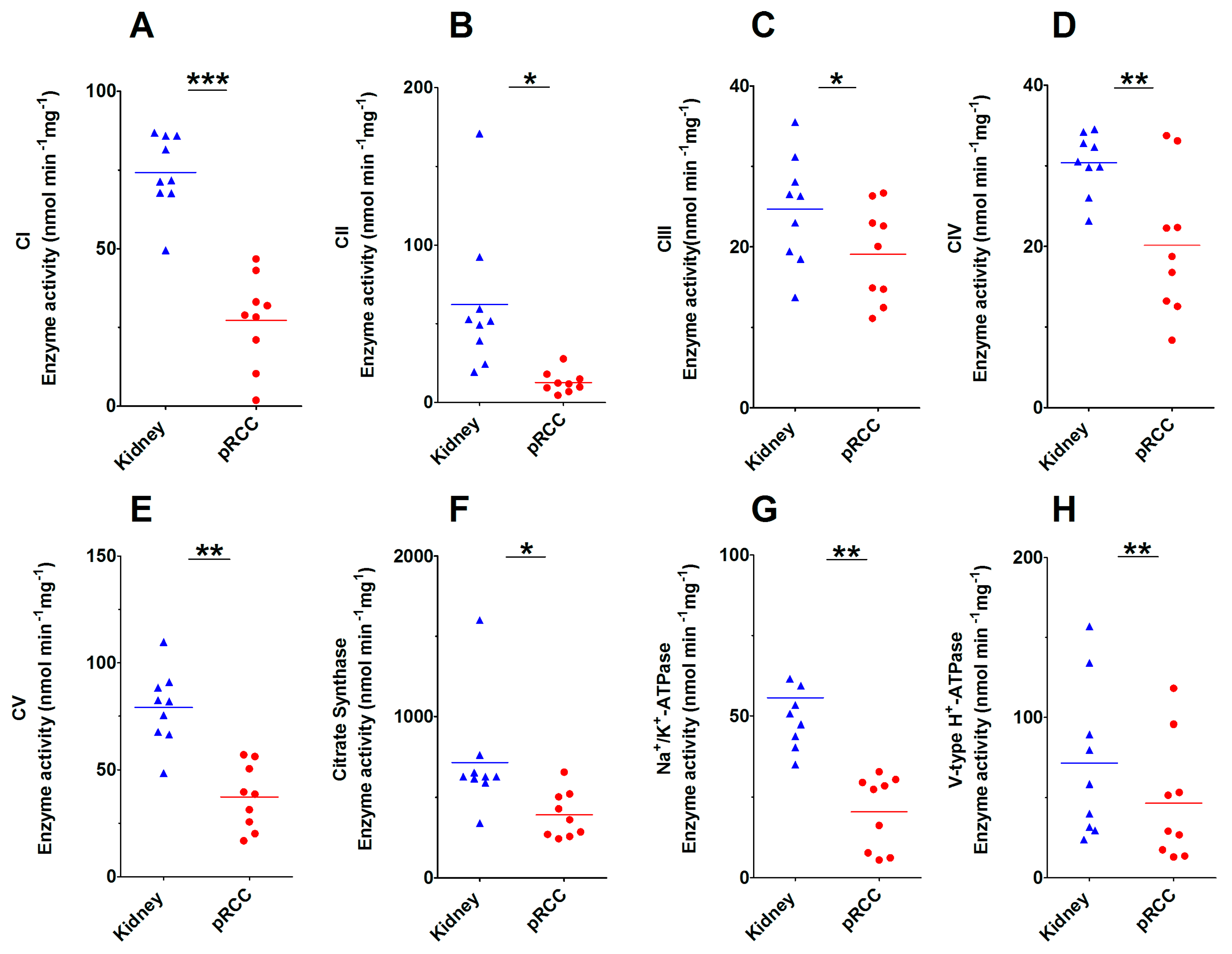
2.3. Significantly Decreased Enzymatic Activity of the Respiratory Chain in pRCC
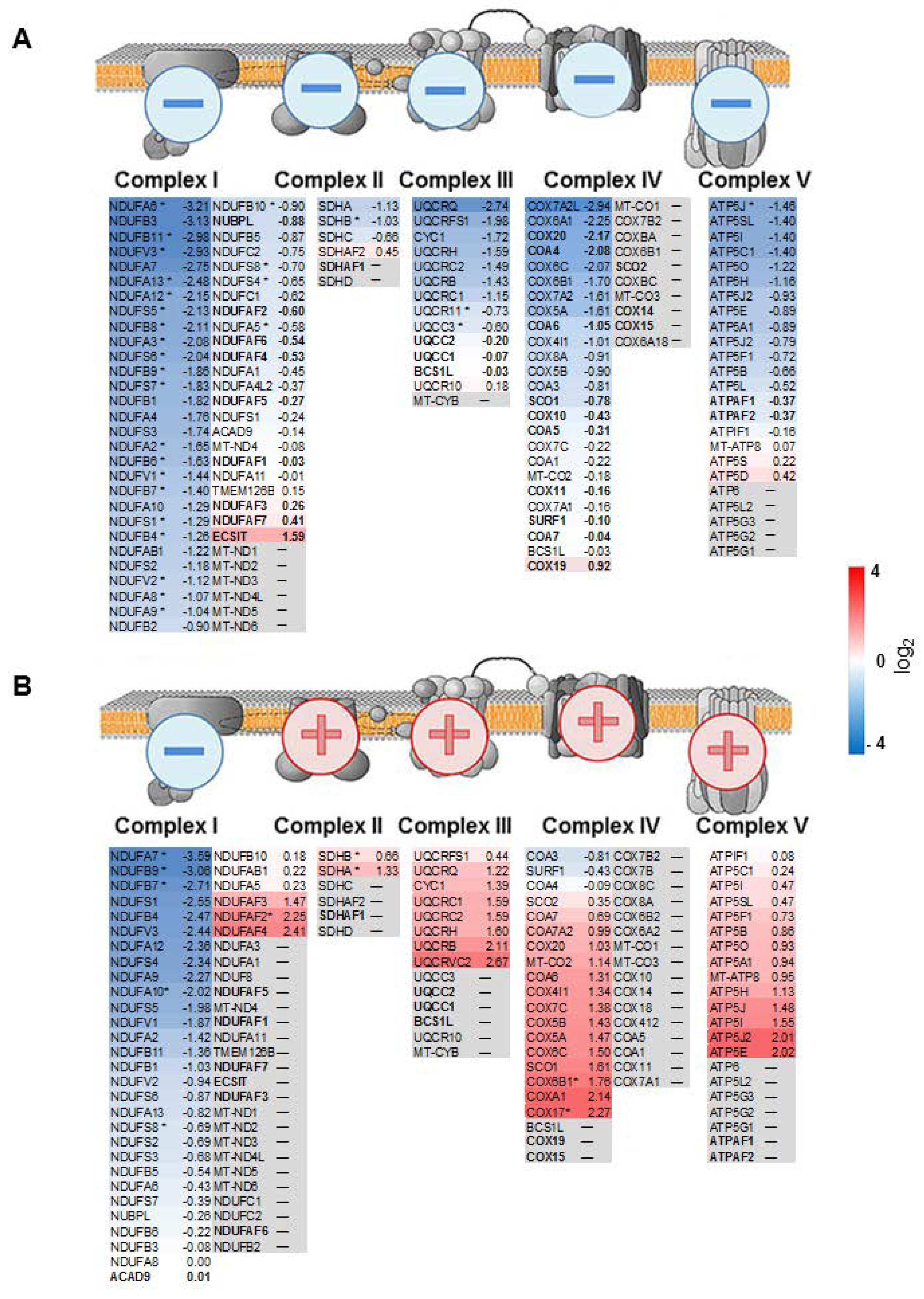
2.4. Anti-Correlation of Transcripts and Proteins of the Respiratory Chain in pRCC
2.5. Comparison of the Abundance of Proteins Involved in the Respiratory Chain between Malignant pRCC and Benign Renal Oncocytomas
2.6. pRCCs have Significantly Decreased Levels of V- and P-ATPases
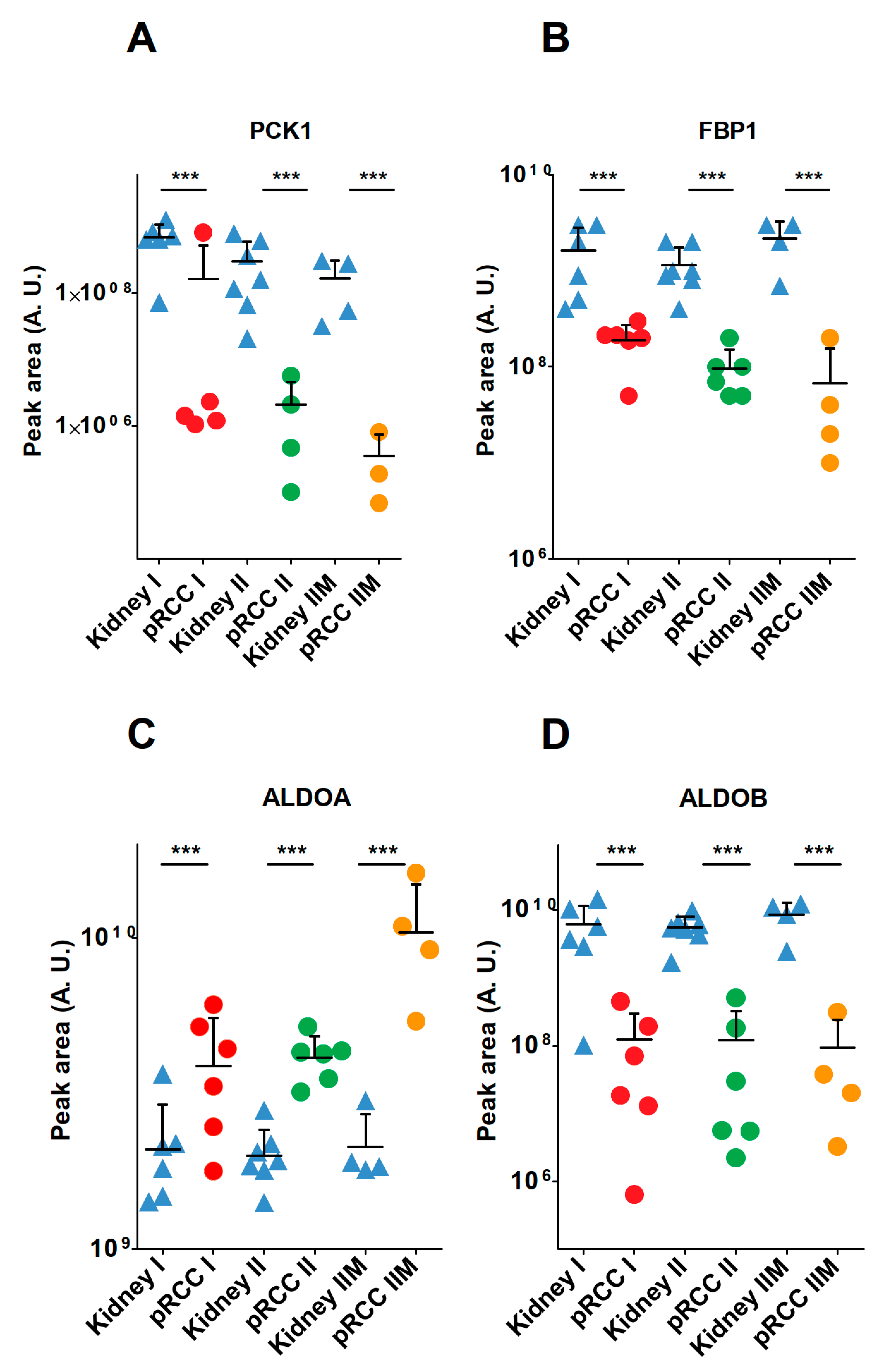
2.7. Anabolic Glucose Synthesis Was Abated in pRCC
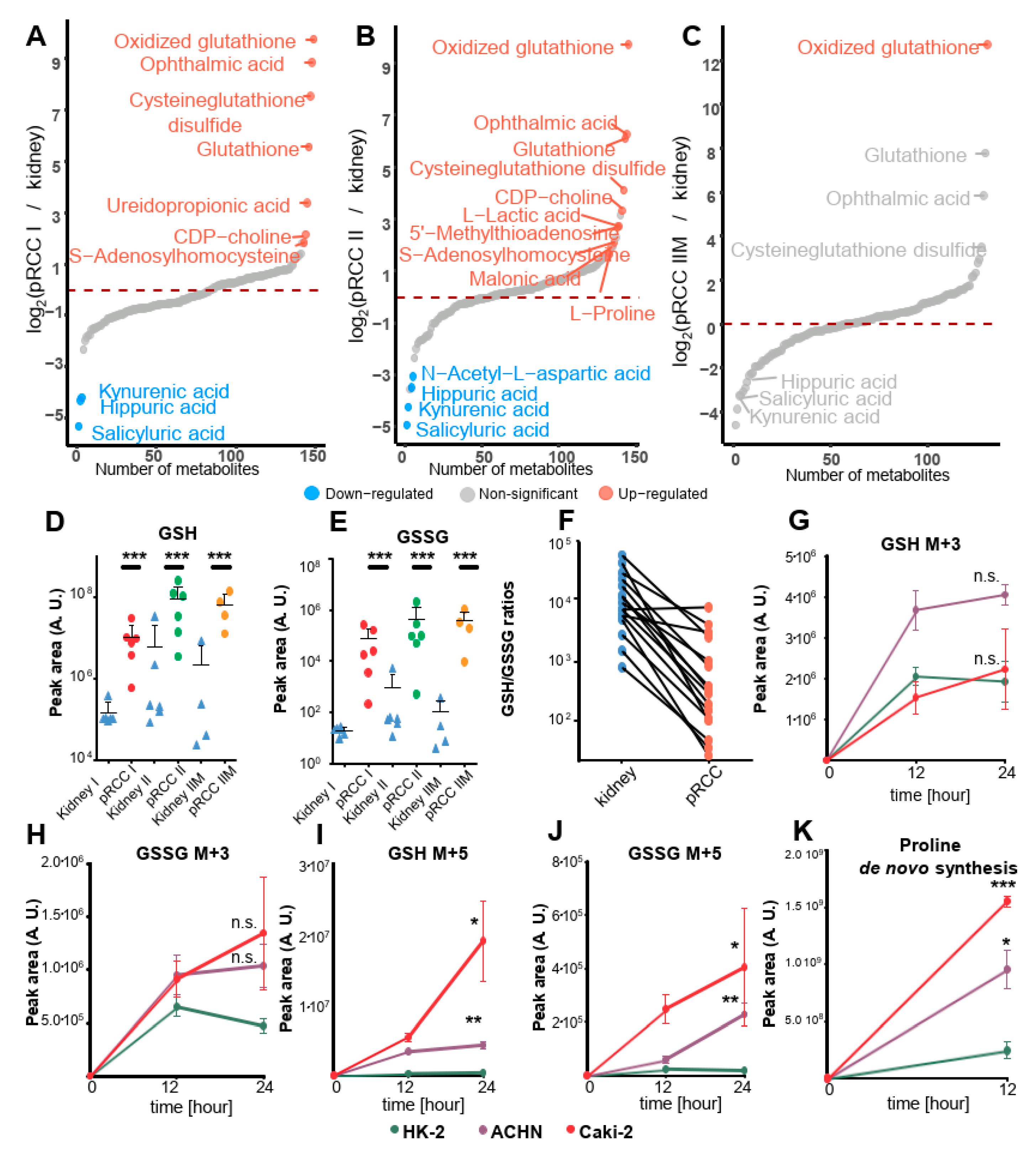
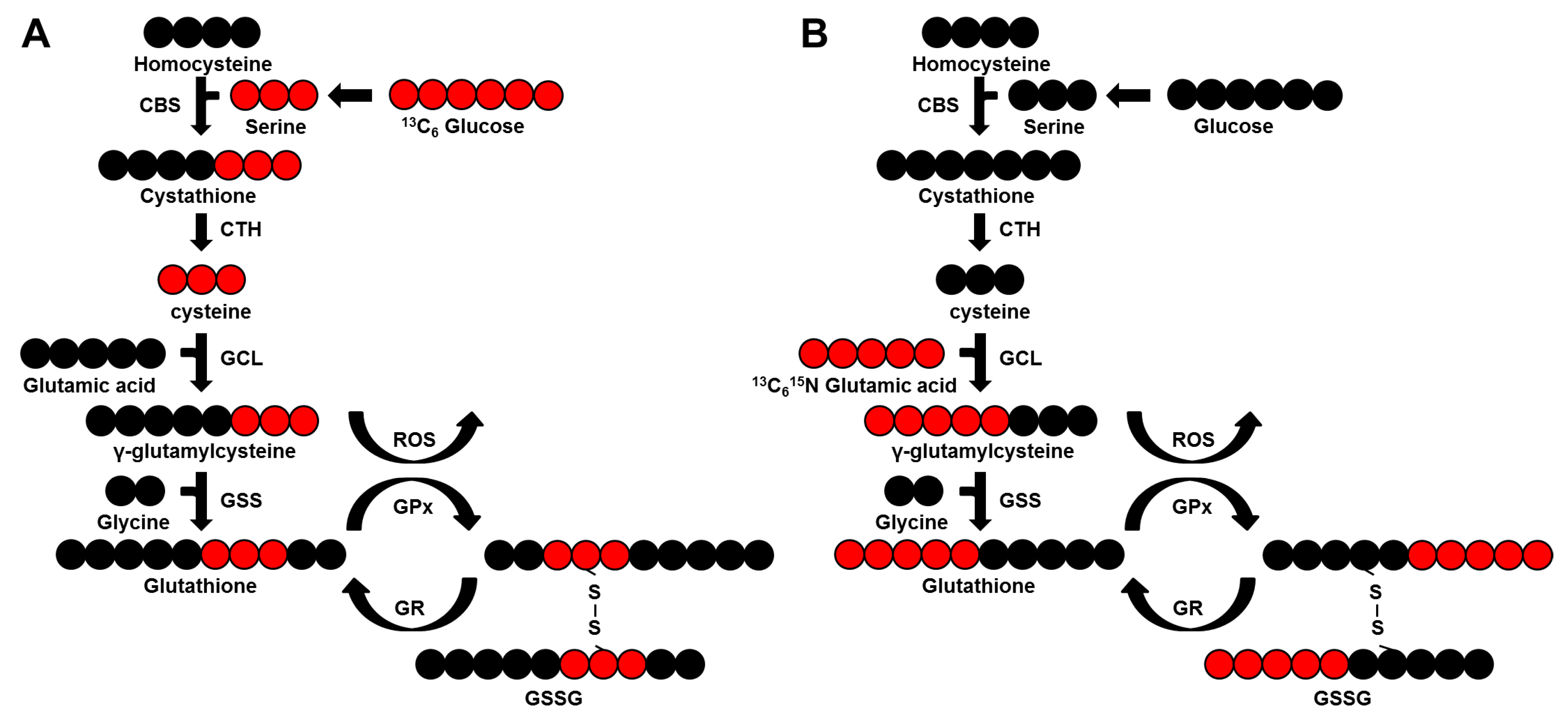
2.8. Dramatically Increased Glutathione Levels in pRCC Are Based on Glutamine Consumption
2.9. Glutamine Is the Main Nutrient Source in pRCC
3. Discussion
4. Experimental Procedures
4.1. Tissue Dissection and Verification of Papillary RCC
4.2. Whole Exome Sequencing (WES)
4.3. Analysis of mtDNA Mutations
4.4. Sample Preparation for Proteomics
4.5. LC-MS Instrument Settings for Shotgun Proteome Profiling and Data Analysis
4.6. Metabolite Extraction and Profiling by Targeted LC-MS
4.7. Cell Culture Conditions for Glutathione and Proline de novo Synthesis
4.8. Experimental Design, Statistical Rationale, and Pathway Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Patard, J.J.; Leray, E.; Rioux-Leclercq, N.; Cindolo, L.; Ficarra, V.; Zisman, A.; De La Taille, A.; Tostain, J.; Artibani, W.; Abbou, C.C.; et al. Prognostic value of histologic subtypes in renal cell carcinoma: A multicenter experience. J. Clin. Oncol. 2005, 23, 2763–2771. [Google Scholar] [CrossRef]
- Pai, A.; Brunson, A.; Brown, M.; Pan, C.X.; Lara, P.N., Jr. Evolving epidemiologic trends in nonclear cell renal cell cancer: An analysis of the california cancer registry. Urology 2013, 82, 840–845. [Google Scholar] [CrossRef]
- Delahunt, B.; Eble, J.N. Papillary renal cell carcinoma: A clinicopathologic and immunohistochemical study of 105 tumors. Mod. Pathol. 1997, 10, 537–544. [Google Scholar]
- Klatte, T.; Pantuck, A.J.; Said, J.W.; Seligson, D.B.; Rao, N.P.; LaRochelle, J.C.; Shuch, B.; Zisman, A.; Kabbinavar, F.F.; Belldegrun, A.S. Cytogenetic and molecular tumor profiling for type 1 and type 2 papillary renal cell carcinoma. Clin. Cancer Res. 2009, 15, 1162–1169. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, Network; Linehan, W.M.; Spellman, P.T.; Ricketts, C.J.; Creighton, C.J.; Fei, S.S.; Davis, C.; Wheeler, D.A.; Murray, B.A.; Schmidt, L.; et al. Comprehensive molecular characterization of papillary renal-cell carcinoma. N. Engl. J. Med. 2016, 374, 135–145. [Google Scholar]
- Chen, F.; Zhang, Y.; Senbabaoglu, Y.; Ciriello, G.; Yang, L.; Reznik, E.; Shuch, B.; Micevic, G.; De Velasco, G.; Shinbrot, E.; et al. Multilevel genomics-based taxonomy of renal cell carcinoma. Cell Rep. 2016, 14, 2476–2489. [Google Scholar] [CrossRef]
- Kauffman, E.C.; Ricketts, C.J.; Rais-Bahrami, S.; Yang, Y.; Merino, M.J.; Bottaro, D.P.; Srinivasan, R.; Linehan, W.M. Molecular genetics and cellular features of tfe3 and tfeb fusion kidney cancers. Nat. Rev. Urol. 2014, 11, 465–475. [Google Scholar] [CrossRef]
- Shuch, B.; Hahn, A.W.; Agarwal, N. Current treatment landscape of advanced papillary renal cancer. J. Clin. Oncol. 2017, 35, 2981–2983. [Google Scholar] [CrossRef]
- Haake, S.M.; Weyandt, J.D.; Rathmell, W.K. Insights into the genetic basis of the renal cell carcinomas from the cancer genome atlas. Mol. Cancer Res. 2016, 14, 589–598. [Google Scholar] [CrossRef]
- Li, S.; Shuch, B.M.; Gerstein, M.B. Whole-genome analysis of papillary kidney cancer finds significant noncoding alterations. PLoS Genet. 2017, 13, e1006685. [Google Scholar] [CrossRef]
- Ricketts, C.J.; De Cubas, A.A.; Fan, H.; Smith, C.C.; Lang, M.; Reznik, E.; Bowlby, R.; Gibb, E.A.; Akbani, R.; Beroukhim, R.; et al. The cancer genome atlas comprehensive molecular characterization of renal cell carcinoma. Cell Rep. 2018, 23, 313–326.e5. [Google Scholar] [CrossRef]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell 2018, 173, 291–304.e6. [Google Scholar] [CrossRef]
- Doehn, C.; Grunwald, V.; Steiner, T.; Follmann, M.; Rexer, H.; Krege, S. The diagnosis, treatment, and follow-up of renal cell carcinoma. Dtsch. Arztebl. Int. 2016, 113, 590–596. [Google Scholar] [CrossRef]
- Schaeffeler, E.; Buttner, F.; Reustle, A.; Klumpp, V.; Winter, S.; Rausch, S.; Fisel, P.; Hennenlotter, J.; Kruck, S.; Stenzl, A.; et al. Metabolic and lipidomic reprogramming in renal cell carcinoma subtypes reflects regions of tumor origin. Eur. Urol. Focus 2018. [Google Scholar] [CrossRef]
- Meierhofer, D.; Mayr, J.A.; Foetschl, U.; Berger, A.; Fink, K.; Schmeller, N.; Hacker, G.W.; Hauser-Kronberger, C.; Kofler, B.; Sperl, W. Decrease of mitochondrial DNA content and energy metabolism in renal cell carcinoma. Carcinogenesis 2004, 25, 1005–1010. [Google Scholar] [CrossRef] [Green Version]
- Pelletier, J.; Thomas, G.; Volarevic, S. Ribosome biogenesis in cancer: New players and therapeutic avenues. Nat. Rev. Cancer 2018, 18, 51–63. [Google Scholar] [CrossRef]
- Sulima, S.O.; Hofman, I.J.F.; De Keersmaecker, K.; Dinman, J.D. How ribosomes translate cancer. Cancer Discov. 2017, 7, 1069–1087. [Google Scholar] [CrossRef]
- Kurschner, G.; Zhang, Q.; Clima, R.; Xiao, Y.; Busch, J.F.; Kilic, E.; Jung, K.; Berndt, N.; Bulik, S.; Holzhutter, H.G.; et al. Renal oncocytoma characterized by the defective complex i of the respiratory chain boosts the synthesis of the ros scavenger glutathione. Oncotarget 2017, 8, 105882–105904. [Google Scholar] [CrossRef]
- Stransky, L.; Cotter, K.; Forgac, M. The function of v-atpases in cancer. Physiol. Rev. 2016, 96, 1071–1091. [Google Scholar] [CrossRef]
- Penhoet, E.; Rajkumar, T.; Rutter, W.J. Multiple forms of fructose diphosphate aldolase in mammalian tissues. Proc. Natl. Acad. Sci. USA 1966, 56, 1275–1282. [Google Scholar] [CrossRef]
- Saez, D.E.; Slebe, J.C. Subcellular localization of aldolase B. J. Cell. Biochem. 2000, 78, 62–72. [Google Scholar] [CrossRef]
- Droppelmann, C.A.; Saez, D.E.; Asenjo, J.L.; Yanez, A.J.; Garcia-Rocha, M.; Concha, I.I.; Grez, M.; Guinovart, J.J.; Slebe, J.C. A new level of regulation in gluconeogenesis: Metabolic state modulates the intracellular localization of aldolase b and its interaction with liver fructose-1,6-bisphosphatase. Biochem. J. 2015, 472, 225–237. [Google Scholar] [CrossRef]
- Liu, Y.; Hyde, A.S.; Simpson, M.A.; Barycki, J.J. Emerging regulatory paradigms in glutathione metabolism. Adv. Cancer Res. 2014, 122, 69–101. [Google Scholar]
- Soga, T.; Baran, R.; Suematsu, M.; Ueno, Y.; Ikeda, S.; Sakurakawa, T.; Kakazu, Y.; Ishikawa, T.; Robert, M.; Nishioka, T.; et al. Differential metabolomics reveals ophthalmic acid as an oxidative stress biomarker indicating hepatic glutathione consumption. J. Biol. Chem. 2006, 281, 16768–16776. [Google Scholar] [CrossRef]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta 2013, 1830, 3143–3153. [Google Scholar] [CrossRef]
- Reynaert, N.L.; Ckless, K.; Guala, A.S.; Wouters, E.F.; van der Vliet, A.; Janssen-Heininger, Y.M. In situ detection of s-glutathionylated proteins following glutaredoxin-1 catalyzed cysteine derivatization. Biochim. Biophys. Acta 2006, 1760, 380–387. [Google Scholar] [CrossRef]
- Schilling, S.; Wasternack, C.; Demuth, H.U. Glutaminyl cyclases from animals and plants: A case of functionally convergent protein evolution. Biol. Chem. 2008, 389, 983–991. [Google Scholar] [CrossRef]
- Xiao, Y.; Clima, R.; Busch, J.F.; Rabien, A.; Kilic, E.; Villegas, S.; Türkmen, S.; Timmermann, B.; Attimonelli, M.; Jung, K.; et al. Metabolic reprogramming and elevation of glutathione in chromophobe renal cell carcinomas. bioRxiv 2019, 649046. [Google Scholar] [CrossRef]
- Li, B.; Qiu, B.; Lee, D.S.; Walton, Z.E.; Ochocki, J.D.; Mathew, L.K.; Mancuso, A.; Gade, T.P.; Keith, B.; Nissim, I.; et al. Fructose-1,6-bisphosphatase opposes renal carcinoma progression. Nature 2014, 513, 251–255. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Tu, B.; Wang, H.; Cao, Z.; Tang, M.; Zhang, C.; Gu, B.; Li, Z.; Wang, L.; Yang, Y.; et al. Tumor suppressor p53 cooperates with sirt6 to regulate gluconeogenesis by promoting foxo1 nuclear exclusion. Proc. Natl. Acad. Sci. USA 2014, 111, 10684–10689. [Google Scholar] [CrossRef]
- Schulze, A.; Harris, A.L. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature 2012, 491, 364–373. [Google Scholar] [CrossRef]
- Gerich, J.E.; Meyer, C.; Woerle, H.J.; Stumvoll, M. Renal gluconeogenesis: Its importance in human glucose homeostasis. Diabetes Care 2001, 24, 382–391. [Google Scholar] [CrossRef]
- Nakajima, R.; Nozaki, S.; Kondo, T.; Nagashima, Y.; Abe, K.; Sakai, S. Evaluation of renal cell carcinoma histological subtype and fuhrman grade using (18)f-fluorodeoxyglucose-positron emission tomography/computed tomography. Eur. Radiol. 2017, 27, 4866–4873. [Google Scholar] [CrossRef]
- Khan, M.W.; Biswas, D.; Ghosh, M.; Mandloi, S.; Chakrabarti, S.; Chakrabarti, P. Mtorc2 controls cancer cell survival by modulating gluconeogenesis. Cell Death Discov. 2015, 1, 15016. [Google Scholar] [CrossRef]
- Zhang, C.S.; Hawley, S.A.; Zong, Y.; Li, M.; Wang, Z.; Gray, A.; Ma, T.; Cui, J.; Feng, J.W.; Zhu, M.; et al. Fructose-1,6-bisphosphate and aldolase mediate glucose sensing by ampk. Nature 2017, 548, 112–116. [Google Scholar] [CrossRef]
- Bu, P.; Chen, K.Y.; Xiang, K.; Johnson, C.; Crown, S.B.; Rakhilin, N.; Ai, Y.; Wang, L.; Xi, R.; Astapova, I.; et al. Aldolase b-mediated fructose metabolism drives metabolic reprogramming of colon cancer liver metastasis. Cell Metab. 2018, 27, 1249–1262.e4. [Google Scholar] [CrossRef]
- Tian, Y.F.; Hsieh, P.L.; Lin, C.Y.; Sun, D.P.; Sheu, M.J.; Yang, C.C.; Lin, L.C.; He, H.L.; Solorzano, J.; Li, C.F.; et al. High expression of aldolase b confers a poor prognosis for rectal cancer patients receiving neoadjuvant chemoradiotherapy. J. Cancer 2017, 8, 1197–1204. [Google Scholar] [CrossRef]
- Li, Q.; Li, Y.; Xu, J.; Wang, S.; Xu, Y.; Li, X.; Cai, S. Aldolase b overexpression is associated with poor prognosis and promotes tumor progression by epithelial-mesenchymal transition in colorectal adenocarcinoma. Cell. Physiol. Biochem. 2017, 42, 397–406. [Google Scholar] [CrossRef]
- He, J.; Jin, Y.; Chen, Y.; Yao, H.B.; Xia, Y.J.; Ma, Y.Y.; Wang, W.; Shao, Q.S. Downregulation of aldob is associated with poor prognosis of patients with gastric cancer. Onco Targets Ther. 2016, 9, 6099–6109. [Google Scholar] [CrossRef]
- Lessa, R.C.; Campos, A.H.; Freitas, C.E.; Silva, F.R.; Kowalski, L.P.; Carvalho, A.L.; Vettore, A.L. Identification of upregulated genes in oral squamous cell carcinomas. Head Neck 2013, 35, 1475–1481. [Google Scholar] [CrossRef]
- Hamaguchi, T.; Iizuka, N.; Tsunedomi, R.; Hamamoto, Y.; Miyamoto, T.; Iida, M.; Tokuhisa, Y.; Sakamoto, K.; Takashima, M.; Tamesa, T.; et al. Glycolysis module activated by hypoxia-inducible factor 1alpha is related to the aggressive phenotype of hepatocellular carcinoma. Int. J. Oncol. 2008, 33, 725–731. [Google Scholar]
- Huang, Z.; Hua, Y.; Tian, Y.; Qin, C.; Qian, J.; Bao, M.; Liu, Y.; Wang, S.; Cao, Q.; Ju, X.; et al. High expression of fructose-bisphosphate aldolase a induces progression of renal cell carcinoma. Oncol. Rep. 2018, 39, 2996–3006. [Google Scholar]
- Na, N.; Li, H.; Xu, C.; Miao, B.; Hong, L.; Huang, Z.; Jiang, Q. High expression of aldolase a predicts poor survival in patients with clear-cell renal cell carcinoma. Ther. Clin. Risk Manag. 2017, 13, 279–285. [Google Scholar] [CrossRef]
- Chen, X.; Yang, T.T.; Zhou, Y.; Wang, W.; Qiu, X.C.; Gao, J.; Li, C.X.; Long, H.; Ma, B.A.; Ma, Q.; et al. Proteomic profiling of osteosarcoma cells identifies aldoa and sult1a3 as negative survival markers of human osteosarcoma. Mol. Carcinog. 2014, 53, 138–144. [Google Scholar] [CrossRef]
- Ojika, T.; Imaizumi, M.; Watanabe, H.; Abe, T.; Kato, K. An immunohistochemical study on three aldolase isozymes in human lung cancer. Nihon Kyobu Geka Gakkai Zasshi 1992, 40, 382–386. [Google Scholar]
- Collins, M.P.; Forgac, M. Regulation of v-atpase assembly in nutrient sensing and function of v-atpases in breast cancer metastasis. Front. Physiol. 2018, 9, 902. [Google Scholar] [CrossRef]
- Cotter, K.; Capecci, J.; Sennoune, S.; Huss, M.; Maier, M.; Martinez-Zaguilan, R.; Forgac, M. Activity of plasma membrane v-atpases is critical for the invasion of mda-mb231 breast cancer cells. J. Biol. Chem. 2015, 290, 3680–3692. [Google Scholar] [CrossRef]
- Zhang, C.S.; Jiang, B.; Li, M.; Zhu, M.; Peng, Y.; Zhang, Y.L.; Wu, Y.Q.; Li, T.Y.; Liang, Y.; Lu, Z.; et al. The lysosomal v-atpase-ragulator complex is a common activator for ampk and mtorc1, acting as a switch between catabolism and anabolism. Cell Metab. 2014, 20, 526–540. [Google Scholar] [CrossRef]
- Lu, M.; Ammar, D.; Ives, H.; Albrecht, F.; Gluck, S.L. Physical interaction between aldolase and vacuolar h+-atpase is essential for the assembly and activity of the proton pump. J. Biol. Chem. 2007, 282, 24495–24503. [Google Scholar] [CrossRef]
- Lu, M.; Sautin, Y.Y.; Holliday, L.S.; Gluck, S.L. The glycolytic enzyme aldolase mediates assembly, expression, and activity of vacuolar h+-atpase. J. Biol. Chem. 2004, 279, 8732–8739. [Google Scholar] [CrossRef]
- Zhao, Y.H.; Zhou, M.; Liu, H.; Ding, Y.; Khong, H.T.; Yu, D.; Fodstad, O.; Tan, M. Upregulation of lactate dehydrogenase a by erbb2 through heat shock factor 1 promotes breast cancer cell glycolysis and growth. Oncogene 2009, 28, 3689–3701. [Google Scholar] [CrossRef]
- Wang, Z.Y.; Loo, T.Y.; Shen, J.G.; Wang, N.; Wang, D.M.; Yang, D.P.; Mo, S.L.; Guan, X.Y.; Chen, J.P. Ldh-a silencing suppresses breast cancer tumorigenicity through induction of oxidative stress mediated mitochondrial pathway apoptosis. Breast Cancer Res. Treat. 2012, 131, 791–800. [Google Scholar] [CrossRef]
- Dong, T.; Liu, Z.; Xuan, Q.; Wang, Z.; Ma, W.; Zhang, Q. Tumor ldh-a expression and serum ldh status are two metabolic predictors for triple negative breast cancer brain metastasis. Sci.Rep. 2017, 7, 6069. [Google Scholar] [CrossRef]
- Mayr, J.A.; Meierhofer, D.; Zimmermann, F.; Feichtinger, R.; Kogler, C.; Ratschek, M.; Schmeller, N.; Sperl, W.; Kofler, B. Loss of complex i due to mitochondrial DNA mutations in renal oncocytoma. Clin. Cancer Res. 2008, 14, 2270–2275. [Google Scholar] [CrossRef]
- Gopal, R.K.; Calvo, S.E.; Shih, A.R.; Chaves, F.L.; McGuone, D.; Mick, E.; Pierce, K.A.; Li, Y.; Garofalo, A.; Van Allen, E.M.; et al. Early loss of mitochondrial complex i and rewiring of glutathione metabolism in renal oncocytoma. Proc. Natl. Acad. Sci. USA 2018, 115, E6283–E6290. [Google Scholar] [CrossRef]
- Priolo, C.; Khabibullin, D.; Reznik, E.; Filippakis, H.; Ogorek, B.; Kavanagh, T.R.; Nijmeh, J.; Herbert, Z.T.; Asara, J.M.; Kwiatkowski, D.J.; et al. Impairment of gamma-glutamyl transferase 1 activity in the metabolic pathogenesis of chromophobe renal cell carcinoma. Proc. Natl. Acad. Sci. USA 2018, 115, E6274–E6282. [Google Scholar] [CrossRef] [Green Version]
- Circu, M.L.; Aw, T.Y. Glutathione and apoptosis. Free Radic. Res. 2008, 42, 689–706. [Google Scholar] [CrossRef] [Green Version]
- Abu Aboud, O.; Habib, S.L.; Trott, J.; Stewart, B.; Liang, S.; Chaudhari, A.J.; Sutcliffe, J.; Weiss, R.H. Glutamine addiction in kidney cancer suppresses oxidative stress and can be exploited for real-time imaging. Cancer Res. 2017, 77, 6746–6758. [Google Scholar] [CrossRef]
- Thyagarajan, A.; Sahu, R.P. Potential contributions of antioxidants to cancer therapy: Immunomodulation and radiosensitization. Integr. Cancer Ther. 2018, 17, 210–216. [Google Scholar] [CrossRef]
- Hsieh, C.L.; Peng, C.C.; Cheng, Y.M.; Lin, L.Y.; Ker, Y.B.; Chang, C.H.; Chen, K.C.; Peng, R.Y. Quercetin and ferulic acid aggravate renal carcinoma in long-term diabetic victims. J. Agric. Food Chem. 2010, 58, 9273–9280. [Google Scholar] [CrossRef]
- Xiao, Y.; Meierhofer, D. Glutathione Metabolism in Renal Cell Carcinoma Progression and Implications for Therapies. Int. J. Mol. Sci. 2019, 20, 3672. [Google Scholar] [CrossRef]
- Calabrese, C.; Simone, D.; Diroma, M.A.; Santorsola, M.; Gutta, C.; Gasparre, G.; Picardi, E.; Pesole, G.; Attimonelli, M. Mtoolbox: A highly automated pipeline for heteroplasmy annotation and prioritization analysis of human mitochondrial variants in high-throughput sequencing. Bioinformatics 2014, 30, 3115–3117. [Google Scholar] [CrossRef]
- Santorsola, M.; Calabrese, C.; Girolimetti, G.; Diroma, M.A.; Gasparre, G.; Attimonelli, M. A multi-parametric workflow for the prioritization of mitochondrial DNA variants of clinical interest. Hum. Genet. 2016, 135, 121–136. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. Maxquant enables high peptide identification rates, individualized p.P.B.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Vizcaino, J.A.; Cote, R.G.; Csordas, A.; Dianes, J.A.; Fabregat, A.; Foster, J.M.; Griss, J.; Alpi, E.; Birim, M.; Contell, J.; et al. The proteomics identifications (pride) database and associated tools: Status in 2013. Nucleic Acids Res. 2013, 41, D1063–D1069. [Google Scholar] [CrossRef]
- Meierhofer, D.; Halbach, M.; Sen, N.E.; Gispert, S.; Auburger, G. Ataxin-2 (atxn2)-knock-out mice show branched chain amino acids and fatty acids pathway alterations. Mol. Cell. Proteom. 2016, 15, 1728–1739. [Google Scholar] [CrossRef]
- Gielisch, I.; Meierhofer, D. Metabolome and proteome profiling of complex i deficiency induced by rotenone. J. Proteome Res. 2015, 14, 224–235. [Google Scholar] [CrossRef]
- Brodaczewska, K.K.; Szczylik, C.; Fiedorowicz, M.; Porta, C.; Czarnecka, A.M. Choosing the right cell line for renal cell cancer research. Mol. Cancer 2016, 15, 83. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Franceschini, A.; Szklarczyk, D.; Frankild, S.; Kuhn, M.; Simonovic, M.; Roth, A.; Lin, J.; Minguez, P.; Bork, P.; von Mering, C.; et al. String v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013, 41, D808–D815. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case ID | Age at Surgery | Gender | Pathologic T Grade | Fuhrman Grade | Tumor Size (cm) |
|---|---|---|---|---|---|
| C1 type I | 68 | Male | pT3a | 3 | 5 |
| C2 type I | 83 | Male | pT1b | 1 | 6.5 |
| C3 type I | 60 | Male | pT1a | 2 | 3.9 |
| C4 type I | 74 | Male | pT1b | 2 | 6 |
| C5 type I | 35 | Male | pT3a | 2 | 3.5 |
| C6 type I | 49 | Male | pT1b | 2 | 4.2 |
| C7 type I | 59 | Male | pT1b | 2 | 4.5 |
| C1 type II | 54 | Male | pT3b | 3 | 16 |
| C2 type II | 70 | Male | pT1a | 2 | 2.5 |
| C3 type II | 57 | Male | pT2b | 3 | 12.5 |
| C4 type II | 80 | Female | pT1b | 2 | 6.2 |
| C5 type II | 77 | Male | pT3b | 2 | 6 |
| C6 type II | 81 | Female | pT3a | 3 | 8.5 |
| C7 type II | 71 | Male | pT2a | 3 | 7.5 |
| C1 type IIM | 42 | Male | pT3a | 3 | 6.5 |
| C2 type IIM | 72 | Male | pT3a | 3 | 17 |
| C3 type IIM | 67 | Female | pT3a | 3 | 5.5 |
| C4 type IIM | 80 | Male | pT2b | 3 | 11 |
| C5 type IIM | 32 | Male | pT3a | 3 | 10.3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, A.A.; Paffrath, V.; Clima, R.; Busch, J.F.; Rabien, A.; Kilic, E.; Villegas, S.; Timmermann, B.; Attimonelli, M.; Jung, K.; et al. Papillary Renal Cell Carcinomas Rewire Glutathione Metabolism and Are Deficient in Both Anabolic Glucose Synthesis and Oxidative Phosphorylation. Cancers 2019, 11, 1298. https://doi.org/10.3390/cancers11091298
Ahmad AA, Paffrath V, Clima R, Busch JF, Rabien A, Kilic E, Villegas S, Timmermann B, Attimonelli M, Jung K, et al. Papillary Renal Cell Carcinomas Rewire Glutathione Metabolism and Are Deficient in Both Anabolic Glucose Synthesis and Oxidative Phosphorylation. Cancers. 2019; 11(9):1298. https://doi.org/10.3390/cancers11091298
Chicago/Turabian StyleAhmad, Ayham Al, Vanessa Paffrath, Rosanna Clima, Jonas Felix Busch, Anja Rabien, Ergin Kilic, Sonia Villegas, Bernd Timmermann, Marcella Attimonelli, Klaus Jung, and et al. 2019. "Papillary Renal Cell Carcinomas Rewire Glutathione Metabolism and Are Deficient in Both Anabolic Glucose Synthesis and Oxidative Phosphorylation" Cancers 11, no. 9: 1298. https://doi.org/10.3390/cancers11091298