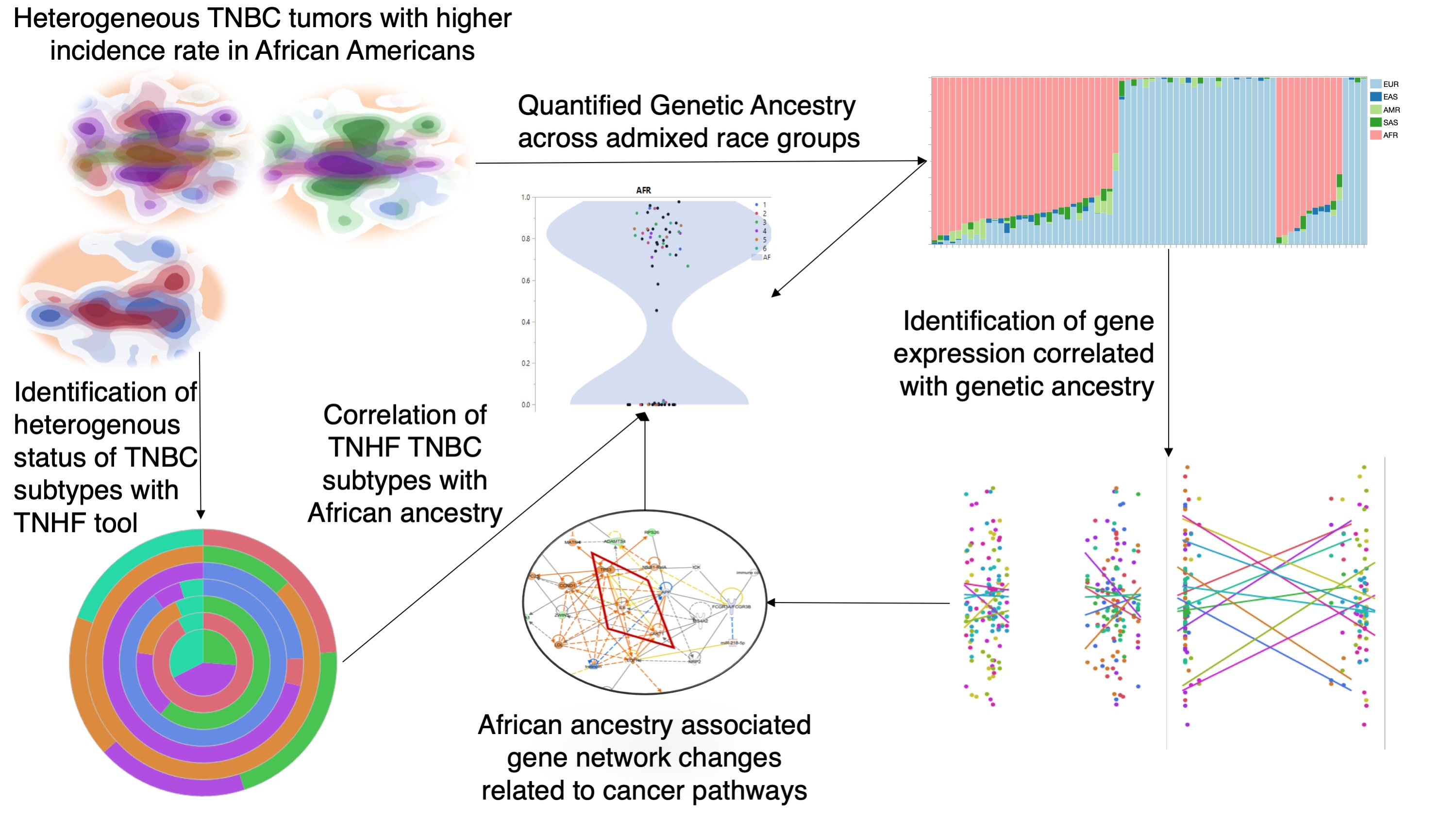
Identification of Distinct Heterogenic Subtypes and Molecular Signatures Associated with African Ancestry in Triple Negative Breast Cancer Using Quantified Genetic Ancestry Models in Admixed Race Populations

 , and
, and 
Abstract

1. Introduction
2. Results
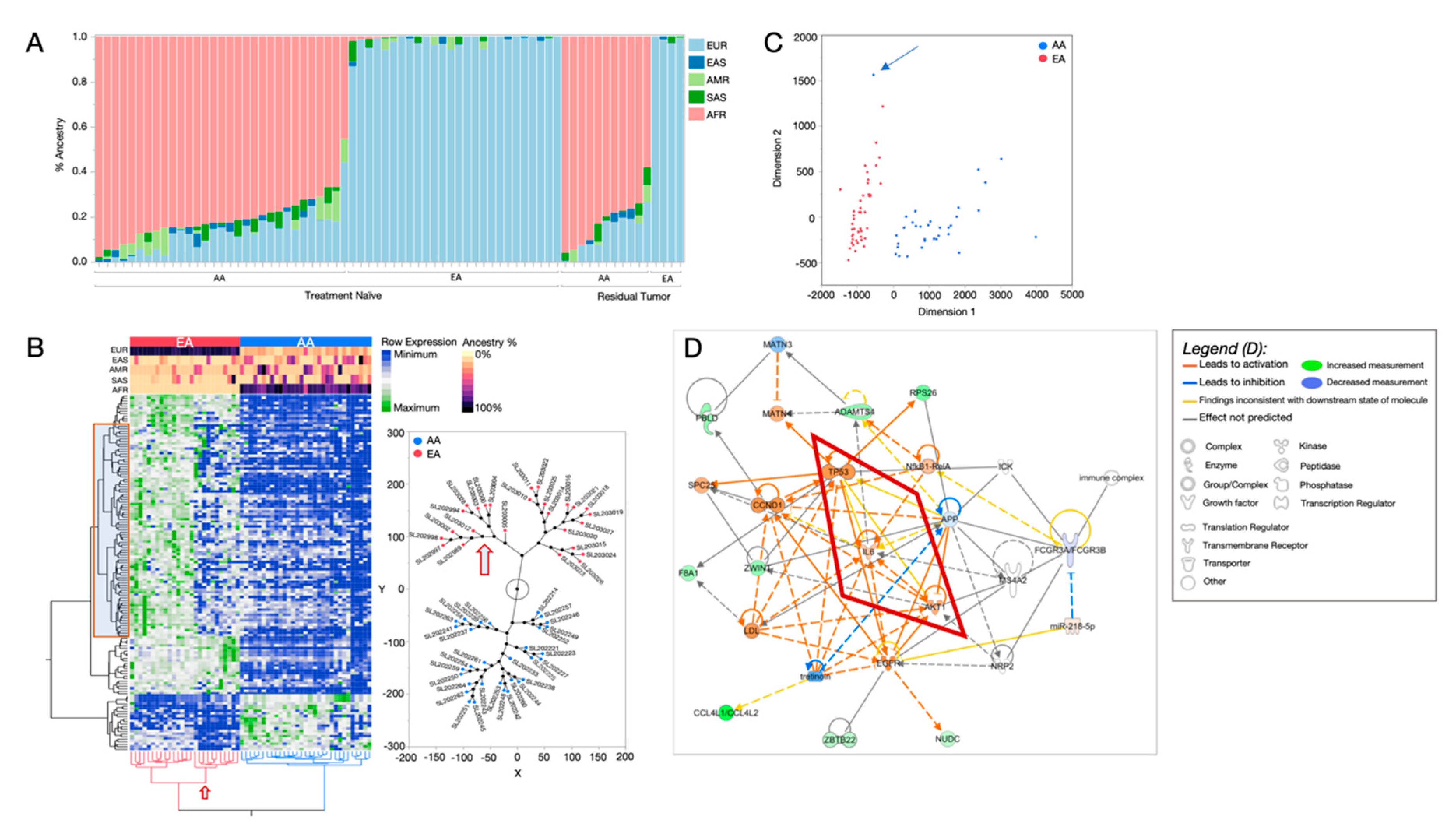
2.1. African Ancestry-Associated Gene Signatures in Treatment-Naïve and Post-Treatment TNBCs
2.2. Distinct Biological Networks of African Ancestry and Differentially Expressed Genes
2.3. Prevalence of TNBC Subtypes across Race and Ancestry Groups
2.4. Differences in Immune Responses by RNAseq Deconvolution
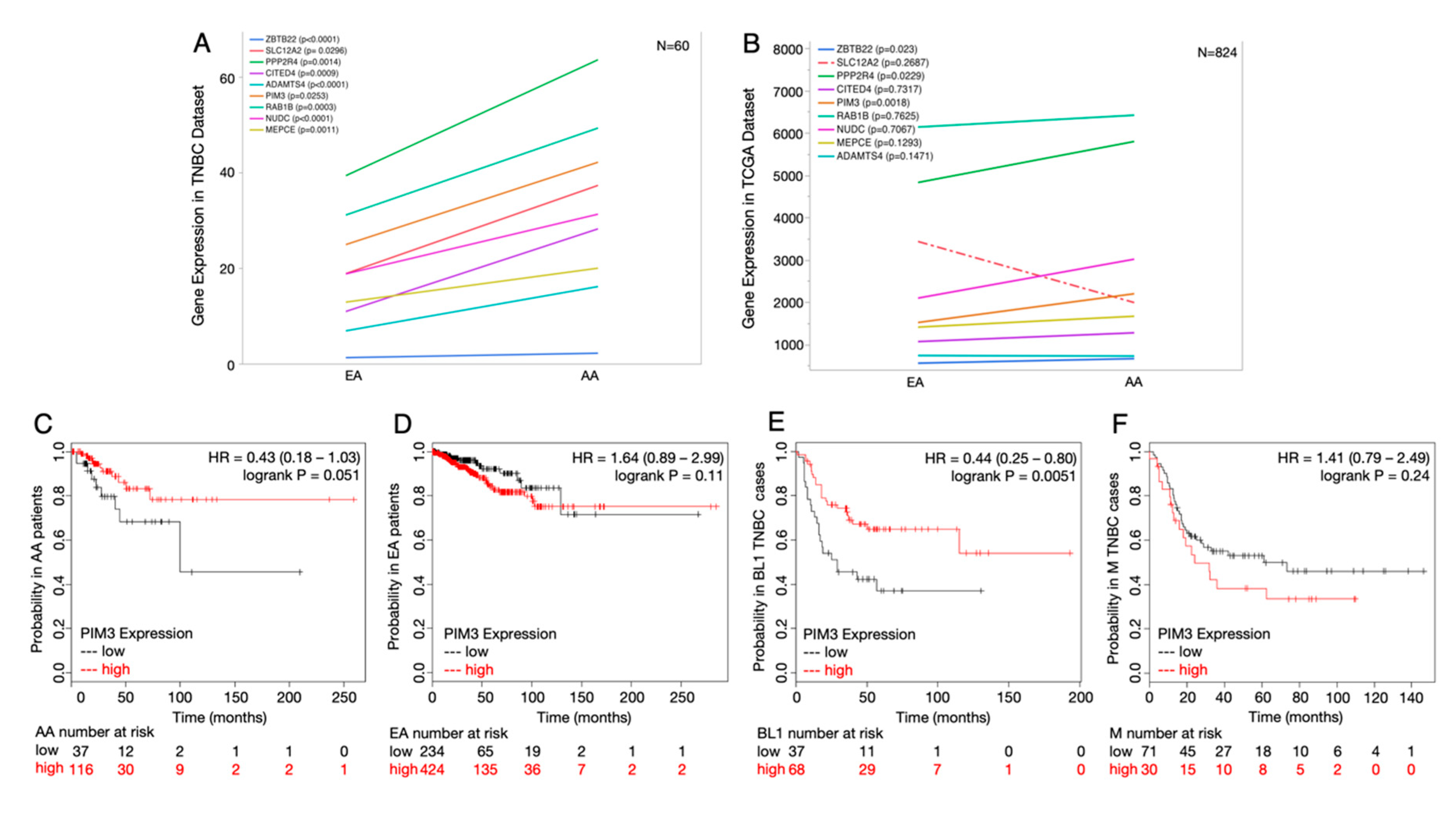
2.5. Potential Druggable Targets from Ancestry-Associated Gene Signatures
3. Discussion
4. Materials and Methods
4.1. TNBC Patient Cohort and Sample Collection
4.2. RNA Extraction, Library Preparation, and Primary Analysis
4.3. Quality Control and Sequence Alignment
4.4. Gene Expression Quantification and Differential Gene Expression Analyses
4.5. TNHF TNBC Subtyping
4.6. Genetic Ancestry and Admixture Estimations from RNAseq Single Nucleotide Variants (SNV)
4.7. Gene Network Analyses
4.8. Estimation of Tumor-Associated Leukocyte Populations
4.9. Survival Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Alcaraz, K.I.; Wiedt, T.L.; Daniels, E.C.; Yabroff, K.R.; Guerra, C.E.; Wender, R.C. Understanding and addressing social determinants to advance cancer health equity in the United States: A blueprint for practice, research, and policy. CA Cancer J. Clin. 2020, 70, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Warnecke, R.B.; Campbell, R.T.; Vijayasiri, G.; Barrett, R.E.; Rauscher, G.H. Multilevel Examination of Health Disparity: The Role of Policy Implementation in Neighborhood Context, in Patient Resources, and in Healthcare Facilities on Later Stage of Breast Cancer Diagnosis. Cancer Epidemiol. Biomark. Prev. 2019, 28, 59–66. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, C.E.; Ma, J.; Gaudet, M.M.; Newman, L.A.; Miller, K.D.; Goding Sauer, A.; Jemal, A.; Siegel, R.L. Breast cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 438–451. [Google Scholar] [CrossRef] [PubMed]
- Newman, L.A. Parsing the Etiology of Breast Cancer Disparities. J. Clin. Oncol. 2016, 34, 1013–1014. [Google Scholar] [CrossRef]
- Newman, L.A.; Kaljee, L.M. Health Disparities and Triple-Negative Breast Cancer in African American Women: A Review. JAMA Surg. 2017, 152, 485–493. [Google Scholar] [CrossRef]
- Newman, L.A. Breast Cancer Disparities: Socioeconomic Factors versus Biology. Ann. Surg. Oncol. 2017, 24, 2869–2875. [Google Scholar] [CrossRef]
- DeSantis, C.E.; Ma, J.; Goding Sauer, A.; Newman, L.A.; Jemal, A. Breast cancer statistics, 2017, racial disparity in mortality by state. CA Cancer J. Clin. 2017, 67, 439–448. [Google Scholar] [CrossRef]
- Newman, L.A. Disparities in breast cancer and african ancestry: A global perspective. Breast J. 2015, 21, 133–139. [Google Scholar] [CrossRef]
- Lukong, K.E.; Ogunbolude, Y.; Kamdem, J.P. Breast cancer in Africa: Prevalence, treatment options, herbal medicines, and socioeconomic determinants. Breast Cancer Res. Treat. 2017, 166, 351–365. [Google Scholar] [CrossRef]
- Vidal, G.; Bursac, Z.; Miranda-Carboni, G.; White-Means, S.; Starlard-Davenport, A. Racial disparities in survival outcomes by breast tumor subtype among African American women in Memphis, Tennessee. Cancer Med. 2017, 6, 1776–1786. [Google Scholar] [CrossRef] [PubMed]
- Vadaparampil, S.T.; Christie, J.; Donovan, K.A.; Kim, J.; Augusto, B.; Kasting, M.L.; Holt, C.L.; Ashing, K.; Halbert, C.H.; Pal, T. Health-related quality of life in Black breast cancer survivors with and without triple-negative breast cancer (TNBC). Breast Cancer Res. Treat. 2017, 163, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Deloumeaux, J.; Gaumond, S.; Bhakkan, B.; Manip M’Ebobisse, N.; Lafrance, W.; Lancelot, P.; Vacque, D.; Negesse, Y.; Diedhiou, A.; Kadhel, P. Incidence, mortality and receptor status of breast cancer in African Caribbean women: Data from the cancer registry of Guadeloupe. Cancer Epidemiol. 2017, 47, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.; Tripathi, S.; Hughley, R.; He, Q.; Bae, S.; Karanam, B.; Martini, R.; Newman, L.; Colomb, W.; Grizzle, W.; et al. AR negative triple negative or “quadruple negative” breast cancers in African American women have an enriched basal and immune signature. PLoS ONE 2018, 13, e0196909. [Google Scholar] [CrossRef]
- Jiagge, E.; Chitale, D.; Newman, L.A. Triple-Negative Breast Cancer, Stem Cells, and African Ancestry. Am. J. Pathol. 2018, 188, 271–279. [Google Scholar] [CrossRef]
- Jiagge, E.; Oppong, J.K.; Bensenhaver, J.; Aitpillah, F.; Gyan, K.; Kyei, I.; Osei-Bonsu, E.; Adjei, E.; Ohene-Yeboah, M.; Toy, K.; et al. Breast Cancer and African Ancestry: Lessons Learned at the 10-Year Anniversary of the Ghana-Michigan Research Partnership and International Breast Registry. J. Glob. Oncol. 2016, 2, 302–310. [Google Scholar] [CrossRef]
- Davis, M.B.; Newman, L.A. Breast Cancer Disparities: How Can We Leverage Genomics to Improve Outcomes? Surg. Oncol. Clin. 2018, 27, 217–234. [Google Scholar] [CrossRef]
- Dietze, E.C.; Sistrunk, C.; Miranda-Carboni, G.; O’Regan, R.; Seewaldt, V.L. Triple-negative breast cancer in African-American women: Disparities versus biology. Nat. Rev. Cancer 2015, 15, 248–254. [Google Scholar] [CrossRef]
- Der, E.M.; Gyasi, R.K.; Tettey, Y.; Edusei, L.; Bayor, M.T.; Jiagge, E.; Gyakobo, M.; Merajver, S.D.; Newman, L.A. Triple-Negative Breast Cancer in Ghanaian Women: The Korle Bu Teaching Hospital Experience. Breast J. 2015, 21, 627–633. [Google Scholar] [CrossRef]
- Sturtz, L.A.; Melley, J.; Mamula, K.; Shriver, C.D.; Ellsworth, R.E. Outcome disparities in African American women with triple negative breast cancer: A comparison of epidemiological and molecular factors between African American and Caucasian women with triple negative breast cancer. BMC Cancer 2014, 14, 62. [Google Scholar] [CrossRef]
- Singh, M.; Ding, Y.; Zhang, L.Y.; Song, D.; Gong, Y.; Adams, S.; Ross, D.S.; Wang, J.H.; Grover, S.; Doval, D.C.; et al. Distinct breast cancer subtypes in women with early-onset disease across races. Am. J. Cancer Res. 2014, 4, 337–352. [Google Scholar] [PubMed]
- Hebert-Magee, S.; Yu, H.; Behring, M.; Jadhav, T.; Shanmugam, C.; Frost, A.; Eltoum, I.E.; Varambally, S.; Manne, U. The combined survival effect of codon 72 polymorphisms and p53 somatic mutations in breast cancer depends on race and molecular subtype. PLoS ONE 2019, 14, e0211734. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, V.B.; Cavalli, L.R.; Dash, C.; Kanaan, Y.M.; Dilawari, A.A.; Horton, S.; Makambi, K.H. Correlates of Triple Negative Breast Cancer and Chemotherapy Patterns in Black and White Women With Breast Cancer. Clin. Breast Cancer 2017, 17, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Parise, C.A.; Caggiano, V. Risk factors associated with the triple-negative breast cancer subtype within four race/ethnicities. Breast Cancer Res. Treat. 2017, 163, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.C.; Mobley, L.R.; Kuo, T.M.; Il’yasova, D. Update on triple-negative breast cancer disparities for the United States: A population-based study from the United States Cancer Statistics database, 2010 through 2014. Cancer 2019, 125, 3412–3417. [Google Scholar] [CrossRef] [PubMed]
- Guan, A.; Lichtensztajn, D.; Oh, D.; Jain, J.; Tao, L.; Hiatt, R.A.; Gomez, S.L.; Fejerman, L.; San Francisco Cancer Initiative Breast Cancer Task Force. Breast Cancer in San Francisco: Disentangling Disparities at the Neighborhood Level. Cancer Epidemiol. Biomark. Prev. 2019, 28, 1968–1976. [Google Scholar] [CrossRef]
- Hossain, F.; Danos, D.; Prakash, O.; Gilliland, A.; Ferguson, T.F.; Simonsen, N.; Leonardi, C.; Yu, Q.; Wu, X.C.; Miele, L.; et al. Neighborhood Social Determinants of Triple Negative Breast Cancer. Front. Public Health 2019, 7, 18. [Google Scholar] [CrossRef]
- Siddharth, S.; Sharma, D. Racial Disparity and Triple-Negative Breast Cancer in African-American Women: A Multifaceted Affair between Obesity, Biology, and Socioeconomic Determinants. Cancers 2018, 10, 514. [Google Scholar] [CrossRef]
- Amirikia, K.C.; Mills, P.; Bush, J.; Newman, L.A. Higher population-based incidence rates of triple-negative breast cancer among young African-American women: Implications for breast cancer screening recommendations. Cancer 2011, 117, 2747–2753. [Google Scholar] [CrossRef]
- Foy, K.C.; Fisher, J.L.; Lustberg, M.B.; Gray, D.M.; DeGraffinreid, C.R.; Paskett, E.D. Disparities in breast cancer tumor characteristics, treatment, time to treatment, and survival probability among African American and white women. NPJ Breast Cancer 2018, 4, 7. [Google Scholar] [CrossRef]
- Williams, F.; Thompson, E. Disparities in Breast Cancer Stage at Diagnosis: Importance of Race, Poverty, and Age. J. Health Dispar. Res. Pract. 2017, 10, 34–45. [Google Scholar] [PubMed]
- Passmore, S.R.; Williams-Parry, K.F.; Casper, E.; Thomas, S.B. Message Received: African American Women and Breast Cancer Screening. Health Promot. Pract. 2017, 18, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Mobley, L.R.; Kuo, T.M. Demographic Disparities in Late-Stage Diagnosis of Breast and Colorectal Cancers Across the USA. J. Racial. Ethn. Health Disparities 2017, 4, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Markossian, T.W.; Hines, R.B. Disparities in late stage diagnosis, treatment, and breast cancer-related death by race, age, and rural residence among women in Georgia. Women Health 2012, 52, 317–335. [Google Scholar] [CrossRef] [PubMed]
- Ring, B.Z.; Hout, D.R.; Morris, S.W.; Lawrence, K.; Schweitzer, B.L.; Bailey, D.B.; Lehmann, B.D.; Pietenpol, J.A.; Seitz, R.S. Generation of an algorithm based on minimal gene sets to clinically subtype triple negative breast cancer patients. BMC Cancer 2016, 16, 143. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Pietenpol, J.A.; Tan, A.R. Triple-negative breast cancer: Molecular subtypes and new targets for therapy. Am. Soc. Clin. Oncol. Educ. Book 2015, e31–e39. [Google Scholar] [CrossRef]
- da Silva, J.L.; Cardoso Nunes, N.C.; Izetti, P.; de Mesquita, G.G.; de Melo, A.C. Triple negative breast cancer: A thorough review of biomarkers. Crit. Rev. Oncol. Hematol. 2019, 145, 102855. [Google Scholar] [CrossRef]
- Millis, S.Z.; Gatalica, Z.; Winkler, J.; Vranic, S.; Kimbrough, J.; Reddy, S.; O’Shaughnessy, J.A. Predictive Biomarker Profiling of > 6000 Breast Cancer Patients Shows Heterogeneity in TNBC, With Treatment Implications. Clin. Breast Cancer 2015, 15, 473–481.e3. [Google Scholar] [CrossRef]
- Global Burden of Disease Cancer Collaboration; Fitzmaurice, C.; Abate, D.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdel-Rahman, O.; Abdelalim, A.; Abdoli, A.; Abdollahpour, I.; et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-Years for 29 Cancer Groups, 1990 to 2017: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2019. [Google Scholar] [CrossRef]
- Kramer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef]
- Chen, X.; Li, J.; Gray, W.H.; Lehmann, B.D.; Bauer, J.A.; Shyr, Y.; Pietenpol, J.A. TNBCtype: A Subtyping Tool for Triple-Negative Breast Cancer. Cancer Inform. 2012, 11, 147–156. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Jovanovic, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Angajala, A.; Mothershed, E.; Davis, M.B.; Tripathi, S.; He, Q.; Bedi, D.; Dean-Colomb, W.; Yates, C. Quadruple Negative Breast Cancers (QNBC) Demonstrate Subtype Consistency among Primary and Recurrent or Metastatic Breast Cancer. Transl. Oncol. 2019, 12, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, B.D.; Martini, R.N.; Hire, R.; Brown, A.; Bennett, B.; Brown, I.; Howerth, E.W.; Egan, M.; Hodgson, J.; Yates, C.; et al. Atypical Chemokine Receptor 1 (DARC/ACKR1) in Breast Tumors Is Associated with Survival, Circulating Chemokines, Tumor-Infiltrating Immune Cells, and African Ancestry. Cancer Epidemiol. Biomark. Prev. 2019, 28, 690–700. [Google Scholar] [CrossRef] [PubMed]
- Keenan, T.; Moy, B.; Mroz, E.A.; Ross, K.; Niemierko, A.; Rocco, J.W.; Isakoff, S.; Ellisen, L.W.; Bardia, A. Comparison of the Genomic Landscape Between Primary Breast Cancer in African American Versus White Women and the Association of Racial Differences With Tumor Recurrence. J. Clin. Oncol. 2015, 33, 3621–3627. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.B.; Newman, L.A. Oncologic Anthropology in Triple Negative Breast Cancer. 2020. in review. [Google Scholar]
- Grizzle, W.E.; Kittles, R.A.; Rais-Bahrami, S.; Shah, E.; Adams, G.W.; DeGuenther, M.S.; Kolettis, P.N.; Nix, J.W.; Bryant, J.E.; Chinsky, R.; et al. Self-Identified African Americans and prostate cancer risk: West African genetic ancestry is associated with prostate cancer diagnosis and with higher Gleason sum on biopsy. Cancer Med. 2019, 8, 6915–6922. [Google Scholar] [CrossRef]
- Chen, Y.; Sadasivan, S.; Ruicong, S.; Datta, I.; Taneja, K.; Chitale, D.; Gupta, N.; Davis, M.B.; Newman, L.A.; Rogers, C.G.; et al. Breast and prostate cancers harbor common somatic copy number alterations that consistently differ by race and are associated with survival. BMC Genom. 2020. in revision. [Google Scholar]
- Morceau, F.; Chateauvieux, S.; Gaigneaux, A.; Dicato, M.; Diederich, M. Long and short non-coding RNAs as regulators of hematopoietic differentiation. Int. J. Mol. Sci. 2013, 14, 14744–14770. [Google Scholar] [CrossRef] [PubMed]
- Koduru, S.V.; Leberfinger, A.N.; Ravnic, D.J. Small Non-coding RNA Abundance in Adrenocortical Carcinoma: A Footprint of a Rare Cancer. J. Genom. 2017, 5, 99–118. [Google Scholar] [CrossRef]
- Wang, B.D.; Ceniccola, K.; Hwang, S.; Andrawis, R.; Horvath, A.; Freedman, J.A.; Olender, J.; Knapp, S.; Ching, T.; Garmire, L.; et al. Alternative splicing promotes tumour aggressiveness and drug resistance in African American prostate cancer. Nat. Commun. 2017, 8, 15921. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Freedman, J.A.; Liu, H.; Moorman, P.G.; Hyslop, T.; George, D.J.; Lee, N.H.; Patierno, S.R.; Wei, Q. Associations between RNA splicing regulatory variants of stemness-related genes and racial disparities in susceptibility to prostate cancer. Int. J. Cancer 2017, 141, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Shuch, B.; Mikhail, M.; Satagopan, J.; Lee, P.; Yee, H.; Chang, C.; Cordon-Cardo, C.; Taneja, S.S.; Osman, I. Racial disparity of epidermal growth factor receptor expression in prostate cancer. J. Clin. Oncol. 2004, 22, 4725–4729. [Google Scholar] [CrossRef]
- Cheang, M.C.; Voduc, D.; Bajdik, C.; Leung, S.; McKinney, S.; Chia, S.K.; Perou, C.M.; Nielsen, T.O. Basal-like breast cancer defined by five biomarkers has superior prognostic value than triple-negative phenotype. Clin. Cancer Res. 2008, 14, 1368–1376. [Google Scholar] [CrossRef]
- Liedtke, C.; Mazouni, C.; Hess, K.R.; Andre, F.; Tordai, A.; Mejia, J.A.; Symmans, W.F.; Gonzalez-Angulo, A.M.; Hennessy, B.; Green, M.; et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J. Clin. Oncol. 2008, 26, 1275–1281. [Google Scholar] [CrossRef]
- Cortazar, P.; Zhang, L.; Untch, M.; Mehta, K.; Costantino, J.P.; Wolmark, N.; Bonnefoi, H.; Cameron, D.; Gianni, L.; Valagussa, P.; et al. Pathological complete response and long-term clinical benefit in breast cancer: The CTNeoBC pooled analysis. Lancet 2014, 384, 164–172. [Google Scholar] [CrossRef]
- McAndrew, N.; DeMichele, A. Neoadjuvant Chemotherapy Considerations in Triple-Negative Breast Cancer. J. Target Ther. Cancer 2018, 7, 52–69. [Google Scholar]
- Prat, A.; Lluch, A.; Albanell, J.; Barry, W.T.; Fan, C.; Chacon, J.I.; Parker, J.S.; Calvo, L.; Plazaola, A.; Arcusa, A.; et al. Predicting response and survival in chemotherapy-treated triple-negative breast cancer. Br. J. Cancer 2014, 111, 1532–1541. [Google Scholar] [CrossRef]
- Isakoff, S.J.; Mayer, E.L.; He, L.; Traina, T.A.; Carey, L.A.; Krag, K.J.; Rugo, H.S.; Liu, M.C.; Stearns, V.; Come, S.E.; et al. TBCRC009: A Multicenter Phase II Clinical Trial of Platinum Monotherapy With Biomarker Assessment in Metastatic Triple-Negative Breast Cancer. J. Clin. Oncol. 2015, 33, 1902–1909. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, B.; Mayer, I.A.; Mayer, E.L.; Abramson, V.G.; Bardia, A.; Sanders, M.E.; Kuba, M.G.; Estrada, M.V.; Beeler, J.S.; Shaver, T.M.; et al. A Randomized Phase II Neoadjuvant Study of Cisplatin, Paclitaxel With or Without Everolimus in Patients with Stage II/III Triple-Negative Breast Cancer (TNBC): Responses and Long-term Outcome Correlated with Increased Frequency of DNA Damage Response Gene Mutations, TNBC Subtype, AR Status, and Ki67. Clin. Cancer Res. 2017, 23, 4035–4045. [Google Scholar] [CrossRef] [PubMed]
- Lindner, R.; Sullivan, C.; Offor, O.; Lezon-Geyda, K.; Halligan, K.; Fischbach, N.; Shah, M.; Bossuyt, V.; Schulz, V.; Tuck, D.P.; et al. Molecular phenotypes in triple negative breast cancer from African American patients suggest targets for therapy. PLoS ONE 2013, 8, e71915. [Google Scholar] [CrossRef] [PubMed]
- Elnaggar, J.; Tsien, F.; Yates, C.; Davis, M.; Miele, L.; Hicks, C. An Integrative Genomics Approach for Associated Genetic Susceptibility with the Tumor Immune Microenvironment in Triple Negative Breast Cancer. Biomed. J. Sci. Tech. Res. 2019, 15, 1–12. [Google Scholar] [CrossRef]
- Powell, I.J.; Dyson, G.; Land, S.; Ruterbusch, J.; Bock, C.H.; Lenk, S.; Herawi, M.; Everson, R.; Giroux, C.N.; Schwartz, A.G.; et al. Genes associated with prostate cancer are differentially expressed in African American and European American men. Cancer Epidemiol. Biomark. Prev. 2013, 22, 891–897. [Google Scholar] [CrossRef]
- Porter, P.L.; Lund, M.J.; Lin, M.G.; Yuan, X.; Liff, J.M.; Flagg, E.W.; Coates, R.J.; Eley, J.W. Racial differences in the expression of cell cycle-regulatory proteins in breast carcinoma. Cancer 2004, 100, 2533–2542. [Google Scholar] [CrossRef]
- Martin, D.N.; Boersma, B.J.; Yi, M.; Reimers, M.; Howe, T.M.; Yfantis, H.G.; Tsai, Y.C.; Williams, E.H.; Lee, D.H.; Stephens, R.M.; et al. Differences in the tumor microenvironment between African-American and European-American breast cancer patients. PLoS ONE 2009, 4, e4531. [Google Scholar] [CrossRef]
- Morris, G.J.; Naidu, S.; Topham, A.K.; Guiles, F.; Xu, Y.; McCue, P.; Schwartz, G.F.; Park, P.K.; Rosenberg, A.L.; Brill, K.; et al. Differences in breast carcinoma characteristics in newly diagnosed African-American and Caucasian patients: A single-institution compilation compared with the National Cancer Institute’s Surveillance, Epidemiology, and End Results database. Cancer 2007, 110, 876–884. [Google Scholar] [CrossRef]
- Caleffi, M.; Teague, M.W.; Jensen, R.A.; Vnencak-Jones, C.L.; Dupont, W.D.; Parl, F.F. p53 gene mutations and steroid receptor status in breast cancer. Clinicopathologic correlations and prognostic assessment. Cancer 1994, 73, 2147–2156. [Google Scholar] [CrossRef]
- Perou, C.M. Molecular stratification of triple-negative breast cancers. Oncologist 2011, 16 (Suppl. 1), 61–70. [Google Scholar] [CrossRef]
- Yeyeodu, S.T.; Kidd, L.R.; Kimbro, K.S. Protective Innate Immune Variants in Racial/Ethnic Disparities of Breast and Prostate Cancer. Cancer Immunol. Res. 2019, 7, 1384–1389. [Google Scholar] [CrossRef]
- O’Meara, T.; Safonov, A.; Casadevall, D.; Qing, T.; Silber, A.; Killelea, B.; Hatzis, C.; Pusztai, L. Immune microenvironment of triple-negative breast cancer in African-American and Caucasian women. Breast Cancer Res. Treat. 2019, 175, 247–259. [Google Scholar] [CrossRef]
- Sikandar, B.; Qureshi, M.A.; Naseem, S.; Khan, S.; Mirza, T. Increased Tumour Infiltration of CD4+ and CD8+ T-Lymphocytes in Patients with Triple Negative Breast Cancer Suggests Susceptibility to Immune Therapy. Asian Pac. J. Cancer Prev. 2017, 18, 1827–1832. [Google Scholar] [CrossRef]
- Forero, A.; Li, Y.; Chen, D.; Grizzle, W.E.; Updike, K.L.; Merz, N.D.; Downs-Kelly, E.; Burwell, T.C.; Vaklavas, C.; Buchsbaum, D.J.; et al. Expression of the MHC Class II Pathway in Triple-Negative Breast Cancer Tumor Cells Is Associated with a Good Prognosis and Infiltrating Lymphocytes. Cancer Immunol. Res. 2016, 4, 390–399. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Thoughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 9 May 2020).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Genomes Project, C.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef]
- Gyorffy, B.; Lanczky, A.; Eklund, A.C.; Denkert, C.; Budczies, J.; Li, Q.; Szallasi, Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1809 patients. Breast Cancer Res. Treat. 2010, 123, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Nagy, A.; Lanczky, A.; Menyhart, O.; Gyorffy, B. Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Sci. Rep. 2018, 8, 9227. [Google Scholar] [CrossRef] [PubMed]
- Regnante, J.M.; Richie, N.A.; Fashoyin-Aje, L.; Vichnin, M.; Ford, M.; Roy, U.B.; Turner, K.; Hall, L.L.; Gonzalez, E.; Esnaola, N.; et al. US Cancer Centers of Excellence Strategies for Increased Inclusion of Racial and Ethnic Minorities in Clinical Trials. J. Oncol. Pract. 2019, 15, e289–e299. [Google Scholar] [CrossRef] [PubMed]
- Clark, L.T.; Watkins, L.; Pina, I.L.; Elmer, M.; Akinboboye, O.; Gorham, M.; Jamerson, B.; McCullough, C.; Pierre, C.; Polis, A.B.; et al. Increasing Diversity in Clinical Trials: Overcoming Critical Barriers. Curr. Probl. Cardiol. 2019, 44, 148–172. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Name | Drugs Tested in Cancer | Disease (Cancer or Other) | Organism | Pubmed ID (PMID) |
|---|---|---|---|---|---|
| AKT1 | AKT Serine/Threonine Kinase 1 | Arsenic Trioxide, Carboplatin, Everolimus, Cisplatin, Nelfinavir | Various Cancers | Human | 12480548 |
| CCND1 | Cyclin D1 | Arsenic Trioxide, Cetuximab, Aspirin, Trametinib, Palbociclib | Various Cancers and other diseases | Human | 12480548 |
| SLC12A2 | Solute Carrier Family 12 Member 2 | Bumetanide and Furosemide | Neonatal Seizures, Autism, Heart Failure | Human | 11698253 |
| PPP2R4 | Protein Phosphatase 2 Phosphatase Activator | Ceramide | Breast Cancer, Diabetes, Obesity | Human | 29261144 |
| RELA | RELA Proto-Oncogene, NF-KB Subunit | Dimethyl fumarate | Multiple Sclerosis | Human | 26683377 |
| CITED4 | Cbp/P300 Interacting Transactivator With Glu/Asp Rich Carboxy-Terminal Domain 4 | Fluorouracil | Cardiac ischaemia/reperfusion (I/R) injury | Mouse | 28304151 |
| PIM3 | Pim-3 Proto-Oncogene, Serine/Threonine Kinase | Fostamatinib, Gefitinib, Sunitinib, Ruboxistaurin | Cancer and others | Human | 26516587 |
| EGFR | Epidermal Growth Factor Receptor | Gefitinib, Erlotinib, Lapatinib and Cetuximab | Non-small cell lung cancer | Human | 15118073 |
| LPL | Lipoprotein Lipase | Orlistat, Fenofibrate | Obesity and Diabetes | Human | 24016212 |
| NUDC | Nuclear Distribution C, Dynein Complex Regulator | Phenethyl Isothiocyanate | Various Cancers and Cardiovascular Disease | Human | 21838287 |
| MEPCE | Methylphosphate Capping Enzyme | S-Adenosyl methionine | Various | Human | 23985780 |
| IL6 | Interleukin 6 | Siltuximab, Vitamin C and E, Adalimumab | Various | Human | 8823310 |
| NFKB1 | Nuclear Factor Kappa B Subunit 1 | Thalidomide, Donepezil, Glycyrrhizin, Triflusal | Various | Human | 15723633 |
| ADAMTS4 | ADAM Metallopeptidase with Thrombospondin Type 1 Motif 4 | Tinzaparin | Brain Tumors, Thromboembolism, Thrombosis | Human | 15723278 |
| TP53 | Tumor Protein P53 | Venetoclax, Cyclophosphamide, Fluorouracil, Cisplatin | Various | Human | 27069256 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davis, M.; Martini, R.; Newman, L.; Elemento, O.; White, J.; Verma, A.; Datta, I.; Adrianto, I.; Chen, Y.; Gardner, K.; et al. Identification of Distinct Heterogenic Subtypes and Molecular Signatures Associated with African Ancestry in Triple Negative Breast Cancer Using Quantified Genetic Ancestry Models in Admixed Race Populations. Cancers 2020, 12, 1220. https://doi.org/10.3390/cancers12051220
Davis M, Martini R, Newman L, Elemento O, White J, Verma A, Datta I, Adrianto I, Chen Y, Gardner K, et al. Identification of Distinct Heterogenic Subtypes and Molecular Signatures Associated with African Ancestry in Triple Negative Breast Cancer Using Quantified Genetic Ancestry Models in Admixed Race Populations. Cancers. 2020; 12(5):1220. https://doi.org/10.3390/cancers12051220
Chicago/Turabian StyleDavis, Melissa, Rachel Martini, Lisa Newman, Olivier Elemento, Jason White, Akanksha Verma, Indrani Datta, Indra Adrianto, Yalei Chen, Kevin Gardner, and et al. 2020. "Identification of Distinct Heterogenic Subtypes and Molecular Signatures Associated with African Ancestry in Triple Negative Breast Cancer Using Quantified Genetic Ancestry Models in Admixed Race Populations" Cancers 12, no. 5: 1220. https://doi.org/10.3390/cancers12051220
APA StyleDavis, M., Martini, R., Newman, L., Elemento, O., White, J., Verma, A., Datta, I., Adrianto, I., Chen, Y., Gardner, K., Kim, H.-G., Colomb, W. D., Eltoum, I.-E., Frost, A. R., Grizzle, W. E., Sboner, A., Manne, U., & Yates, C. (2020). Identification of Distinct Heterogenic Subtypes and Molecular Signatures Associated with African Ancestry in Triple Negative Breast Cancer Using Quantified Genetic Ancestry Models in Admixed Race Populations. Cancers, 12(5), 1220. https://doi.org/10.3390/cancers12051220