Interfering with ROS Metabolism in Cancer Cells: The Potential Role of Quercetin

Abstract
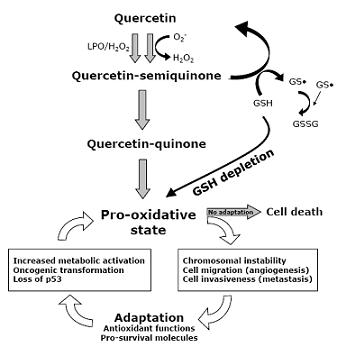
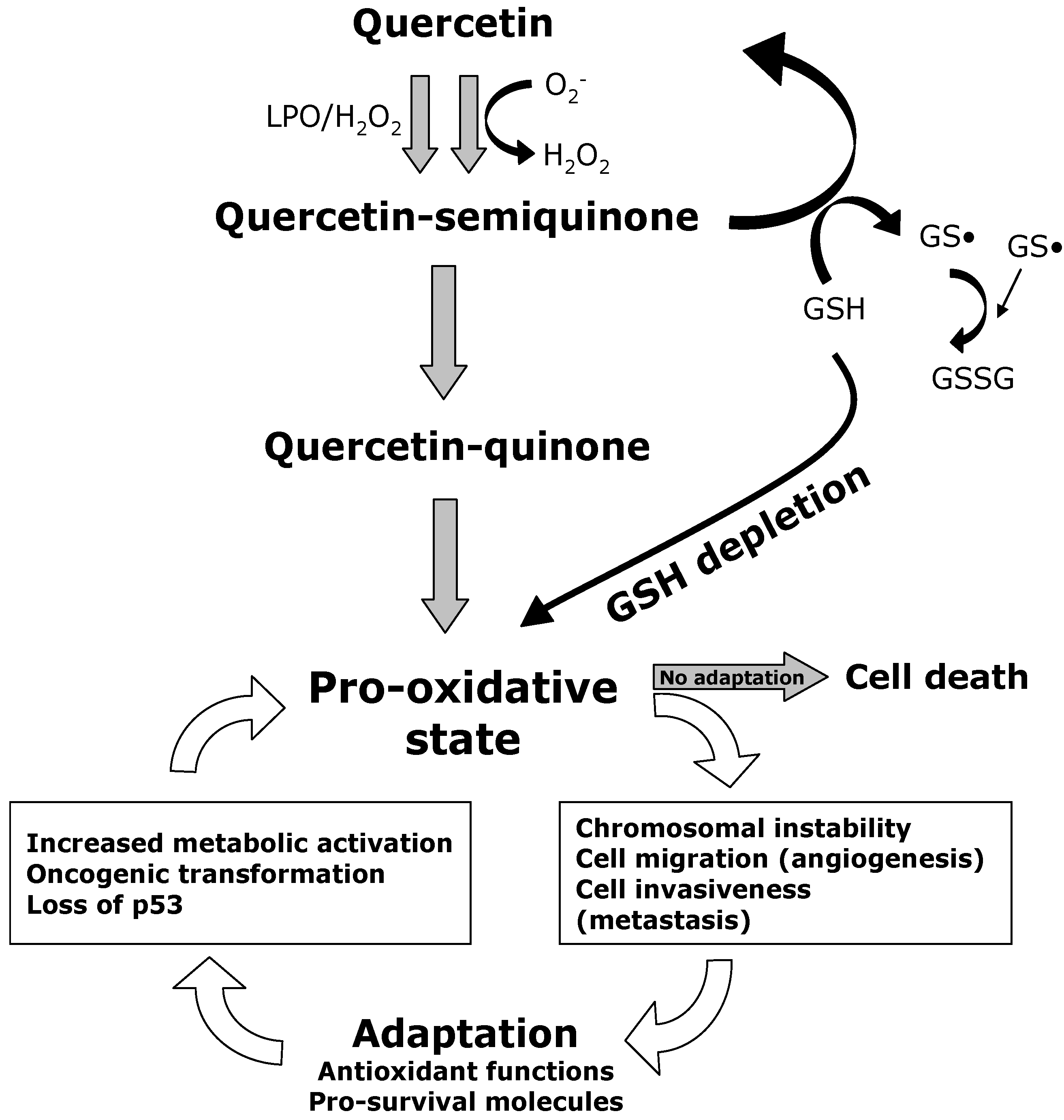
:
{kind=link}
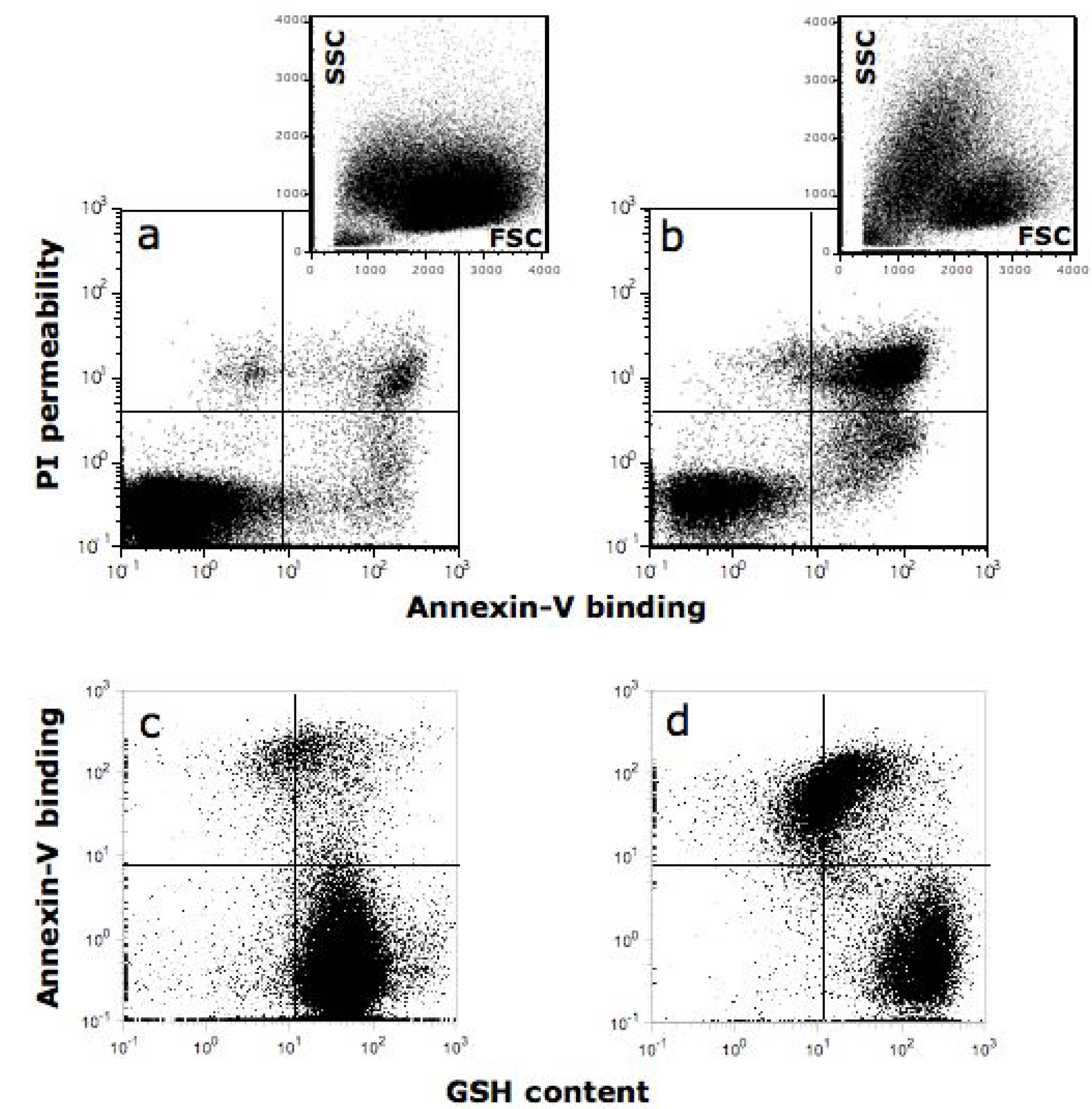
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. ROS Balance in Cancer Cells
3. Cancer Cells Adapt Unbalanced ROS Levels
4. ROS Exert Several Effects on Cancer Cells
5. Excessive ROS Levels Cause Cell Damage and Cell Death
6. Targeting Intrinsic Oxidative Stress in Cancer Cells
7. GSH Depletion in Cancer Cells
8. Flavonoids Interfere with ROS Metabolism
9. Quercetin Interferes with ROS Metabolism
10. Quercetin Induces Apoptosis in Cancer Cells

11. Relevance of Quercetin for Human Health

12. Concluding Remarks
Acknowledgements
References
- Rhee, S.G. Cell signaling. H2O2, a necessary evil for cell signaling. Science 2006, 312, 1882–1883. [Google Scholar] [CrossRef]
- Lambeth, J.D. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004, 4, 181–189. [Google Scholar]
- Schafer, F.Q.; Buettner, G.R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 2001, 30, 1191–1212. [Google Scholar] [CrossRef]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef]
- Meyer, Y.; Buchanan, B.B.; Vignols, F.; Reichheld, J.P. Thioredoxins and glutaredoxins: unifying elements in redox biology. Annu. Rev. Genet. 2009, 43, 335–367. [Google Scholar] [CrossRef]
- Szatrowski, T.P.; Nathan, C.F. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991, 51, 794–798. [Google Scholar]
- Toyokuni, S.; Okamoto, K.; Yodoi, J.; Hiai, H. Persistent oxidative stress in cancer. FEBS Lett. 1995, 358, 1–3. [Google Scholar] [CrossRef]
- Zhou, Y.; Hileman, E.O.; Plunkett, W.; Keating, M.J.; Huang, P. Free radical stress in chronic lymphocytic leukemia cells and its role in cellular sensitivity to ROS-generating anticancer agents. Blood 2003, 101, 4098–4104. [Google Scholar] [CrossRef]
- Pelicano, H.; Carney, D.; Huang, P. ROS stress in cancer cells and therapeutic implications. Drug Resist. Updat. 2004, 7, 97–110. [Google Scholar] [CrossRef]
- Tennant, D.A.; Duran, R.V.; Boulahbel, H.; Gottlieb, E. Metabolic transformation in cancer. Carcinogenesis 2009, 30, 1269–1280. [Google Scholar] [CrossRef]
- Pfeiffer, T.; Schuster, S.; Bonhoeffer, S. Cooperation and competition in the evolution of ATP-producing pathways. Science 2001, 292, 504–507. [Google Scholar] [CrossRef]
- Lin, P.C.; Lin, J.K.; Yang, S. H.; Wang, H.S.; Li, A.F.; Chang, S.C. Expression of beta-F1-ATPase and mitochondrial transcription factor A and the change in mitochondrial DNA content in colorectal cancer: clinical data analysis and evidence from an in vitro study. Int. J. Colorectal. Dis. 2008, 23, 1223–1232. [Google Scholar] [CrossRef]
- Lopez-Rios, F.; Sanchez-Arago, M.; Garcia-Garcia, E.; Ortega, A.D.; Berrendero, J.R.; Pozo-Rodriguez, F.; Lopez-Encuentra, A.; Ballestin, C.; Cuezva, J.M. Loss of the mitochondrial bioenergetic capacity underlies the glucose avidity of carcinomas. Cancer Res. 2007, 67, 9013–9017. [Google Scholar] [CrossRef]
- Dang, C.V.; Kim, J.W.; Gao, P.; Yustein, J. The interplay between MYC and HIF in cancer. Nat. Rev. Cancer 2008, 8, 51–56. [Google Scholar] [CrossRef]
- Bertout, J.A.; Patel, S.A.; Simon, M.C. The impact of O2 availability on human cancer. Nat. Rev. Cancer 2008, 8, 967–975. [Google Scholar] [CrossRef]
- Sattler, M.; Verma, S.; Shrikhande, G.; Byrne, C.H.; Pride, Y.B.; Winkler, T.; Greenfield, E.A.; Salgia, R.; Griffin, J.D. The BCR/ABL tyrosine kinase induces production of reactive oxygen species in hematopoietic cells. J. Biol. Chem. 2000, 275, 24273–24278. [Google Scholar] [CrossRef]
- Vafa, O.; Wade, M.; Kern, S.; Beeche, M.; Pandita, T.K.; Hampton, G.M.; Wahl, G.M. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: a mechanism for oncogene-induced genetic instability. Mol. Cell 2002, 9, 1031–1044. [Google Scholar] [CrossRef]
- Irani, K.; Xia, Y.; Zweier, J.L.; Sollott, S.J.; Der, C.J.; Fearon, E.R.; Sundaresan, M.; Finkel, T.; Goldschmidt-Clermont, P.J. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science 1997, 275, 1649–1652. [Google Scholar] [CrossRef]
- Mitsushita, J.; Lambeth, J.D.; Kamata, T. The superoxide-generating oxidase Nox1 is functionally required for Ras oncogene transformation. Cancer Res. 2004, 64, 3580–3585. [Google Scholar] [CrossRef]
- Fu, X.; Beer, D.G.; Behar, J.; Wands, J.; Lambeth, D.; Cao, W. cAMP-response element-binding protein mediates acid-induced NADPH oxidase NOX5-S expression in Barrett esophageal adenocarcinoma cells. J. Biol. Chem. 2006, 281, 20368–20382. [Google Scholar] [CrossRef]
- Mochizuki, T.; Furuta, S.; Mitsushita, J.; Shang, W.H.; Ito, M.; Yokoo, Y.; Yamaura, M.; Ishizone, S.; Nakayama, J.; Konagai, A.; Hirose, K.; Kiyosawa, K.; Kamata, T. Inhibition of NADPH oxidase 4 activates apoptosis via the AKT/apoptosis signal-regulating kinase 1 pathway in pancreatic cancer PANC-1 cells. Oncogene 2006, 25, 3699–3707. [Google Scholar] [CrossRef]
- Yamaura, M.; Mitsushita, J.; Furuta, S.; Kiniwa, Y.; Ashida, A.; Goto, Y.; Shang, W.H.; Kubodera, M.; Kato, M.; Takata, M.; Saida, T.; Kamata, T. NADPH oxidase 4 contributes to transformation phenotype of melanoma cells by regulating G2-M cell cycle progression. Cancer Res. 2009, 69, 2647–2654. [Google Scholar] [CrossRef]
- Vousden, K.H.; Lane, D.P. p53 in health and disease. Nat. Rev. Mol. Cell. Biol. 2007, 8, 275–283. [Google Scholar] [CrossRef]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef]
- Cano, C.E.; Gommeaux, J.; Pietri, S.; Culcasi, M.; Garcia, S.; Seux, M.; Barelier, S.; Vasseur, S.; Spoto, R.P.; Pebusque, M.J.; Dusetti, N.J.; Iovanna, J.L.; Carrier, A. Tumor protein 53-induced nuclear protein 1 is a major mediator of p53 antioxidant function. Cancer Res. 2009, 69, 219–226. [Google Scholar] [CrossRef]
- Sablina, A.A.; Budanov, A.V.; Ilyinskaya, G.V.; Agapova, L.S.; Kravchenko, J.E.; Chumakov, P.M. The antioxidant function of the p53 tumor suppressor. Nat. Med. 2005, 11, 1306–1313. [Google Scholar] [CrossRef]
- Farber, E.; Rubin, H. Cellular adaptation in the origin and development of cancer. Cancer Res. 1991, 51, 2751–2761. [Google Scholar]
- Park, H.J.; Carr, J.R.; Wang, Z.; Nogueira, V.; Hay, N.; Tyner, A.L.; Lau, L.F.; Costa, R.H.; Raychaudhuri, P. FoxM1, a critical regulator of oxidative stress during oncogenesis. EMBO J. 2009, 28, 2908–2918. [Google Scholar] [CrossRef]
- Bae, I.; Fan, S.; Meng, Q.; Rih, J.K.; Kim, H.J.; Kang, H.J.; Xu, J.; Goldberg, I.D.; Jaiswal, A.K.; Rosen, E.M. BRCA1 induces antioxidant gene expression and resistance to oxidative stress. Cancer Res. 2004, 64, 7893–7909. [Google Scholar] [CrossRef]
- Saha, T.; Rih, J.K.; Rosen, E.M. BRCA1 down-regulates cellular levels of reactive oxygen species. FEBS Lett. 2009, 583, 1535–1543. [Google Scholar] [CrossRef]
- Cao, L.; Xu, X.; Cao, L.L.; Wang, R.H.; Coumoul, X.; Kim, S.S.; Deng, C.X. Absence of full-length Brca1 sensitizes mice to oxidative stress and carcinogen-induced tumorigenesis in the esophagus and forestomach. Carcinogenesis 2007, 28, 1401–1407. [Google Scholar] [CrossRef]
- Sundaresan, N.R.; Gupta, M.; Kim, G.; Rajamohan, S.B.; Isbatan, A.; Gupta, M.P. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J. Clin. Invest. 2009, 119, 2758–2771. [Google Scholar]
- Hu, Y.; Rosen, D.G.; Zhou, Y.; Feng, L.; Yang, G.; Liu, J.; Huang, P. Mitochondrial manganese-superoxide dismutase expression in ovarian cancer: role in cell proliferation and response to oxidative stress. J. Biol. Chem. 2005, 280, 39485–39492. [Google Scholar]
- Nakata, T.; Suzuki, K.; Fujii, J.; Ishikawa, M.; Tatsumi, H.; Sugiyama, T.; Nishida, T.; Shimizu, T.; Yakushiji, M.; Taniguchi, N. High expression of manganese superoxide dismutase in 7,12-dimethylbenz[a]anthracene-induced ovarian cancer and increased serum levels in the tumor-bearing rats. Carcinogenesis 1992, 13, 1941–1943. [Google Scholar] [CrossRef]
- Hileman, E.O.; Liu, J.; Albitar, M.; Keating, M.J.; Huang, P. Intrinsic oxidative stress in cancer cells: a biochemical basis for therapeutic selectivity. Cancer Chemother. Pharmacol. 2004, 53, 209–219. [Google Scholar] [CrossRef]
- Janssen, A.M.; Bosman, C.B.; van Duijn, W.; Oostendorp-van de Ruit, M.M.; Kubben, F.J.; Griffioen, G.; Lamers, C.B.; van Krieken, J.H.; van de Velde, C.J.; Verspaget, H.W. Superoxide dismutases in gastric and esophageal cancer and the prognostic impact in gastric cancer. Clin. Cancer Res. 2000, 6, 3183–3192. [Google Scholar]
- Recktenwald, C.V.; Kellner, R.; Lichtenfels, R.; Seliger, B. Altered detoxification status and increased resistance to oxidative stress by K-ras transformation. Cancer Res. 2008, 68, 10086–10093. [Google Scholar] [CrossRef]
- Benassi, B.; Fanciulli, M.; Fiorentino, F.; Porrello, A.; Chiorino, G.; Loda, M.; Zupi, G.; Biroccio, A. c-Myc phosphorylation is required for cellular response to oxidative stress. Mol. Cell 2006, 21, 509–519. [Google Scholar] [CrossRef]
- Radisky, D.C.; Levy, D.D.; Littlepage, L.E.; Liu, H.; Nelson, C.M.; Fata, J.E.; Leake, D.; Godden, E.L.; Albertson, D.G.; Nieto, M.A.; Werb, Z.; Bissell, M.J. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature 2005, 436, 123–127. [Google Scholar] [CrossRef]
- Samper, E.; Nicholls, D.G.; Melov, S. Mitochondrial oxidative stress causes chromosomal instability of mouse embryonic fibroblasts. Aging Cell 2003, 2, 277–285. [Google Scholar] [CrossRef]
- Burdon, R.H. Superoxide and hydrogen peroxide in relation to mammalian cell proliferation. Free Radic. Biol. Med. 1995, 18, 775–794. [Google Scholar] [CrossRef]
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Nilsa, R.D.; Huang, P. Redox regulation of cell survival. Antioxid. Redox Signal. 2008, 10, 1343–1374. [Google Scholar] [CrossRef]
- Karin, M.; Lin, A. NF-kappaB at the crossroads of life and death. Nat. Immunol. 2002, 3, 221–227. [Google Scholar] [CrossRef]
- Ishii, T.; Itoh, K.; Takahashi, S.; Sato, H.; Yanagawa, T.; Katoh, Y.; Bannai, S.; Yamamoto, M. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J. Biol. Chem. 2000, 275, 16023–16029. [Google Scholar]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; Stivala, F.; Libra, M.; Basecke, J.; Evangelisti, C.; Martelli, A.M.; Franklin, R.A. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta 2007, 1773, 1263–1284. [Google Scholar]
- Mori, K.; Shibanuma, M.; Nose, K. Invasive potential induced under long-term oxidative stress in mammary epithelial cells. Cancer Res. 2004, 64, 7464–7472. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Matrix metalloproteinases and the development of cancer. Chem. Biol. 1996, 3, 895–904. [Google Scholar] [CrossRef]
- Lochter, A.; Galosy, S.; Muschler, J.; Freedman, N.; Werb, Z.; Bissell, M.J. Matrix metalloproteinase stromelysin-1 triggers a cascade of molecular alterations that leads to stable epithelial-to-mesenchymal conversion and a premalignant phenotype in mammary epithelial cells. J. Cell. Biol. 1997, 139, 1861–1872. [Google Scholar] [CrossRef]
- Shinohara, M.; Adachi, Y.; Mitsushita, J.; Kuwabara, M.; Nagasawa, A.; Harada, S.; Furuta, S.; Zhang, Y.; Seheli, K.; Miyazaki, H.; Kamata, T. Reactive oxygen generated by NADPH oxidase 1 (nox1) contributes to cell invasion by regulating matrix metalloprotease-9 production and cell migration. J. Biol. Chem. 2009, 285, 4481–4488. [Google Scholar]
- Binker, M.G.; Binker-Cosen, A.A.; Richards, D.; Oliver, B.; Cosen-Binker, L.I. EGF promotes invasion by PANC-1 cells through Rac1/ROS-dependent secretion and activation of MMP-2. Biochem. Biophys. Res. Commun. 2009, 379, 445–450. [Google Scholar] [CrossRef]
- Sung, S.Y.; Kubo, H.; Shigemura, K.; Arnold, R.S.; Logani, S.; Wang, R.; Konaka, H.; Nakagawa, M.; Mousses, S.; Amin, M.; Anderson, C.; Johnstone, P.; Petros, J.A.; Marshall, F.F.; Zhau, H.E.; Chung, L.W. Oxidative stress induces ADAM9 protein expression in human prostate cancer cells. Cancer Res. 2006, 66, 9519–9526. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef]
- Maulik, N. Redox signaling of angiogenesis. Antioxid. Redox Signal. 2002, 4, 805–815. [Google Scholar] [CrossRef]
- Liu, L.Z.; Hu, X.W.; Xia, C.; He, J.; Zhou, Q.; Shi, X.; Fang, J.; Jiang, B.H. Reactive oxygen species regulate epidermal growth factor-induced vascular endothelial growth factor and hypoxia-inducible factor-1alpha expression through activation of AKT and P70S6K1 in human ovarian cancer cells. Free Radic. Biol. Med. 2006, 41, 1521–1533. [Google Scholar] [CrossRef]
- Zhou, Q.; Liu, L.Z.; Fu, B.; Hu, X.; Shi, X.; Fang, J.; Jiang, B.H. Reactive oxygen species regulate insulin-induced VEGF and HIF-1alpha expression through the activation of p70S6K1 in human prostate cancer cells. Carcinogenesis 2007, 28, 28–37. [Google Scholar] [CrossRef]
- Arbiser, J.L.; Petros, J.; Klafter, R.; Govindajaran, B.; McLaughlin, E.R.; Brown, L.F.; Cohen, C.; Moses, M.; Kilroy, S.; Arnold, R.S.; Lambeth, J.D. Reactive oxygen generated by Nox1 triggers the angiogenic switch. Proc. Natl. Acad. Sci. USA 2002, 99, 715–720. [Google Scholar] [CrossRef]
- Lim, S.D.; Sun, C.; Lambeth, J.D.; Marshall, F.; Amin, M.; Chung, L.; Petros, J.A.; Arnold, R.S. Increased Nox1 and hydrogen peroxide in prostate cancer. Prostate 2005, 62, 200–207. [Google Scholar] [CrossRef]
- Xia, C.; Meng, Q.; Liu, L.Z.; Rojanasakul, Y.; Wang, X.R.; Jiang, B.H. Reactive oxygen species regulate angiogenesis and tumor growth through vascular endothelial growth factor. Cancer Res. 2007, 67, 10823–10830. [Google Scholar] [CrossRef]
- Dalle-Donne, I.; Giustarini, D.; Colombo, R.; Rossi, R.; Milzani, A. Protein carbonylation in human diseases. Trends Mol. Med. 2003, 9, 169–176. [Google Scholar] [CrossRef]
- Nystrom, T. Role of oxidative carbonylation in protein quality control and senescence. EMBO J. 2005, 24, 1311–1317. [Google Scholar] [CrossRef]
- Dalle-Donne, I.; Aldini, G.; Carini, M.; Colombo, R.; Rossi, R.; Milzani, A. Protein carbonylation, cellular dysfunction, and disease progression. J. Cell. Mol. Med. 2006, 10, 389–406. [Google Scholar] [CrossRef]
- Dalle-Donne, I.; Rossi, R.; Giustarini, D.; Gagliano, N.; Lusini, L.; Milzani, A.; Di Simplicio, P.; Colombo, R. Actin carbonylation: from a simple marker of protein oxidation to relevant signs of severe functional impairment. Free Radic. Biol. Med. 2001, 31, 1075–1083. [Google Scholar] [CrossRef]
- Fang, J.; Holmgren, A. Inhibition of thioredoxin and thioredoxin reductase by 4-hydroxy-2-nonenal in vitro and in vivo. J. Am. Chem. Soc. 2006, 128, 1879–1885. [Google Scholar] [CrossRef]
- England, K.; Cotter, T.G. Direct oxidative modifications of signaling proteins in mammalian cells and their effects on apoptosis. Redox Rep. 2005, 10, 237–245. [Google Scholar] [CrossRef]
- Niki, E. Lipid peroxidation: physiological levels and dual biological effects. Free Radic. Biol. Med. 2009, 47, 469–484. [Google Scholar] [CrossRef]
- Fruhwirth, G.O.; Loidl, A.; Hermetter, A. Oxidized phospholipids: from molecular properties to disease. Biochim. Biophys. Acta 2007, 1772, 718–736. [Google Scholar]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat. Rev. Drug. Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Barbieri, D.; Grassilli, E.; Monti, D.; Salvioli, S.; Franceschini, M.G.; Franchini, A.; Bellesia, E.; Salomoni, P.; Negro, P.; Capri, M.; et al. D-ribose and deoxy-D-ribose induce apoptosis in human quiescent peripheral blood mononuclear cells. Biochem. Biophys. Res. Commun. 1994, 201, 1109–1116. [Google Scholar] [CrossRef]
- Ceruti, S.; Barbieri, D.; Veronese, E.; Cattabeni, F.; Cossarizza, A.; Giammarioli, A.M.; Malorni, W.; Franceschi, C.; Abbracchio, M.P. Different pathways of apoptosis revealed by 2-chloro-adenosine and deoxy-D-ribose in mammalian astroglial cells. J. Neurosci. Res. 1997, 47, 372–383. [Google Scholar] [CrossRef]
- Pelicano, H.; Feng, L.; Zhou, Y.; Carew, J.S.; Hileman, E.O.; Plunkett, W.; Keating, M.J.; Huang, P. Inhibition of mitochondrial respiration: a novel strategy to enhance drug-induced apoptosis in human leukemia cells by a reactive oxygen species-mediated mechanism. J. Biol. Chem. 2003, 278, 37832–37839. [Google Scholar]
- Jing, Y.; Dai, J.; Chalmers-Redman, R.M.; Tatton, W.G.; Waxman, S. Arsenic trioxide selectively induces acute promyelocytic leukemia cell apoptosis via a hydrogen peroxide-dependent pathway. Blood 1999, 94, 2102–2111. [Google Scholar]
- Huang, H.S.; Chang, W.C.; Chen, C.J. Involvement of reactive oxygen species in arsenite-induced downregulation of phospholipid hydroperoxide glutathione peroxidase in human epidermoid carcinoma A431 cells. Free Radic. Biol. Med. 2002, 33, 864–873. [Google Scholar] [CrossRef]
- Han, Y.H.; Kim, S.Z.; Kim, S.H.; Park, W.H. Induction of apoptosis in arsenic trioxide-treated lung cancer A549 cells by buthionine sulfoximine. Mol. Cells 2008, 26, 158–164. [Google Scholar]
- Kang, Y.H.; Lee, S.J. The role of p38 MAPK and JNK in Arsenic trioxide-induced mitochondrial cell death in human cervical cancer cells. J. Cell. Physiol. 2008, 217, 23–33. [Google Scholar] [CrossRef]
- Evens, A.M.; Lecane, P.; Magda, D.; Prachand, S.; Singhal, S.; Nelson, J.; Miller, R.A.; Gartenhaus, R.B.; Gordon, L.I. Motexafin gadolinium generates reactive oxygen species and induces apoptosis in sensitive and highly resistant multiple myeloma cells. Blood 2005, 105, 1265–1273. [Google Scholar]
- Hashemy, S.I.; Ungerstedt, J.S.; Zahedi Avval, F.; Holmgren, A. Motexafin gadolinium, a tumor-selective drug targeting thioredoxin reductase and ribonucleotide reductase. J. Biol. Chem. 2006, 281, 10691–10697. [Google Scholar] [CrossRef]
- Kato, S.; Sadarangani, A.; Lange, S.; Delpiano, A.M.; Vargas, M.; Branes, J.; Carvajal, J.; Lipkowitz, S.; Owen, G.I.; Cuello, M.A. 2-methoxyestradiol mediates apoptosis through caspase-dependent and independent mechanisms in ovarian cancer cells but not in normal counterparts. Reprod. Sci. 2008, 15, 878–894. [Google Scholar] [CrossRef]
- Lee, Y.M.; Ting, C.M.; Cheng, Y.K.; Fan, T.P.; Wong, R.N.; Lung, M.L.; Mak, N.K. Mechanisms of 2-methoxyestradiol-induced apoptosis and G2/M cell-cycle arrest of nasopharyngeal carcinoma cells. Cancer Lett. 2008, 268, 295–307. [Google Scholar] [CrossRef]
- Bechtel, W.; Bauer, G. Catalase protects tumor cells from apoptosis induction by intercellular ROS signaling. Anticancer Res. 2009, 29, 4541–4557. [Google Scholar]
- Griffith, O.W.; Meister, A. Potent and specific inhibition of glutathione synthesis by buthionine sulfoximine (S-n-butyl homocysteine sulfoximine). J. Biol. Chem. 1979, 254, 7558–7560. [Google Scholar]
- Anderson, C.P.; Tsai, J.M.; Meek, W.E.; Liu, R.M.; Tang, Y.; Forman, H.J.; Reynolds, C.P. Depletion of glutathione by buthionine sulfoxine is cytotoxic for human neuroblastoma cell lines via apoptosis. Exp. Cell Res. 1999, 246, 183–192. [Google Scholar] [CrossRef]
- Schnelldorfer, T.; Gansauge, S.; Gansauge, F.; Schlosser, S.; Beger, H.G.; Nussler, A.K. Glutathione depletion causes cell growth inhibition and enhanced apoptosis in pancreatic cancer cells. Cancer 2000, 89, 1440–1447. [Google Scholar] [CrossRef]
- Rudin, C.M.; Yang, Z.; Schumaker, L.M.; VanderWeele, D.J.; Newkirk, K.; Egorin, M.J.; Zuhowski, E.G.; Cullen, K.J. Inhibition of glutathione synthesis reverses Bcl-2-mediated cisplatin resistance. Cancer Res. 2003, 63, 312–318. [Google Scholar]
- Davison, K.; Cote, S.; Mader, S.; Miller, W.H. Glutathione depletion overcomes resistance to arsenic trioxide in arsenic-resistant cell lines. Leukemia 2003, 17, 931–940. [Google Scholar] [CrossRef]
- Ortega, A.; Ferrer, P.; Carretero, J.; Obrador, E.; Asensi, M.; Pellicer, J.A.; Estrela, J.M. Down-regulation of glutathione and Bcl-2 synthesis in mouse B16 melanoma cells avoids their survival during interaction with the vascular endothelium. J. Biol. Chem. 2003, 278, 39591–39599. [Google Scholar]
- Hanigan, M.H.; Ricketts, W.A. Extracellular glutathione is a source of cysteine for cells that express gamma-glutamyl transpeptidase. Biochemistry 1993, 32, 6302–6306. [Google Scholar] [CrossRef]
- Mena, S.; Benlloch, M.; Ortega, A.; Carretero, J.; Obrador, E.; Asensi, M.; Petschen, I.; Brown, B.D.; Estrela, J.M. Bcl-2 and glutathione depletion sensitizes B16 melanoma to combination therapy and eliminates metastatic disease. Clin. Cancer Res. 2007, 13, 2658–2666. [Google Scholar] [CrossRef]
- Taylor, S.A.; Crowley, J.; Pollock, T.W.; Eyre, H.J.; Jaeckle, C.; Hynes, H.E.; Stephens, R.L. Objective antitumor activity of acivicin in patients with recurrent CNS malignancies: a Southwest Oncology Group trial. J. Clin. Oncol. 1991, 9, 1476–1479. [Google Scholar]
- Benlloch, M.; Ortega, A.; Ferrer, P.; Segarra, R.; Obrador, E.; Asensi, M.; Carretero, J.; Estrela, J.M. Acceleration of glutathione efflux and inhibition of gamma-glutamyltranspeptidase sensitize metastatic B16 melanoma cells to endothelium-induced cytotoxicity. J. Biol. Chem. 2005, 280, 6950–6959. [Google Scholar] [CrossRef]
- Lo, M.; Wang, Y.Z.; Gout, P.W. The x(c)- cystine/glutamate antiporter: a potential target for therapy of cancer and other diseases. J. Cell. Physiol. 2008, 215, 593–602. [Google Scholar] [CrossRef]
- Lo, M.; Ling, V.; Wang, Y.Z.; Gout, P.W. The xc- cystine/glutamate antiporter: a mediator of pancreatic cancer growth with a role in drug resistance. Br. J. Cancer. 2008, 99, 464–472. [Google Scholar] [CrossRef]
- Guan, J.; Lo, M.; Dockery, P.; Mahon, S.; Karp, C.M.; Buckley, A.R.; Lam, S.; Gout, P.W.; Wang, Y.Z. The xc- cystine/glutamate antiporter as a potential therapeutic target for small-cell lung cancer: use of sulfasalazine. Cancer Chemother. Pharmacol. 2009, 64, 463–472. [Google Scholar] [CrossRef]
- Park, B.C.; Park, S.H.; Paek, S.H.; Park, S.Y.; Kwak, M.K.; Choi, H.G.; Yong, C.S.; Yoo, B.K.; Kim, J.A. Chloroquine-induced nitric oxide increase and cell death is dependent on cellular GSH depletion in A172 human glioblastoma cells. Toxicol. Lett. 2008, 178, 52–60. [Google Scholar] [CrossRef]
- Ramos, S. Effects of dietary flavonoids on apoptotic pathways related to cancer chemoprevention. J. Nutr. Biochem. 2007, 18, 427–442. [Google Scholar] [CrossRef]
- Bravo, L. Polyphenols: chemistry, dietary sources, metabolism, and nutritional significance. Nutr. Rev. 1998, 56, 317–333. [Google Scholar] [CrossRef]
- Rice-Evans, C. Flavonoid antioxidants. Curr. Med. Chem. 2001, 8, 797–807. [Google Scholar] [CrossRef]
- Sharma, V.; Joseph, C.; Ghosh, S.; Agarwal, A.; Mishra, M.K.; Sen, E. Kaempferol induces apoptosis in glioblastoma cells through oxidative stress. Mol. Cancer Ther. 2007, 6, 2544–2553. [Google Scholar] [CrossRef]
- Lu, J.; Papp, L.V.; Fang, J.; Rodriguez-Nieto, S.; Zhivotovsky, B.; Holmgren, A. Inhibition of Mammalian thioredoxin reductase by some flavonoids: implications for myricetin and quercetin anticancer activity. Cancer Res. 2006, 66, 4410–4418. [Google Scholar] [CrossRef]
- Laughton, M.J.; Evans, P.J.; Moroney, M.A.; Hoult, J.R.; Halliwell, B. Inhibition of mammalian 5-lipoxygenase and cyclo-oxygenase by flavonoids and phenolic dietary additives. Relationship to antioxidant activity and to iron ion-reducing ability. Biochem. Pharmacol. 1991, 42, 1673–1681. [Google Scholar] [CrossRef]
- Raso, G.M.; Meli, R.; Di Carlo, G.; Pacilio, M.; Di Carlo, R. Inhibition of inducible nitric oxide synthase and cyclooxygenase-2 expression by flavonoids in macrophage J774A.1. Life Sci. 2001, 68, 921–931. [Google Scholar] [CrossRef]
- Elliott, A.J.; Scheiber, S.A.; Thomas, C.; Pardini, R.S. Inhibition of glutathione reductase by flavonoids. A structure-activity study. Biochem. Pharmacol. 1992, 44, 1603–1608. [Google Scholar] [CrossRef]
- Kachadourian, R.; Day, B.J. Flavonoid-induced glutathione depletion: potential implications for cancer treatment. Free Radic. Biol. Med. 2006, 41, 65–76. [Google Scholar] [CrossRef]
- Formica, J.V.; Regelson, W. Review of the biology of Quercetin and related bioflavonoids. Food Chem. Toxicol. 1995, 33, 1061–1080. [Google Scholar] [CrossRef]
- Jeong, J.H.; An, J.Y.; Kwon, Y.T.; Rhee, J.G.; Lee, Y.J. Effects of low dose quercetin: cancer cell-specific inhibition of cell cycle progression. J. Cell. Biochem. 2009, 106, 73–82. [Google Scholar] [CrossRef]
- Lee, Y.K.; Hwang, J.T.; Kwon, D.Y.; Surh, Y.J.; Park, O.J. Induction of apoptosis by quercetin is mediated through AMPKalpha1/ASK1/p38 pathway. Cancer Lett. 2010, 292, 228–236. [Google Scholar] [CrossRef]
- Hattori, K.; Naguro, I.; Runchel, C.; Ichijo, H. The roles of ASK family proteins in stress responses and diseases. Cell Commun. Signal. 2009, 7, 9. [Google Scholar] [CrossRef]
- Matsuzawa, A.; Ichijo, H. Redox control of cell fate by MAP kinase: physiological roles of ASK1-MAP kinase pathway in stress signaling. Biochim. Biophys. Acta 2008, 1780, 1325–1336. [Google Scholar]
- Lee, Y.K.; Park, S.Y.; Kim, Y.M.; Lee, W.S.; Park, O.J. AMP kinase/cyclooxygenase-2 pathway regulates proliferation and apoptosis of cancer cells treated with quercetin. Exp. Mol. Med. 2009, 41, 201–207. [Google Scholar] [CrossRef]
- Ferraresi, R.; Troiano, L.; Roat, E.; Lugli, E.; Nemes, E.; Nasi, M.; Pinti, M.; Fernandez, M.I.; Cooper, E.L.; Cossarizza, A. Essential requirement of reduced glutathione (GSH) for the anti-oxidant effect of the flavonoid quercetin. Free Radic. Res. 2005, 39, 1249–1258. [Google Scholar]
- Ramos, A.M.; Aller, P. Quercetin decreases intracellular GSH content and potentiates the apoptotic action of the antileukemic drug arsenic trioxide in human leukemia cell lines. Biochem. Pharmacol. 2008, 75, 1912–1923. [Google Scholar] [CrossRef]
- Cossarizza, A.; Ferraresi, R.; Troiano, L.; Roat, E.; Gibellini, L.; Bertoncelli, L.; Nasi, M.; Pinti, M. Simultaneous analysis of reactive oxygen species and reduced glutathione content in living cells by polychromatic flow cytometry. Nat. Protoc. 2009, 4, 1790–1797. [Google Scholar] [CrossRef]
- Metodiewa, D.; Jaiswal, A.K.; Cenas, N.; Dickancaite, E.; Segura-Aguilar, J. Quercetin may act as a cytotoxic prooxidant after its metabolic activation to semiquinone and quinoidal product. Free Radic. Biol. Med. 1999, 26, 107–116. [Google Scholar] [CrossRef]
- Boots, A.W.; Kubben, N.; Haenen, G.R.; Bast, A. Oxidized quercetin reacts with thiols rather than with ascorbate: implication for quercetin supplementation. Biochem. Biophys. Res. Commun. 2003, 308, 560–565. [Google Scholar] [CrossRef]
- Sahu, S.C.; Gray, G.C. Pro-oxidant activity of flavonoids: effects on glutathione and glutathione S-transferase in isolated rat liver nuclei. Cancer Lett. 1996, 104, 193–196. [Google Scholar] [CrossRef]
- Cossarizza, A.; Pinti, M.; Moretti, L.; Bricalli, D.; Bianchi, R.; Troiano, L.; Fernandez, M.G.; Balli, F.; Brambilla, P.; Mussini, C.; Vigano, A. Mitochondrial functionality and mitochondrial DNA content in lymphocytes of vertically infected human immunodeficiency virus-positive children with highly active antiretroviral therapy-related lipodystrophy. J. Infect. Dis. 2002, 185, 299–305. [Google Scholar] [CrossRef]
- Foli, A.; Benvenuto, F.; Piccinini, G.; Bareggi, A.; Cossarizza, A.; Lisziewicz, J.; Lori, F. Direct analysis of mitochondrial toxicity of antiretroviral drugs. AIDS 2001, 15, 1687–1694. [Google Scholar] [CrossRef]
- Lugli, E.; Troiano, L.; Ferraresi, R.; Roat, E.; Prada, N.; Nasi, M.; Pinti, M.; Cooper, E.L.; Cossarizza, A. Characterization of cells with different mitochondrial membrane potential during apoptosis. Cytometry A 2005, 68, 28–35. [Google Scholar]
- Salvioli, S.; Dobrucki, J.; Moretti, L.; Troiano, L.; Fernandez, M.G.; Pinti, M.; Pedrazzi, J.; Franceschi, C.; Cossarizza, A. Mitochondrial heterogeneity during staurosporine-induced apoptosis in HL60 cells: analysis at the single cell and single organelle level. Cytometry 2000, 40, 189–197. [Google Scholar] [CrossRef]
- Salvioli, S.; Storci, G.; Pinti, M.; Quaglino, D.; Moretti, L.; Merlo-Pich, M.; Lenaz, G.; Filosa, S.; Fico, A.; Bonafè, M.; Monti, D.; Troiano, L.; Nasi, M.; Cossarizza, A.; Franceschi, C. Apoptosis-resistant phenotype in HL-60-derived cells HCW-2 is related to changes in expression of stress-induced proteins that impact on redox status and mitochondrial metabolism. Cell Death Differ. 2003, 10, 163–174. [Google Scholar] [CrossRef]
- Tropea, F.; Troiano, L.; Monti, D.; Lovato, E.; Malorni, W.; Rainaldi, G.; Mattana, P.; Viscomi, G.; Ingletti, M.C.; Portolani, M.; et al. Sendai virus and herpes virus type 1 induce apoptosis in human peripheral blood mononuclear cells. Exp. Cell. Res. 1995, 218, 63–70. [Google Scholar] [CrossRef]
- Troiano, L.; Ferraresi, R.; Lugli, E.; Nemes, E.; Roat, E.; Nasi, M.; Pinti, M.; Cossarizza, A. Multiparametric analysis of cells with different mitochondrial membrane potential during apoptosis by polychromatic flow cytometry. Nat. Protoc. 2007, 2, 2719–2727. [Google Scholar] [CrossRef]
- Lo, J.F.; Wang, H.F.; Tam, M.F.; Lee, T.C. Glutathione S-transferase pi in an arsenic-resistant Chinese hamster ovary cell line. Biochem. J. 1992, 288, 977–982. [Google Scholar]
- Trachootham, D.; Zhou, Y.; Zhang, H.; Demizu, Y.; Chen, Z.; Pelicano, H.; Chiao, P.J.; Achanta, G.; Arlinghaus, R.B.; Liu, J.; Huang, P. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell 2006, 10, 241–252. [Google Scholar] [CrossRef]
- Trachootham, D.; Zhang, H.; Zhang, W.; Feng, L.; Du, M.; Zhou, Y.; Chen, Z.; Pelicano, H.; Plunkett, W.; Wierda, W.G.; Keating, M.J.; Huang, P. Effective elimination of fludarabine-resistant CLL cells by PEITC through a redox-mediated mechanism. Blood 2008, 112, 1912–1922. [Google Scholar] [CrossRef]
- Zhang, H.; Trachootham, D.; Lu, W.; Carew, J.; Giles, F.J.; Keating, M.J.; Arlinghaus, R.B.; Huang, P. Effective killing of Gleevec-resistant CML cells with T315I mutation by a natural compound PEITC through redox-mediated mechanism. Leukemia 2008, 22, 1191–1199. [Google Scholar] [CrossRef]
- Lugli, E.; Ferraresi, R.; Roat, E.; Troiano, L.; Pinti, M.; Nasi, M.; Nemes, E.; Bertoncelli, L.; Gibellini, L.; Salomoni, P.; Cooper, E.L.; Cossarizza, A. Quercetin inhibits lymphocyte activation and proliferation without inducing apoptosis in peripheral mononuclear cells. Leuk. Res. 2009, 33, 140–150. [Google Scholar] [CrossRef]
- Chien, S.Y.; Wu, Y.C.; Chung, J.G.; Yang, J.S.; Lu, H.F.; Tsou, M.F.; Wood, W.G.; Kuo, S.J.; Chen, D.R. Quercetin-induced apoptosis acts through mitochondrial- and caspase-3-dependent pathways in human breast cancer MDA-MB-231 cells. Hum. Exp. Toxicol. 2009, 28, 493–503. [Google Scholar] [CrossRef]
- Gupta, K.; Panda, D. Perturbation of microtubule polymerization by quercetin through tubulin binding: a novel mechanism of its antiproliferative activity. Biochemistry 2002, 41, 13029–13038. [Google Scholar] [CrossRef]
- Jung, Y.H.; Heo, J.; Lee, Y.J.; Kwon, T.K.; Kim, Y.H. Quercetin enhances TRAIL-induced apoptosis in prostate cancer cells via increased protein stability of death receptor 5. Life Sci. 2010, 86, 351–357. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kim, E.H.; Park, S.S.; Lim, J.H.; Kwon, T.K.; Choi, K.S. Quercetin sensitizes human hepatoma cells to TRAIL-induced apoptosis via Sp1-mediated DR5 up-regulation and proteasome-mediated c-FLIPS down-regulation. J. Cell. Biochem. 2008, 105, 1386–1398. [Google Scholar] [CrossRef]
- Kuo, P.C.; Liu, H.F.; Chao, J.I. Survivin and p53 modulate quercetin-induced cell growth inhibition and apoptosis in human lung carcinoma cells. J. Biol. Chem. 2004, 279, 55875–55885. [Google Scholar] [CrossRef]
- Ma, H.; Nguyen, C.; Lee, K.S.; Kahn, M. Differential roles for the coactivators CBP and p300 on TCF/beta-catenin-mediated survivin gene expression. Oncogene 2005, 24, 3619–3631. [Google Scholar] [CrossRef]
- Shan, B.E.; Wang, M.X.; Li, R.Q. Quercetin inhibit human SW480 colon cancer growth in association with inhibition of cyclin D1 and survivin expression through Wnt/beta-catenin signaling pathway. Cancer Invest. 2009, 27, 604–612. [Google Scholar] [CrossRef]
- Lee, T.J.; Kim, O.H.; Kim, Y.H.; Lim, J.H.; Kim, S.; Park, J.W.; Kwon, T.K. Quercetin arrests G2/M phase and induces caspase-dependent cell death in U937 cells. Cancer Lett. 2006, 240, 234–242. [Google Scholar] [CrossRef]
- Yang, J.H.; Hsia, T.C.; Kuo, H.M.; Chao, P.D.; Chou, C.C.; Wei, Y.H.; Chung, J.G. Inhibition of lung cancer cell growth by quercetin glucuronides via G2/M arrest and induction of apoptosis. Drug Metab. Dispos. 2006, 34, 296–304. [Google Scholar]
- Granado-Serrano, A.B.; Martin, M.A.; Bravo, L.; Goya, L.; Ramos, S. Quercetin induces apoptosis via caspase activation, regulation of Bcl-2, and inhibition of PI-3-kinase/Akt and ERK pathways in a human hepatoma cell line (HepG2). J. Nutr. 2006, 136, 2715–2721. [Google Scholar]
- Singhal, R.L.; Yeh, Y.A.; Praja, N.; Olah, E.; Sledge, G.W., Jr.; Weber, G. Quercetin down-regulates signal transduction in human breast carcinoma cells. Biochem. Biophys. Res. Commun. 1995, 208, 425–431. [Google Scholar] [CrossRef]
- Tan, J.; Wang, B.; Zhu, L. Regulation of survivin and Bcl-2 in HepG2 cell apoptosis induced by quercetin. Chem. Biodivers. 2009, 6, 1101–1110. [Google Scholar] [CrossRef]
- Siegelin, M.D.; Reuss, D.E.; Habel, A.; Rami, A.; von Deimling, A. Quercetin promotes degradation of survivin and thereby enhances death-receptor-mediated apoptosis in glioma cells. Neuro Oncol. 2009, 11, 122–131. [Google Scholar]
- Chen, W.; Wang, X.; Zhuang, J.; Zhang, L.; Lin, Y. Induction of death receptor 5 and suppression of survivin contribute to sensitization of TRAIL-induced cytotoxicity by quercetin in non-small cell lung cancer cells. Carcinogenesis 2007, 28, 2114–2121. [Google Scholar] [CrossRef]
- Ambrosini, G.; Adida, C.; Altieri, D.C. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat. Med. 1997, 3, 917–921. [Google Scholar] [CrossRef]
- Aalinkeel, R.; Bindukumar, B.; Reynolds, J.L.; Sykes, D.E.; Mahajan, S.D.; Chadha, K.C.; Schwartz, S.A. The dietary bioflavonoid, quercetin, selectively induces apoptosis of prostate cancer cells by down-regulating the expression of heat shock protein 90. Prostate 2008, 68, 1773–1789. [Google Scholar] [CrossRef]
- Ye, B.; Yang, J.L.; Chen, L.J.; Wu, X.X.; Yang, H.S.; Zhao, J.M.; Yuan, Z.P.; Li, J.; Wen, Y.J.; Mao, Y.Q.; Lei, S.; Kan, B.; Fan, L.Y.; Yao, W.X.; Wang, R.; Wang, G.Q.; Du, X.B.; Liu, H.Y.; Wu, H.B.; Xu, J.R.; Li, H.X.; Zhang, W.; Zhao, X.; Wei, Y.Q.; Cheng, L. Induction of apoptosis by phenylisocyanate derivative of quercetin: involvement of heat shock protein. Anticancer Drugs 2007, 18, 1165–1171. [Google Scholar] [CrossRef]
- Barbieri, D.; Abbracchio, M.P.; Salvioli, S.; Monti, D.; Cossarizza, A.; Ceruti, S.; Brambilla, R.; Cattabeni, F.; Jacobson, K.A.; Franceschi, C. Apoptosis by 2-chloro-2'-deoxy-adenosine and 2-chloro-adenosine in human peripheral blood mononuclear cells. Neurochem. Int. 1998, 32, 493–504. [Google Scholar] [CrossRef]
- Monti, D.; Salvioli, S.; Capri, M.; Malorni, W.; Straface, E.; Cossarizza, A.; Botti, B.; Piacentini, M.; Baggio, G.; Barbi, C.; Valensin, S.; Bonafe, M.; Franceschi, C. Decreased susceptibility to oxidative stress-induced apoptosis of peripheral blood mononuclear cells from healthy elderly and centenarians. Mech. Ageing Dev. 2000, 121, 239–250. [Google Scholar]
- Caltagirone, S.; Rossi, C.; Poggi, A.; Ranelletti, F.O.; Natali, P.G.; Brunetti, M.; Aiello, F.B.; Piantelli, M. Flavonoids apigenin and quercetin inhibit melanoma growth and metastatic potential. Int. J. Cancer 2000, 87, 595–600. [Google Scholar] [CrossRef]
- Nair, H.K.; Rao, K.V.; Aalinkeel, R.; Mahajan, S.; Chawda, R.; Schwartz, S.A. Inhibition of prostate cancer cell colony formation by the flavonoid quercetin correlates with modulation of specific regulatory genes. Clin. Diagn. Lab. Immunol. 2004, 11, 63–69. [Google Scholar]
- Kamaraj, S.; Vinodhkumar, R.; Anandakumar, P.; Jagan, S.; Ramakrishnan, G.; Devaki, T. The effects of quercetin on antioxidant status and tumor markers in the lung and serum of mice treated with benzo(a)pyrene. Biol. Pharm. Bull. 2007, 30, 2268–2273. [Google Scholar] [CrossRef]
- Seufi, A.M.; Ibrahim, S.S.; Elmaghraby, T.K.; Hafez, E.E. Preventive effect of the flavonoid, quercetin, on hepatic cancer in rats via oxidant/antioxidant activity: molecular and histological evidences. J. Exp. Clin. Cancer Res. 2009, 28, 80. [Google Scholar] [CrossRef]
- Volate, S.R.; Davenport, D.M.; Muga, S.J.; Wargovich, M.J. Modulation of aberrant crypt foci and apoptosis by dietary herbal supplements (quercetin, curcumin, silymarin, ginseng and rutin). Carcinogenesis 2005, 26, 1450–1456. [Google Scholar] [CrossRef]
- Devipriya, S.; Ganapathy, V.; Shyamaladevi, C.S. Suppression of tumor growth and invasion in 9,10 dimethyl benz(a) anthracene induced mammary carcinoma by the plant bioflavonoid quercetin. Chem. Biol. Interact. 2006, 162, 106–113. [Google Scholar] [CrossRef]
- Mulholland, P.J.; Ferry, D.R.; Anderson, D.; Hussain, S.A.; Young, A.M.; Cook, J.E.; Hodgkin, E.; Seymour, L.W.; Kerr, D.J. Pre-clinical and clinical study of QC12, a water-soluble, pro-drug of quercetin. Ann. Oncol. 2001, 12, 245–248. [Google Scholar] [CrossRef]
- Jaruga, E.; Salvioli, S.; Dobrucki, J.; Chrul, S.; Bandorowicz-Pikula, J.; Sikora, E.; Franceschi, C.; Cossarizza, A.; Bartosz, G. Apoptosis-like, reversible changes in plasma membrane asymmetry and permeability, and transient modifications in mitochondrial membrane potential induced by curcumin in rat thymocytes. FEBS Lett. 1998, 433, 287–293. [Google Scholar] [CrossRef]
- Cruz-Correa, M.; Shoskes, D.A.; Sanchez, P.; Zhao, R.; Hylind, L.M.; Wexner, S.D.; Giardiello, F.M. Combination treatment with curcumin and quercetin of adenomas in familial adenomatous polyposis. Clin. Gastroenterol. Hepatol. 2006, 4, 1035–1038. [Google Scholar] [CrossRef]
- Ma, Z.S.; Huynh, T.H.; Ng, C.P.; Do, P.T.; Nguyen, T.H.; Huynh, H. Reduction of CWR22 prostate tumor xenograft growth by combined tamoxifen-quercetin treatment is associated with inhibition of angiogenesis and cellular proliferation. Int. J. Oncol. 2004, 24, 1297–1304. [Google Scholar]
- Harwood, M.; Danielewska-Nikiel, B.; Borzelleca, J.F.; Flamm, G.W.; Williams, G.M.; Lines, T.C. A critical review of the data related to the safety of quercetin and lack of evidence of in vivo toxicity, including lack of genotoxic/carcinogenic properties. Food Chem. Toxicol. 2007, 45, 2179–2205. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an Open Access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gibellini, L.; Pinti, M.; Nasi, M.; De Biasi, S.; Roat, E.; Bertoncelli, L.; Cossarizza, A. Interfering with ROS Metabolism in Cancer Cells: The Potential Role of Quercetin. Cancers 2010, 2, 1288-1311. https://doi.org/10.3390/cancers2021288
Gibellini L, Pinti M, Nasi M, De Biasi S, Roat E, Bertoncelli L, Cossarizza A. Interfering with ROS Metabolism in Cancer Cells: The Potential Role of Quercetin. Cancers. 2010; 2(2):1288-1311. https://doi.org/10.3390/cancers2021288
Chicago/Turabian StyleGibellini, Lara, Marcello Pinti, Milena Nasi, Sara De Biasi, Erika Roat, Linda Bertoncelli, and Andrea Cossarizza. 2010. "Interfering with ROS Metabolism in Cancer Cells: The Potential Role of Quercetin" Cancers 2, no. 2: 1288-1311. https://doi.org/10.3390/cancers2021288