The "Two-Faced" Effects of Reactive Oxygen Species and the Lipid Peroxidation Product 4-Hydroxynonenal in the Hallmarks of Cancer

 ,
,
Abstract
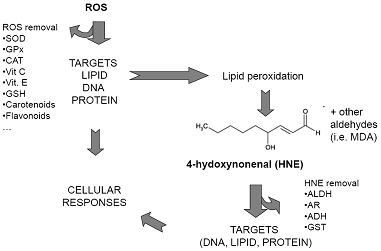
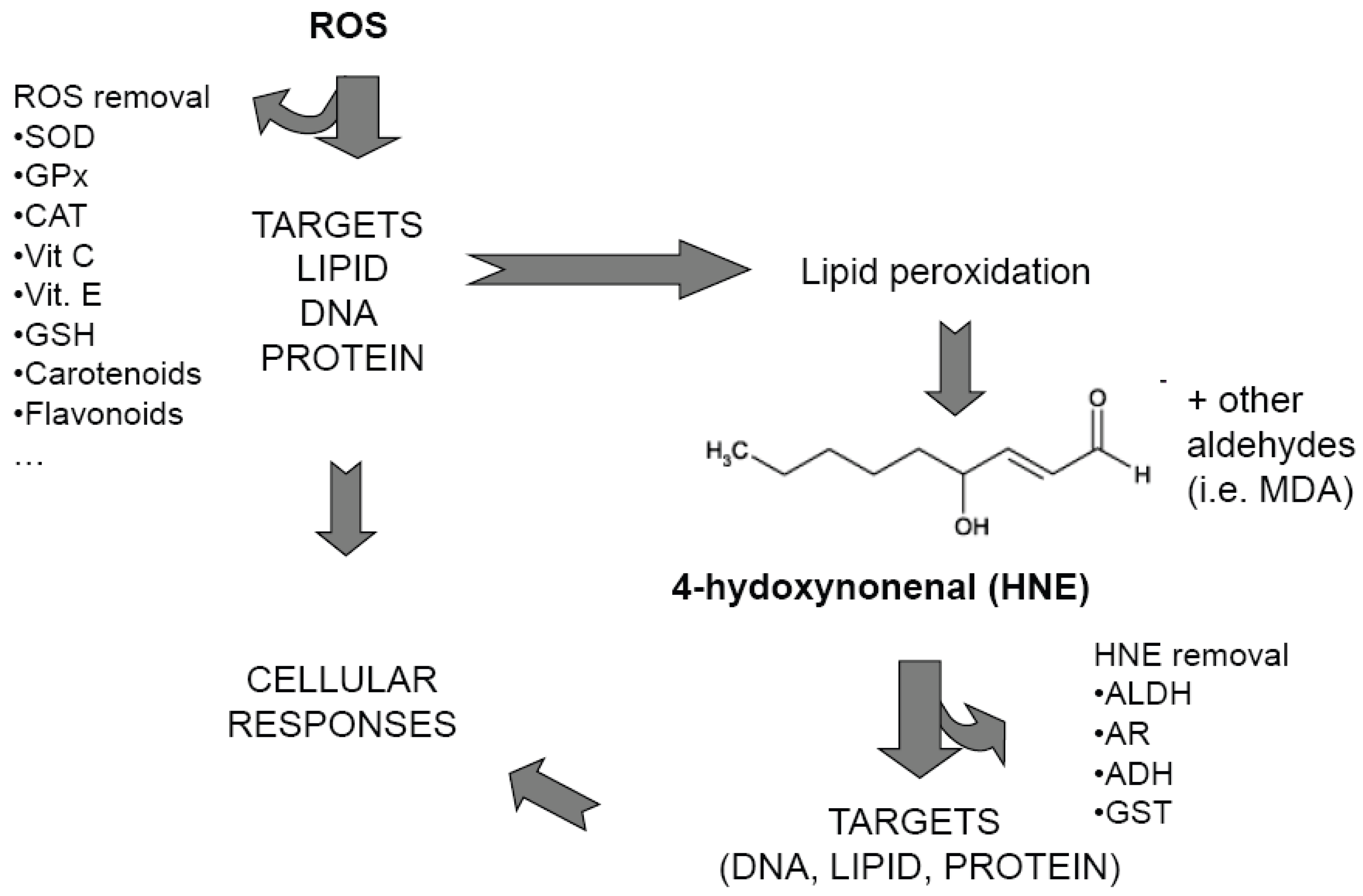
:
{kind=link}
{kind=link}
1. Oxidative Stress and Lipid Peroxidation, an Introduction

2. ROS and HNE in Carcinogenesis
3. Role of ROS and HNE in the Hallmarks of Cancer
3.1. Regulation of Cell Proliferation
3.3. Regulation of Replicative Potential of Cells
3.4. Regulation of Angiogenesis
3.5. Regulation of Cell Adhesion, Tumor Invasion and Metastasis
3.6. Regulation of Genomic Instability-DNA Repair
4. Concluding Remarks
Acknowledgements
References
- Klaunig, J.E.; Kamendulis, L.M. The role of oxidative stress in carcinogenesis. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 239–267. [Google Scholar] [CrossRef]
- Poli, G.; Leonarduzzi, G.; Biasi, F.; Chiarpotto, E. Oxidative stress and cell signalling. Curr. Med. Chem. 2004, 11, 1163–1182. [Google Scholar] [CrossRef]
- Ercal, N.; Gurer-Orhan, H.; Aykin-Burns, N. Toxic metals and oxidative stress part I: mechanisms involved in metal-induced oxidative damage. Curr. Top. Med. Chem. 2001, 1, 529–539. [Google Scholar] [CrossRef]
- Kovacic, P.; Osuna, J.A., Jr. Mechanisms of anti-cancer agents: emphasis on oxidative stress and electron transfer. Curr. Pharm. Des. 2000, 6, 277–309. [Google Scholar] [CrossRef]
- Clarkson, P.M.; Thompson, H.S. Antioxidants: What role do they play in physical activity and health? Am. J. Clin. Nutr. 2000, 72, 637S–646S. [Google Scholar]
- Mann, G.E.; Bonacasa, B.; Ishii, T.; Siow, R.C. Targeting the redox sensitive Nrf2-Keap1 defense pathway in cardiovascular disease: protection afforded by dietary isoflavones. Curr. Opin. Pharmacol. 2009, 9, 139–145. [Google Scholar] [CrossRef]
- Gao, L.; Mann, G.E. Vascular NAD(P)H oxidase activation in diabetes: a double-edged sword in redox signalling. Cardiovasc. Res. 2009, 82, 9–20. [Google Scholar]
- Chakravarti, B.; Chakravarti, D.N. Oxidative modification of proteins: age-related changes. Gerontology 2007, 53, 128–139. [Google Scholar] [CrossRef]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: mechanisms, mutation and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef]
- Evans, M.D.; Dizdaroglu, M.; Cooke, M.S. Oxidative DNA damage and disease: induction, repair and significance. Mutat. Res. 2004, 567, 1–61. [Google Scholar] [CrossRef]
- Filipcik, P.; Cente, M.; Ferencik, M.; Hulin, I.; Novak, M. The role of oxidative stress in the pathogenesis of Alzheimer's disease. Bratisl. Lek. Listy 2006, 107, 384–394. [Google Scholar]
- Wu, W.S. The signalling mechanism of ROS in tumor progression. Cancer Metastasis Rev. 2006, 25, 695–705. [Google Scholar] [CrossRef]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar]
- Dianzani, M.U.; Barrera, G.; Parola, M. 4-Hydroxy-2,3-nonenal as a signal for cell function and differentiation. Acta Biochim. Pol. 1999, 46, 61–75. [Google Scholar]
- Uchida, K. 4-Hydroxy-2-nonenal: a product and mediator of oxidative stress. Prog. Lipid Res. 2003, 42, 318–343. [Google Scholar] [CrossRef]
- Forman, H.J.; Fukuto, J.M.; Miller, T.; Zhang, H.; Rinna, A.; Levy, S. The chemistry of cell signaling by reactive oxygen and nitrogen species and 4-hydroxynonenal. Arch. Biochem. Biophys. 2008, 477, 183–195. [Google Scholar] [CrossRef]
- Poli, G.; Schaur, RJ.; Siems, WG.; Leonarduzzi, G. 4-hydroxynonenal: a membrane lipid oxidation product of medicinal interest. Med. Res. Rev. 2008, 28, 569–631. [Google Scholar]
- Esterbauer, H.; Zollner, H.; Lang, J. Metabolism of the lipid peroxidation product 4-hydroxynonenal by isolated hepatocytes and by liver cytosolic fractions. Biochem J. 1985, 228, 363–373. [Google Scholar]
- Barrera, G.; Biasi, F.; Fazio, V.M.; Paradisi, L.; Dianzani, M.U. Repeated treatments with a low HNE concentration affect K562 cell proliferation. In Chemical Carcinogenesis; Plenum Press: New York, NY, USA, 1991. [Google Scholar]
- Parola, M.; Bellomo, G.; Robino, G.; Barrera, G.; Dianzani, M.U. 4-Hydroxynonenal as a biological signal: molecular basis and pathophysiological implications. Antioxid. Redox Signal. 1999, 1, 255–284. [Google Scholar]
- Dianzani, M.U. 4-hydroxynonenal from pathology to physiology. Mol. Aspects Med. 2003, 24, 263–272. [Google Scholar]
- Leonarduzzi, G.; Robbesyn, F.; Poli, G. Signaling kinases modulated by 4-hydroxynonenal. Free Radic. Biol. Med. 2004, 37, 1694–1702. [Google Scholar] [CrossRef]
- Barrera, G.; Pizzimenti, S.; Dianzani, M.U. Lipid peroxidation: control of cell proliferation cell differentiation and cell death. Mol. Aspects Med. 2008, 29, 1–8. [Google Scholar] [CrossRef]
- Lau, A.T.; Wang, Y.; Chiu, J.F. Reactive oxygen species: current knowledge and applications in cancer research and therapeutic. J. Cell. Biochem. 2008, 104, 657–667. [Google Scholar] [CrossRef]
- Wu, L.L.; Chiou, C.C.; Chang, P.Y.; Wu, J.T. Urinary 8-OHdG: a marker of oxidative stress to DNA and a risk factor for cancer, atherosclerosis and diabetics. Clin. Chim. Acta 2004, 339, 1–9. [Google Scholar] [CrossRef]
- Fruehauf, J.P.; Meyskens, F.L., Jr. Reactive oxygen species: a breath of life or death? Clin. Cancer Res. 2007, 13, 789–794. [Google Scholar] [CrossRef]
- Halliwell, B. Oxidative stress and cancer. Have we moved forward? Biochem. J. 2007, 401, 1–11. [Google Scholar]
- Halliwell, B. Effect of diet on cancer development: is oxidative DNA damage a biomarker? Free Radic. Biol. Med. 2002, 32, 968–974. [Google Scholar] [CrossRef]
- Eckl, P.M. Genotoxicity of HNE. Mol. Aspects Med. 2003, 24, 161–165. [Google Scholar] [CrossRef]
- Feng, Z.; Hu, W.; Tang, M.S. Trans-4-hydroxy-2-nonenal inhibits nucleotide excision repair in human cells: a possible mechanism for lipid peroxidation-induced carcinogenesis. Proc. Natl. Acad. Sci. USA. 2004, 101, 8598–8602. [Google Scholar] [CrossRef]
- Cajelli, E.; Ferraris, A.; Brambilla, G. Mutagenicity of 4-hydroxynonenal in V79 Chinese hamster cells. Mutat. Res. 1987, 190, 169–171. [Google Scholar] [CrossRef]
- Singh, S.P.; Chen, T.; Chen, L.; Mei, N.; McLain, E.; Samokyszyn, V.; Thaden, J.J.; Moore, M.M.; Zimniak, P. Mutagenic effects of 4-hydroxynonenal triacetate, a chemically protected form of the lipid peroxidation product 4-hydroxynonenal, as assayed in L5178Y/Tk+/- mouse lymphoma cells. J. Pharmacol. Exp. Ther. 2005, 313, 855–861. [Google Scholar]
- Glei, M.; Schaeferhenrich, A.; Claussen, U.; Kuechler, A.; Liehr, T.; Weise, A.; Marian, B.; Sendt, W.; Pool-Zobel, B.L. Comet fluorescence in situ hybridization analysis for oxidative stress-induced DNA damage in colon cancer relevant genes. Toxicol. Sci. 2007, 96, 279–284. [Google Scholar]
- Chung, F.L.; Chen, H.J.; Nath, R.G. Lipid peroxidation as a potential endogenous source for the formation of exocyclic DNA adducts. Carcinogenesis 1996, 17, 2105–2111. [Google Scholar] [CrossRef]
- Douki, T.; Ames, B.N. An HPLC-EC assay for 1,N2-propano adducts of 2'-deoxyguanosine with 4-hydroxynonenal and other alpha,beta-unsaturated aldehydes. Chem. Res. Toxicol. 1994, 7, 511–518. [Google Scholar] [CrossRef]
- Bartsch, H.; Nair, J. Accumulation of lipid peroxidation-derived DNA lesions: potential lead markers for chemoprevention of inflammation-driven malignancies. Mutat. Res. 2005, 591, 34–44. [Google Scholar] [CrossRef]
- Hussain, S.P.; Amstad, P.; Raja, K.; Sawyer, M.; Hofseth, L.; Shields, P.G.; Hewer, A.; Phillips, D.H.; Ryberg, D.; Haugen, A.; Harris, C.C. Increased p53 mutation load in nontumorous human liver of Wilson disease and hemochromatosis: oxyradical overload diseases. Proc. Natl. Acad. Sci. 2000, 97, 12770–12775. [Google Scholar] [CrossRef]
- Hsu, I.C.; Metcalf, R.A.; Sun, T.; Welsh, J.A.; Wang, N.J.; Harris, C.C. Mutational hotspot in the p53 gene in human hepatocellular carcinomas. Nature 1991, 350, 427–428. [Google Scholar] [CrossRef]
- Chung, F.L.; Nath, R.G.; Ocando, J.; Nishikawa, A.; Zhang, L. Deoxyguanosine adducts of t-4-hydroxy-2-nonenal are endogenous DNA lesions in rodents and humans: detection and potential sources. Cancer Res. 2000, 60, 1507–1511. [Google Scholar]
- Hu, W.; Feng, Z.; Eveleigh, J.; Lyer, G.; Pan, J.; Amin, S.; Chung, F.T.; Tang, M.S. The major lipid peroxidation product, trans-4-hydroxy-2-nonenal, preferentially forms DNA adducts at codon 249 of human p53 gene, a unique mutational hotspot in hepatocellular carcinoma. Carcinogenesis 2002, 23, 1781–1789. [Google Scholar]
- Koster, J.F.; Slee, R.G.; Montfoort, A.; Lang, J.; Esterbauer, H. Comparison of the inactivation of microsomal glucose-6-phosphatase by in situ lipid peroxidation-derived 4-hydroxynonenal and exogenous 4-hydroxynonenal. Free Radic. Res. Commun. 1986, 1, 273–287. [Google Scholar] [CrossRef]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef]
- Martindale, J.L.; Holbrook, N.J. Cellular response to oxidative stress: signalling for suicide and survival. J. Cell. Physiol. 2002, 192, 1–15. [Google Scholar] [CrossRef]
- Benhar, M.; Engelberg, D.; Levitzki, A. ROS, stress-activated kinases and stress signaling in cancer. EMBO Rep. 2002, 3, 420–425. [Google Scholar]
- Leonard, S.S.; Harris, G.K.; Shi, X. Metal-induced oxidative stress and signal transduction. Free Radic. Biol. Med. 2004, 37, 1921–1942. [Google Scholar] [CrossRef]
- Storz, P. Reactive Oxigen species in tumor progression. Front. Biosci. 2005, 10, 1881–1896. [Google Scholar] [CrossRef]
- Barrera, G.; Di Mauro, C.; Muraca, R.; Ferrero, D.; Cavalli, G.; Fazio, V.M.; Paradisi, L.; Dianzani, M.U. Induction of differentiation in human HL-60 cells by 4-hydroxynonenal.; a product of lipid peroxidation. Exp. Cell. Res. 1991, 197, 148–152. [Google Scholar] [CrossRef]
- Barrera, G.; Martinotti, S.; Fazio, V.; Manzari, V.; Paradisi, L.; Parola, M.; Frati, L.; Dianzani, M.U. Effect of 4-hydroxynonenal on c-myc expression. Toxicol. Pathol. 1987, 15, 238–240. [Google Scholar]
- Barrera, G.; Muraca, R.; Pizzimenti, S.; Serra, A.; Rosso, C.; Saglio, G.; Farace, M.G.; Fazio, V.M.; Dianzani, M.U. Inhibition of c-myc expression induced by 4-hydroxynonenal, a product of lipid peroxidation, in the HL-60 human leukemic cell line. Biochem. Biophys. Res. Commun. 1994, 203, 553–561. [Google Scholar]
- Pizzimenti, S.; Briatore, F.; Laurora, S.; Toaldo, C.; Maggio, M.; De Grandi, M.; Meaglia, L.; Menegatti, E.; Giglioni, B.; Dianzani, M.U.; Barrera, G. 4-Hydroxynonenal inhibits telomerase activity and hTERT expression in human leukemic cell lines. Free Radic. Biol. Med. 2006, 40, 1578–1591. [Google Scholar] [CrossRef]
- Rinaldi, M.; Barrera, G.; Aquino, A.; Spinsanti, P.; Pizzimenti, S.; Farace, M.G.; Dianzani, M.U.; Fazio, V.M. 4-Hydroxynonenal-induced MEL cell differentiation involves PKC activity translocation Biochem. Biophys. Res. Commun. 2000, 272, 75–80. [Google Scholar] [CrossRef]
- Barrera, G.; Pizzimenti, S.; Muraca, R.; Barbiero, G.; Bonelli, G.; Baccino, FM.; Fazio, V.M.; Dianzani, M.U. Effect of 4-Hydroxynonenal on cell cycle progression and expression of differentiation-associated antigens in HL-60 cells. Free Radic. Biol. Med. 1996, 20, 455–462. [Google Scholar] [CrossRef]
- Pizzimenti, S.; Barrera, G.; Dianzani, M.U.; Brüsselbach, S. Inhibition of D1, D2, and A-cyclin expression in HL-60 cells by the lipid peroxydation product 4-hydroxynonenal. Free Radic. Biol. Med. 1999, 26, 1578–1586. [Google Scholar] [CrossRef]
- Barrera, G.; Pizzimenti, S.; Laurora, S.; Moroni, E.; Giglioni, B.; Dianzani, M.U. 4-Hydroxynonenal affects pRb/E2F pathway in HL-60 human leukemic cells. Biochem. Biophys. Res. Commun. 2002, 295, 267–275. [Google Scholar]
- Laurora, S.; Tamagno, E.; Briatore, F.; Bardini, P.; Pizzimenti, S.; Toaldo, C.; Reffo, P.; Costelli, P.; Dianzani, MU.; Danni, O.; Barrera, G. 4-Hydroxynonenal modulation of p53 family gene expression in the SK-N-BE neuroblastoma cell line. Free Radic. Biol. Med. 2005, 38, 215–225. [Google Scholar] [CrossRef]
- Muzio, G.; Canuto, R.A.; Trombetta, A.; Maggiora, M. Inhibition of cytosolic class 3 aldehyde dehydrogenase by antisense oligonucleotides in rat hepatoma cells. Chem. Biol. Interact. 2001, 130, 219–225. [Google Scholar]
- Canuto, R.A.; Muzio, G.; Ferro, M.; Maggiora, M.; Federa, R.; Bassi, A.M.; Lindahl, R.; Dianzani, M.U. Inhibition of class-3 aldehyde dehydrogenase and cell growth by restored lipid peroxidation in hepatoma cell lines. Free Radic. Biol. Med. 1999, 26, 333–340. [Google Scholar]
- Muzio, G.; Trombetta, A.; Martinasso, G.; Canuto, RA.; Maggiora, M. Antisense oligonucleotides against aldehyde dehydrogenase 3 inhibit hepatoma cell proliferation by affecting MAP kinases. Chem. Biol. Interact. 2003, 143–144, 37–43. [Google Scholar] [CrossRef]
- Shiraishi, K.; Naito, K. Effects of 4-hydroxy-2-nonenal, a marker of oxidative stress, on spermatogenesis and expression of p53 protein in male infertility. J. Urol. 2007, 178, 1012–1017. [Google Scholar] [CrossRef]
- Vizio, B.; Poli, G.; Chiarpotto, E.; Biasi, F. 4-hydroxynonenal and TGF-beta1 concur in inducing antiproliferative effects on the CaCo-2 human colon adenocarcinoma cell line. Biofactors 2005, 24, 237–246. [Google Scholar] [CrossRef]
- Zanetti, D.; Poli, G.; Vizio, B.; Zingaro, B.; Chiarpotto, E.; Biasi, F. 4-hydroxynonenal and transforming growth factor-beta1 expression in colon cancer. Mol. Aspects Med. 2003, 24, 273–280. [Google Scholar] [CrossRef]
- Cerbone, A.; Toaldo, C.; Laurora, S.; Briatore, F.; Pizzimenti, S.; Dianzani, M.U.; Ferretti, C.; Barrera, G. 4-Hydroxynonenal and PPARgamma ligands affect proliferation, differentiation, and apoptosis in colon cancer cells. Free Radic. Biol. Med. 2007, 42, 1661–1670. [Google Scholar] [CrossRef]
- Albright, C.D.; Klem, E.; Shah, A.A.; Gallagher, P. Breast cancer cell-targeted oxidative stress: enhancement of cancer cell uptake of conjugated linoleic acid, activation of p53, and inhibition of proliferation. Exp. Mol. Pathol. 2005, 79, 118–125. [Google Scholar] [CrossRef]
- Sunjic, S.B.; Cipak, A.; Rabuzin, F.; Wildburger, R.; Zarkovic, N. The influence of 4-hydroxy-2-nonenal on proliferation, differentiation and apoptosis of human osteosarcoma cells. Biofactors 2005, 24, 141–148. [Google Scholar] [CrossRef]
- Kakishita, H.; Hattori, Y. Vascular smooth muscle cell activation and growth by 4-hydroxynonenal. Life Sci. 2001, 69, 689–697. [Google Scholar] [CrossRef]
- Watanabe, T.; Pakala, R.; Katagiri, T.; Benedict, C.R. Lipid peroxidation product 4-hydroxy-2-nonenal acts synergistically with serotonin in inducing vascular smooth muscle cell proliferation. Atherosclerosis 2001, 155, 37–44. [Google Scholar] [CrossRef]
- Lee, T.J.; Lee, J.T.; Moon, S.K.; Kim, C.H.; Park, J.W.; Kwon, T.K. Age-related differential growth rate and respo61nse to 4-hydroxynonenal in mouse aortic smooth muscle cells. Int. J. Mol. Med. 2006, 17, 29–35. [Google Scholar]
- Vindis, C.; Escargueil-Blanc, I,; Uchida, K.; Elbaz, M.; Salvayre, R.; Negre-Salvayre, A. Lipid oxidation products and oxidized low-density lipoproteins impair platelet-derived growth factor receptor activity in smooth muscle cells: implication in atherosclerosis. Redox Rep. 2007, 12, 96–100. [Google Scholar] [CrossRef]
- Semlitsch, T.; Tillian, H.M.; Zarkovic, N.; Borovic, S.; Purtscher, M.; Hohenwarter, O.; Schaur, R.J. Differential influence of the lipid peroxidation product 4-hydroxynonenal on the growth of human lymphatic leukaemia cells and human periopherial blood lymphocytes. Anticancer Res. 2002, 22, 1689–1697. [Google Scholar]
- Fang, J.; Nakamura, H.; Iyer, A.K. Regulation of replicative potential of cells. J. Drug. Target. 2007, 15, 475–486. [Google Scholar] [CrossRef]
- Ozben, T. Oxidative stress and apoptosis: Impact on cancer therapy. J. Pharm. Sci. 2007, 96, 2181–2196. [Google Scholar] [CrossRef]
- Wang, L.; Azad, N.; Kongkaneramit, L.; Chen, F.; Lu, Y.; Jiang, B.H.; Rojanasakul, Y. The Fas death signaling pathway connecting reactive oxygen species generation and FLICE inhibitory protein down-regulation. J. Immunol. 2008, 180, 3072–3080. [Google Scholar]
- Jantová, S.; Repický, A.; Letasiová, S.; Cipák, L. 4-Amino-3-acetylquinoline-induced apoptosis of murine L1210 leukemia cells involves ROS-mitochondrial-mediated death signaling and activation of p38 MAPK. Cell. Biochem. Funct. 2008, 26, 609–619. [Google Scholar] [CrossRef]
- Lee, K.B.; Lee, J.S.; Park, J.W.; Huh, T.L.; Lee, Y.M. Low energy proton beam induces tumor cell apoptosis through reactive oxygen species and activation of caspases. Exp. Mol. Med. 2008, 40, 118–129. [Google Scholar] [CrossRef]
- Noguchi, T.; Ishii, K.; Fukutomi, H.; Naguro, I.; Matsuzawa, A.; Takeda, K.; Ichijo, H. Requirement of reactive oxygen species-dependent activation of ASK1-p38 MAPK pathway for extracellular ATP-induced apoptosis in macrophage. J. Biol. Chem. 2008, 283, 7657–7665. [Google Scholar]
- Higuchi, Y. Glutathione depletion-induced chromosomal DNA fragmentation associated with apoptosis and necrosis. J. Cell. Mol. Med. 2004, 8, 455–464. [Google Scholar] [CrossRef]
- Awasthi, YC.; Yang, Y.; Tiwari, NK.; Patrick, B.; Sharma, A.; Li, J.; Awasthi, S. Regulation of 4-hydroxynonenal-mediated signaling by glutathione S-transferases. Free Radic. Biol. Med. 2004, 37, 607–619. [Google Scholar] [CrossRef]
- Rabacchi, S.A.; Friedman, W.J.; Shelanski, M.L.; Troy, C.M. Divergence of the apoptotic pathways induced by 4-hydroxynonenal and amyloid beta-protein. Neurobiol. Aging. 2004, 25, 1057–1066. [Google Scholar] [CrossRef]
- Li, J.; Sharma, R.; Patrick, B.; Sharma, A.; Jeyabal, P.V.; Reddy, PM.; Saini, M.K.; Dwivedi, S.; Dhanani, S.; Ansari, N.H.; Zimniak, P.; Awasthi, S.; Awasthi, Y.C. Regulation of CD95 (Fas) expression and Fas-mediated apoptotic signaling in HLE B-3 cells by 4-hydroxynonenal. Biochemistry 2006, 45, 12253–12264. [Google Scholar] [CrossRef]
- de Villiers, W.J.; Song, Z.; Nasser, M.S.; Deaciuc, I.V.; McClain, C.J. 4-Hydroxynonenal-induced apoptosis in rat hepatic stellate cells: mechanistic approach. J. Gastroenterol. Hepatol. 2007, 22, 414–422. [Google Scholar] [CrossRef]
- Kutuk, O.; Basaga, H. Apoptosis signalling by 4-hydroxynonenal: a role for JNK-c-Jun/AP-1 pathway. Redox Rep. 2007, 12, 30–34. [Google Scholar] [CrossRef]
- Biasi, F.; Vizio, B.; Mascia, C.; Gaia, E.; Zarkovic, N.; Chiarpotto, E.; Leonarduzzi, G.; Poli, G. c-Jun N-terminal kinase upregulation as a key event in the proapoptotic interaction between transforming growth factor-beta1 and 4-hydroxynonenal in colon mucosa. Free Radic. Biol. Med. 2006, 41, 443–454. [Google Scholar] [CrossRef]
- Lovell, M.A.; Markesbery, W.R. Amyloid beta peptide, 4-hydroxynonenal and apoptosis. Curr. Alzheimer Res. 2006, 3, 359–364. [Google Scholar] [CrossRef]
- Yang, Y.; Sharma, A.; Sharma, R.; Patrick, B.; Singhal, S.S.; Zimniak, P.; Awasthi, S.; Awasthi, Y.C. Cells preconditioned with mild.; transient UVA irradiation acquire resistance to oxidative stress and UVA-induced apoptosis: role of 4-hydroxynonenal in UVA-mediated signaling for apoptosis. J. Biol. Chem. 2003, 278, 41380–4138. [Google Scholar] [CrossRef]
- Cheng, J.Z.; Singhal, S.S.; Sharma, A.; Saini, M.; Yang, Y.; Awasthi, S.; Zimniak, P.; Awasthi, Y.C. Transfection of mGSTA4 in HL-60 cells protects against 4-hydroxynonenal-induced apoptosis by inhibiting JNK-mediated signaling. Arch. Biochem. Biophys. 2001, 392, 197–207. [Google Scholar]
- Singhal, S.S.; Awasthi, Y.C.; Awasthi, S. Regression of melanoma in a murine model by RLIP76 depletion. Cancer Res. 2006, 66, 2354–2360. [Google Scholar]
- Singhal, S.S.; Singhal, J.; Yadav, S.; Dwivedi, S.; Boor, P.J.; Awasthi, Y.C.; Awasthi, S. Regression of lung and colon cancer xenografts by depleting or inhibiting RLIP76 (RALBP1). Cancer Res. 2007, 67, 4382–4389. [Google Scholar]
- Singhal, S.S.; Singhal, J.; Sharma, R.; Singh, S.V.; Zimniak, P.; Awasthi, Y.C.; Awasthi, S. Role of RLIP76 in lung cancer doxorubicin resistance. I. The ATPase activity of RLIP76 correlates with doxorubicin and 4-hydroxynonenal resistance in lung cancer cells. Int. J. Oncol. 2003, 22, 365–375. [Google Scholar]
- Awasthi, S.; Singhal, S.S.; Singhal, J.; Cheng, J.; Zimniak, P.; Awasthi, Y.C. Role of RLIP76 in lung cancer doxorubicin resistance. II. Doxorubicin transport in lung cancer by RLIP76. Int. J. Oncol. 2003, 22, 713–720. [Google Scholar]
- Awasthi, S.; Singhal, S.S.; Singhal, J.; Yang, Y.; Zimniak, P.; Awasthi, Y.C. Role of RLIP76 in lung cancer doxorubicin resistance. III. Anti-RLIP76 antibodies trigger apoptosis in lung cancer cells and synergistically increase doxorubicin cytotoxicity. Int. J. Oncol. 2003, 22, 721–732. [Google Scholar]
- Hayflick, L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell. Res. 1965, 37, 614–636. [Google Scholar] [CrossRef]
- Chen, Q.; Ames, B.N. Senescence-like growth arrest induced by hydrogen peroxide in human diploid fibroblast F65 cells. Proc. Natl. Acad. Sci. USA 1994, 91, 4130–4134. [Google Scholar] [CrossRef]
- Chen, Q.; Fischer, A.; Reagan, J.D.; Yan, L.J.; Ames, B.N. Oxidative DNA damage and senescence of human diploid fibroblast cells. Proc. Natl. Acad. Sci. USA 1995, 92, 4337–4341. [Google Scholar]
- Fridovich, I. Oxygen toxicity: a radical explanation. J. Exp. Biol. 1998, 201, 1203–1209. [Google Scholar]
- Saretzki, G.; Feng, J.; von Zglinicki, T.; Villeponteau, B. Similar gene expression pattern in senescent and hyperoxic-treated fibroblasts. J. Gerontol. A Biol. Sci. Med. Sci. 1998, 53, B438–B442. [Google Scholar]
- Martin, J.A.; Klingelhutz, A.J.; Moussavi-Harami, F.; Buckwalter, J.A. Effects of oxidative damage and telomerase activity on human articular cartilage chondrocyte senescence. J. Gerontol. A Biol. Sci. Med. Sci. 2004, 59, 324–337. [Google Scholar] [CrossRef]
- Orr, W.C.; Sohal, R.S. Extension of life-span by overexpression of superoxide dismutase and catalase in Drosophila melanogaster. Science 1994, 263, 1128–1130. [Google Scholar]
- Bell, E.L.; Klimova, T.A.; Eisenbart, J.; Schumacker, P.T.; Chandel, N.S. Mitochondrial reactive oxygen species trigger hypoxia-inducible factor-dependent extension of the replicative life span during hypoxia. Mol. Cell. Biol. 2007, 27, 5737–5745. [Google Scholar] [CrossRef]
- Greider, C.W. Telomere length regulation. Annu. Rev. Biochem. 1996, 65, 337–365. [Google Scholar] [CrossRef]
- Watson, J.D. Origin of concatemeric T7 DNA. Nat. New Biol. 1972, 239, 197–201. [Google Scholar] [CrossRef]
- Greider, C.W.; Blackburn, E.H. A telomeric sequence in the RNA of Tetrahymena telomerase required for telomere repeat synthesis. Nature 1989, 337, 331–337. [Google Scholar] [CrossRef]
- Herbert, B.; Pitts, A.E.; Baker, S.I.; Hamilton, S.E.; Wright, W.E.; Shay, J.W.; Corey, D.R. Inhibition of human telomerase in immortal human cells leads to progressive telomere shortening and cell death. Proc. Natl. Acad. Sci. USA 1999, 96, 14276–14281. [Google Scholar]
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific association of human telomerase activity with immortal cells and cancer. Science 1994, 266, 2011–2015. [Google Scholar]
- Shay, J.W.; Bacchetti, S. A survey of telomerase activity in human cancer. Eur. J. Cancer 1997, 33, 787–791. [Google Scholar] [CrossRef]
- Matthews, C.; Gorenne, I.; Scott, S.; Figg, N.; Kirkpatrick, P.; Ritchie, A.; Goddard, M.; Bennett, M. Vascular smooth muscle cells undergo telomere-based senescence in human atherosclerosis: effects of telomerase and oxidative stress. Circ. Res. 2006, 99, 156–164. [Google Scholar] [CrossRef]
- Nishikawa, M. Reactive oxygen species in tumor metastasis. Cancer Letters 2008, 266, 53–59. [Google Scholar] [CrossRef]
- Borras, C.; Esteve, J.M.; Vina, J.C.; Sastre, J.; Vina, J.; Pallardo, F.V. Glutathione regulates telomerase activity in 3T3 fibroblasts. J. Biol. Chem. 2004, 279, 34332–34335. [Google Scholar]
- Pizzimenti, S.; Menegatti, E.; Berardi, D.; Toaldo, C.; Pettazzoni, P.; Minelli, R.; Giglioni, B.; Cerbone, A.; Dianzani, M.U.; Ferretti, C.; Barrera, G. 4-Hydroxynonenal.; a lipid peroxidation product of dietary polyunsaturated fatty acids, has anticarcinogenic properties in coloncarcinoma cell lines, through the inhibition of telomerase activity. J. Nutr. Biochem. 2009, in press. [Google Scholar]
- Cong, Y.S.; Wen, J.; Bacchetti, S. The human telomerase catalytic subunit hTERT: organization of the gene and characterization of the promoter. Hum. Mol. Genet. 1999, 8, 137–142. [Google Scholar] [CrossRef]
- Hurlin, P.J.; Queva, C.; Eisenman, R.N. Mnt, a novel Max-interacting protein is coexpressed with Myc in proliferating cells and mediates repression at Myc binding sites. Genes Dev. 1997, 11, 44–58. [Google Scholar] [CrossRef]
- Bergers, G.; Benjamin, LE. Tumorigenesis and the angiogenic switch. Nat. Rev. Cancer. 2003, 3, 401–410. [Google Scholar] [CrossRef]
- Jain, R.K. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef]
- Ushio-Fukai, M.; Nakamura, Y. Reactive oxygen species and angiogenesis: NADPH oxidase as target for cancer therapy. Cancer Lett. 2008, 266, 37–52. [Google Scholar] [CrossRef]
- Wang, J.; Jing, Yi. Cancer cell killing via ROS. Cancer Biol. Ther. 2008, 7, 1875–1884. [Google Scholar] [CrossRef]
- Maulik, N.; Das, D.K. Redox signalling in vascular angiogenesis. Free Radic. Biol. Med. 2002, 33, 1047–1060. [Google Scholar] [CrossRef]
- Stone, J.R.; Collins, T. The role of hydrogen peroxide in endothelial proliferative responses. Endothelium 2002, 9, 231–238. [Google Scholar] [CrossRef]
- Luczak, K.; Balcerczyk, A.; Soszynski, M.; Bartosz, G. Low concentration of oxidant and nitric oxide donors stimulate proliferation of human endothelial cells in vitro. Cell Biol. Int. 2004, 28, 483–486. [Google Scholar] [CrossRef]
- Yasuda, M.; Ohzeki, Y.; Shimizu, S.; Naito, S.; Ohtsuru, A.; Yamamoto, T.; Kuroiwa, Y. Stimulation of in vitro angiogenesis by hydrogen peroxide and the relation with ETS-1 in endothelial cells. Life Sci. 1999, 64, 249–258. [Google Scholar]
- Chua, C.C.; Hamdy, R.C.; Chua, B.H. Upregulation of vascular endothelial growth factor by H2O2 in rat heart endothelial cells. Free Radic. Biol. Med. 1998, 25, 891–897. [Google Scholar] [CrossRef]
- Vepa, S.; Scribner, W.M.; Parinandi, N.L.; English, D.; Garcia, J.G.; Natarajan, V. Hydrogen peroxide stimulates tyrosine phosphorylation of focal adhesion kinase in vascular endothelial cells. Am. J. Physiol. 1999, 277, L150–L158. [Google Scholar]
- Shono, T.; Ono, M.; Izumi, H.; Jimi, S.I.; Matsushima, K.; Okamoto, T.; Kohno, K.; Kuwano, M. Involvement of the transcription factor NF-κB in tubular morphogenesis of human microvascular endothelial cells by oxidative stress. Mol. Cell. Biol. 1996, 16, 4231–4239. [Google Scholar]
- Kusmartsev, S.; Eruslanov, E.; Kübler, H.; Tseng, T.; Sakai, Y.; Su, Z.; Kaliberov, S.; Heiser, A.; Rosser, C.; Dahm, P.; Siemann, D.; Vieweg, J. Oxidative stress regulates expression of VEGFR1 in myeloid cells: link to tumor-induced immune suppression in renal cell carcinoma. J. Immunol. 2008, 181, 346–353. [Google Scholar]
- Xia, C.; Meng, Q.; Liu, LZ.; Rojanasakul, Y.; Wang, XR.; Jiang, BH. Reactive oxygen species regulate angiogenesis and tumor growth through vascular endothelial growth factor. Cancer Res. 2007, 67, 10823–10830. [Google Scholar] [CrossRef]
- Szatrowski, T.P.; Nathan, C.F. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991, 51, 794–798. [Google Scholar]
- Lim, S.D.; Sun, C.; Lambeth, J.D.; Marshall, F.; Amin, M.; Chung, L.; Petros, J.A.; Arnold, R.S. Increased Nox1 and hydrogen peroxide in prostate cancer. Prostate 2005, 62, 200–207. [Google Scholar] [CrossRef]
- Al-Shabrawey, M.; Bartoli, M.; El-Remessy, A.B.; Platt, D.H.; Matragoon, S.; Behzadian, M.A.; Caldwell, R.W.; Caldwell, R.B. Inhibition of NAD(P)H oxidase activity blocks vascular endothelial growth factor overexpression and neovascularization during ischemic retinopathy. Am. J. Pathol. 2005, 167, 599–607. [Google Scholar] [CrossRef]
- Yamaura, M.; Mitsushita, J.; Furuta, S.; Kiniwa, Y.; Ashida, A.; Goto, Y.; Shang, W.H.; Kubodera, M.; Kato, M.; Takata, M.; Saida, T.; Kamata, T. NADPH oxidase 4 contributes to transformation phenotype of melanoma cells by regulating G2-M cell cycle progression. Cancer Res. 2009, 69, 2647–2654. [Google Scholar] [CrossRef]
- Arbiser, J.L.; Petros, J.; Klafter, R.; Govindajaran, B.; McLaughlin, E.R.; Brown, L.F.; Cohen, C.; Moses, M.; Kilroy, S.; Arnold, R.S.; Lambeth, J.D. Reactive oxygen generated by Nox1 triggers the angiogenic switch. Proc. Natl. Acad. Sci. USA 2002, 99, 715–720. [Google Scholar] [CrossRef]
- Ruef, J.; Rao, G.N.; Li, F.; Bode, C.; Patterson, C.; Bhatnagar, A.; Runge, M.S. Induction of rat aortic smooth muscle cell growth by the lipid peroxidation product 4-hydroxy-2-nonenal. Circulation 1998, 97, 1071–1078. [Google Scholar] [CrossRef]
- Ayalasomayajula, S.P.; Kompella, U.B. Induction of vascular endothelial growth factor by 4-hydroxynonenal and its prevention by glutathione precursors in retinal pigment epithelial cells. Eur. J. Pharmacol. 2002, 449, 213–220. [Google Scholar] [CrossRef]
- Stagos, D.; Zhou, H.; Ross, D.; Vasiliou, V. 4-HNE inhibits tube formation and up-regulates chondromodulin-I in human endothelial cells. Biochem. Biophys. Res. Commun. 2009, 379, 654–658. [Google Scholar] [CrossRef]
- Pizzimenti, S.; Ferracin, M.; Sabbioni, S.; Toaldo, C.; Pettazzoni, P.; Dianzani, M.U.; Negrini, M.; Barrera, G. MicroRNA expression changes during human leukemic HL-60 cell differentiation induced by 4-hydroxynonenal, a product of lipid peroxidation. Free Radic. Biol. Med. 2009, 46, 282–288. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Lee, D.Y.; Deng, Z.; Wang, C.H.; Yang, B.B. MicroRNA-378 promotes cell survival, tumor growth, and angiogenesis by targeting SuFu and Fus-1 expression. Proc. Natl. Acad. Sci. USA 2007, 104, 20350–20355. [Google Scholar] [CrossRef]
- Chambers, A.F.; Groom, A.C.; MacDonald, I.C. Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2002, 2, 563–572. [Google Scholar] [CrossRef]
- Cavallaro, U.; Christofori, G. Multitasking in tumor progression: signaling functions of cell adhesion molecules. Ann. N.Y. Acad. Sci. 2004, 1014, 58–66. [Google Scholar] [CrossRef]
- Hood, J.D.; Cheresh, D.A. Role of integrins in cell invasion and migration. Nat. Rev. Cancer. 2002, 2, 91–100. [Google Scholar] [CrossRef]
- Tilghman, R.W.; Parsons, J.T. Focal adhesion kinase as a regulator of cell tension in the progression of cancer. Semin. Cancer Biol. 2008, 18, 45–52. [Google Scholar] [CrossRef]
- Golubovskaya, V.M.; Kweh, F.A.; Cance, W.G. Focal adhesion kinase and cancer. Histol. Histopathol. 2009, 24, 503–510. [Google Scholar]
- Egeblad, M.; Werb, Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer. 2002, 2, 161–74. [Google Scholar] [CrossRef]
- Martin, M.D.; Matrisian, L.M. The other side of MMPs: protective roles in tumor progression. Cancer Metastasis Rev. 2007, 26, 717–724. [Google Scholar] [CrossRef]
- Baker, A.H.; Edwards, D.R.; Murphy, G. Metalloproteinase inhibitors: biological actions and therapeutic opportunities. J. Cell Sci. 2002, 115, 3719–3727. [Google Scholar] [CrossRef]
- Kobayashi, H.; Boelte, K.C.; Lin, P.C. Endothelial cell adhesion molecules and cancer progression. Curr. Med. Chem. 2007, 14, 377–386. [Google Scholar] [CrossRef]
- Jing, X.; Ueki, N.; Cheng, J.; Imanishi, H.; Hada, T. Induction of apoptosis in hepatocellular carcinoma cell lines by emodin. Jpn. J. Cancer Res. 2002, 93, 874–882. [Google Scholar] [CrossRef]
- Ferraro, D.; Corso, S.; Fasano, E.; Panieri, E.; Santangelo, R.; Borrello, S.; Giordano, S.; Pani, G.; Galeotti, T. Pro-metastatic signaling by c-Met through RAC-1 and reactive oxygen species (ROS). Oncogene 2006, 25, 3689–3698. [Google Scholar] [CrossRef]
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 2008, 320, 661–664. [Google Scholar] [CrossRef] [Green Version]
- Mori, K.; Shibanuma, M.; Nose, K. Invasive potential induced under long-term oxidative stress in mammary epithelial cells. Cancer Res. 2004, 64, 7464–7472. [Google Scholar] [CrossRef]
- Krizbai, I.A.; Bauer, H.; Bresgen, N.; Eckl, P.M.; Farkas, A.; Szatmari, E.; Traweger, A.; Wejksza, K.; Bauer, H.C. Effect of oxidative stress on the junctional proteins of cultured cerebral endothelial cells. Cell. Mol. Neurobiol. 2005, 25, 129–139. [Google Scholar] [CrossRef]
- Rao, R.K.; Basuroy, S.; Rao, V.U.; Karnaky, K.J., Jr.; Gupta, A. Tyrosine phosphorylation and dissociation of occludin-ZO-1 and E-cadherin-ß-catenin complexes from the cytoskeleton by oxidative stress. Biochem. J. 2002, 368, 471–481. [Google Scholar] [CrossRef]
- Bockhorn, M.; Roberge, S.; Sousa, C.; Jain, R.K.; Munn, L.L. Differential gene expression in metastasizing cells shed from kidney tumors. Cancer Res. 2004, 64, 2469–2473. [Google Scholar] [CrossRef]
- Thews, O.; Lambert, C.; Kelleher, D.K.; Biesalski, H.K.; Vaupel, P.; Frank, J. Impact of therapeutically induced reactive oxygen species and radical scavenging by α-tocopherol on tumor cell adhesion. Oncol. Rep. 2007, 18, 965–971. [Google Scholar]
- Ben Mahdi, M.H.; Andrieu, V.; Pasquier, C. Focal adhesion kinase regulation by oxidative stress in different cell types. IUBMB Life 2000, 50, 291–299. [Google Scholar] [CrossRef]
- Rajagopalan, S.; Meng, X.P.; Ramasamy, S.; Harrison, D.G.; Galis, Z.S. Reactive oxygen species produced by macrophage-derived foam cells regulate the activity of vascular matrix metalloproteinases in vitro. Implications for atherosclerotic plaque stability. J. Clin. Invest. 1996, 98, 2572–2579. [Google Scholar] [CrossRef]
- Nelson, K.K.; Melendez, J.A. Mitochondrial redox control of matrix metalloproteinases. Free Radic. Biol. Med. 2004, 37, 768–784. [Google Scholar] [CrossRef]
- Lander, H.M.; Hajjar, D.P.; Hempstead, B.L.; Mirza, U.A.; Chait, B.T.; Campbell, S.; Quilliam, L.A. A molecular redox switch on p21(ras). Structural basis for the nitric oxide-p21(ras) interaction. J. Biol. Chem. 1997, 272, 4323–4326. [Google Scholar]
- Westermarck, J.; Kahari, V.M. Regulation of matrix metalloproteinase expression in tumor invasion. FASEB J. 1999, 13, 781–792. [Google Scholar]
- Haorah, J.; Ramirez, SH.; Schall, K.; Smith, D.; Pandya, R.; Persidsky, Y. Oxidative stress activates protein tyrosine kinase and matrix metalloproteinases leading to blood-brain barrier dysfunction. J. Neurochem. 2007, 101, 566–576. [Google Scholar] [CrossRef]
- Hemmerlein, B.; Johanns, U.; Halbfass, J.; Böttcher, T.; Heuser, M.; Radzun, H.J.; Thelen, P. The balance between MMP-2/-9 and TIMP-1/-2 is shifted towards MMP in renal cell carcinomas and can be further disturbed by hydrogen peroxide. Int. J. Oncol. 2004, 24, 1069–1076. [Google Scholar]
- Jinga, D.C.; Blidaru, A.; Condrea, I.; Ardeleanu, C.; Dragomir, C.; Szegli, G. MMP-9 and MMP-2 gelatinases and TIMP-1 and TIMP-2 inhibitors in breast cancer: correlations with prognostic factors. J. Cell. Mol. Med. 2007, 10, 499–510. [Google Scholar]
- Engers, R.; Springer, E.; Kehren, V.; Simic, T.; Young, D.A.; Beier, J.; Klotz, L.O.; Clark, I.M.; Sies, H.; Gabbert, H.E. Rac upregulates tissue inhibitor of metalloproteinase-1 expression by redox-dependent activation of extracellular signal-regulated kinase signaling. FEBS J. 2006, 273, 4754–4769. [Google Scholar] [CrossRef]
- Engers, R.; Springer, E.; Michiels, F.; Collard, J.G.; Gabbert, H.E. Rac affects invasion of human renal cell carcinomas by up-regulating tissue inhibitor of metalloproteinases (TIMP)-1 and TIMP-2 expression. J. Biol. Chem. 2001, 276, 41889–41897. [Google Scholar]
- Roebuck, K.A. Oxidant stress regulation of IL-8 and ICAM-1 gene expression: differential activation and binding of the transcription factors AP-1 and NF-κB. Int. J. Mol. Med. 1999, 4, 223–230. [Google Scholar]
- Pizzimenti, S.; Barrera, G.; Calzavara, E.; Mirandola, L.; Toaldo, C.; Dianzani, M.U.; Comi, P.; Chiaramonte, R. Down-regulation of Notch1 expression is involved in HL-60 cell growth inhibition induced by 4-hydroxynonenal, a product of lipid peroxidation. Med. Chem. 2008, 4, 551–557. [Google Scholar] [CrossRef]
- Timmerman, L.A.; Grego-Bessa, J.; Raya, A.; Bertrán, E.; Pérez-Pomares, J.M.; Díez, J.; Aranda, S.; Palomo, S.; McCormick, F.; Izpisúa-Belmonte, J.C.; de la Pompa, J.L. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev. 2004, 18, 99–115. [Google Scholar] [CrossRef]
- Leong, K.G.; Niessen, K.; Kulic, I.; Raouf, A.; Eaves, C.; Pollet, I.; Karsan, A. Jagged1-mediated Notch activation induces epithelial-to-mesenchymal transition through Slug-induced repression of E-cadherin. J. Exp. Med. 2007, 204, 2935–2948. [Google Scholar] [CrossRef]
- Usatyuk, P.V.; Parinandi, N.L.; Natarajan, V. Redox regulation of 4-hydroxy-2-nonenal-mediated endothelial barrier dysfunction by focal adhesion, adherens, and tight junction proteins. J. Biol. Chem. 2006, 281, 35554–35566. [Google Scholar] [CrossRef]
- Calonghi, N.; Boga, C.; Cappadone, C.; Pagnotta, E.; Bertucci, C.; Fiori, J.; Masotti, L. Cytotoxic and cytostatic effects induced by 4-hydroxynonenal in human osteosarcoma cells. Biochem. Biophys. Res. Commun. 2002, 293, 1502–1507. [Google Scholar] [CrossRef]
- Schirner, M.; Herzberg, F.; Scmidt, R.; Streit, M.; Schoning, M.; Hummel, M.; Kaufmann, C.; Thiel, E.; Kreuser, E.D. Integrin α5β1: a potent inhibitor of experimental lung metastasis. Clin. Exp. Metastasis 1998, 16, 427–435. [Google Scholar]
- Gong, J.; Ko, T.C.; Brattain, M.G. Disruption of fibronectin binding to the α5β1 integrin stimulates the expression of cyclin-dependent kinases and DNA syntesis through activation of extracellular signal-regulated kinase. J. Biol. Chem. 1998, 273, 1662–1669. [Google Scholar] [CrossRef]
- Lee, S.J.; Seo, K.W.; Yun, M.R.; Bae, S.S.; Lee, W.S.; Hong, K.W.; Kim, C.D. 4-Hydroxynonenal enhances MMP-2 production in vascular smooth muscle cells via mitochondrial ROS-mediated activation of the Akt/NF-κB signaling pathways. Free Radic. Biol. Med. 2008, 45, 1487–1492. [Google Scholar] [CrossRef]
- Akiba, S.; Kumazawa, S.; Yamaguchi, H.; Hontani, N.; Matsumoto, T.; Ikeda, T.; Oka, M.; Sato, T. Acceleration of matrix metalloproteinase-1 production and activation of platelet-derived growth factor receptor beta in human coronary smooth muscle cells by oxidized LDL and 4-hydroxynonenal. Biochim. Biophys. Acta 2006, 1763, 797–804. [Google Scholar] [CrossRef]
- Morquette, B.; Shi, Q.; Lavigne, P.; Ranger, P.; Fernandes, J.C.; Benderdour, M. Production of lipid peroxidation products in osteoarthritic tissues: new evidence linking 4-hydroxynonenal to cartilage degradation. Arthritis Rheum. 2006, 54, 271–281. [Google Scholar] [CrossRef]
- Zamara, E.; Novo, E.; Marra, F.; Gentilini, A.; Romanelli, R.G.; Caligiuri, A.; Robino, G.; Tamagno, E.; Aragno, M.; Danni, O.; Autelli, R.; Colombatto, S.; Dianzani, M.U.; Pinzani, M.; Parola, M. 4–Hydroxynonenal as a selective pro-fibrogenic stimulus for activated human hepatic stellate cells. J. Hepat. 2004, 40, 60–68. [Google Scholar]
- Herbst, U.; Toborek, M.; Kaiser, S.; Mattson, M.P.; Hennig, B. 4-Hydroxynonenal induces dysfunction and apoptosis of cultured endothelial cells. J. Cell. Physiol. 1999, 181, 295–303. [Google Scholar] [CrossRef]
- Minekura, H.; Kumagai, T.; Kawamoto, Y.; Nara, F.; Uchida, K. 4-Hydroxy-2-nonenal is a powerful endogenous inhibitor of endothelial response. Biochem. Biophys. Res. Commun. 2001, 282, 557–561. [Google Scholar] [CrossRef]
- Gentile, F.; Pizzimenti, S.; Arcaro, A.; Pettazzoni, P.; Minelli, R.; D'Angelo, D.; Mamone, G.; Ferranti, P.; Toaldo, C.; Cetrangolo, G.; Formisano, S.; Dianzani, M.U.; Uchida, K.; Dianzani, C.; Barrera, G. Exposure of HL-60 human leukemic cells to 4-hydroxynonenal promotes the formation of adduct(s) with alpha-enolase devoid of plasminogen binding activity. Biochem. J. 2009, 422, 285–294. [Google Scholar] [CrossRef]
- Anderson Garth, R. Genomic instability in cancer. Curr. Sci. 2001, 81, 501–507. [Google Scholar]
- Murphy, K.; Rosen, J. Mutant p53 and genomic instability in a transgenic mouse model of breast cancer. Oncogene 2000, 19, 1045–1051. [Google Scholar] [CrossRef]
- Valgardsdottir, R.; Tryggvadottir, L.; Steinarsdottir , M.; Olafsdottir, K.; Jonasdottir, S.; Jonasson, J.G.; Ogmundsdottir, H.M.; Eyfjörd, J.E. Genomic instability and poor prognosis associated with abnormal TP53 in breast carcinomas. Molecular and immunohistochemical analysis. APMIS 1997, 105, 121–130. [Google Scholar] [CrossRef]
- Sierra A., X. Castellsague, A.; Escobedo Lloveras, B., García-Ramirez; Moreno, A.; Fabra, A. Bcl-2 with loss of apoptosis allows accumulation of genetic alterations: a pathway to metastatic progression in human breast cancer. Int. J. Cancer 2000, 89, 142–147. [Google Scholar] [CrossRef]
- Griffith, J.; Bryant, J.; Fordyce, C.; Gilliland, F.; Joste, N.; Moyzis, R. Reduced telomere DNA content is correlated with genomic instability and metastasis in invasive human breast carcinoma. Breast Cancer Res. Treat. 1999, 54, 59–64. [Google Scholar] [CrossRef]
- Lindahl, T.; Wood, R.D. Quality control by DNA repair. Science 1999, 286, 1897–1905. [Google Scholar] [CrossRef]
- Peltomäki, P. DNA mismatch repair and cancer. Mutat. Res. 2001, 488, 77–85. [Google Scholar] [CrossRef]
- Wood, R.D.; Mitchell, M.; Sgouros, J.; Lindahl, T. Human DNA repair genes. Science 2001, 291, 1284–1289. [Google Scholar] [CrossRef]
- Maynard, S.; Schurman, S.H.; Harboe, C.; de Souza-Pinto, N.C.; Bohr, V.A. Base excision repair of oxidative DNA damage and association with cancer and aging. Carcinogenesis 2009, 30, 2–10. [Google Scholar]
- O'Brien, V.; Brown, R. Signalling cell cycle arrest and cell death through the MMR System. Carcinogenesis 2006, 27, 682–692. [Google Scholar] [CrossRef]
- Lee, S.H.; Chang, D.K.; Goel, A.; Boland, C.R.; Bugbee, W.; Boyle, D.L.; Firestein, G.S. Microsatellite Instability and Suppressed DNA Repair Enzyme Expression in Rheumatoid Arthritis. J. Immunol. 2003, 170, 2214–2220. [Google Scholar]
- Schwartz, D.; Rotter, V. p53-dependent cell cycle control: response to genotoxic stress. Semin. Cancer. Biol. 1998, 8, 325–336. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pizzimenti, S.; Toaldo, C.; Pettazzoni, P.; Dianzani, M.U.; Barrera, G. The "Two-Faced" Effects of Reactive Oxygen Species and the Lipid Peroxidation Product 4-Hydroxynonenal in the Hallmarks of Cancer. Cancers 2010, 2, 338-363. https://doi.org/10.3390/cancers2020338
Pizzimenti S, Toaldo C, Pettazzoni P, Dianzani MU, Barrera G. The "Two-Faced" Effects of Reactive Oxygen Species and the Lipid Peroxidation Product 4-Hydroxynonenal in the Hallmarks of Cancer. Cancers. 2010; 2(2):338-363. https://doi.org/10.3390/cancers2020338
Chicago/Turabian StylePizzimenti, Stefania, Cristina Toaldo, Piergiorgio Pettazzoni, Mario U. Dianzani, and Giuseppina Barrera. 2010. "The "Two-Faced" Effects of Reactive Oxygen Species and the Lipid Peroxidation Product 4-Hydroxynonenal in the Hallmarks of Cancer" Cancers 2, no. 2: 338-363. https://doi.org/10.3390/cancers2020338