Nrf2 and NF-κB and Their Concerted Modulation in Cancer Pathogenesis and Progression

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction

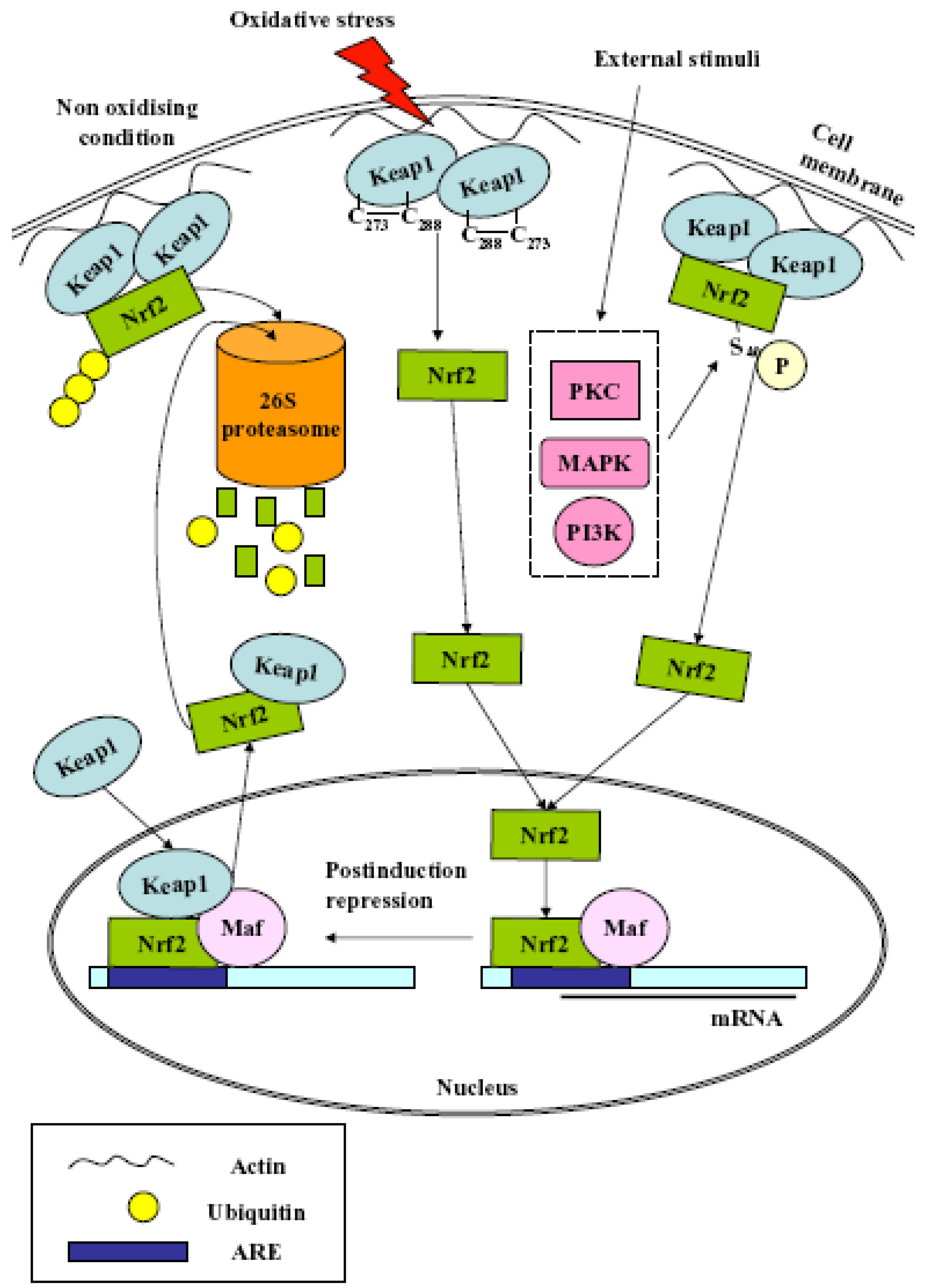
2. Nrf2
- (1)
- Nrf2 release from cytoplasmic anchoring: under normal homeostatic conditions Nrf2 is retained in the cytoplasm by its interaction with Keap1. During redox stress, Nrf2 is released from Keap1 and translocates from the cytoplasm to the nucleus. The release is due to several effectors, i.e., modification of Cys residues in Keap1, Protein Kinase C (PKC)-mediated Nrf2 phosphorylation at Ser40, casein kinase 2 (CK2), extracellular signal-regulated kinase 2 (ERK2), ERK5, glycogen synthase kinase-3β (GSK-3β), c-Jun N-terminal kinase 1 (JNK1), PKR-like ER-localized eIF2α kinase (PERK) and phosphoinositide-3-kinase (PI3K);
- (2)
- Nrf2 release from nuclear sequestration and recruitment to ARE enhancers: Keap1 can undergo nuclear-cytoplasmic shuttling through a potential nuclear export signal (NES). Upon stress stimulation, the nuclear protein prothymosin α binds the Kelch-repeat domain and liberates Nrf2, thus activating its target genes;
- (3)
- Nrf2 protein stabilization: under normal homeostatic conditions, Nrf2 is continuously degraded by the 26S proteasome in a Keap1-dependent fashion. Keap1 also binds Cullin-3 (Cul3) to form a core E3 ubiquitin ligase complex. This complex targets Nrf2 for degradation in a redox-dependent fashion. Recently, the existence of two distinct binding sites in the Nrf2 domain for the Kelch-repeat domain has been proposed. It has been postulated that Keap1 immobilizes these ubiquitin acceptor sites by tethering Nrf2 across the two Kelch-repeat domains, bringing them into close proximity to Cul3–Rbx1 thereby facilitating ubiquitylation. Non-ubiquitylated Nrf2 remains bound to the Keap1–Cul3–Rbx1 complex until Keap1 regains its substrate adaptor activity. Newly translated Nrf2 protein would bypass the Keap1–Cul3–Rbx1 complex and accumulate rapidly in the nucleus;
- (4)
- Antagonism of Nrf2 nuclear-cytoplasmic shuttling: under normal homeostatic conditions, Nrf2 nuclear abundance is restricted by the existence of a redox-dependent nuclear-cytoplasmic shuttling that, in turn, is controlled through the presence of a single nuclear localization sequence (NLS) and two NES motifs in the transcription factor. The NES contains an embedded Cysteine, and it has been proposed that modification of this residue by oxidants or electrophiles prevents recognition of the motif by exportin 1, thus leading to its nuclear accumulation;
- (5)
- Nrf2 gene induction: the Nrf2 promoter contains two ARE sequences and xenobiotic response elements (XREs). Multiple single nucleotide polymorphisms in the promoter of human Nrf2 can regulate its expression.
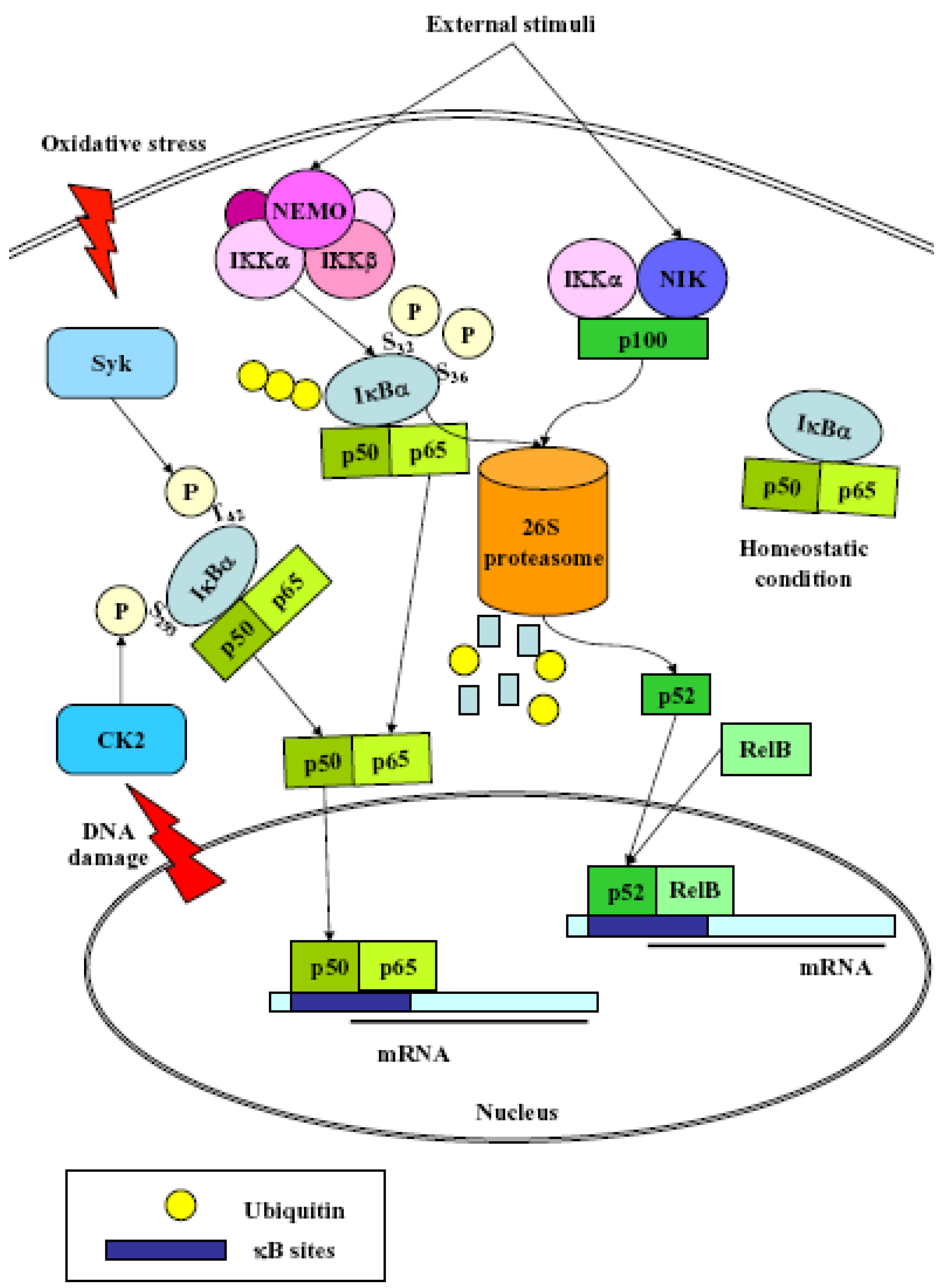
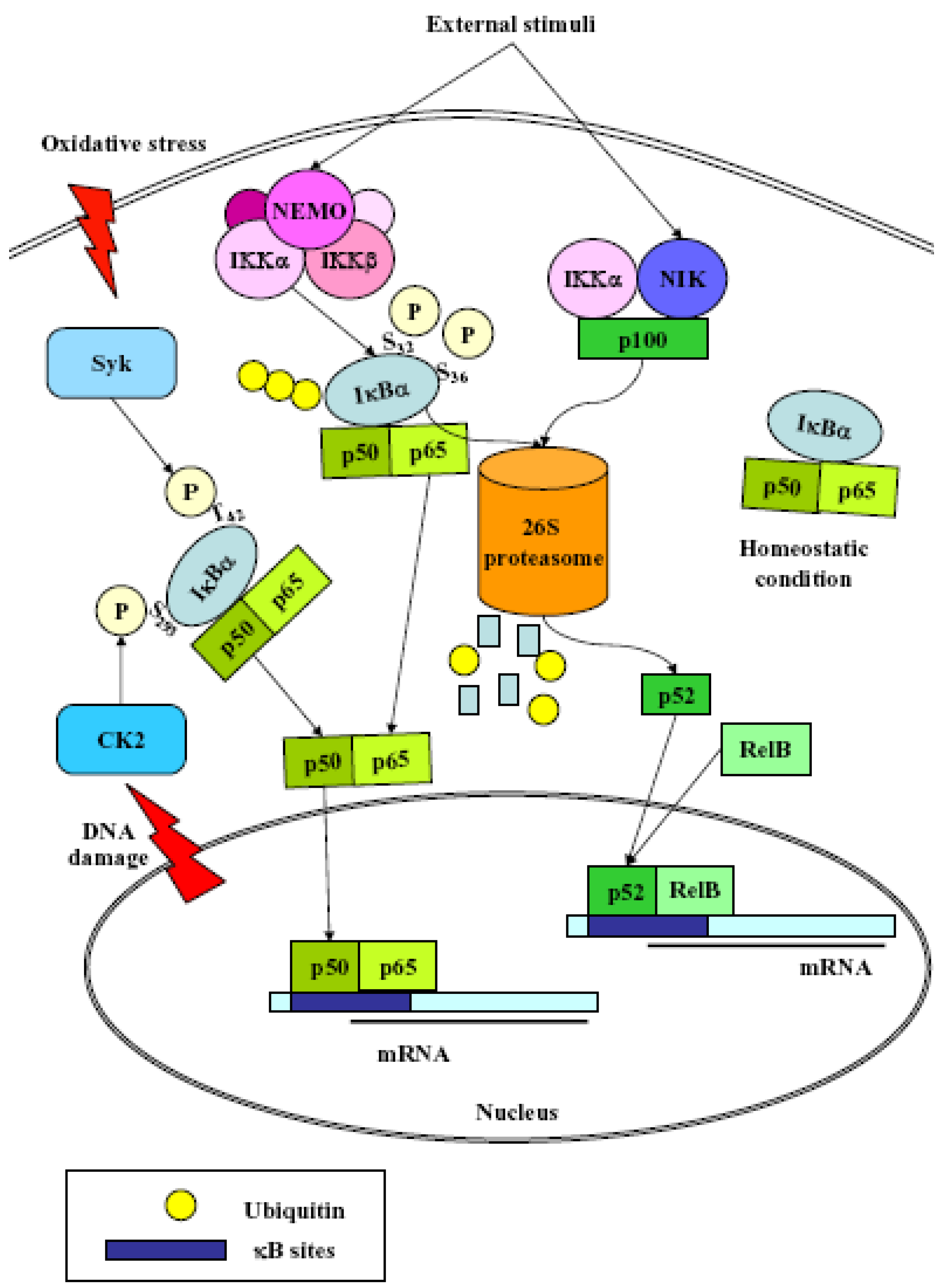
3. NF-κB
- (1)
- Classical: induced by a variety of innate and adaptative immunity mediators, turned on within minutes. In basal conditions NF-κB is sequestered in the cytoplasm by inhibitor proteins, usually IκBα. Upon stimulation, IκBα is rapidly phosphorylated by the IkB kinase complex (IKK), which contains the catalytic subunits IKKα and IKKβ, the regulatory subunits NEMO and ELKS, and the heat shock protein Hsp90/Cdc37 chaperone complex [25]. Phosphorylated IκBα is substrate for ubiquitination and subsequent degradation by the 26 S proteasome [26]. The released NF-κB dimer then translocates to the nucleus and activates target genes by binding with high affinity to κB elements in their promoter;
- (2)
- Alternative/NEMO-independent pathway: important for secondary lymphoid organ development, homeostasis and adaptive immunity, turned on in few hours. It involves the activation of NF-κB inducing kinase (NIK)- and IKKα-dependent proteasomal processing of p100 into p52, which binds DNA in association with its partners, i.e., RelB [24]. NIK mediates p100 phosphorylation/ubiquitination in an IKKα-independent and IKKα-dependent manners. The phospho-p100, recognized and polyubiquitinated by the E3 ligase, is partially degraded by the proteasome to generate p52/RelB and p52/c-Rel or p52/p65 dimers. p52/RelB dimers move to the nucleus whereas p52/c-Rel and p52/p65 dimers, first captured by IκBs for their cytosolic retention, are activated through the classical pathway;
- (3)
- atypical: independent from IKK, triggered by DNA damage such as UV [27] or doxorubicin [28], still requires the proteasome. UV radiation induces IκBα degradation via the proteasome, and the targeted serine residues are located within a C-terminal cluster, which is recognized by the p38-activated CK2;
- (4)
- Oxidative stress-induced: relying on signal-induced phosphorylation events. NF-κB activation is achieved via IkBα tyrosine phosphorylation [29] by Syk protein tyrosine kinase [30]. H2O2 is the central second messenger to NF-κB activation [31] which involves several different mechanisms, since the redox-sensitive pathways triggering this activation are different depending on the cell-type considered [30]. NF-κB is also sensitive to oxidative modifications of Cys62 in p50, essential for DNA binding [32,33]. Its activation and nuclear translocation are stimulated by oxidizing conditions, while DNA binding is inhibited by the redox sensitive Cysteine residue [34,35].
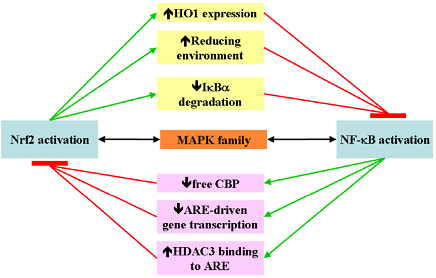
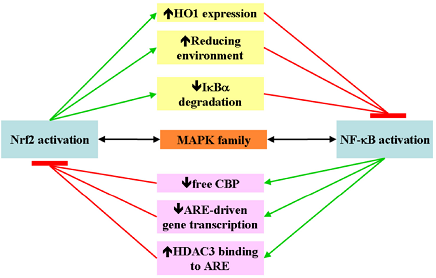
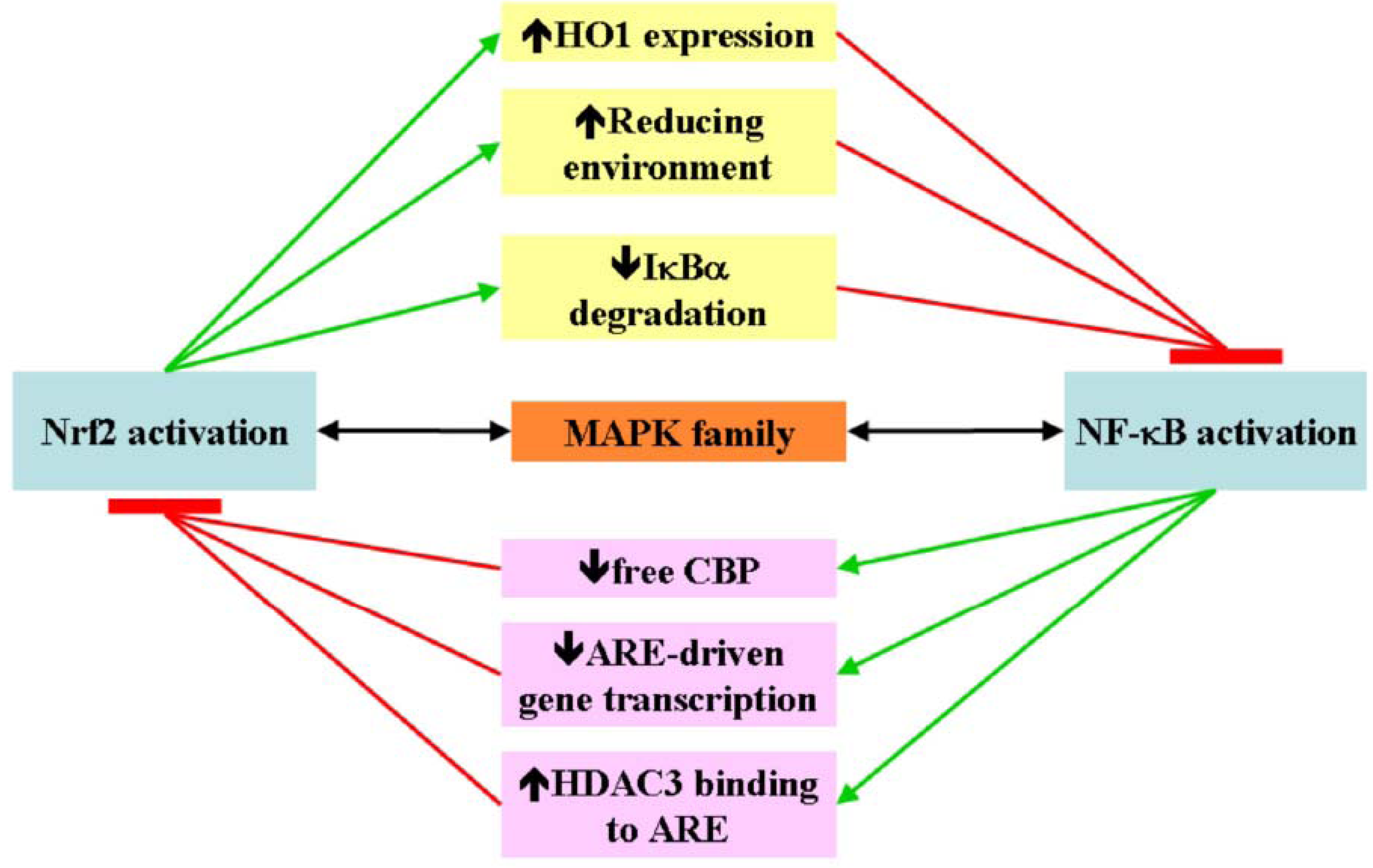
4. Nrf2 and NF-κB Crosstalk

5. Nrf2 and NF-κB in Cancer Pathogenesis
6. Nrf2 and NF-κB in Cancer Progression
7. Conclusions
Acknowledgements
References
- Yu, X.; Kensler, T. Nrf2 as a target for cancer chemoprevention. Mutat. Res. 2005, 591, 93–102. [Google Scholar] [CrossRef]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef]
- Marnett, L.J. Oxyradicals and DNA damage. Carcinogenesis 2000, 21, 361–370. [Google Scholar] [CrossRef]
- Evans, M.D.M.; Dizdaroglu, M.; Cooke, M.S. Oxidative DNA damage and disease: induction, repair and significance. Mutat. Res. 2004, 567, 1–61. [Google Scholar] [CrossRef]
- Nyaga, S.G.; Jaruga, P.; Lohani, A.; Dizdaroglu, M.; Evans, M.K. Accumulation of oxidatively induced DNA damage in human breast cancer cell lines following treatment with hydrogen peroxide. Cell Cycle 2007, 6, 1472–1478. [Google Scholar]
- Lesnefsky, E.J.; Hoppel, C.L. Oxidative phosphorylation and aging. Ageing Res. Rev. 2006, 5, 402–433. [Google Scholar] [CrossRef]
- Pamplona, R.; Barja, G. Mitochondrial oxidative stress, aging and caloric restriction: the protein and methionine connection. Biochim. Biophys. Acta 2006, 1757, 496–508. [Google Scholar] [CrossRef]
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930. [Google Scholar] [CrossRef]
- Chan, K.; Lu, R.; Chang, J.C.; Kan, Y.W. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc. Natl. Acad. Sci. USA 1996, 93, 13943–13948. [Google Scholar] [CrossRef]
- Venugopal, R.; Jaiswal, A.K. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1 gene. Proc. Natl. Acad. Sci. USA 1996, 93, 14960–14965. [Google Scholar] [CrossRef]
- Hayes, J.D.; McMahon, M. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem. Sci. 2009, 34, 176–188. [Google Scholar] [CrossRef]
- McMahon, M.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J. Biol. Chem. 2003, 278, 21592–21600. [Google Scholar]
- Motohashi, H.; Yamamoto, M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 2004, 10, 549–557. [Google Scholar] [CrossRef]
- Lau, A.; Villeneuve, N.F.; Sun, Z.; Wong, P.K.; Zhang, D.D. Dual roles of Nrf2 in cancer. Pharmacol. Res. 2008, 58, 262–270. [Google Scholar] [CrossRef]
- Yamamoto, T.; Suzuki, T.; Kobayashi, A.; Wakabayashi, J.; Maher, J.; Motohashi, H.; Yamamoto, M. Physiological significance of reactive cysteine residues of Keap1 in determining Nrf2 activity. Mol. Cell. Biol. 2008, 28, 2758–2770. [Google Scholar] [CrossRef]
- Nguyen, T.; Yang, C.S.; Pickett, C.B. The pathways and molecular mechanisms regulating Nrf2 activation in response to chemical stress. Free Radic. Biol. Med. 2004, 37, 433–441. [Google Scholar] [CrossRef]
- Levonen, A.L.; Landar, A.; Ramachandran, A.M.; Ceaser, E.K.; Dickinson, D.A.; Zanoni, G.; Morrow, J.D.; Darley-Usmar, V.M. Cellular mechanisms of redox cell signalling: role of cysteine modification in controlling antioxidant defences in response to electrophilic lipid oxidation products. Biochem. J. 2004, 378, 373–382. [Google Scholar] [CrossRef]
- Zhu, M.; Fahl, W.E. Functional characterization of transcription regulators that interact with the electrophile response element. Biochem. Biophys. Res. Commun. 2001, 289, 212–219. [Google Scholar] [CrossRef]
- Sun, Z.; Chin, Y.E.; Zhang, D.D. Acetylation of Nrf2 by p300/CBP augments promoter-specific DNA binding of Nrf2 during the antioxidant response. Mol. Cell. Biol. 2009, 29, 2658–2672. [Google Scholar] [CrossRef]
- Liu, G.H.; Qu, J.; Shen, X. NF-kappaB/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim. Biophys. Acta 2008, 1783, 713–727. [Google Scholar] [CrossRef]
- Ki, S.H.; Cho, I.J.; Choi, D.W.; Kim, S.G. Glucocorticoid receptor (GR)-associated SMRT binding to C/EBPbeta TAD and Nrf2 Neh4/5: role of SMRT recruited to GR in GSTA2 gene repression. Mol. Cell. Biol. 2005, 25, 4150–4165. [Google Scholar] [CrossRef]
- Purdom-Dickinson, S.E.; Sheveleva, E.V.; Sun, H.; Chen, Q.M. Translational control of nrf2 protein in activation of antioxidant response by oxidants. Mol. Pharmacol. 2007, 72, 1074–1081. [Google Scholar] [CrossRef]
- Sha, W.C. Regulation of immune responses by NF-kappa B/Rel transcription factors. J. Exp. Med. 1998, 187, 143–146. [Google Scholar] [CrossRef]
- Dejardin, E. The alternative NF-kappaB pathway from biochemistry to biology: pitfalls and promises for future drug development. Biochem. Pharmacol. 2006, 72, 1161–1179. [Google Scholar] [CrossRef]
- Pahl, H.L.; Baeuerle, P.A. A novel signal transduction pathway from the endoplasmic reticulum to the nucleus is mediated by transcription factor NF-kappa B. EMBO J. 1995, 14, 2580–2588. [Google Scholar]
- Whiteside, S.T.; Israël, A. I kappa B proteins: structure, function and regulation. Semin. Cancer Biol. 1997, 8, 75–82. [Google Scholar] [CrossRef]
- Kato, T., Jr.; Delhase, M.; Hoffmann, A.; Karin, M. CK2 Is a C-Terminal IkappaB Kinase Responsible for NF-kappaB Activation during the UV Response. Mol. Cell. 2003, 12, 829–839. [Google Scholar] [CrossRef]
- Tergaonkar, V.; Bottero, V.; Ikawa, M.; Li, Q.; Verma, I.M. IkappaB kinase-independent IkappaBalpha degradation pathway: functional NF-kappaB activity and implications for cancer therapy. Mol. Cell. Biol. 2003, 23, 8070–8083. [Google Scholar] [CrossRef]
- Takada, Y.; Mukhopadhyay, A.; Kundu, G.C.; Mahabeleshwar, G.H.; Singh, S.; Aggarwal, B.B. Hydrogen peroxide activates NF-kappa B through tyrosine phosphorylation of I kappa B alpha and serine phosphorylation of p65: evidence for the involvement of I kappa B alpha kinase and Syk protein-tyrosine kinase. J. Biol. Chem. 2003, 278, 24233–24241. [Google Scholar]
- Gloire, G.; Legrand-Poels, S.; Piette, J. NF-kappaB activation by reactive oxygen species: fifteen years later. Biochem. Pharmacol. 2006, 72, 1493–1505. [Google Scholar] [CrossRef]
- Schmidt, K.N.; Amstad, P.; Cerutti, P.; Baeuerle, P.A. The roles of hydrogen peroxide and superoxide as messengers in the activation of transcription factor NF-kappa B. Chem. Biol. 1995, 2, 13–22. [Google Scholar] [CrossRef]
- Matthews, J.R.; Wakasugi, N.; Virelizier, J.L.; Yodoi, J.; Hay, R.T. Thioredoxin regulates the DNA binding activity of NF-kappa B by reduction of a disulphide bond involving cysteine 62. Nucleic Acids Res. 1992, 20, 3821–3830. [Google Scholar] [CrossRef]
- Matthews, J.R.; Kaszubska, W.; Turcatti, G.; Wells, T.N.; Hay, R.T. Role of cysteine62 in DNA recognition by the P50 subunit of NF-kappa B. Nucleic Acids Res. 1993, 21, 1727–1734. [Google Scholar] [CrossRef]
- Droge, W.; Schulze-Osthoff, K.; Mihm, S.; Galter, D.; Schenk, H.; Eckm, H.P.; Roth, S.; Gmunder, H. Functions of glutathione and glutathione disulfide in immunology and immunopathology. FASEB J. 1994, 8, 1131–1138. [Google Scholar]
- Bowie, A.; O'Neill, L.A. Oxidative stress and nuclear factor-kappaB activation: a reassessment of the evidence in the light of recent discoveries. Biochem. Pharmacol. 2000, 59, 13–23. [Google Scholar] [CrossRef]
- Viatour, P.; Merville, M.P.; Bours, V.; Chariot, A. Phosphorylation of NF-kappaB and IkappaB proteins: implications in cancer and inflammation. Trends Biochem. Sci. 2005, 30, 43–52. [Google Scholar] [CrossRef]
- Gao, Z.; Chiao, P.; Zhang, X.; Zhang, X.; Lazar, M.A.; Seto, E.; Young, H.A.; Ye, J. Coactivators and corepressors of NF-kappaB in IkappaB alpha gene promoter. J. Biol. Chem. 2005, 280, 21091–21098. [Google Scholar]
- Li, W.; Khor, T.O.; Xu, C.; Shen, G.; Jeong, W.S.; Yu, S.; Kong, A.N. Activation of Nrf2-antioxidant signaling attenuates NFkappaB-inflammatory response and elicits apoptosis. Biochem. Pharmacol. 2008, 76, 1485–1489. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J. Free Radicals in Biology and Medicine; Oxford University Press: Oxford, UK, 1999. [Google Scholar]
- Jin, W.; Wang, H.; Yan, W.; Xu, L.; Wang, X.; Zhao, X.; Yang, X.; Chen, G.; Ji, Y. Disruption of Nrf2 enhances upregulation of nuclear factor-kappaB activity, proinflammatory cytokines, and intercellular adhesion molecule-1 in the brain after traumatic brain injury. Mediators Inflamm. 2008, 2008, 725174. [Google Scholar]
- Song, M.Y.; Kim, E.K.; Moon, W.S.; Park, J.W.; Kim, H.J.; So, H.S.; Park, R.; Kwon, K.B.; Park, B.H. Sulforaphane protects against cytokine- and streptozotocin-induced beta-cell damage by suppressing the NF-kappaB pathway. Toxicol. Appl. Pharmacol. 2009, 235, 57–67. [Google Scholar] [CrossRef]
- Kang, E.S.; Kim, G.H.; Kim, H.J.; Woo, I.S.; Ham, S.A.; Jin, H.; Kim, M.Y.; Kim, H.J.; Lee, J.H.; Chang, K.C.; Seo, H.G.; Hwang, J.Y. Nrf2 regulates curcumin-induced aldose reductase expression indirectly via nuclear factor-kappaB. Pharmacol. Res. 2008, 58, 15–21. [Google Scholar] [CrossRef]
- Liao, B.C.; Hsieh, C.W.; Liu, Y.C.; Tzeng, T.T.; Sun, Y.W.; Wung, B.S. Cinnamaldehyde inhibits the tumor necrosis factor-alpha-induced expression of cell adhesion molecules in endothelial cells by suppressing NF-kappaB activation: effects upon IkappaB and Nrf2. Toxicol. Appl. Pharmacol. 2008, 229, 161–171. [Google Scholar] [CrossRef]
- Banning, A.; Brigelius-Flohé, R. NF-kappaB, Nrf2, and HO-1 interplay in redox-regulated VCAM-1 expression. Antioxid. Redox Signal. 2005, 7, 889–899. [Google Scholar] [CrossRef]
- Rushworth, S.A.; MacEwan, D.J. HO-1 underlies resistance of AML cells to TNF-induced apoptosis. Blood 2008, 111, 3793–3801. [Google Scholar] [CrossRef]
- Nair, S.; Doh, S.T.; Chan, J.Y.; Kong, A.N.; Cai, L. Regulatory potential for concerted modulation of Nrf2- and Nfkb1-mediated gene expression in inflammation and carcinogenesis. Br. J. Cancer. 2008, 99, 2070–2082. [Google Scholar] [CrossRef]
- Giudice, A.; Montella, M. Activation of the Nrf2-ARE signaling pathway: a promising strategy in cancer prevention. Bioessays 2006, 28, 169–181. [Google Scholar] [CrossRef]
- Surh, Y.J. Cancer chemoprevention with dietary phytochemicals. Nat. Rev. Cancer 2003, 3, 768–780. [Google Scholar] [CrossRef]
- Kundu, J.K.; Surh, Y.J. Molecular basis of chemoprevention by resveratrol: NF-κB and AP-1 as potential targets. Mutat. Res. 2004, 555, 65–80. [Google Scholar] [CrossRef]
- Greten, F.R.; Karin, M. The IKK/NF-kappaB activation pathway-a target for prevention and treatment of cancer. Cancer Lett. 2004, 206, 193–199. [Google Scholar] [CrossRef]
- Pahl, H.L. Activators and target genes of Rel/NF-kB transcription factors. Oncogene 1999, 18, 6853–6866. [Google Scholar] [CrossRef]
- Ryan, K.M.; Ernst, M.K.; Rice, N.R.; Vousden, K.H. Role of NF-kappaB in p53-mediated programmed cell death. Nature 2000, 404, 892–897. [Google Scholar] [CrossRef]
- Rocha, S.; Martin, A.M.; Meek, D.W.; Perkins, N.D. p53 represses cyclin D1 transcription through down regulation of Bcl-3 and inducing increased association of the p52 NF-kappaB subunit with histone deacetylase 1. Mol. Cell. Biol. 2003, 23, 4713–4727. [Google Scholar] [CrossRef]
- Rocha, S.; Campbell, K.J.; Perkins, N.D. p53- and Mdm2-independent repression of NF-kappa B transactivation by the ARF tumor suppressor. Mol. Cell 2003, 12, 15–25. [Google Scholar] [CrossRef]
- Perkins, N.D. NF-kappaB: tumor promoter or suppressor? Trends Cell. Biol. 2004, 14, 64–69. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Barkett, M.; Gilmore, T.D. Control of apoptosis by Rel/NF-kappaB transcription factors. Oncogene 1999, 18, 6910–6924. [Google Scholar] [CrossRef]
- Baldwin, A.S. Control of oncogenesis and cancer therapy resistance by the transcription factor NF-kappaB. J. Clin. Invest. 2001, 107, 241–246. [Google Scholar] [CrossRef]
- Garg, A.; Aggarwal, B.B. Nuclear transcription factor-kappaB as a target for cancer drug development. Leukemia 2002, 16, 1053–1068. [Google Scholar] [CrossRef]
- Karin, M.; Cao, Y.; Greten, F.R.; Li, Z.W. NF-kappaB in cancer: from innocent bystander to major culprit. Nat. Rev. Cancer. 2002, 2, 301–310. [Google Scholar] [CrossRef]
- Chen, F.; Castranova, V.; Shi, X. New insights into the role of nuclear factor-kappaB in cell growth regulation. Am. J. Pathol. 2001, 159, 387–397. [Google Scholar] [CrossRef]
- Ohta, T.; Iijima, K.; Miyamoto, M.; Nakahara, I.; Tanaka, H.; Ohtsuji, M. Loss of Keap1 function activates Nrf2 and provides advantages for lung cancer cell growth. Cancer Res. 2008, 68, 1303–1309. [Google Scholar] [CrossRef]
- Nioi, P.; Nguyen, T. A mutation of Keap1 found in breast cancer impairs its ability to repress Nrf2 activity. Biochem. Biophys. Res. Commun. 2007, 362, 816–821. [Google Scholar] [CrossRef]
- Kim, Y.J.; Ahn, J.Y.; Liang, P.; Ip, C.; Zhang, Y.; Park, Y.M. Human prx1 gene is a target of Nrf2 and is up-regulated by hypoxia/reoxygenation: implication to tumor biology. Cancer Res. 2007, 67, 546–554. [Google Scholar] [CrossRef]
- Wang, R.; An, J.; Ji, F.; Jiao, H.; Sun, H.; Zhou, D. Hypermethylation of the Keap1 gene in human lung cancer cell lines and lung cancer tissues. Biochem. Biophys. Res. Commun. 2008, 373, 151–154. [Google Scholar] [CrossRef]
- Wang, X.J.; Sun, Z.; Villeneuve, N.F.; Zhang, S.; Zhao, F; Li, Y. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis 2008, 29, 1235–1243. [Google Scholar] [CrossRef]
- Pi, J.; Diwan, B.A.; Sun, Y.; Liu, J.; Qu, W.; He, Y.; Styblo, M.; Waalkes, M.P. Arsenic-induced malignant transformation of human keratinocytes: involvement of Nrf2. Free Radic. Biol. Med. 2008, 45, 651–658. [Google Scholar] [CrossRef]
- Minelli, A.; Bellezza, I.; Conte, C.; Culig, Z. Oxidative stress-related aging: A role for prostate cancer? Biochim. Biophys. Acta 2009, 1795, 83–91. [Google Scholar]
- Suzuki, Y.; Kondo, Y.; Himeno, S.; Nemoto, K.; Akimoto, M.; Imura, N. Role of antioxidant systems in human androgen-independent prostate cancer cells. Prostate 2000, 43, 144–149. [Google Scholar] [CrossRef]
- Lim, H.W.; Hong, S.; Jin, W.; Lim, S.; Kim, S.J.; Kang, H.J.; Park, E.H.; Ahn, K.; Lim, C.J. Up-regulation of defense enzymes is responsible for low reactive oxygen species in malignant prostate cancer cells. Exp. Mol. Med. 2005, 37, 497–506. [Google Scholar] [CrossRef]
- Ouyang, X.; DeWeese, T.L.; Nelson, W.G.; Abate-Shen, C. Loss-of-function of Nkx3.1 promotes increased oxidative damage in prostate carcinogenesis. Cancer Res. 2005, 65, 6773–6779. [Google Scholar] [CrossRef]
- Tam, N.N.; Nyska, A.; Maronpot, R.R.; Kissling, G.; Lomnitski, L.; Suttie, A.; Bakshi, S.; Bergman, M.; Grossman, S.; Ho, S.M. Differential attenuation of oxidative/nitrosative injuries in early prostatic neoplastic lesions in TRAMP mice by dietary antioxidants. Prostate 2006, 66, 57–69. [Google Scholar] [CrossRef]
- Tam, N.N.; Leav, I.; Ho, S.M. Sex hormones induce direct epithelial and inflammation-mediated oxidative/nitrosative stress that favors prostatic carcinogenesis in the noble rat. Am. J. Pathol. 2007, 171, 1334–1341. [Google Scholar] [CrossRef]
- Bostwick, D.G.; Meiers, I.; Shanks, J.H. Glutathione S-transferase: differential expression of alpha, mu, and pi isoenzymes in benign prostate, prostatic intraepithelial neoplasia, and prostatic adenocarcinoma. Hum. Pathol. 2007, 38, 1394–1401. [Google Scholar] [CrossRef]
- Frohlich, D.A.; McCabe, M.T.; Arnold, R.S.; Day, M.L. The role of Nrf2 in increased reactive oxygen species and DNA damage in prostate tumorigenesis. Oncogene 2008, 27, 4353–4362. [Google Scholar] [CrossRef]
- Kumar, B.; Koul, S.; Khandrika, L.; Meacham, R.B.; Koul, H.K. Oxidative stress is inherent in prostate cancer cells and is required for aggressive phenotype. Cancer Res. 2008, 68, 1777–1785. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bellezza, I.; Mierla, A.L.; Minelli, A. Nrf2 and NF-κB and Their Concerted Modulation in Cancer Pathogenesis and Progression. Cancers 2010, 2, 483-497. https://doi.org/10.3390/cancers2020483
Bellezza I, Mierla AL, Minelli A. Nrf2 and NF-κB and Their Concerted Modulation in Cancer Pathogenesis and Progression. Cancers. 2010; 2(2):483-497. https://doi.org/10.3390/cancers2020483
Chicago/Turabian StyleBellezza, Ilaria, Anna Lisa Mierla, and Alba Minelli. 2010. "Nrf2 and NF-κB and Their Concerted Modulation in Cancer Pathogenesis and Progression" Cancers 2, no. 2: 483-497. https://doi.org/10.3390/cancers2020483