5-FU Metabolism in Cancer and Orally-Administrable 5-FU Drugs

Abstract
:1. Introduction
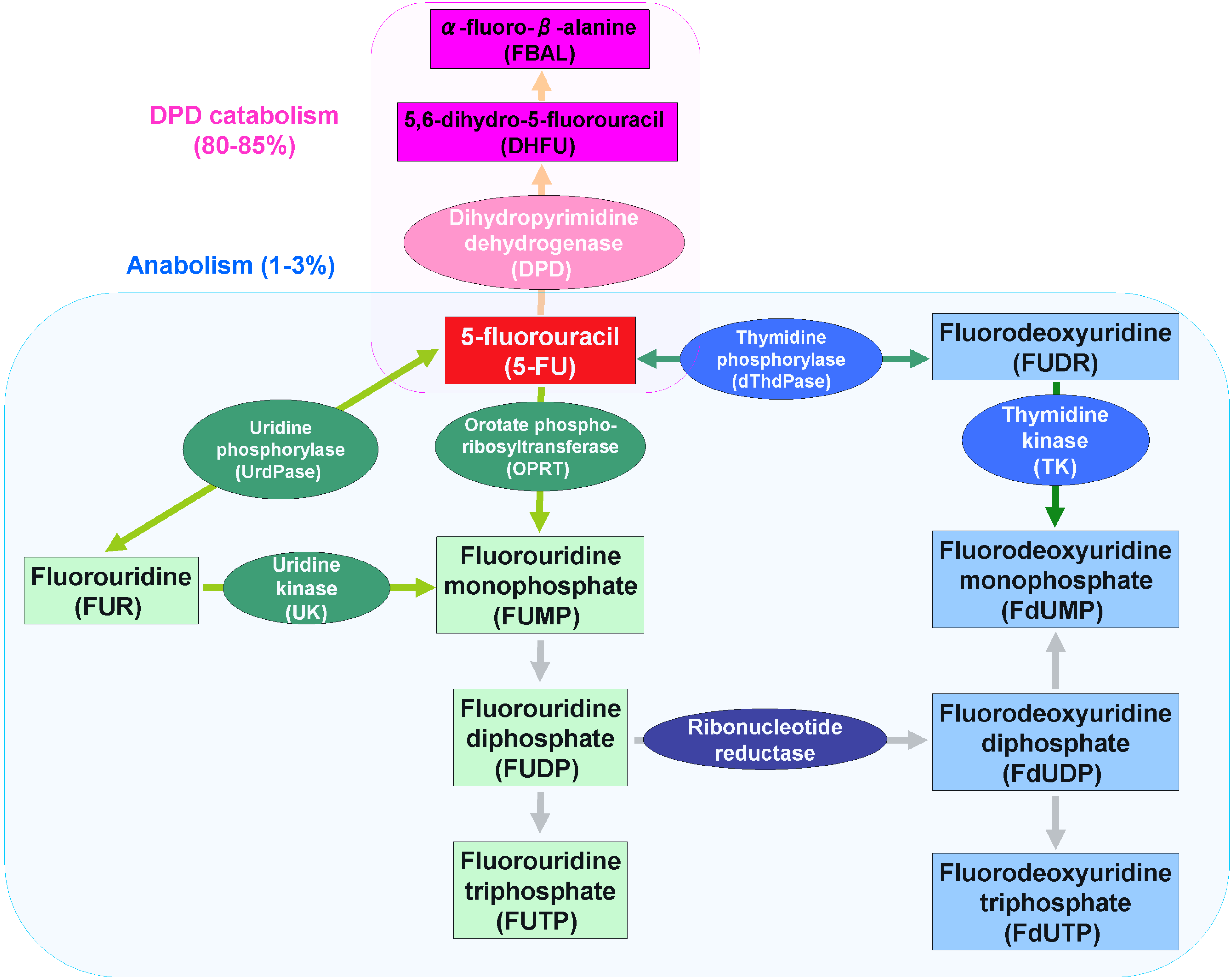
2. 5-FU Metabolism
2.1. 5-FU Anabolism

2.2. 5-FU Catabolism
2.3. Ternary Complex
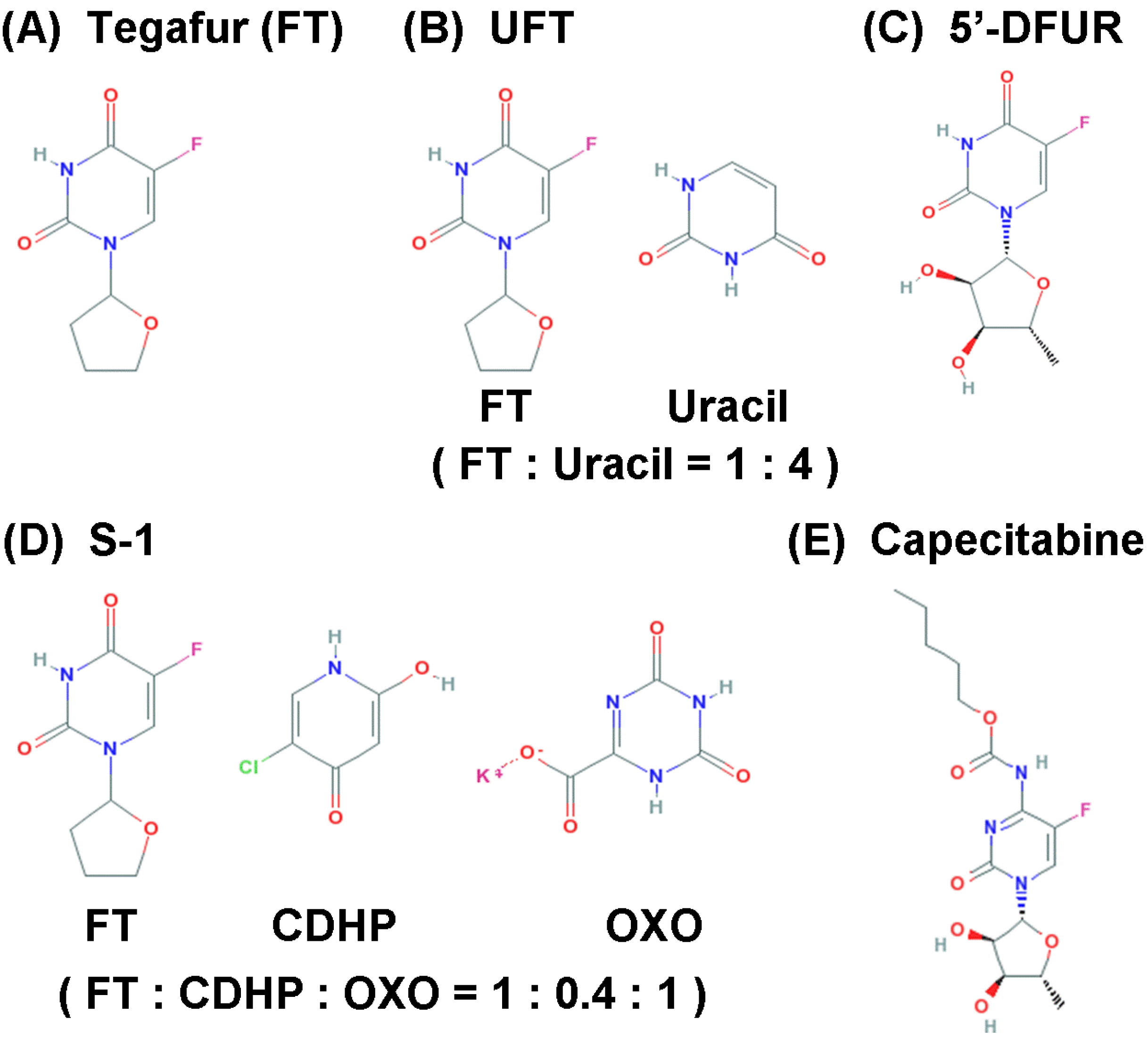
3. Oral 5-FU Drugs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug name | Structure (Composition) | Concept | Developer | Refs. |
|---|---|---|---|---|
| Tegafur | 1-(2-Tetrahydrofuryl)-5-fluorouracil | Prodrug | National Institute for Organic Syntheses (Latvia) | [28] |
| UFT | FT:Uracil = 1:4 | Prodrug, DPD inhibitor | Osaka University (Japan) | [30] |
| 5’-DFUR | 5’-Deoxy-5-fluorouridine | Prodrug | Hoffmann-La Roche (Switzerland); Nippon Roche Research Center (Japan) | [38,39] |
| S-1 | FT:CDHP:OXO = 1:0.4:1 | DPD inhibitor, OPRT inhibitor | Taiho Pharmaceuticals (Japan) | [40] |
| Capecitabine | N4-Pentyloxycarbonyl-5′-deoxy-5-fluorocytidine | Prodrug | Nippon Roche Research Center (Japan) | [44] |

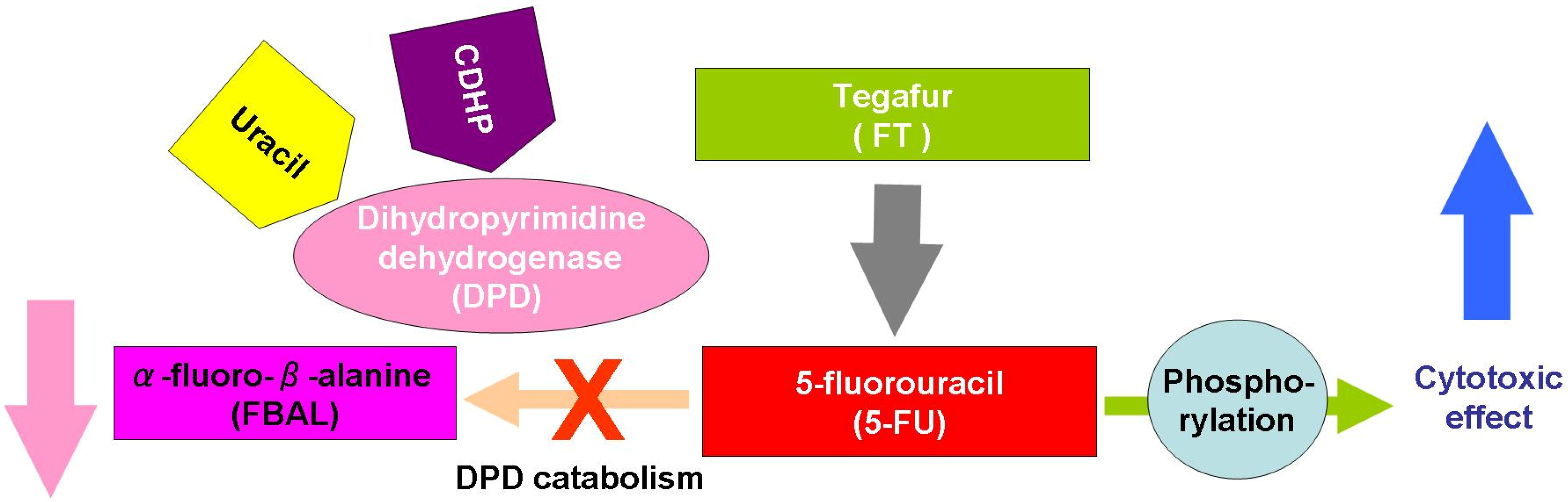
3.1. Tegafur
3.2. UFT
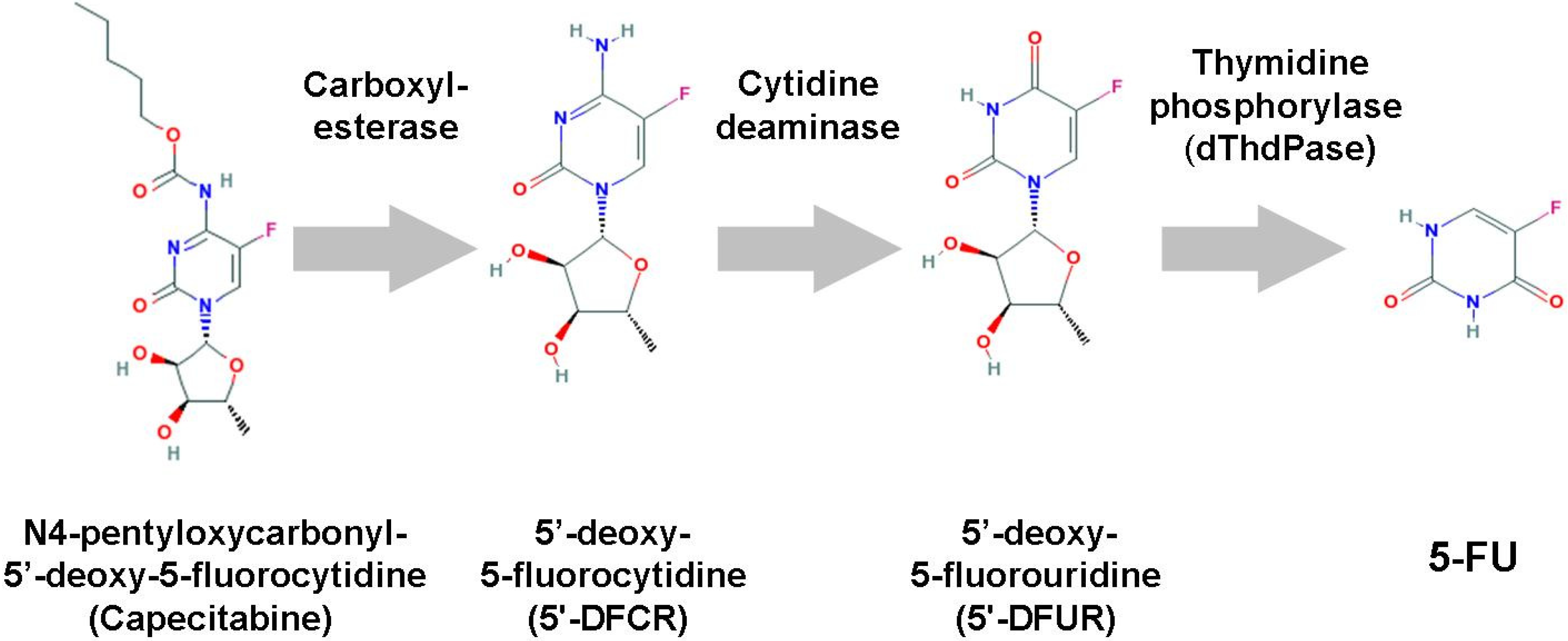
3.3. 5’-DFUR
3.4. S-1

3.5. Capecitabine

4. Conclusions
Acknowledgements
References
- Heidelberger, C.; Chaudhuri, N.K.; Danneberg, P.; Mooren, D.; Griesbach, L.; Duschinsky, R.; Schnitzer, R.J.; Pleven, E.; Scheiner, J. Fluorinated pyrimidines, a new class of tumour-inhibitory compounds. Nature 1957, 179, 663–666. [Google Scholar] [CrossRef]
- Rutman, R.J.; Cantarow, A.; Paschkis, K.E. The catabolism of uracil in vivo and in vitro. J. Biol. Chem. 1954, 210, 321–329. [Google Scholar]
- Handschumacher, R.E.; Welch, A.D. Microbial studies of 6-azauracil, an antagonist of uracil. Cancer Res. 1956, 16, 965–969. [Google Scholar]
- Skipper, H.E.; Schabel, F.M., Jr.; Wilcox, W.S. Experimental evaluation of potential anticancer agents. XIII. On the criteria and kinetics associated with "curability" of experimental leukemia. Cancer Chemother. Rep. 1964, 35, 1–111. [Google Scholar]
- Meta-analysis Group In Cancer. Efficacy of intravenous continuous infusion of fluorouracil compared with bolus administration in advanced colorectal cancer. J. Clin. Oncol. 1998, 16, 301–308.
- Saif, M.W.; Syrigos, K.N.; Katirtzoglou, N.A. S-1: A promising new oral fluoropyrimidine derivative. Expert Opin. Investig. Drugs 2009, 18, 335–348. [Google Scholar] [CrossRef]
- Wohlhueter, R.M.; McIvor, R.S.; Plagemann, P.G. Facilitated transport of uracil and 5-fluorouracil, and permeation of orotic acid into cultured mammalian cells. J. Cell. Physiol. 1980, 104, 309–319. [Google Scholar] [CrossRef]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-fluorouracil: mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef]
- Houghton, J.A.; Houghton, P.J.; Wooten, R.S. Mechanism of induction of gastrointestinal toxicity in the mouse by 5-fluorouracil, 5-fluorouridine, and 5-fluoro-2'-deoxyuridine. Cancer Res. 1979, 39, 2406–2413. [Google Scholar]
- Schuetz, J.D.; Wallace, H.J.; Diasio, R.B. 5-fluorouracil incorporation into DNA of CF-1 mouse bone marrow cells as a possible mechanism of toxicity. Cancer Res. 1984, 44, 1358–1363. [Google Scholar]
- Meta-Analysis Group In Cancer. Toxicity of fluorouracil in patients with advanced colorectal cancer: effect of administration schedule and prognostic factors. J. Clin. Oncol. 1998, 16, 3537–3541.
- Diasio, R.B.; Harris, B.E. Clinical pharmacology of 5-fluorouracil. Clin. Pharmacokinet. 1989, 16, 215–237. [Google Scholar] [CrossRef]
- Heggie, G.D.; Sommadossi, J.P.; Cross, D.S.; Huster, W.J.; Diasio, R.B. Clinical pharmaco-kinetics of 5-fluorouracil and its metabolites in plasma, urine, and bile. Cancer Res. 1987, 47, 2203–2206. [Google Scholar]
- Koenig, H.; Patel, A. Biochemical basis for fluorouracil neurotoxicity. The role of Krebs cycle inhibition by fluoroacetate. Arch. Neurol. 1970, 23, 155–160. [Google Scholar] [CrossRef]
- Okeda, R.; Shibutani, M.; Matsuo, T.; Kuroiwa, T.; Shimokawa, R.; Tajima, T. Experimental neurotoxicity of 5-fluorouracil and its derivatives is due to poisoning by the monofluorinated organic metabolites, monofluoroacetic acid and alpha-fluoro-beta-alanine. Acta Neuropathol. 1990, 81, 66–73. [Google Scholar] [CrossRef]
- Matsubara, I.; Kamiya, J.; Imai, S. Cardiotoxic effects of 5-fluorouracil in the guinea pig. Jpn. J. Pharmacol. 1980, 30, 871–879. [Google Scholar] [CrossRef]
- Santi, D.V.; McHenry, C.S. 5-Fluoro-2'-deoxyuridylate: covalent complex with thymidylate synthetase. Proc. Natl. Acad. Sci. USA 1972, 69, 1855–1857. [Google Scholar] [CrossRef]
- Jackson, R.C.; Grindley, G.B. The biochemical basis for methotrexate cytotoxicity. In Folate Antagonists as Therapeutic Agents, 2nd edition; Sirotnak, F.M., Burchell, J.J., Ensminger, W.D., Eds.; Academic Press: New York, NY, USA, 1984; Volume 1, pp. 289–315. [Google Scholar]
- Yoshioka, A.; Tanaka, S.; Hiraoka, O.; Koyama, Y.; Hirota, Y.; Ayusawa, D.; Seno, T.; Garrett, C.; Wataya, Y. Deoxyribonucleoside triphosphate imbalance. 5-Fluorodeoxyuridine-induced DNA double strand breaks in mouse FM3A cells and the mechanism of cell death. J. Biol. Chem. 1987, 262, 8235–8241. [Google Scholar]
- Mitrovski, B.; Pressacco, J.; Mandelbaum, S.; Erlichman, C. Biochemical effects of folate-based inhibitors of thymidylate synthase in MGH-U1 cells. Cancer Chemother. Pharmacol. 1994, 35, 109–114. [Google Scholar] [CrossRef]
- Grem, J.L.; Fischer, P.H. Enhancement of 5-fluorouracil's anticancer activity by dipyridamole. Pharmacol. Ther. 1989, 40, 349–371. [Google Scholar] [CrossRef]
- Showalter, S.L.; Showalter, T.N.; Witkiewicz, A.; Havens, R.; Kennedy, E.P.; Hucl, T.; Kern, S.E.; Yeo, C.J.; Brody, J.R. Evaluating the drug-target relationship between thymidylate synthase expression and tumor response to 5-fluorouracil. Is it time to move forward? Cancer Biol. Ther. 2008, 7, 986–994. [Google Scholar] [CrossRef]
- Lembersky, B.C.; Wieand, H.S.; Petrelli, N.J.; O'Connell, M.J.; Colangelo, L.H.; Smith, R.E.; Seay, T.E.; Giguere, J.K.; Marshall, M.E.; Jacobs, A.D.; et al. Oral uracil and tegafur plus leucovorin compared with intravenous fluorouracil and leucovorin in stage II and III carcinoma of the colon: results from National Surgical Adjuvant Breast and Bowel Project Protocol C-06. J. Clin. Oncol. 2006, 24, 2059–2064. [Google Scholar] [CrossRef]
- Boku, N.; Yamamoto, S.; Fukuda, H.; Shirao, K.; Doi, T.; Sawaki, A.; Koizumi, W.; Saito, H.; Yamaguchi, K.; Takiuchi, H.; et al. Fluorouracil versus combination of irinotecan plus cisplatin versus S-1 in metastatic gastric cancer: A randomised phase 3 study. Lancet Oncol. 2009, 10, 1063–1069. [Google Scholar] [CrossRef]
- Mansfield, P.F.; Hohn, D.C.; Fornage, B.D.; Gregurich, M.A.; Ota, D.M. Complications and failures of subclavian-vein catheterization. N. Engl. J. Med. 1994, 331, 1735–1738. [Google Scholar] [CrossRef]
- Agnelli, G.; Verso, M. Therapy Insight: venous-catheter-related thrombosis in cancer patients. Nat. Clin. Pract. Oncol. 2006, 3, 214–222. [Google Scholar] [CrossRef]
- Lokich, J.J.; Moore, C.L.; Anderson, N.R. Comparison of costs for infusion versus bolus chemotherapy administration–Part two. Use of charges versus reimbursement for cost basis. Cancer 1996, 78, 300–303. [Google Scholar] [CrossRef]
- Giller, S.A.; Zhuk, R.A.; Lidak, M.Iu. Analogs of pyrimidine nucleosides. I. N1-(alpha-furanidyl) derivatives of natural pyrimidine bases and their antimetabolities. Dokl. Akad. Nauk. SSSR. 1967, 176, 332–335, (article in Russian). [Google Scholar]
- Toide, H.; Akiyoshi, H.; Minato, Y.; Okuda, H.; Fujii, S. Comparative studies on the metabolism of 2-(tetrahydrofuryl)-5-fluorouracil and 5-fluorouracil. Gann 1977, 68, 553–560. [Google Scholar]
- Fujii, S.; Ikenaka, K.; Fukushima, M.; Shirasaka, T. Effect of uracil and its derivatives on antitumor activity of 5-fluorouracil and 1-(2-tetrahydrofuryl)-5-fluorouracil. Gann 1978, 69, 763–772. [Google Scholar]
- El Sayed, Y.M.; Sadée, W. Metabolic activation of R,S-1-(tetrahydro-2-furanyl)-5-fluorouracil (ftorafur) to 5-fluorouracil by soluble enzymes. Cancer Res. 1983, 43, 4039–4044. [Google Scholar]
- Rustum, Y.M. Mechanism-based improvement in the therapeutic selectivity of 5-FU prodrug alone and under conditions of metabolic modulation. Oncology 1997, 54 (Suppl. 1), 7–11. [Google Scholar] [CrossRef]
- Diasio, R.B. The role of dihydropyrimidine dehydrogenase (DPD) modulation in 5-FU pharmacology. Oncology 1998, 12, 23–27. [Google Scholar]
- Fujii, S.; Kitano, S.; Ikenaka, K.; Shirasaka, T. Effect of coadministration of uracil or cytosine on the anti-tumor activity of clinical doses of 1-(2-tetrahydrofuryl)-5-fluorouracil and level of 5-fluorouracil in rodents. Gann 1979, 70, 209–214. [Google Scholar]
- Hoff, P.M.; Cassidy, J.; Schmoll, H.J. The evolution of fluoropyrimidine therapy: From intravenous to oral. Oncologist 2001, 6 (Suppl. 4), 3–11. [Google Scholar]
- Poon, M.A.; O'Connell, M.J.; Wieand, H.S.; Krook, J.E.; Gerstner, J.B.; Tschetter, L.K.; Levitt, R.; Kardinal, C.G.; Mailliard, J.A. Biochemical modulation of fluorouracil with leucovorin: confirmatory evidence of improved therapeutic efficacy in advanced colorectal cancer. J. Clin. Oncol. 1991, 9, 1967–1972. [Google Scholar]
- Ichikura, T.; Tomimatsu, S.; Okusa, Y.; Yahara, T.; Uefuji, K.; Tamakuma, S. Thymidylate synthase inhibition by an oral regimen consisting of tegafur-uracil (UFT) and low-dose leucovorin for patients with gastric cancer. Cancer Chemother. Pharmacol. 1996, 38, 401–405. [Google Scholar] [CrossRef]
- Cook, A.F.; Holman, M.J.; Kramer, M.J.; Trown, P.W. Fluorinated pyrimidine nucleosides. 3. Synthesis and antitumor activity of a series of 5'-deoxy-5-fluoropyrimidine nucleosides. J. Med. Chem. 1979, 22, 1330–1335. [Google Scholar] [CrossRef]
- Ishitsuka, H.; Miwa, M.; Takemoto, K.; Fukuoka, K.; Itoga, A.; Maruyama, HB. Role of uridine phosphorylase for antitumor activity of 5'-deoxy-5-fluorouridine. Gann 1980, 71, 112–123. [Google Scholar]
- Shirasaka, T.; Shimamato, Y.; Ohshimo, H.; Yamaguchi, M.; Kato, T.; Yonekura, K.; Fukushima, M. Development of a novel form of an oral 5-fluorouracil derivative (S-1) directed to the potentiation of the tumor selective cytotoxicity of 5-fluorouracil by two biochemical modulators. Anticancer Drugs 1996, 7, 548–557. [Google Scholar] [CrossRef]
- Tatsumi, K.; Fukushima, M.; Shirasaka, T.; Fujii, S. Inhibitory effects of pyrimidine, barbituric acid and pyridine derivatives on 5-fluorouracil degradation in rat liver extracts. Jpn. J. Cancer Res. 1987, 78, 748–755. [Google Scholar]
- Shirasaka, T.; Shimamoto, Y.; Fukushima, M. Inhibition by oxonic acid of gastrointestinal toxicity of 5-fluorouracil without loss of its antitumor activity in rats. Cancer Res. 1993, 53, 4004–4009. [Google Scholar]
- Muneoka, K.; Shirai, Y.; Yokoyama, N.; Wakai, T.; Hatakeyama, K. 5-Fluorouracil cardiotoxicity induced by alpha-fluoro-beta-alanine. Int. J. Clin. Oncol. 2005, 10, 441–443. [Google Scholar] [CrossRef]
- Miwa, M.; Ura, M.; Nishida, M.; Sawada, N.; Ishikawa, T.; Mori, K.; Shimma, N.; Umeda, I.; Ishitsuka, H. Design of a novel oral fluoropyrimidine carbamate, capecitabine, which generates 5-fluorouracil selectively in tumours by enzymes concentrated in human liver and cancer tissue. Eur. J. Cancer 1998, 34, 1274–1281. [Google Scholar] [CrossRef]
- Shimma, N.; Umeda, I.; Arasaki, M.; Murasaki, C.; Masubuchi, K.; Kohchi, Y.; Miwa, M.; Ura, M.; Sawada, N.; Tahara, H.; et al. The design and synthesis of a new tumor-selective fluoropyrimidine carbamate, capecitabine. Bioorg. Med. Chem. 2000, 8, 1697–1706. [Google Scholar] [CrossRef]
- Ishikawa, T.; Utoh, M.; Sawada, N.; Nishida, M.; Fukase, Y.; Sekiguchi, F.; Ishitsuka, H. Tumor selective delivery of 5-fluorouracil by capecitabine, a new oral fluoropyrimidine carbamate, in human cancer xenografts. Biochem. Pharmacol. 1998, 55, 1091–1097. [Google Scholar] [CrossRef]
- Yen-Revollo, J.L.; Goldberg, R.M.; McLeod, H.L. Can inhibiting dihydropyrimidine dehydrogenase limit hand-foot syndrome caused by fluoropyrimidines? Clin. Cancer Res. 2008, 14, 8–13. [Google Scholar] [CrossRef]
- Milano, G.; Etienne-Grimaldi, M.C.; Mari, M.; Lassalle, S.; Formento, J.L.; Francoual, M.; Lacour, J.P.; Hofman, P. Candidate mechanisms for capecitabine-related hand-foot syndrome. Br. J. Clin. Pharmacol. 2008, 66, 88–95. [Google Scholar] [CrossRef]
- Hattori, K.; Kohchi, Y.; Oikawa, N.; Suda, H.; Ura, M.; Ishikawa, T.; Miwa, M.; Endoh, M.; Eda, H.; Tanimura, H.; et al. Design and synthesis of the tumor-activated prodrug of dihydropyrimidine dehydrogenase (DPD) inhibitor, RO0094889 for combination therapy with capecitabine. Bioorg. Med. Chem. Lett. 2003, 13, 867–872. [Google Scholar] [CrossRef]
- Lee, J.L.; Kang, Y.K.; Kang, H.J.; Lee, K.H.; Zang, D.Y.; Ryoo, B.Y.; Kim, J.G.; Park, S.R.; Kang, W.K.; Shin, D.B.; et al. A randomised multicentre phase II trial of capecitabine vs S-1 as first-line treatment in elderly patients with metastatic or recurrent unresectable gastric cancer. Br. J. Cancer 2008, 99, 584–590. [Google Scholar] [CrossRef]
- Seol, Y.M.; Song, M.K.; Choi, Y.J.; Kim, G.H.; Shin, H.J.; Song, G.A.; Chung, J.S.; Cho, G.J. Oral fluoropyrimidines (capecitabine or S-1) and cisplatin as first line treatment in elderly patients with advanced gastric cancer: a retrospective study. Jpn. J. Clin. Oncol. 2009, 39, 43–48. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Miura, K.; Kinouchi, M.; Ishida, K.; Fujibuchi, W.; Naitoh, T.; Ogawa, H.; Ando, T.; Yazaki, N.; Watanabe, K.; Haneda, S.; et al. 5-FU Metabolism in Cancer and Orally-Administrable 5-FU Drugs. Cancers 2010, 2, 1717-1730. https://doi.org/10.3390/cancers2031717
Miura K, Kinouchi M, Ishida K, Fujibuchi W, Naitoh T, Ogawa H, Ando T, Yazaki N, Watanabe K, Haneda S, et al. 5-FU Metabolism in Cancer and Orally-Administrable 5-FU Drugs. Cancers. 2010; 2(3):1717-1730. https://doi.org/10.3390/cancers2031717
Chicago/Turabian StyleMiura, Koh, Makoto Kinouchi, Kazuyuki Ishida, Wataru Fujibuchi, Takeshi Naitoh, Hitoshi Ogawa, Toshinori Ando, Nobuki Yazaki, Kazuhiro Watanabe, Sho Haneda, and et al. 2010. "5-FU Metabolism in Cancer and Orally-Administrable 5-FU Drugs" Cancers 2, no. 3: 1717-1730. https://doi.org/10.3390/cancers2031717