Contribution of Epithelial-Mesenchymal Transition to Pancreatic Cancer Progression

Abstract
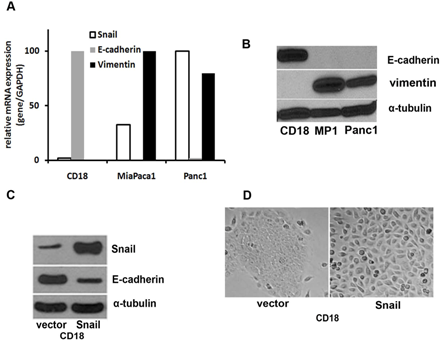
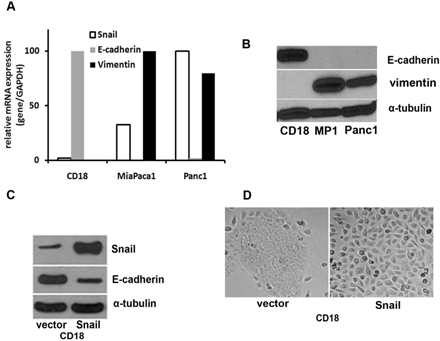
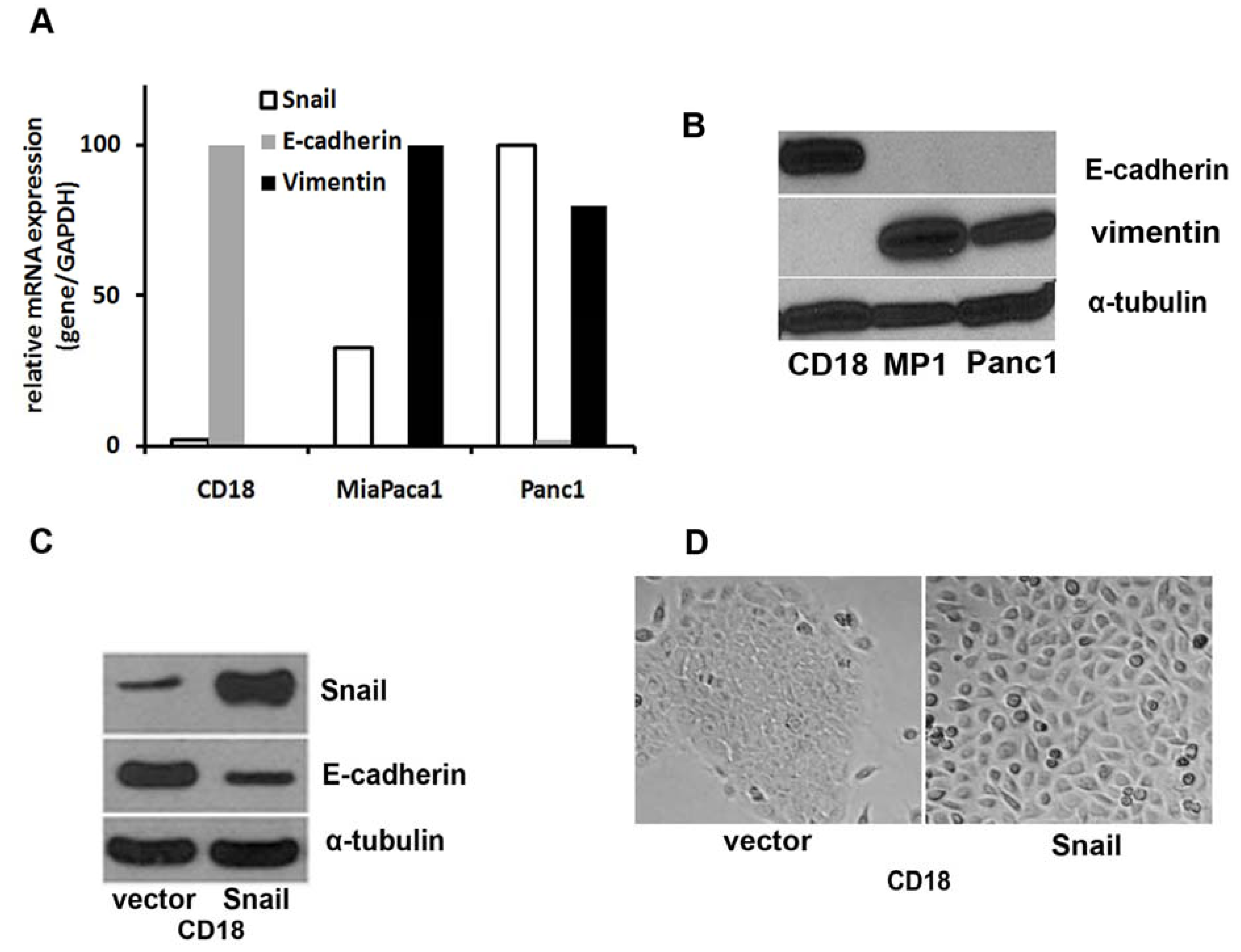
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
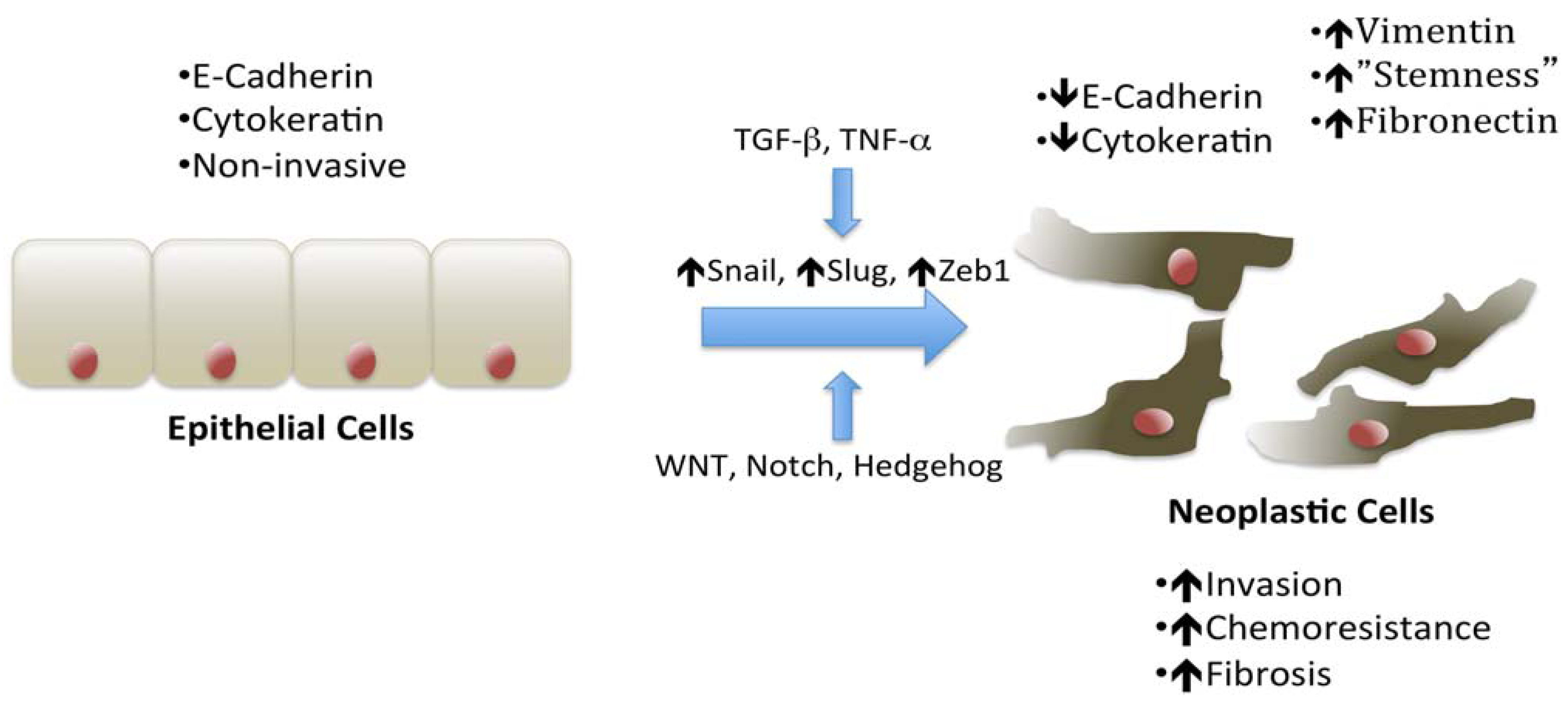
2. Epithelial to Mesenchymal Transition and Pancreatic Cancer


3. Role of microRNAs in Modulating EMT in Pancreatic Cancer
4. Contribution of EMT to Stem Cells in Pancreatic Cancer

5. Importance of EMT in Enhancing Drug Resistance in Pancreatic Cancer
6. Targeting EMT to Reduce the Morbidity and Mortality of Pancreatic Cancer
7. Conclusions
Acknowledgements
References
- Jemal, A.; Siegel, R.; Xu, J.; Ward, E. Cancer statistics, 2010. CA. Cancer. J. Clin. 2010, 60, 277–300. [Google Scholar] [CrossRef]
- Bilimoria, K.Y.; Bentrem, D.J.; Ko, C.Y.; Ritchey, J.; Stewart, A.K.; Winchester, D.P.; Talamonti, M.S. Validation of the 6th edition AJCC Pancreatic Cancer Staging System: Report from the National Cancer Database. Cancer 2007, 110, 738–744. [Google Scholar] [CrossRef]
- Altekruse, S.F.; Kosary, C.L.; Krapcho, M.; Neyman, N.; Aminou, R.; Waldron, W.; Ruhl, J.; Howlader, N.; Tatalovich, Z.; Cho, H.; Mariotto, A.; Eisner, M.P.; Lewis, D.R.; Cronin, K.; Chen, H.S.; Feuer, E.; Stinchcomb, D.G.; Edwards, B.K. SEER Cancer Statistics Review, 1975-2007,NCI. Available online: http://seer.cancer.gov/csr/1975_2007/ (1 October 2010).
- Stojadinovic, A.; Hoos, A.; Brennan, M.F.; Conlon, K.C.P. Randomized clinical trials in pancreatic cancer. Surg. Oncol. Clin. N. Am. 2002, 11, 207–229. [Google Scholar] [CrossRef]
- Winter, J.M.; Cameron, J.L.; Campbell, K.A.; Arnold, M.A.; Chang, D.C.; Coleman, J.; Hodgin, M.B.; Sauter, P.K.; Hruban, R.H.; Riall, T.S.; Schulick, R.D.; Choti, M.A.; Lillemoe, K.D.; Yeo, C.J. 1423 pancreaticoduodenectomies for pancreatic cancer: A single-institution experience. J. Gastrointest. Surg. 2006, 10, 1199–1210. [Google Scholar] [CrossRef]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; Frese, K.K.; Denicola, G.; Feig, C.; Combs, C.; Winter, S.P.; Ireland-Zecchini, H.; Reichelt, S.; Howat, W.J.; Chang, A.; Dhara, M.; Wang, L.; Rückert, F.; Grützmann, R.; Pilarsky, C.; Izeradjene, K.; Hingorani, S.R.; Huang, P.; Davies, S.E.; Plunkett, W.; Egorin, M.; Hruban, R.H.; Whitebread, N.; McGovern, K.; Adams, J.; Iacobuzio-Donahue, C.; Griffiths, J.; Tuveson, D.A. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef]
- Ottaviano, A.J.; Sun, L.; Ananthanarayanan, V.; Munshi, H.G. Extracellular matrix-mediated membrane-type 1 matrix metalloproteinase expression in pancreatic ductal cells is regulated by transforming growth factor-beta1. Cancer Res. 2006, 66, 7032–7040. [Google Scholar] [CrossRef]
- Hidalgo, M. Pancreatic cancer. N. Engl. J. Med. 2010, 362, 1605–1617. [Google Scholar] [CrossRef]
- Lee, J.M.; Dedhar, S.; Kalluri, R.; Thompson, E.W. The epithelial-mesenchymal transition: New insights in signaling, development, and disease. J. Cell. Biol. 2006, 172, 973–981. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Invest. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Yang, A.D.; Camp, E.R.; Fan, F.; Shen, L.; Gray, M.J.; Liu, W.; Somcio, R.; Bauer, T.W.; Wu, Y.; Hicklin, D.; Ellis, L.M. Vascular endothelial growth factor receptor-1 activation mediates epithelial to mesenchymal transition in human pancreatic carcinoma cells. Cancer Res. 2006, 66, 46–51. [Google Scholar] [CrossRef]
- Javle, M.M.; Gibbs, J.F.; Iwata, K.K.; Pak, Y.; Rutledge, P.; Yu, J.; Black, J.D.; Tan, D.; Khoury, T. Epithelial-mesenchymal transition (EMT) and activated extracellular signal-regulated kinase (p-Erk) in surgically resected pancreatic cancer. Ann. Surg. Oncol. 2007, 14, 3527–3533. [Google Scholar] [CrossRef]
- Rasheed, Z.A.; Yang, J.; Wang, Q.; Kowalski, J.; Freed, I.; Murter, C.; Hong, S.-M.; Koorstra, J.-B.; Rajeshkumar, N.V.; He, X.; Goggins, M.; Iacobuzio-Donahue, C.; Berman, D.M.; Laheru, D.; Jimeno, A.; Hidalgo, M.; Maitra, A.; Matsui, W. Prognostic significance of tumorigenic cells with mesenchymal features in pancreatic adenocarcinoma. J. Natl. Cancer Inst. 2010, 102, 340–351. [Google Scholar] [CrossRef]
- Tarin, D.; Thompson, E.W.; Newgreen, D.F. The fallacy of epithelial mesenchymal transition in neoplasia. Cancer Res. 2005, 65, 5996–6000. [Google Scholar] [CrossRef]
- Hotz, B.; Arndt, M.; Dullat, S.; Bhargava, S.; Buhr, H.-J.; Hotz, H.G. Epithelial to mesenchymal transition: Expression of the regulators snail, slug, and twist in pancreatic cancer. Clin. Cancer Res. 2007, 13, 4769–4776. [Google Scholar] [CrossRef]
- Buck, E.; Eyzaguirre, A.; Barr, S.; Thompson, S.; Sennello, R.; Young, D.; Iwata, K.K.; Gibson, N.W.; Cagnoni, P.; Haley, J.D. Loss of homotypic cell adhesion by epithelial-mesenchymal transition or mutation limits sensitivity to epidermal growth factor receptor inhibition. Mol. Cancer Ther. 2007, 6, 532–541. [Google Scholar] [CrossRef]
- Arumugam, T.; Ramachandran, V.; Fournier, K.F.; Wang, H.; Marquis, L.; Abbruzzese, J.L.; Gallick, G.E.; Logsdon, C.D.; Mcconkey, D.J.; Choi, W. Epithelial to Mesenchymal Transition Contributes to Drug Resistance in Pancreatic Cancer. Cancer Res. 2009, 69, 5820–5828. [Google Scholar] [CrossRef]
- Trimboli, A.J.; Fukino, K.; de Bruin, A.; Wei, G.; Shen, L.; Tanner, S.M.; Creasap, N.; Rosol, T.J.; Robinson, M.L.; Eng, C.; Ostrowski, M.C.; Leone, G. Direct evidence for epithelial-mesenchymal transitions in breast cancer. Cancer Res. 2008, 68, 937–945. [Google Scholar] [CrossRef]
- Flier, S.N.; Tanjore, H.; Kokkotou, E.G.; Sugimoto, H.; Zeisberg, M.; Kalluri, R. Identification of epithelial to mesenchymal transition as a novel source of fibroblasts in intestinal fibrosis. J. Biol. Chem. 2010, 285, 20202–20212. [Google Scholar]
- Maier, H.J.; Schmidt-Strassburger, U.; Huber, M.A.; Wiedemann, E.M.; Beug, H.; Wirth, T. NF-kappaB promotes epithelial-mesenchymal transition, migration and invasion of pancreatic carcinoma cells. Cancer Lett. 2010, 295, 214–228. [Google Scholar] [CrossRef]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef]
- Shi, Y.; Massagué, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Massagué, J. How cells read TGF-beta signals. Nat. Rev. Mol. Cell Biol. 2000, 1, 169–178. [Google Scholar] [CrossRef]
- Izeradjene, K.; Combs, C.; Best, M.; Gopinathan, A.; Wagner, A.; Grady, W.M.; Deng, C.-X.; Hruban, R.H.; Adsay, N.V.; Tuveson, D.A.; Hingorani, S.R. Kras(G12D) and Smad4/Dpc4 haploinsufficiency cooperate to induce mucinous cystic neoplasms and invasive adenocarcinoma of the pancreas. Cancer Cell 2007, 11, 229–243. [Google Scholar] [CrossRef]
- Bardeesy, N.; Cheng, K.-H.; Berger, J.H.; Chu, G.C.; Pahler, J.; Olson, P.; Hezel, A.F.; Horner, J.; Lauwers, G.Y.; Hanahan, D.; Depinho, R.A. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev. 2006, 20, 3130–3146. [Google Scholar] [CrossRef]
- Almoguera, C.; Shibata, D.; Forrester, K.; Martin, J.; Arnheim, N.; Perucho, M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell 1988, 53, 549–554. [Google Scholar]
- Singh, A.; Greninger, P.; Rhodes, D.; Koopman, L.; Violette, S.; Bardeesy, N.; Settleman, J. A gene expression signature associated with "K-Ras addiction" reveals regulators of EMT and tumor cell survival. Cancer Cell 2009, 15, 489–500. [Google Scholar] [CrossRef]
- Gidekel Friedlander, S.Y.; Chu, G.C.; Snyder, E.L.; Girnius, N.; Dibelius, G.; Crowley, D.; Vasile, E.; Depinho, R.A.; Jacks, T. Context-dependent transformation of adult pancreatic cells by oncogenic K-Ras. Cancer Cell 2009, 16, 379–389. [Google Scholar] [CrossRef]
- Guerra, C.; Schuhmacher, A.J.; Cañamero, M.; Grippo, P.J.; Verdaguer, L.; Pérez-Gallego, L.; Dubus, P.; Sandgren, E.P.; Barbacid, M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 2007, 11, 291–302. [Google Scholar] [CrossRef]
- Wu, Y.; Deng, J.; Rychahou, P.G.; Qiu, S.; Evers, B.M.; Zhou, B.P. Stabilization of Snail by NF-κB Is Required for Inflammation-Induced Cell Migration and Invasion. Cancer Cell 2009, 15, 416–428. [Google Scholar] [CrossRef]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef]
- Lim, L.P.; Lau, N.C.; Garrett-Engele, P.; Grimson, A.; Schelter, J.M.; Castle, J.; Bartel, D.P.; Linsley, P.S.; Johnson, J.M. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 2005, 433, 769–773. [Google Scholar] [CrossRef]
- McManus, M.T.; Sharp, P.A. Gene silencing in mammals by small interfering RNAs. Nat. Rev. Genet. 2002, 3, 737–747. [Google Scholar] [CrossRef]
- Dillhoff, M.; Wojcik, S.E.; Bloomston, M. MicroRNAs in solid tumors. J. Surg. Res. 2009, 154, 349–354. [Google Scholar] [CrossRef]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef]
- Peter, M.E. Let-7 and miR-200 microRNAs: Guardians against pluripotency and cancer progression. Cell Cycle 2009, 8, 843–852. [Google Scholar] [CrossRef]
- Kent, O.A.; Mullendore, M.; Wentzel, E.A.; López-Romero, P.; Tan, A.C.; Alvarez, H.; West, K.; Ochs, M.F.; Hidalgo, M.; Arking, D.E.; Maitra, A.; Mendell, J.T. A resource for analysis of microRNA expression and function in pancreatic ductal adenocarcinoma cells. Cancer Biol. Ther. 2009, 8, 2013–2024. [Google Scholar] [CrossRef]
- Yu, J.; Ohuchida, K.; Mizumoto, K.; Sato, N.; Kayashima, T.; Fujita, H.; Nakata, K.; Tanaka, M. MicroRNA, hsa-miR-200c, is an independent prognostic factor in pancreatic cancer and its upregulation inhibits pancreatic cancer invasion but increases cell proliferation. Mol. Cancer 2010, 9, 169. [Google Scholar] [CrossRef]
- Burk, U.; Schubert, J.; Wellner, U.; Schmalhofer, O.; Vincan, E.; Spaderna, S.; Brabletz, T. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008, 9, 582–589. [Google Scholar] [CrossRef]
- Gupta, P.B.; Onder, T.T.; Jiang, G.; Tao, K.; Kuperwasser, C.; Weinberg, R.A.; Lander, E.S. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 2009, 138, 645–659. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. P.Natl. Acad. Sci. USA. 2003, 100, 3983–3988. [Google Scholar]
- Korkaya, H.; Wicha, M.S. Cancer stem cells: Nature versus nurture. Nat. Cell. Biol. 2010, 12, 419–421. [Google Scholar] [CrossRef]
- Santisteban, M.; Reiman, J.M.; Asiedu, M.K.; Behrens, M.D.; Nassar, A.; Kalli, K.R.; Haluska, P.; Ingle, J.N.; Hartmann, L.C.; Manjili, M.H.; Radisky, D.C.; Ferrone, S.; Knutson, K.L. Immune-induced epithelial to mesenchymal transition in vivo generates breast cancer stem cells. Cancer Res. 2009, 69, 2887–2895. [Google Scholar] [CrossRef]
- Gupta, P.B.; Chaffer, C.L.; Weinberg, R.A. Cancer stem cells: Mirage or reality? Nat. Med. 2009, 15, 1010–1012. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; Campbell, L.L.; Polyak, K.; Brisken, C.; Yang, J.; Weinberg, R.A. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef]
- Lee, C.J.; Dosch, J.; Simeone, D.M. Pancreatic cancer stem cells. J. Clin. Oncol. 2008, 26, 2806–2812. [Google Scholar] [CrossRef]
- Wellner, U.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; zur Hausen, A.; Brunton, V.G.; Morton, J.; Sansom, O.; Schüler, J.; Stemmler, M.P.; Herzberger, C.; Hopt, U.; Keck, T.; Brabletz, S.; Brabletz, T. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat. Cell Biol. 2009, 11, 1487–1495. [Google Scholar] [CrossRef]
- Shimono, Y.; Zabala, M.; Cho, R.W.; Lobo, N.; Dalerba, P.; Qian, D.; Diehn, M.; Liu, H.; Panula, S.P.; Chiao, E.; Dirbas, F.M.; Somlo, G.; Pera, R.A.R.; Lao, K.; Clarke, M.F. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell 2009, 138, 592–603. [Google Scholar] [CrossRef]
- Li, Y.; VandenBoom, T.G.; Kong, D.; Wang, Z.; Ali, S.; Philip, P.A.; Sarkar, F.H. Up-regulation of miR-200 and let-7 by natural agents leads to the reversal of epithelial-to-mesenchymal transition in gemcitabine-resistant pancreatic cancer cells. Cancer Res. 2009, 69, 6704–6712. [Google Scholar] [CrossRef]
- Yang, A.D.; Fan, F.; Camp, E.R.; van Buren, G.; Liu, W.; Somcio, R.; Gray, M.J.; Cheng, H.; Hoff, P.M.; Ellis, L.M. Chronic oxaliplatin resistance induces epithelial-to-mesenchymal transition in colorectal cancer cell lines. Clin. Cancer Res. 2006, 12, 4147–4153. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Y.; Kong, D.; Banerjee, S.; Ahmad, A.; Azmi, A.; Ali, S.; Abbruzzese, J.L.; Gallick, G.E.; Sarkar, F. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res. 2009, 69, 2400–2407. [Google Scholar] [CrossRef]
- Shah, A.N.; Summy, J.M.; Zhang, J.; Park, S.; Parikh, N.; Gallick, G.E. Development and characterization of gemcitabine-resistant pancreatic tumor cells. Ann. Surg. Oncol. 2007, 14, 3629–3637. [Google Scholar] [CrossRef]
- Hwang, R.F.; Moore, T.; Arumugam, T.; Ramachandran, V.; Amos, K.D.; Rivera, A.; Ji, B.; Evans, D.B.; Logsdon, C.D. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008, 68, 918–926. [Google Scholar] [CrossRef]
- Apte, M.V.; Park, S.; Phillips, P.A.; Santucci, N.; Goldstein, D.; Kumar, R.K.; Ramm, G.A.; Buchler, M.; Friess, H.; McCarroll, J.A.; Keogh, G.; Merrett, N.; Pirola, R.; Wilson, J.S. Desmoplastic reaction in pancreatic cancer: Role of pancreatic stellate cells. Pancreas 2004, 29, 179–187. [Google Scholar] [CrossRef]
- Menke, A.; Adler, G. TGFbeta-induced fibrogenesis of the pancreas. Int. J. Gastrointest. Cancer 2002, 31, 41–46. [Google Scholar] [CrossRef]
- Lewis, M.P.; Lygoe, K.A.; Nystrom, M.L.; Anderson, W.P.; Speight, P.M.; Marshall, J.F.; Thomas, G.J. Tumour-derived TGF-beta1 modulates myofibroblast differentiation and promotes HGF/SF-dependent invasion of squamous carcinoma cells. Br. J. Cancer 2004, 90, 822–832. [Google Scholar] [CrossRef]
- Iwano, M.; Plieth, D.; Danoff, T.M.; Xue, C.; Okada, H.; Neilson, E.G. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Inveset. 2002, 110, 341–350. [Google Scholar]
- Boutet, A.; De Frutos, C.A.; Maxwell, P.H.; Mayol, M.J.; Romero, J.; Nieto, M.A. Snail activation disrupts tissue homeostasis and induces fibrosis in the adult kidney. EMBO J. 2006, 25, 5603–5613. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Y.; Kong, D.; Sarkar, F.H. The role of Notch signaling pathway in epithelial-mesenchymal transition (EMT) during development and tumor aggressiveness. Curr. Drug Targets 2010, 11, 745–751. [Google Scholar] [CrossRef]
- Brown, B.D.; Naldini, L. Exploiting and antagonizing microRNA regulation for therapeutic and experimental applications. Nat. Rev. Genet. 2009, 10, 578–585. [Google Scholar] [CrossRef]
- Jackson, A.L.; Linsley, P.S. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat. Rev. Drug Discov. 2010, 9, 57–67. [Google Scholar] [CrossRef]
- Ali, S.; Ahmad, A.; Banerjee, S.; Padhye, S.; Dominiak, K.; Schaffert, J.M.; Wang, Z.; Philip, P. A.; Sarkar, F.H. Gemcitabine sensitivity can be induced in pancreatic cancer cells through modulation of miR-200 and miR-21 expression by curcumin or its analogue CDF. Cancer Res. 2010, 70, 3606–3617. [Google Scholar] [CrossRef]
- Wu, K.; Zeng, J.; Li, L.; Fan, J.; Zhang, D.; Xue, Y.; Zhu, G.; Yang, L.; Wang, X.; He, D. Silibinin reverses epithelial-to-mesenchymal transition in metastatic prostate cancer cells by targeting transcription factors. Oncol. Rep. 2010, 23, 1545–1552. [Google Scholar]
- Vazquez-Martin, A.; Oliveras-Ferraros, C.; Cufí, S.; Del Barco, S.; Martin-Castillo, B.; Menendez, J.A. Metformin regulates breast cancer stem cell ontogeny by transcriptional regulation of the epithelial-mesenchymal transition (EMT) status. Cell Cycle 2010, 9, 3807–3814. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Krantz, S.B.; Shields, M.A.; Dangi-Garimella, S.; Bentrem, D.J.; Munshi, H.G. Contribution of Epithelial-Mesenchymal Transition to Pancreatic Cancer Progression. Cancers 2010, 2, 2084-2097. https://doi.org/10.3390/cancers2042084
Krantz SB, Shields MA, Dangi-Garimella S, Bentrem DJ, Munshi HG. Contribution of Epithelial-Mesenchymal Transition to Pancreatic Cancer Progression. Cancers. 2010; 2(4):2084-2097. https://doi.org/10.3390/cancers2042084
Chicago/Turabian StyleKrantz, Seth B., Mario A. Shields, Surabhi Dangi-Garimella, David J. Bentrem, and Hidayatullah G. Munshi. 2010. "Contribution of Epithelial-Mesenchymal Transition to Pancreatic Cancer Progression" Cancers 2, no. 4: 2084-2097. https://doi.org/10.3390/cancers2042084