Macrophage-Mediated Lymphangiogenesis: The Emerging Role of Macrophages as Lymphatic Endothelial Progenitors

Abstract
:Abbreviations
| TAM(s) | tumor-associated macrophage(s) |
| M-LECP | macrophage-derived lymphatic endothelial cell progenitor(s) |
| LN(s) | lymph node(s) |
| BM | bone marrow |
| LEC(s) | lymphatic endothelial cell(s) |
| IFP | interstitial fluid pressure |
| Prox1 | prospero-related homeobox-1 |
| VEGFR | vascular endothelial growth factor receptor |
| LECP | lymphatic endothelial cell progenitor(s) |
| LVD | lymphatic vessel density |
| VEGF | vascular endothelial growth factor |
| NF-κB | nuclear factor-kappaB |
| BEC | blood vascular endothelial cell(s) |
| IBC | inflammatory breast cancer |
| MMP | matrix metalloproteinase(s) |
| CDL | clodronate liposomes |
| GFP | fluorescent protein |
1. Introduction
1.1. Structure, Function, and Organization of the Lymphatic Vasculature
1.2. Markers of Lymphatic Endothelial Cells (LECs)
1.3. Development of the Lymphatic Vascular Network During Embryogenesis
2. Generation of New Lymphatic Vessels in the Adult
2.1. Inflammation-Induced Lymphangiogenesis

{kind=link}
| Model/Condition | Quantitative measure | Qualitative change | Ref. |
|---|---|---|---|
| Psoriasis (H) | 2–6 fold increased LVD and ~2 fold increased Ki-67 index | N/A | [54] |
| Inflammatory bowel disease (H) | ~2–3 fold increase in LVD | N/A | [56] |
| Irradiated skin (H) | 18% increase in total LVD and 44% increase in vessels <10 µm in diameter | N/A | [59] |
| Kidney transplant rejection (H) | >50 fold increased LVD in grafts undergoing rejection | N/A | [61] |
| Breast cancer (H) | LVD was 12 fold higher in tumors compared to benign lesions | N/A | [67] |
| UVB irradiation of skin (m) | 2–3 fold increased LV area and size but no increase in LV number | Hyperplastic vessels associated with increased macrophage infiltration | [63] |
| LPS induced peritonitis (m) | ~2–4 fold increased LVD in diaphragm; 2.4 fold more proliferating LECs; 17 fold more vessel sprouts | LVs were enlarged and LV network patterning was atypical; increase in randomly oriented branching; new LVs were dysfunctional | [64] |
| TG stimulated peritonitis (m) | 1.9 fold increased LVD in diaphragm | N/A | [68] |
| Chronic airway inflammation (m) | LVD and LV sprouts increased many folds (roughly 10 fold) in trachea | N/A | [65] |
| Chronic airway inflammation (m) | LVD increased >10 fold in trachea | VE-cadherin LEC junctions are remodeled, intermittent buttons give way to continuous zippers | [66] |
2.2. Molecular Mediators of Inflammatory Lymphangiogenesis
2.3. Tumor-Induced Lymphangiogenesis
2.3.1. Induction of Lymphangiogenesis in Tumors
2.3.2. Correlation between Tumor-Induced Lymphangiogenesis and Metastasis
3. Role of Macrophages in Postnatal Formation of New Lymphatic Vessels
3.1. Subtypes of TAMs Displaying Pro- or Anti-Tumorigenic Behavior
| Name | Produced by TAMs | Evidence for lymphangiogenic activity | Ref. | Correlates with LN metastasis? | Ref. |
|---|---|---|---|---|---|
| VEGF-A | [158] | Activates LEC and directly induces lymphangiogenesis in various inflammation and tumor models | [91,159] | yes | [60] |
| VEGF-C | [22] | A ligand for VEGFR-3, a key inducer of lymphangiogenesis | [4,160] | yes | [60] |
| VEGF-D | [22] | A ligand for VEGFR-3, a key inducer of lymphangiogenesis | [4,161] | yes | [60,147] |
| PDGF | [125] | Direct lymphangiogenic factor in mouse cornea and PDGF-overexpressing T241 tumors | [110] | yes | [162] |
| Adrenomedullin | [163] | Direct lymphangiogenic factor acting through the calcitonin receptor-like receptor | [164] | yes | [165] |
| HGF/SF | [149] | Direct lymphangiogenic factor; the corresponding receptor c-Met is upregulated on LEC during inflammation | [166] | yes | [167] |
| COX-2 | [74] | Induces lymphangiogenesis indirectly through PGE2 that upregulates VEGF-C | [74,116] | yes | [168] |
| βFGF (FGF-2) | [169] | Induces lymphangiogenesis indirectly through upregulation of VEGF-C and VEGF-D | [62,170] | yes | [171] |
| TNF-α | [172] | Potentially regulates lymphangiogenesis by increasing VEGF-C transcription in fibroblasts | [73] | yes | [173] |
| MMP-2 & MMP-9 | [174] | Suppression of MMP-2-/9 inhibits LEC invasion through matrigel | [175] | yes | [176] |
| Heparanase | [177] | Indirectly lymphangiogenic by increasing VEGF-C expression in cancer cells | [178] | yes | [179] |
| Urokinase plasminogen activator system | [180] | Plasmin is indirectly lymphangiogenic by increasing maturation of VEGF-C/-D propeptides | [181] | yes | [182] |
| Angiopoietin-2 * (Ang-2) | [183] | Directly lymphangiogenic; activates LEC via Tie-2 receptor; overexpression of Ang-2 induces lymphangiogenesis in vivo | [184,185] | yes | [186] |
3.2. Association of Tumor Lymphangiogenesis and Lymphatic Metastasis with Macrophage Infiltrates
3.3. Experimental Evidence Demonstrating Correlation Between TAMs, Increased LVD and Lymphatic Metastasis
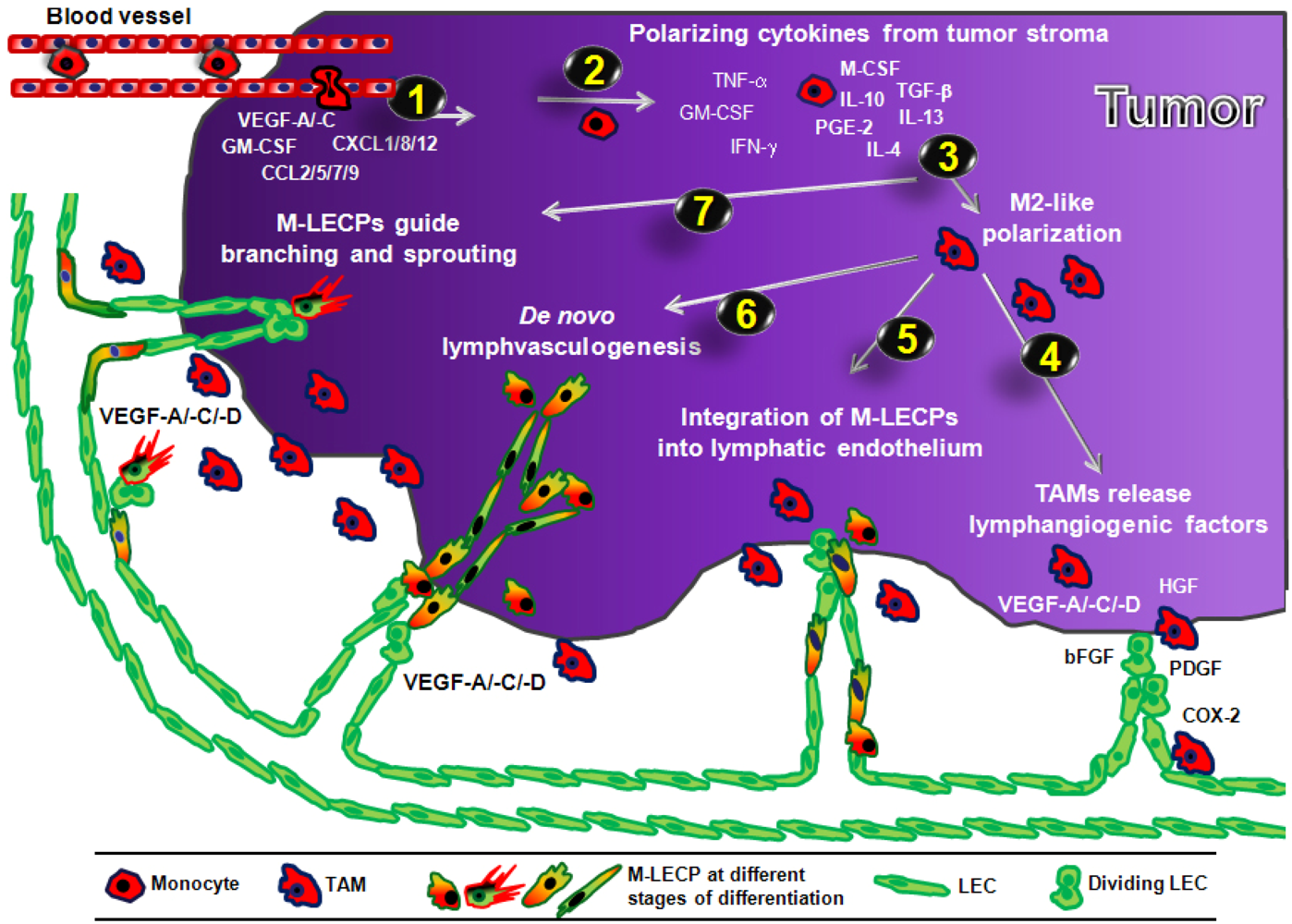
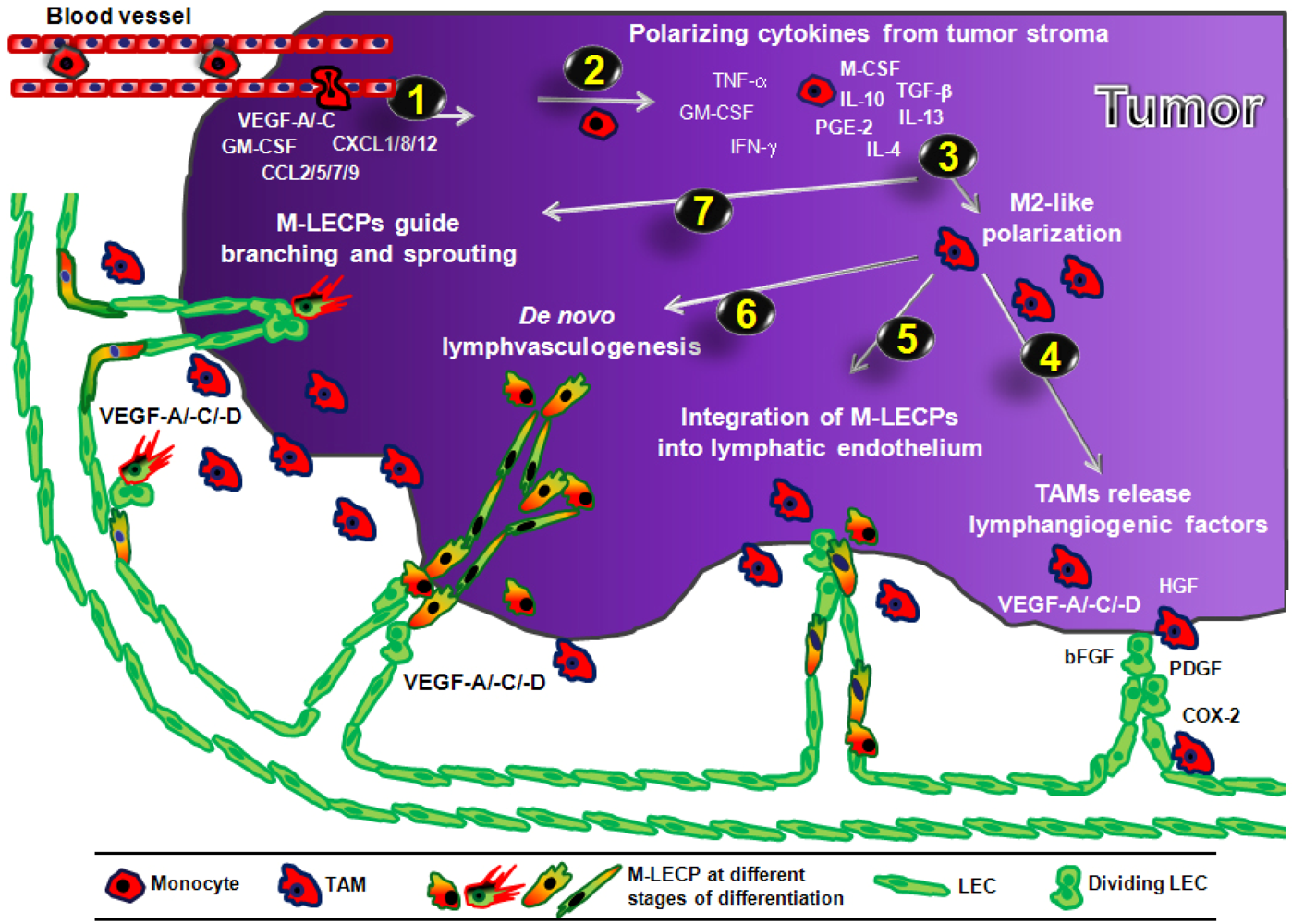
3.4. Mechanisms of Macrophage-Mediated Contribution to Tumor Lymphangiogenesis
3.4.1. Role of Pro-Lymphangiogenic Factors and Proteolytic Enzymes Produced by TAMs
3.4.2. Role of Macrophage-Derived LEC Progenitors (M-LECP)
| Gene name | Comments | Detection method | Ref. |
|---|---|---|---|
| VEGFR-3 | Expressed by TG-stimulated peritoneal macrophages in culture | RT-PCR | [16] |
| Detected in TG-induced peritoneal macrophages in culture | RT-qPCR | [19] | |
| Detected in bone marrow-derived macrophages in culture | RT-qPCR | [23] | |
| Expressed on culture CD11b+ bone marrow-derived cells that integrated into LV after reintroduction into mice | FACS, IHC | [53] | |
| Detected in activated peritoneal macrophages in vivo and in RAW264.7 macrophages in vitro | RT-qPCR, FACS | [30] | |
| Expressed by monocytes freshly purified from human blood | IHC, RT-PCR | [20] | |
| Podoplanin | Detected on TG-stimulated peritoneal macrophages in culture | FACS, IHC | [16] |
| Co-expressed with F4/80+ cells incorporated into LV in vivo | IHC | [19] | |
| Expressed by myeloid cells incorporated into LV in vivo | IHC | [23] | |
| Expressed on CD11b+ bone marrow-derived cells that integrated into LV in vivo | IHC, FACS | [53] | |
| Co-expressed on CD11b+ cells incorporated into LV in vivo and in activated peritoneal macrophages in vivo | IHC, RT-qPCR | [30] | |
| Expressed by cultured monocytes purified from human blood | IHC, RT-PCR | [20] | |
| LYVE-1 | Co-expressed on CD11b+ cells in LV in vivo and by TG-stimulated peritoneal macrophages in culture | IHC, FACS, IHC | [16] |
| Co-expressed on F4/80+ cells incorporated into LV in vivo | IHC | [19] | |
| Co-expressed on F4/80+ cells incorporated into embryonic LS and LV | IHC | [52] | |
| LYVE-1 | Co-expressed on F4/80+ cells incorporated into LV in vivo | IHC | [23] |
| Expressed on culture CD11b+ bone marrow-derived cells that integrated into LV after reintroduction into mice | FACS, IHC | [53] | |
| Co-expressed on CD11b+ cells incorporated into LV in vivo and inactivated peritoneal macrophages in vivo | IHC, RT-qPCR | [30] | |
| Expressed by monocytes freshly purified from human blood | IHC, RT-PCR | [20] | |
| Prox-1 | Co-expressed on CD11b+ cells in LV in vivo and by TG-stimulated peritoneal macrophages in culture | IHC, FACS, IHC | [16] |
| Co-expressed with F4/80+ cells incorporated into embryonic LS and LV | IHC | [52] | |
| Expressed by myeloid cells incorporated into LV | IHC | [23] | |
| Expressed on cultured CD11b+ bone marrow-derived cells that integrated into LV after reintroduction into mice | FACS | [53] | |
| Co-expressed on CD11b+ cells incorporated into LV in vivo; Activated peritoneal macrophages in vivo expressed reduced levels compared to control group | IHC, RT-qPCR | [30] | |
| Tie2 | Activated peritoneal macrophages in vivo expressed reduced levels compared to control group | RT-qPCR | [30] |
| Model | Cell origin or type | Tag | Markers | Time point of analysis | Integration of LECP into LV | Ref. |
|---|---|---|---|---|---|---|
| LPS induced peritonitis (m) | Native macrophages | none | CD11b, F4/80, LYVE-1 | 2 days a | ~50% of LV contained macrophages | [30] |
| LPS induced peritonitis (m) | RAW264.7 macrophages | GFP | CD11b, F4/80, LYVE-1, Podo | 7 days a | ~20% of LV contained macrophages | [30] |
| Corneal micropocket (m) | CD34+/VEGFR-3+ BM-LECP | GFP | LYVE-1 | 1–4 days b | ~1.5% of lymphatic endothelium | [15] |
| Corneal micropocket (m) | CD34+/VEGFR-2+ BM-LECP | GFP | LYVE-1 | 1–4 days b | ~0.5% of lymphatic endothelium | [15] |
| Corneal micropocket (m) | Cultured Podo+ BM-MNC | DiI | LYVE-1 | 7 days b | 5.2% of LV contained DiI+ cells | [53] |
| Skin and ear wound (m) | Cultured Podo+ BM-MNC | DiI | LYVE-1 | 7 days b | 5.5% of LV contained DiI+ cells | [53] |
| Liver of irradiated mice c | Hematopoietic stem cells | GFP | LYVE-1, VEGFR-3 | 1 month b & >1 year b | 2.4% & 3.2% of LV contained GFP+ cells | [217] |
| Gastro-intestinal tissue of irradiated mice | Hematopoietic stem cells | GFP | LYVE-1, VEGFR-3 | >1 year b | 1.0–1.4% of LV contained GFP+ cells | [217] |
| Skin and ear wound (m) | Fresh Podo+ BM-MNC | DiI | LYVE-1 | 7 days b | detected, not quantified | [53] |
| Corneal inflammation (m) | BM-MNC | GFP | CD11b, LYVE-1, Prox-1 | 3 or 7 days a | detected, not quantified | [16] |
| Skin wound (m) | Native myeloid cells | none | F4/80, LYVE-1 | 5 days a | detected, not quantified | [19] |
| Kidney transplant rejection (H) | Presumably BM | none | Y-chromosome, LYVE-1, Podo | N/A | 4.5% of LEC were Y-chromosome+ | [20] |
| Interstitial lung disease (H) | Native macrophages | none | CD68, Podo, VEGFR-3 | N/A | ~1.6 cells/mm of LV | [218] |
| Oncocerciasis (H) | Native macrophages | none | CD68, LYVE-1 | N/A | detected, not quantified | [29] |
| Model | Cell origin or type | Tag | Markers | Time point of analysis | Integration of LECP into LV | Ref. |
|---|---|---|---|---|---|---|
| Rip1Tag2 insulinoma (m) | BM Cells (-T cells) | GFP | Podo, LYVE-1, Prox1 | 5–7 weeks a | 3.5% GFP+/Prox1+b 3.5% GFP+/LYVE-1+b 3% GFP+/Podo+b | [23] |
| TRAMPC-1 prostate cancer (m) | BM cells (-T cells) | GFP | Podo, LYVE-1, Prox1 | 3–4 weeks c | minimal GFP+/Prox1+ 2.8% GFP+/LYVE-1+b4.1% GFP+/Podo+b | [23] |
| B16-F1 melanoma (m) | Cultured Podo+ BM-MNC | DiI | LYVE-1 | 7 days a | 8.5% of LV contained DiI+ cells | [53] |
| T241 fibrosarcoma (m) | CD34+/VEGFR-3+ BM-LECP | GFP | LYVE-1 | 1–4 days a | detected, not quantified | [15] |
| T241 fibrosarcoma (m) | CD34+/VEGFR-2+ BM-LECP | GFP | LYVE-1 | 1–4 days a | detected, not quantified | [15] |
| Multiple intestinal neoplasia (m) | Hematopoietic stem cells | GFP | LYVE-1 | 6 weeks a | detected, not quantified | [217] |
| Rip1Tag2 insulinoma (m) | BM Cells (-T cells) | GFP | LYVE-1, F4/80 | 5–7 weeks a | detected, not quantified | [23] |
| Rip1Tag2 insulinoma (m) | CD11b+ cells | GFP | LYVE-1, Prox1 | 3 weeks a | detected, not quantified | [23] |
| Rip1Tag2 insulinoma (m) | Common myeloid progenitor cells | GFP | Podo, LYVE-1 | 3 weeks a | detected, not quantified | [23] |
| TRAMPC-1 prostate cancer (m) | Native CD11b+ cells | GFP | Podo, LYVE-1, Prox1 | 3–4 weeks c | detected, not quantified | [23] |
| EL4 lymphoma & Lewis lung carcinoma (m) | Native myeloid cells | β-gal | CD31, Prox1 | 10–14 days c | detected but lacked Prox1, not quantified | [219] |
3.4.2.1. Incorporation of M-LECP into Inflammation-Induced and tumor Lymphatic Vessels
3.4.2.2. Transdifferentiation of Macrophages into M-LECP
3.4.2.3. Evidence of Lymphvasculogenesis Induced by Adult M-LECP
4. Other BM-Derived Progenitors that Might Contribute to Tumor and Inflammatory Lymphangiogenesis
5. Conclusions
Acknowledgments
References
- Tammela, T.; Alitalo, K. Lymphangiogenesis: Molecular mechanisms and future promise. Cell 2010, 140, 460–476. [Google Scholar] [CrossRef]
- Dejana, E.; Orsenigo, F.; Molendini, C.; Baluk, P.; McDonald, D.M. Organization and signaling of endothelial cell-to-cell junctions in various regions of the blood and lymphatic vascular trees. Cell Tissue Res. 2009, 335, 17–25. [Google Scholar]
- Baluk, P.; Fuxe, J.; Hashizume, H.; Romano, T.; Lashnits, E.; Butz, S.; Vestweber, D.; Corada, M.; Molendini, C.; Dejana, E.; et al. Functionally specialized junctions between endothelial cells of lymphatic vessels. J. Exp. Med. 2007, 204, 2349–2362. [Google Scholar]
- Pepper, M.S.; Skobe, M. Lymphatic endothelium: Morphological, molecular and functional properties. J. Cell Biol. 2003, 163, 209–213. [Google Scholar] [CrossRef]
- Zawieja, D.C. Contractile physiology of lymphatics. Lymphat. Res. Biol. 2009, 7, 87–96. [Google Scholar] [CrossRef]
- Muthuchamy, M.; Zawieja, D. Molecular regulation of lymphatic contractility. Ann. NY Acad. Sci. 2008, 1131, 89–99. [Google Scholar]
- Harvey, N.L. The link between lymphatic function and adipose biology. Ann. NY Acad. Sci. 2008, 1131, 82–88. [Google Scholar]
- Miller, N.E.; Michel, C.C.; Nanjee, M.N.; Olszewski, W.L.; Miller, I.P.; Hazell, M.; Olivecrona, G.; Sutton, P.; Humphreys, S.M.; Frayn, K.N. Secretion of adipokines by human adipose tissue in vivo: Partitioning between capillary and lymphatic transport. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E659–E667. [Google Scholar] [CrossRef]
- Angeli, V.; Randolph, G.J. Inflammation, lymphatic function, and dendritic cell migration. Lymphat. Res. Biol. 2006, 4, 217–228. [Google Scholar] [CrossRef]
- Johnson, L.A.; Jackson, D.G. Cell traffic and the lymphatic endothelium. Ann. NY Acad. Sci. 2008, 1131, 119–133. [Google Scholar]
- Kaipainen, A.; Korhonen, J.; Mustonen, T.; van Hinsbergh, V.W.; Fang, G.H.; Dumont, D.; Breitman, M.; Alitalo, K. Expression of the fms-like tyrosine kinase 4 gene becomes restricted to lymphatic endothelium during development. Proc. Natl. Acad. Sci. USA 1995, 92, 3566–3570. [Google Scholar]
- Pytowski, B.; Goldman, J.; Persaud, K.; Wu, Y.; Witte, L.; Hicklin, D.J.; Skobe, M.; Boardman, K.C.; Swartz, M.A. Complete and specific inhibition of adult lymphatic regeneration by a novel VEGFR-3 neutralizing antibody. J. Natl. Cancer Inst. 2005, 97, 14–21. [Google Scholar]
- Paavonen, K.; Puolakkainen, P.; Jussila, L.; Jahkola, T.; Alitalo, K. Vascular endothelial growth factor receptor-3 in lymphangiogenesis in wound healing. Am. J. Pathol. 2000, 156, 1499–1504. [Google Scholar] [CrossRef]
- Tammela, T.; Zarkada, G.; Wallgard, E.; Murtomaki, A.; Suchting, S.; Wirzenius, M.; Waltari, M.; Hellstrom, M.; Schomber, T.; Peltonen, R.; et al. Blocking VEGFR-3 suppresses angiogenic sprouting and vascular network formation. Nature 2008, 454, 656–660. [Google Scholar] [CrossRef]
- Religa, P.; Cao, R.; Bjorndahl, M.; Zhou, Z.; Zhu, Z.; Cao, Y. Presence of bone marrow-derived circulating progenitor endothelial cells in the newly formed lymphatic vessels. Blood 2005, 106, 4184–4190. [Google Scholar]
- Maruyama, K.; Ii, M.; Cursiefen, C.; Jackson, D.G.; Keino, H.; Tomita, M.; van Rooijen, N.; Takenaka, H.; D’Amore, P.A.; Stein-Streilein, J.; et al. Inflammation-induced lymphangiogenesis in the cornea arises from CD11b-positive macrophages. J. Clin. Invest. 2005, 115, 2363–2372. [Google Scholar]
- Skobe, M.; Hamberg, L.M.; Hawighorst, T.; Schirner, M.; Wolf, G.L.; Alitalo, K.; Detmar, M. Concurrent induction of lymphangiogenesis, angiogenesis, and macrophage recruitment by vascular endothelial growth factor-C in melanoma. Am. J. Pathol. 2001, 159, 893–903. [Google Scholar] [CrossRef]
- Saaristo, A.; Tammela, T.; Farkkila, A.; Karkkainen, M.; Suominen, E.; Yla-Herttuala, S.; Alitalo, K. Vascular endothelial growth factor-C accelerates diabetic wound healing. Am. J. Pathol. 2006, 169, 1080–1087. [Google Scholar] [CrossRef]
- Maruyama, K.; Asai, J.; Ii, M.; Thorne, T.; Losordo, D.W.; D’Amore, P.A. Decreased macrophage number and activation lead to reduced lymphatic vessel formation and contribute to impaired diabetic wound healing. Am. J. Pathol. 2007, 170, 1178–1191. [Google Scholar] [CrossRef]
- Kerjaschki, D.; Huttary, N.; Raab, I.; Regele, H.; Bojarski-Nagy, K.; Bartel, G.; Krober, S.M.; Greinix, H.; Rosenmaier, A.; Karlhofer, F.; et al. Lymphatic endothelial progenitor cells contribute to de novo lymphangiogenesis in human renal transplants. Nat. Med. 2006, 12, 230–234. [Google Scholar]
- Hamrah, P.; Chen, L.; Cursiefen, C.; Zhang, Q.; Joyce, N.C.; Dana, M.R. Expression of vascular endothelial growth factor receptor-3 (VEGFR-3) on monocytic bone marrow-derived cells in the conjunctiva. Exp. Eye Res. 2004, 79, 553–561. [Google Scholar] [CrossRef]
- Schoppmann, S.F.; Birner, P.; Stockl, J.; Kalt, R.; Ullrich, R.; Caucig, C.; Kriehuber, E.; Nagy, K.; Alitalo, K.; Kerjaschki, D. Tumor-associated macrophages express lymphatic endothelial growth factors and are related to peritumoral lymphangiogenesis. Am. J. Pathol. 2002, 161, 947–956. [Google Scholar] [CrossRef]
- Zumsteg, A.; Baeriswyl, V.; Imaizumi, N.; Schwendener, R.; Ruegg, C.; Christofori, G. Myeloid cells contribute to tumor lymphangiogenesis. PLoS ONE 2009, 4, e7067. [Google Scholar]
- Schmeisser, A.; Christoph, M.; Augstein, A.; Marquetant, R.; Kasper, M.; Braun-Dullaeus, R.C.; Strasser, R.H. Apoptosis of human macrophages by Flt-4 signaling: Implications for atherosclerotic plaque pathology. Cardiovasc. Res. 2006, 71, 774–784. [Google Scholar] [CrossRef]
- Folpe, A.L.; Veikkola, T.; Valtola, R.; Weiss, S.W. Vascular endothelial growth factor receptor-3 (VEGFR-3): A marker of vascular tumors with presumed lymphatic differentiation, including Kaposi's sarcoma, kaposiform and Dabska-type hemangioendotheliomas, and a subset of angiosarcomas. Mod. Pathol. 2000, 13, 180–185. [Google Scholar] [CrossRef]
- Banerji, S.; Ni, J.; Wang, S.X.; Clasper, S.; Su, J.; Tammi, R.; Jones, M.; Jackson, D.G. LYVE-1, a new homologue of the CD44 glycoprotein, is a lymph-specific receptor for hyaluronan. J. Cell Biol. 1999, 144, 789–801. [Google Scholar] [CrossRef]
- Mouta, C.C.; Nasser, S.M.; di Tomaso, E.; Padera, T.P.; Boucher, Y.; Tomarev, S.I.; Jain, R.K. LYVE-1 is not restricted to the lymph vessels: Expression in normal liver blood sinusoids and down-regulation in human liver cancer and cirrhosis. Cancer Res. 2001, 61, 8079–8084. [Google Scholar]
- Schledzewski, K.; Falkowski, M.; Moldenhauer, G.; Metharom, P.; Kzhyshkowska, J.; Ganss, R.; Demory, A.; Falkowska-Hansen, B.; Kurzen, H.; Ugurel, S.; et al. Lymphatic endothelium-specific hyaluronan receptor LYVE-1 is expressed by stabilin-1+, F4/80+, CD11b+ macrophages in malignant tumours and wound healing tissue in vivo and in bone marrow cultures in vitro: Implications for the assessment of lymphangiogenesis. J. Pathol. 2006, 209, 67–77. [Google Scholar] [CrossRef]
- Attout, T.; Hoerauf, A.; Denece, G.; Debrah, A.Y.; Marfo-Debrekyei, Y.; Boussinesq, M.; Wanji, S.; Martinez, V.; Mand, S.; Adjei, O.; et al. Lymphatic vascularisation and involvement of Lyve-1+ macrophages in the human onchocerca nodule. PLoS ONE 2009, 4, e8234. [Google Scholar]
- Hall, K.L.; Volk-Draper, L.D.; Flister, M.J.; Ran, S. New model of macrophage acquisition of the lymphatic endothelial phenotype. PLoS ONE 2012, 7, e31794. [Google Scholar]
- Flister, M.J.; Volk, L.D.; Ran, S. Characterization of Prox1 and VEGFR-3 expression and lymphatic phenotype in normal organs of mice lacking p50 subunit of NF-kappaB. Microcirculation 2011, 18, 85–101. [Google Scholar] [CrossRef]
- Breiteneder-Geleff, S.; Matsui, K.; Soleiman, A.; Meraner, P.; Poczewski, H.; Kalt, R.; Schaffner, G.; Kerjaschki, D. Podoplanin, novel 43-kd membrane protein of glomerular epithelial cells, is down-regulated in puromycin nephrosis. Am. J. Pathol. 1997, 151, 1141–1152. [Google Scholar]
- Petrova, T.V.; Makinen, T.; Makela, T.P.; Saarela, J.; Virtanen, I.; Ferrell, R.E.; Finegold, D.N.; Kerjaschki, D.; Yla-Herttuala, S.; Alitalo, K. Lymphatic endothelial reprogramming of vascular endothelial cells by the Prox-1 homeobox transcription factor. EMBO J. 2002, 21, 4593–4599. [Google Scholar]
- Vlahakis, N.E.; Young, B.A.; Atakilit, A.; Sheppard, D. The lymphangiogenic vascular endothelial growth factors VEGF-C and -D are ligands for the integrin alpha9beta1. J. Biol. Chem. 2005, 280, 4544–4552. [Google Scholar]
- Palmer, E.L.; Ruegg, C.; Ferrando, R.; Pytela, R.; Sheppard, D. Sequence and tissue distribution of the integrin alpha 9 subunit, a novel partner of beta 1 that is widely distributed in epithelia and muscle. J. Cell Biol. 1993, 123, 1289–1297. [Google Scholar] [CrossRef]
- Chen, H.; Chedotal, A.; He, Z.; Goodman, C.S.; Tessier-Lavigne, M. Neuropilin-2, a novel member of the neuropilin family, is a high affinity receptor for the semaphorins Sema E and Sema IV but not Sema III. Neuron 1997, 19, 547–559. [Google Scholar] [CrossRef]
- Yuan, L.; Moyon, D.; Pardanaud, L.; Breant, C.; Karkkainen, M.J.; Alitalo, K.; Eichmann, A. Abnormal lymphatic vessel development in neuropilin 2 mutant mice. Development 2002, 129, 4797–4806. [Google Scholar]
- Karpanen, T.; Heckman, C.A.; Keskitalo, S.; Jeltsch, M.; Ollila, H.; Neufeld, G.; Tamagnone, L.; Alitalo, K. Functional interaction of VEGF-C and VEGF-D with neuropilin receptors. FASEB J. 2006, 20, 1462–1472. [Google Scholar]
- Xu, Y.; Yuan, L.; Mak, J.; Pardanaud, L.; Caunt, M.; Kasman, I.; Larrivee, B.; del Toro, R.; Suchting, S.; Medvinsky, A.; et al. Neuropilin-2 mediates VEGF-C-induced lymphatic sprouting together with VEGFR3. J. Cell Biol. 2010, 188, 115–130. [Google Scholar] [CrossRef]
- Shin, W.S.; Rockson, S.G. Animal models for the molecular and mechanistic study of lymphatic biology and disease. Ann. NY Acad. Sci. 2008, 1131, 50–74. [Google Scholar]
- Oliver, G.; Srinivasan, R.S. Endothelial cell plasticity: How to become and remain a lymphatic endothelial cell. Development 2010, 137, 363–372. [Google Scholar] [CrossRef]
- Choi, I.; Lee, S.; Hong, Y.K. The new era of the lymphatic system: No longer secondary to the blood vascular system. Cold Spring Harb. Perspect. Med. 2012, 2, a006445. [Google Scholar]
- Srinivasan, R.S.; Dillard, M.E.; Lagutin, O.V.; Lin, F.J.; Tsai, S.; Tsai, M.J.; Samokhvalov, I.M.; Oliver, G. Lineage tracing demonstrates the venous origin of the mammalian lymphatic vasculature. Genes Dev. 2007, 21, 2422–2432. [Google Scholar] [CrossRef]
- Srinivasan, R.S.; Geng, X.; Yang, Y.; Wang, Y.; Mukatira, S.; Studer, M.; Porto, M.P.; Lagutin, O.; Oliver, G. The nuclear hormone receptor Coup-TFII is required for the initiation and early maintenance of Prox1 expression in lymphatic endothelial cells. Genes Dev. 2010, 24, 696–707. [Google Scholar] [CrossRef]
- Francois, M.; Caprini, A.; Hosking, B.; Orsenigo, F.; Wilhelm, D.; Browne, C.; Paavonen, K.; Karnezis, T.; Shayan, R.; Downes, M.; et al. Sox18 induces development of the lymphatic vasculature in mice. Nature 2008, 456, 643–647. [Google Scholar]
- Hong, Y.K.; Harvey, N.; Noh, Y.H.; Schacht, V.; Hirakawa, S.; Detmar, M.; Oliver, G. Prox1 is a master control gene in the program specifying lymphatic endothelial cell fate. Dev. Dyn. 2002, 225, 351–357. [Google Scholar] [CrossRef]
- Ny, A.; Koch, M.; Schneider, M.; Neven, E.; Tong, R.T.; Maity, S.; Fischer, C.; Plaisance, S.; Lambrechts, D.; Heligon, C.; et al. A genetic Xenopus laevis tadpole model to study lymphangiogenesis. Nat. Med. 2005, 11, 998–1004. [Google Scholar]
- Papoutsi, M.; Tomarev, S.I.; Eichmann, A.; Prols, F.; Christ, B.; Wilting, J. Endogenous origin of the lymphatics in the avian chorioallantoic membrane. Dev. Dyn. 2001, 222, 238–251. [Google Scholar] [CrossRef]
- Wilting, J.; Papoutsi, M.; Othman-Hassan, K.; Rodriguez-Niedenfuhr, M.; Prols, F.; Tomarev, S.I.; Eichmann, A. Development of the avian lymphatic system. Microsc. Res. Tech. 2001, 55, 81–91. [Google Scholar] [CrossRef]
- Wilting, J.; Aref, Y.; Huang, R.; Tomarev, S.I.; Schweigerer, L.; Christ, B.; Valasek, P.; Papoutsi, M. Dual origin of avian lymphatics. Dev. Biol. 2006, 292, 165–173. [Google Scholar] [CrossRef]
- Buttler, K.; Kreysing, A.; von Kaisenberg, C.S.; Schweigerer, L.; Gale, N.; Papoutsi, M.; Wilting, J. Mesenchymal cells with leukocyte and lymphendothelial characteristics in murine embryos. Dev. Dyn. 2006, 235, 1554–1562. [Google Scholar]
- Buttler, K.; Ezaki, T.; Wilting, J. Proliferating mesodermal cells in murine embryos exhibiting macrophage and lymphendothelial characteristics. BMC Dev. Biol. 2008, 8, 43. [Google Scholar] [CrossRef]
- Lee, J.Y.; Park, C.; Cho, Y.P.; Lee, E.; Kim, H.; Kim, P.; Yun, S.H.; Yoon, Y.S. Podoplanin-expressing cells derived from bone marrow play a crucial role in postnatal lymphatic neovascularization. Circulation 2010, 122, 1413–1425. [Google Scholar] [CrossRef]
- Henno, A.; Blacher, S.; Lambert, C.; Colige, A.; Seidel, L.; Noel, A.; Lapiere, C.; de la Brassinne, M.; Nusgens, B.V. Altered expression of angiogenesis and lymphangiogenesis markers in the uninvolved skin of plaque-type psoriasis. Br. J. Dermatol. 2009, 160, 581–590. [Google Scholar] [CrossRef]
- Kaiserling, E.; Krober, S.; Geleff, S. Lymphatic vessels in the colonic mucosa in ulcerative colitis. Lymphology 2003, 36, 52–61. [Google Scholar]
- Geleff, S.; Schoppmann, S.F.; Oberhuber, G. Increase in podoplanin-expressing intestinal lymphatic vessels in inflammatory bowel disease. Virchows Arch. 2003, 442, 231–237. [Google Scholar]
- Zhang, Q.; Lu, Y.; Proulx, S.T.; Guo, R.; Yao, Z.; Schwarz, E.M.; Boyce, B.F.; Xing, L. Increased lymphangiogenesis in joints of mice with inflammatory arthritis. Arthritis Res. Ther. 2007, 9, R118. [Google Scholar] [CrossRef]
- Kholova, I.; Dragneva, G.; Cermakova, P.; Laidinen, S.; Kaskenpaa, N.; Hazes, T.; Cermakova, E.; Steiner, I.; Yla-Herttuala, S. Lymphatic vasculature is increased in heart valves, ischaemic and inflamed hearts and in cholesterol-rich and calcified atherosclerotic lesions. Eur. J. Clin. Invest. 2011, 41, 487–497. [Google Scholar] [CrossRef]
- Jackowski, S.; Janusch, M.; Fiedler, E.; Marsch, W.C.; Ulbrich, E.J.; Gaisbauer, G.; Dunst, J.; Kerjaschki, D.; Helmbold, P. Radiogenic lymphangiogenesis in the skin. Am. J. Pathol. 2007, 171, 338–348. [Google Scholar] [CrossRef]
- Ran, S.; Volk, L.; Hall, K.; Flister, M.J. Lymphangiogenesis and lymphatic metastasis in breast cancer. Pathophysiology 2009, 17, 229–251. [Google Scholar]
- Kerjaschki, D.; Regele, H.M.; Moosberger, I.; Nagy-Bojarski, K.; Watschinger, B.; Soleiman, A.; Birner, P.; Krieger, S.; Hovorka, A.; Silberhumer, G.; et al. Lymphatic neoangiogenesis in human kidney transplants is associated with immunologically active lymphocytic infiltrates. J. Am. Soc. Nephrol. 2004, 15, 603–612. [Google Scholar] [CrossRef]
- Kubo, H.; Cao, R.; Brakenhielm, E.; Makinen, T.; Cao, Y.; Alitalo, K. Blockade of vascular endothelial growth factor receptor-3 signaling inhibits fibroblast growth factor-2-induced lymphangiogenesis in mouse cornea. Proc. Natl. Acad. Sci. USA 2002, 99, 8868–8873. [Google Scholar]
- Kajiya, K.; Sawane, M.; Huggenberger, R.; Detmar, M. Activation of the VEGFR-3 pathway by VEGF-C attenuates UVB-induced edema formation and skin inflammation by promoting lymphangiogenesis. J. Invest. Dermatol. 2009, 129, 1292–1298. [Google Scholar] [CrossRef]
- Kim, K.E.; Koh, Y.J.; Jeon, B.H.; Jang, C.; Han, J.; Kataru, R.P.; Schwendener, R.A.; Kim, J.M.; Koh, G.Y. Role of CD11b+ macrophages in intraperitoneal lipopolysaccharide-induced aberrant lymphangiogenesis and lymphatic function in the diaphragm. Am. J. Pathol. 2009, 175, 1733–1745. [Google Scholar] [CrossRef]
- Baluk, P.; Tammela, T.; Ator, E.; Lyubynska, N.; Achen, M.G.; Hicklin, D.J.; Jeltsch, M.; Petrova, T.V.; Pytowski, B.; Stacker, S.A.; et al. Pathogenesis of persistent lymphatic vessel hyperplasia in chronic airway inflammation. J. Clin. Invest. 2005, 115, 247–257. [Google Scholar]
- Yao, L.C.; Baluk, P.; Srinivasan, R.S.; Oliver, G.; McDonald, D.M. Plasticity of button-like junctions in the endothelium of airway lymphatics in development and inflammation. Am. J. Pathol. 2012, 180, 2561–2575. [Google Scholar] [CrossRef]
- Gu, Y.; Qi, X.; Guo, S. Lymphangiogenesis induced by VEGF-C and VEGF-D promotes metastasis and a poor outcome in breast carcinoma: A retrospective study of 61 cases. Clin. Exp. Metastasis 2008, 25, 717–725. [Google Scholar] [CrossRef]
- Flister, M.J.; Wilber, A.; Hall, K.L.; Iwata, C.; Miyazono, K.; Nisato, R.E.; Pepper, M.S.; Zawieja, D.C.; Ran, S. Inflammation induces lymphangiogenesis through up-regulation of VEGFR-3 mediated by NF-kappaB and Prox1. Blood 2010, 115, 418–429. [Google Scholar] [CrossRef]
- Karin, M. Nuclear factor-kappaB in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef]
- Beinke, S.; Ley, S.C. Functions of NF-kappaB1 and NF-kappaB2 in immune cell biology. Biochem. J. 2004, 382, 393–409. [Google Scholar] [CrossRef]
- Kiriakidis, S.; Andreakos, E.; Monaco, C.; Foxwell, B.; Feldmann, M.; Paleolog, E. VEGF expression in human macrophages is NF-kappaB-dependent: Studies using adenoviruses expressing the endogenous NF-kappaB inhibitor IkappaBalpha and a kinase-defective form of the IkappaB kinase 2. J. Cell Sci. 2003, 116, 665–674. [Google Scholar] [CrossRef]
- Tsai, P.W.; Shiah, S.G.; Lin, M.T.; Wu, C.W.; Kuo, M.L. Up-regulation of vascular endothelial growth factor C in breast cancer cells by heregulin-beta 1. A critical role of p38/nuclear factor-kappa B signaling pathway. J. Biol. Chem. 2003, 278, 5750–5759. [Google Scholar]
- Ristimaki, A.; Narko, K.; Enholm, B.; Joukov, V.; Alitalo, K. Proinflammatory cytokines regulate expression of the lymphatic endothelial mitogen vascular endothelial growth factor-C. J. Biol. Chem. 1998, 273, 8413–8418. [Google Scholar]
- Iwata, C.; Kano, M.R.; Komuro, A.; Oka, M.; Kiyono, K.; Johansson, E.; Morishita, Y.; Yashiro, M.; Hirakawa, K.; Kaminishi, M.; et al. Inhibition of cyclooxygenase-2 suppresses lymph node metastasis via reduction of lymphangiogenesis. Cancer Res. 2007, 67, 10181–10189. [Google Scholar]
- Roberts, N.; Kloos, B.; Cassella, M.; Podgrabinska, S.; Persaud, K.; Wu, Y.; Pytowski, B.; Skobe, M. Inhibition of VEGFR-3 activation with the antagonistic antibody more potently suppresses lymph node and distant metastases than inactivation of VEGFR-2. Cancer Res. 2006, 66, 2650–2657. [Google Scholar]
- Mishima, K.; Watabe, T.; Saito, A.; Yoshimatsu, Y.; Imaizumi, N.; Masui, S.; Hirashima, M.; Morisada, T.; Oike, Y.; Araie, M.; et al. Prox1 induces lymphatic endothelial differentiation via integrin alpha9 and other signaling cascades. Mol. Biol. Cell 2007, 18, 1421–1429. [Google Scholar] [CrossRef]
- Trompezinski, S.; Berthier-Vergnes, O.; Denis, A.; Schmitt, D.; Viac, J. Comparative expression of vascular endothelial growth factor family members, VEGF-B, -C and -D, by normal human keratinocytes and fibroblasts. Exp. Dermatol. 2004, 13, 98–105. [Google Scholar] [CrossRef]
- Mazar, A.P.; Henkin, J.; Goldfarb, R.H. The urokinase plasminogen activator system in cancer: Implications for tumor angiogenesis and metastasis. Angiogenesis 1999, 3, 15–32. [Google Scholar] [CrossRef]
- Bassi, D.E.; Mahloogi, H.; Al-Saleem, L.; Lopez De, C.R.; Ridge, J.A.; Klein-Szanto, A.J. Elevated furin expression in aggressive human head and neck tumors and tumor cell lines. Mol. Carcinog. 2001, 31, 224–232. [Google Scholar] [CrossRef]
- Bahram, F.; Claesson-Welsh, L. VEGF-mediated signal transduction in lymphatic endothelial cells. Pathophysiology. 2010, 17, 253–261. [Google Scholar]
- Goldman, J.; Rutkowski, J.M.; Shields, J.D.; Pasquier, M.C.; Cui, Y.; Schmokel, H.G.; Willey, S.; Hicklin, D.J.; Pytowski, B.; Swartz, M.A. Cooperative and redundant roles of VEGFR-2 and VEGFR-3 signaling in adult lymphangiogenesis. FASEB J. 2007, 21, 1003–1012. [Google Scholar] [CrossRef]
- Alam, A.; Herault, J.P.; Barron, P.; Favier, B.; Fons, P.; Delesque-Touchard, N.; Senegas, I.; Laboudie, P.; Bonnin, J.; Cassan, C.; et al. Heterodimerization with vascular endothelial growth factor receptor-2 (VEGFR-2) is necessary for VEGFR-3 activity. Biochem. Biophys. Res. Commun. 2004, 324, 909–915. [Google Scholar] [CrossRef]
- Dvorak, H.F. Vascular permeability factor/vascular endothelial growth factor: A critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J. Clin. Oncol. 2002, 20, 4368–4380. [Google Scholar] [CrossRef]
- Ferrara, N. Vascular endothelial growth factor as a target for anticancer therapy. Oncologist 2004, 9, 2–10. [Google Scholar] [CrossRef]
- Detmar, M.; Brown, L.F.; Claffey, K.P.; Yeo, K.T.; Kocher, O.; Jackman, R.W.; Berse, B.; Dvorak, H.F. Overexpression of vascular permeability factor/vascular endothelial growth factor and its receptors in psoriasis. J. Exp. Med. 1994, 180, 1141–1146. [Google Scholar] [CrossRef]
- Fava, R.A.; Olsen, N.J.; Spencer-Green, G.; Yeo, K.T.; Yeo, T.K.; Berse, B.; Jackman, R.W.; Senger, D.R.; Dvorak, H.F.; Brown, L.F. Vascular permeability factor/endothelial growth factor (VPF/VEGF): Accumulation and expression in human synovial fluids and rheumatoid synovial tissue. J. Exp. Med. 1994, 180, 341–346. [Google Scholar] [CrossRef]
- Kanazawa, S.; Tsunoda, T.; Onuma, E.; Majima, T.; Kagiyama, M.; Kikuchi, K. VEGF, basic-FGF, and TGF-beta in Crohn’s disease and ulcerative colitis: A novel mechanism of chronic intestinal inflammation. Am. J. Gastroenterol. 2001, 96, 822–828. [Google Scholar]
- Lee, C.G.; Link, H.; Baluk, P.; Homer, R.J.; Chapoval, S.; Bhandari, V.; Kang, M.J.; Cohn, L.; Kim, Y.K.; McDonald, D.M.; et al. Vascular endothelial growth factor (VEGF) induces remodeling and enhances TH2-mediated sensitization and inflammation in the lung. Nat. Med. 2004, 10, 1095–1103. [Google Scholar]
- Ryden, L.; Linderholm, B.; Nielsen, N.H.; Emdin, S.; Jonsson, P.E.; Landberg, G. Tumor specific VEGF-A and VEGFR2/KDR protein are co-expressed in breast cancer. Breast Cancer Res. Treat. 2003, 82, 147–154. [Google Scholar] [CrossRef]
- Nagy, J.A.; Vasile, E.; Feng, D.; Sundberg, C.; Brown, L.F.; Detmar, M.J.; Lawitts, J.A.; Benjamin, L.; Tan, X.; Manseau, E.J.; et al. Vascular permeability factor/vascular endothelial growth factor induces lymphangiogenesis as well as angiogenesis. J. Exp. Med. 2002, 196, 1497–1506. [Google Scholar] [CrossRef]
- Cursiefen, C.; Chen, L.; Borges, L.P.; Jackson, D.; Cao, J.; Radziejewski, C.; D’Amore, P.A.; Dana, M.R.; Wiegand, S.J.; Streilein, J.W. VEGF-A stimulates lymphangiogenesis and hemangiogenesis in inflammatory neovascularization via macrophage recruitment. J. Clin. Invest. 2004, 113, 1040–1050. [Google Scholar]
- Cursiefen, C.; Cao, J.; Chen, L.; Liu, Y.; Maruyama, K.; Jackson, D.; Kruse, F.E.; Wiegand, S.J.; Dana, M.R.; Streilein, J.W. Inhibition of hemangiogenesis and lymphangiogenesis after normal-risk corneal transplantation by neutralizing VEGF promotes graft survival. Invest. Ophthalmol. Vis. Sci. 2004, 45, 2666–2673. [Google Scholar] [CrossRef]
- Hirakawa, S.; Kodama, S.; Kunstfeld, R.; Kajiya, K.; Brown, L.F.; Detmar, M. VEGF-A induces tumor and sentinel lymph node lymphangiogenesis and promotes lymphatic metastasis. J. Exp. Med. 2005, 201, 1089–1099. [Google Scholar] [CrossRef]
- Bjorndahl, M.A.; Cao, R.; Burton, J.B.; Brakenhielm, E.; Religa, P.; Galter, D.; Wu, L.; Cao, Y. Vascular endothelial growth factor-a promotes peritumoral lymphangiogenesis and lymphatic metastasis. Cancer Res. 2005, 65, 9261–9268. [Google Scholar] [CrossRef]
- Whitehurst, B.; Flister, M.J.; Bagaitkar, J.; Volk, L.; Bivens, C.M.; Pickett, B.; Castro-Rivera, E.; Brekken, R.A.; Gerard, R.D.; Ran, S. Anti-VEGF-A therapy reduces lymphatic vessel density and expression of VEGFR-3 in an orthotopic breast tumor model. Int. J. Cancer 2007, 121, 2181–2191. [Google Scholar] [CrossRef]
- Hong, Y.K.; Lange-Asschenfeldt, B.; Velasco, P.; Hirakawa, S.; Kunstfeld, R.; Brown, L.F.; Bohlen, P.; Senger, D.R.; Detmar, M. VEGF-A promotes tissue repair-associated lymphatic vessel formation via VEGFR-2 and the alpha1beta1 and alpha2beta1 integrins. FASEB J. 2004, 18, 1111–1113. [Google Scholar]
- Sawano, A.; Iwai, S.; Sakurai, Y.; Ito, M.; Shitara, K.; Nakahata, T.; Shibuya, M. Flt-1, vascular endothelial growth factor receptor 1, is a novel cell surface marker for the lineage of monocyte-macrophages in humans. Blood 2001, 97, 785–791. [Google Scholar] [CrossRef]
- Mallory, B.P.; Mead, T.J.; Wiginton, D.A.; Kulkarni, R.M.; Greenberg, J.M.; Akeson, A.L. Lymphangiogenesis in the developing lung promoted by VEGF-A. Microvasc. Res. 2006, 72, 62–73. [Google Scholar] [CrossRef]
- Hall, K.; Ran, S. Regulation of tumor angiogenesis by the local environment. Front. Biosci. 2010, 15, 195–212. [Google Scholar] [CrossRef]
- Morisada, T.; Oike, Y.; Yamada, Y.; Urano, T.; Akao, M.; Kubota, Y.; Maekawa, H.; Kimura, Y.; Ohmura, M.; Miyamoto, T.; et al. Angiopoietin-1 promotes LYVE-1-positive lymphatic vessel formation. Blood 2005, 105, 4649–4656. [Google Scholar] [CrossRef]
- Maisonpierre, P.C.; Suri, C.; Jones, P.F.; Bartunkova, S.; Wiegand, S.J.; Radziejewski, C.; Compton, D.; McClain, J.; Aldrich, T.H.; Papadopoulos, N.; et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 1997, 277, 55–60. [Google Scholar]
- Tammela, T.; Saaristo, A.; Lohela, M.; Morisada, T.; Tornberg, J.; Norrmen, C.; Oike, Y.; Pajusola, K.; Thurston, G.; Suda, T.; et al. Angiopoietin-1 promotes lymphatic sprouting and hyperplasia. Blood 2005, 105, 4642–4648. [Google Scholar]
- Gale, N.W.; Thurston, G.; Hackett, S.F.; Renard, R.; Wang, Q.; McClain, J.; Martin, C.; Witte, C.; Witte, M.H.; Jackson, D.; et al. Angiopoietin-2 is required for postnatal angiogenesis and lymphatic patterning, and only the latter role is rescued by Angiopoietin-1. Dev. Cell 2002, 3, 411–423. [Google Scholar] [CrossRef]
- Dellinger, M.; Hunter, R.; Bernas, M.; Gale, N.; Yancopoulos, G.; Erickson, R.; Witte, M. Defective remodeling and maturation of the lymphatic vasculature in Angiopoietin-2 deficient mice. Dev. Biol. 2008, 319, 309–320. [Google Scholar]
- Veikkola, T.; Lohela, M.; Ikenberg, K.; Makinen, T.; Korff, T.; Saaristo, A.; Petrova, T.; Jeltsch, M.; Augustin, H.G.; Alitalo, K. Intrinsic versus microenvironmental regulation of lymphatic endothelial cell phenotype and function. FASEB J. 2003, 17, 2006–2013. [Google Scholar] [CrossRef]
- Coffelt, S.B.; Tal, A.O.; Scholz, A.; de Palma, M.; Patel, S.; Urbich, C.; Biswas, S.K.; Murdoch, C.; Plate, K.H.; Reiss, Y.; et al. Angiopoietin-2 regulates gene expression in TIE2-expressing monocytes and augments their inherent proangiogenic functions. Cancer Res. 2010, 70, 5270–5280. [Google Scholar]
- Al Rawi, M.A.; Watkins, G.; Mansel, R.E.; Jiang, W.G. The effects of interleukin-7 on the lymphangiogenic properties of human endothelial cells. Int. J. Oncol. 2005, 27, 721–730. [Google Scholar]
- Al-Rawi, M.A.; Watkins, G.; Mansel, R.E.; Jiang, W.G. Interleukin 7 upregulates vascular endothelial growth factor D in breast cancer cells and induces lymphangiogenesis in vivo. Br. J. Surg. 2005, 92, 305–310. [Google Scholar]
- Al-Rawi, M.A.; Mansel, R.E.; Jiang, W.G. Interleukin-7 (IL-7) and IL-7 receptor (IL-7R) signalling complex in human solid tumours. Histol. Histopathol. 2003, 18, 911–923. [Google Scholar]
- Cao, R.; Bjorndahl, M.A.; Religa, P.; Clasper, S.; Garvin, S.; Galter, D.; Meister, B.; Ikomi, F.; Tritsaris, K.; Dissing, S.; et al. PDGF-BB induces intratumoral lymphangiogenesis and promotes lymphatic metastasis. Cancer Cell 2004, 6, 333–345. [Google Scholar] [CrossRef]
- Bjorndahl, M.; Cao, R.; Nissen, L.J.; Clasper, S.; Johnson, L.A.; Xue, Y.; Zhou, Z.; Jackson, D.; Hansen, A.J.; Cao, Y. Insulin-like growth factors 1 and 2 induce lymphangiogenesis in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 15593–15598. [Google Scholar]
- Cao, R.; Bjorndahl, M.A.; Gallego, M.I.; Chen, S.; Religa, P.; Hansen, A.J.; Cao, Y. Hepatocyte growth factor is a lymphangiogenic factor with an indirect mechanism of action. Blood 2006, 107, 3531–3536. [Google Scholar] [CrossRef]
- Banziger-Tobler, N.E.; Halin, C.; Kajiya, K.; Detmar, M. Growth hormone promotes lymphangiogenesis. Am. J. Pathol. 2008, 173, 586–597. [Google Scholar] [CrossRef]
- Backhed, F.; Crawford, P.A.; O’Donnell, D.; Gordon, J.I. Postnatal lymphatic partitioning from the blood vasculature in the small intestine requires fasting-induced adipose factor. Proc. Natl. Acad. Sci. USA 2007, 104, 606–611. [Google Scholar]
- Yoon, C.M.; Hong, B.S.; Moon, H.G.; Lim, S.; Suh, P.G.; Kim, Y.K.; Chae, C.B.; Gho, Y.S. Sphingosine-1-phosphate promotes lymphangiogenesis by stimulating S1P1/Gi/PLC/Ca2+ signaling pathways. Blood 2008, 112, 1129–1138. [Google Scholar] [CrossRef]
- Timoshenko, A.V.; Chakraborty, C.; Wagner, G.F.; Lala, P.K. COX-2-mediated stimulation of the lymphangiogenic factor VEGF-C in human breast cancer. Br. J. Cancer 2006, 94, 1154–1163. [Google Scholar]
- Balkwill, F.; Charles, K.A.; Mantovani, A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell 2005, 7, 211–217. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef]
- Allavena, P.; Germano, G.; Marchesi, F.; Mantovani, A. Chemokines in cancer related inflammation. Exp. Cell Res. 2011, 317, 664–673. [Google Scholar] [CrossRef]
- Knowles, H.J.; Harris, A.L. Macrophages and the hypoxic tumour microenvironment. Front. Biosci. 2007, 12, 4298–4314. [Google Scholar] [CrossRef]
- Leek, R.D.; Landers, R.J.; Harris, A.L.; Lewis, C.E. Necrosis correlates with high vascular density and focal macrophage infiltration in invasive carcinoma of the breast. Br. J. Cancer 1999, 79, 991–995. [Google Scholar]
- Brown, L.F.; Dvorak, A.M.; Dvorak, H.F. Leaky vessels, fibrin deposition, and fibrosis: A sequence of events common to solid tumors and to many other types of disease. Am. Rev. Respir. Dis. 1989, 140, 1104–1107. [Google Scholar]
- Karin, M.; Greten, F.R. NF-kappaB: Linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005, 5, 749–759. [Google Scholar] [CrossRef]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef]
- Allavena, P.; Sica, A.; Solinas, G.; Porta, C.; Mantovani, A. The inflammatory micro-environment in tumor progression: The role of tumor-associated macrophages. Crit Rev. Oncol. Hematol. 2008, 66, 1–9. [Google Scholar] [CrossRef]
- Talmadge, J.E.; Donkor, M.; Scholar, E. Inflammatory cell infiltration of tumors: Jekyll or Hyde. Cancer Metastasis Rev. 2007, 26, 373–400. [Google Scholar] [CrossRef]
- Rinderknecht, M.; Detmar, M. Tumor lymphangiogenesis and melanoma metastasis. J. Cell Physiol. 2008, 216, 347–354. [Google Scholar] [CrossRef]
- Grabau, D.; Jensen, M.B.; Rank, F.; Blichert-Toft, M. Axillary lymph node micrometastases in invasive breast cancer: National figures on incidence and overall survival. APMIS 2007, 115, 828–837. [Google Scholar] [CrossRef]
- Sivridis, E.; Giatromanolaki, A.; Galazios, G.; Koukourakis, M.I. Node-related factors and survival in node-positive breast carcinomas. Breast 2006, 15, 382–389. [Google Scholar] [CrossRef]
- Colleoni, M.; Rotmensz, N.; Maisonneuve, P.; Sonzogni, A.; Pruneri, G.; Casadio, C.; Luini, A.; Veronesi, P.; Intra, M.; Galimberti, V.; et al. Prognostic role of the extent of peritumoral vascular invasion in operable breast cancer. Ann. Oncol. 2007, 18, 1632–1640. [Google Scholar]
- Viale, G.; Zurrida, S.; Maiorano, E.; Mazzarol, G.; Pruneri, G.; Paganelli, G.; Maisonneuve, P.; Veronesi, U. Predicting the status of axillary sentinel lymph nodes in 4351 patients with invasive breast carcinoma treated in a single institution. Cancer 2005, 103, 492–500. [Google Scholar] [CrossRef]
- Truong, P.T.; Vinh-Hung, V.; Cserni, G.; Woodward, W.A.; Tai, P.; Vlastos, G. The number of positive nodes and the ratio of positive to excised nodes are significant predictors of survival in women with micrometastatic node-positive breast cancer. Eur. J. Cancer 2008, 44, 1670–1677. [Google Scholar] [CrossRef]
- Woo, C.S.; Silberman, H.; Nakamura, S.K.; Ye, W.; Sposto, R.; Colburn, W.; Waisman, J.R.; Silverstein, M.J. Lymph node status combined with lymphovascular invasion creates a more powerful tool for predicting outcome in patients with invasive breast cancer. Am. J. Surg. 2002, 184, 337–340. [Google Scholar] [CrossRef]
- Skobe, M.; Hawighorst, T.; Jackson, D.G.; Prevo, R.; Janes, L.; Velasco, P.; Riccardi, L.; Alitalo, K.; Claffey, K.; Detmar, M. Induction of tumor lymphangiogenesis by VEGF-C promotes breast cancer metastasis. Nat. Med. 2001, 7, 192–198. [Google Scholar] [CrossRef]
- Mattila, M.M.; Ruohola, J.K.; Karpanen, T.; Jackson, D.G.; Alitalo, K.; Harkonen, P.L. VEGF-C induced lymphangiogenesis is associated with lymph node metastasis in orthotopic MCF-7 tumors. Int. J. Cancer 2002, 98, 946–951. [Google Scholar] [CrossRef]
- He, Y.; Kozaki, K.; Karpanen, T.; Koshikawa, K.; Yla-Herttuala, S.; Takahashi, T.; Alitalo, K. Suppression of tumor lymphangiogenesis and lymph node metastasis by blocking vascular endothelial growth factor receptor 3 signaling. J. Natl. Cancer Inst. 2002, 94, 819–825. [Google Scholar] [CrossRef]
- Burton, J.B.; Priceman, S.J.; Sung, J.L.; Brakenhielm, E.; An, D.S.; Pytowski, B.; Alitalo, K.; Wu, L. Suppression of prostate cancer nodal and systemic metastasis by blockade of the lymphangiogenic axis. Cancer Res. 2008, 68, 7828–7837. [Google Scholar]
- Hoshida, T.; Isaka, N.; Hagendoorn, J.; di Tomaso, E.; Chen, Y.L.; Pytowski, B.; Fukumura, D.; Padera, T.P.; Jain, R.K. Imaging steps of lymphatic metastasis reveals that vascular endothelial growth factor-C increases metastasis by increasing delivery of cancer cells to lymph nodes: Therapeutic implications. Cancer Res. 2006, 66, 8065–8075. [Google Scholar]
- Yanai, Y.; Furuhata, T.; Kimura, Y.; Yamaguchi, K.; Yasoshima, T.; Mitaka, T.; Mochizuki, Y.; Hirata, K. Vascular endothelial growth factor C promotes human gastric carcinoma lymph node metastasis in mice. J. Exp. Clin. Cancer Res. 2001, 20, 419–428. [Google Scholar]
- Kawakami, M.; Yanai, Y.; Hata, F.; Hirata, K. Vascular endothelial growth factor C promotes lymph node metastasis in a rectal cancer orthotopic model. Surg. Today 2005, 35, 131–138. [Google Scholar] [CrossRef]
- Shimizu, K.; Kubo, H.; Yamaguchi, K.; Kawashima, K.; Ueda, Y.; Matsuo, K.; Awane, M.; Shimahara, Y.; Takabayashi, A.; Yamaoka, Y.; et al. Suppression of VEGFR-3 signaling inhibits lymph node metastasis in gastric cancer. Cancer Sci. 2004, 95, 328–333. [Google Scholar] [CrossRef]
- Chen, Z.; Varney, M.L.; Backora, M.W.; Cowan, K.; Solheim, J.C.; Talmadge, J.E.; Singh, R.K. Down-regulation of vascular endothelial cell growth factor-C expression using small interfering RNA vectors in mammary tumors inhibits tumor lymphangiogenesis and spontaneous metastasis and enhances survival. Cancer Res. 2005, 65, 9004–9011. [Google Scholar]
- Wong, S.Y.; Haack, H.; Crowley, D.; Barry, M.; Bronson, R.T.; Hynes, R.O. Tumor-secreted vascular endothelial growth factor-C is necessary for prostate cancer lymphangiogenesis, but lymphangiogenesis is unnecessary for lymph node metastasis. Cancer Res. 2005, 65, 9789–9798. [Google Scholar] [CrossRef]
- Shibata, M.A.; Morimoto, J.; Shibata, E.; Otsuki, Y. Combination therapy with short interfering RNA vectors against VEGF-C and VEGF-A suppresses lymph node and lung metastasis in a mouse immunocompetent mammary cancer model. Cancer Gene Ther. 2008, 15, 776–786. [Google Scholar] [CrossRef]
- Thelen, A.; Scholz, A.; Benckert, C.; von Marschall, Z.; Schroder, M.; Wiedenmann, B.; Neuhaus, P.; Rosewicz, S.; Jonas, S. VEGF-D promotes tumor growth and lymphatic spread in a mouse model of hepatocellular carcinoma. Int. J. Cancer 2008, 122, 2471–2481. [Google Scholar]
- Von, M.Z.; Scholz, A.; Stacker, S.A.; Achen, M.G.; Jackson, D.G.; Alves, F.; Schirner, M.; Haberey, M.; Thierauch, K.H.; Wiedenmann, B.; et al. Vascular endothelial growth factor-D induces lymphangiogenesis and lymphatic metastasis in models of ductal pancreatic cancer. Int. J. Oncol. 2005, 27, 669–679. [Google Scholar]
- Koch, M.; Dettori, D.; van Nuffelen, A.; Souffreau, J.; Marconcini, L.; Wallays, G.; Moons, L.; Bruyere, F.; Oliviero, S.; Noel, A.; et al. VEGF-D deficiency in mice does not affect embryonic or postnatal lymphangiogenesis but reduces lymphatic metastasis. J. Pathol. 2009, 219, 356–364. [Google Scholar] [CrossRef]
- Condeelis, J.; Pollard, J.W. Macrophages: Obligate partners for tumor cell migration, invasion, and metastasis. Cell 2006, 124, 263–266. [Google Scholar] [CrossRef]
- Leek, R.D.; Harris, A.L. Tumor-associated macrophages in breast cancer. J. Mammary Gland Biol. Neoplasia 2002, 7, 177–189. [Google Scholar] [CrossRef]
- Murphy, G.; Gavrilovic, J. Proteolysis and cell migration: Creating a path? Curr. Opin. Cell Biol. 1999, 11, 614–621. [Google Scholar] [CrossRef]
- Rolli, M.; Fransvea, E.; Pilch, J.; Saven, A.; Felding-Habermann, B. Activated integrin alphavbeta3 cooperates with metalloproteinase MMP-9 in regulating migration of metastatic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 9482–9487. [Google Scholar]
- Jadhav, U.; Chigurupati, S.; Lakka, S.S.; Mohanam, S. Inhibition of matrix metalloproteinase-9 reduces in vitro invasion and angiogenesis in human microvascular endothelial cells. Int. J. Oncol. 2004, 25, 1407–1414. [Google Scholar]
- Patenaude, A.; Parker, J.; Karsan, A. Involvement of endothelial progenitor cells in tumor vascularization. Microvasc. Res. 2010, 79, 217–223. [Google Scholar] [CrossRef]
- Gordon, S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003, 3, 23–35. [Google Scholar] [CrossRef]
- Solinas, G.; Germano, G.; Mantovani, A.; Allavena, P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J. Leukoc. Biol. 2009, 86, 1065–1073. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer 2009, 9, 239–252. [Google Scholar] [CrossRef]
- Jeon, B.H.; Jang, C.; Han, J.; Kataru, R.P.; Piao, L.; Jung, K.; Cha, H.J.; Schwendener, R.A.; Jang, K.Y.; Kim, K.S.; et al. Profound but dysfunctional lymphangiogenesis via vascular endothelial growth factor ligands from CD11b+ macrophages in advanced ovarian cancer. Cancer Res. 2008, 68, 1100–1109. [Google Scholar]
- Halin, C.; Tobler, N.E.; Vigl, B.; Brown, L.F.; Detmar, M. VEGF-A produced by chronically inflamed tissue induces lymphangiogenesis in draining lymph nodes. Blood 2007, 110, 3158–3167. [Google Scholar] [CrossRef]
- Enholm, B.; Karpanen, T.; Jeltsch, M.; Kubo, H.; Stenback, F.; Prevo, R.; Jackson, D.G.; Yla-Herttuala, S.; Alitalo, K. Adenoviral expression of vascular endothelial growth factor-C induces lymphangiogenesis in the skin. Circ. Res. 2001, 88, 623–629. [Google Scholar] [CrossRef]
- Achen, M.G.; Jeltsch, M.; Kukk, E.; Makinen, T.; Vitali, A.; Wilks, A.F.; Alitalo, K.; Stacker, S.A. Vascular endothelial growth factor D (VEGF-D) is a ligand for the tyrosine kinases VEGF receptor 2 (Flk1) and VEGF receptor 3 (Flt4). Proc. Natl. Acad. Sci. USA 1998, 95, 548–553. [Google Scholar]
- Zhu, L.; Loo, W.T.; Cheng, C.W.; Chow, L.W. Possible predictive markers related to micro-metastasis in breast cancer patients. Oncol. Rep. 2006, 15, 1217–1223. [Google Scholar]
- Chen, P.; Huang, Y.; Bong, R.; Ding, Y.; Song, N.; Wang, X.; Song, X.; Luo, Y. Tumor-associated macrophages promote angiogenesis and melanoma growth via adrenomedullin in a paracrine and autocrine manner. Clin. Cancer Res. 2011, 17, 7230–7239. [Google Scholar] [CrossRef]
- Fritz-Six, K.L.; Dunworth, W.P.; Li, M.; Caron, K.M. Adrenomedullin signaling is necessary for murine lymphatic vascular development. J. Clin. Invest. 2008, 118, 40–50. [Google Scholar] [CrossRef]
- Oehler, M.K.; Fischer, D.C.; Orlowska-Volk, M.; Herrle, F.; Kieback, D.G.; Rees, M.C.; Bicknell, R. Tissue and plasma expression of the angiogenic peptide adrenomedullin in breast cancer. Br. J. Cancer 2003, 89, 1927–1933. [Google Scholar] [CrossRef]
- Kajiya, K.; Hirakawa, S.; Ma, B.; Drinnenberg, I.; Detmar, M. Hepatocyte growth factor promotes lymphatic vessel formation and function. EMBO J. 2005, 24, 2885–2895. [Google Scholar] [CrossRef]
- Uchida, D.; Kawamata, H.; Omotehara, F.; Nakashiro, K.; Kimura-Yanagawa, T.; Hino, S.; Begum, N.M.; Hoque, M.O.; Yoshida, H.; Sato, M.; et al. Role of HGF/c-met system in invasion and metastasis of oral squamous cell carcinoma cells in vitro and its clinical significance. Int. J. Cancer 2001, 93, 489–496. [Google Scholar] [CrossRef]
- Denkert, C.; Winzer, K.J.; Muller, B.M.; Weichert, W.; Pest, S.; Kobel, M.; Kristiansen, G.; Reles, A.; Siegert, A.; Guski, H.; et al. Elevated expression of cyclooxygenase-2 is a negative prognostic factor for disease free survival and overall survival in patients with breast carcinoma. Cancer 2003, 97, 2978–2987. [Google Scholar]
- Leek, R.D.; Harris, A.L.; Lewis, C.E. Cytokine networks in solid human tumors: Regulation of angiogenesis. J. Leukoc. Biol. 1994, 56, 423–435. [Google Scholar]
- Chang, L.K.; Garcia-Cardena, G.; Farnebo, F.; Fannon, M.; Chen, E.J.; Butterfield, C.; Moses, M.A.; Mulligan, R.C.; Folkman, J.; Kaipainen, A. Dose-dependent response of FGF-2 for lymphangiogenesis. Proc. Natl. Acad. Sci. USA 2004, 101, 11658–11663. [Google Scholar]
- Elagoz, S.; Egilmez, R.; Koyuncu, A.; Muslehiddinoglu, A.; Arici, S. The intratumoral microvessel density and expression of bFGF and nm23-H1 in colorectal cancer. Pathol. Oncol. Res. 2006, 12, 21–27. [Google Scholar] [CrossRef]
- Lewis, C.E.; Leek, R.; Harris, A.; McGee, J.O. Cytokine regulation of angiogenesis in breast cancer: The role of tumor-associated macrophages. J. Leukoc. Biol. 1995, 57, 747–751. [Google Scholar]
- Leek, R.D.; Landers, R.; Fox, S.B.; Ng, F.; Harris, A.L.; Lewis, C.E. Association of tumour necrosis factor alpha and its receptors with thymidine phosphorylase expression in invasive breast carcinoma. Br. J. Cancer 1998, 77, 2246–2251. [Google Scholar] [CrossRef]
- Davies, B.; Waxman, J.; Wasan, H.; Abel, P.; Williams, G.; Krausz, T.; Neal, D.; Thomas, D.; Hanby, A.; Balkwill, F. Levels of matrix metalloproteases in bladder cancer correlate with tumor grade and invasion. Cancer Res. 1993, 53, 5365–5369. [Google Scholar]
- Nakamura, E.S.; Koizumi, K.; Kobayashi, M.; Saiki, I. Inhibition of lymphangiogenesis-related properties of murine lymphatic endothelial cells and lymph node metastasis of lung cancer by the matrix metalloproteinase inhibitor MMI270. Cancer Sci. 2004, 95, 25–31. [Google Scholar] [CrossRef]
- Hao, L.; Zhang, C.; Qiu, Y.; Wang, L.; Luo, Y.; Jin, M.; Zhang, Y.; Guo, T.B.; Matsushima, K.; Zhang, Y. Recombination of CXCR4, VEGF, and MMP-9 predicting lymph node metastasis in human breast cancer. Cancer Lett. 2007, 253, 34–42. [Google Scholar] [CrossRef]
- Friedmann, Y.; Vlodavsky, I.; Aingorn, H.; Aviv, A.; Peretz, T.; Pecker, I.; Pappo, O. Expression of heparanase in normal, dysplastic, and neoplastic human colonic mucosa and strom. Evidence for its role in colonic tumorigenesis. Am. J. Pathol. 2000, 157, 1167–1175. [Google Scholar] [CrossRef]
- Cohen-Kaplan, V.; Naroditsky, I.; Zetser, A.; Ilan, N.; Vlodavsky, I.; Doweck, I. Heparanase induces VEGF C and facilitates tumor lymphangiogenesis. Int. J. Cancer 2008, 123, 2566–2573. [Google Scholar] [CrossRef]
- Maxhimer, J.B.; Quiros, R.M.; Stewart, R.; Dowlatshahi, K.; Gattuso, P.; Fan, M.; Prinz, R.A.; Xu, X. Heparanase-1 expression is associated with the metastatic potential of breast cancer. Surgery 2002, 132, 326–333. [Google Scholar] [CrossRef]
- Hildenbrand, R.; Wolf, G.; Bohme, B.; Bleyl, U.; Steinborn, A. Urokinase plasminogen activator receptor (CD87) expression of tumor-associated macrophages in ductal carcinoma in situ, breast cancer, and resident macrophages of normal breast tissue. J. Leukoc. Biol. 1999, 66, 40–49. [Google Scholar]
- McColl, B.K.; Baldwin, M.E.; Roufail, S.; Freeman, C.; Moritz, R.L.; Simpson, R.J.; Alitalo, K.; Stacker, S.A.; Achen, M.G. Plasmin Activates the Lymphangiogenic Growth Factors VEGF-C and VEGF-D. J. Exp. Med. 2003, 198, 863–868. [Google Scholar] [CrossRef]
- Sumiyoshi, K.; Serizawa, K.; Urano, T.; Takada, Y.; Takada, A.; Baba, S. Plasminogen activator system in human breast cancer. Int. J. Cancer 1992, 50, 345–348. [Google Scholar] [CrossRef]
- Hubbard, N.E.; Lim, D.; Mukutmoni, M.; Cai, A.; Erickson, K.L. Expression and regulation of murine macrophage angiopoietin-2. Cell Immunol. 2005, 234, 102–109. [Google Scholar] [CrossRef]
- Nguyen, V.P.; Chen, S.H.; Trinh, J.; Kim, H.; Coomber, B.L.; Dumont, D.J. Differential response of lymphatic, venous and arterial endothelial cells to angiopoietin-1 and angiopoietin-2. BMC Cell Biol. 2007, 8, 10. [Google Scholar] [CrossRef]
- Fagiani, E.; Lorentz, P.; Kopfstein, L.; Christofori, G. Angiopoietin-1 and -2 exert antagonistic functions in tumor angiogenesis, yet both induce lymphangiogenesis. Cancer Res. 2011, 71, 5717–5727. [Google Scholar] [CrossRef]
- Sfiligoi, C.; de Luca, A.; Cascone, I.; Sorbello, V.; Fuso, L.; Ponzone, R.; Biglia, N.; Audero, E.; Arisio, R.; Bussolino, F.; et al. Angiopoietin-2 expression in breast cancer correlates with lymph node invasion and short survival. Int. J. Cancer 2003, 103, 466–474. [Google Scholar] [CrossRef]
- Ben Baruch, A. Host microenvironment in breast cancer development: Inflammatory cells, cytokines and chemokines in breast cancer progression: Reciprocal tumor-microenvironment interactions. Breast Cancer Res. 2003, 5, 31–36. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.L.; Rak, J.W. Host microenvironment in breast cancer development: Inflammatory and immune cells in tumour angiogenesis and arteriogenesis. Breast Cancer Res. 2003, 5, 83–88. [Google Scholar] [CrossRef]
- Dobrovolskaia, M.A.; Kozlov, S.V. Inflammation and cancer: When NF-kappaB amalgamates the perilous partnership. Curr. Cancer Drug Targets 2005, 5, 325–344. [Google Scholar] [CrossRef]
- Ditsworth, D.; Zong, W.X. NF-kappaB: Key mediator of inflammation-associated cancer. Cancer Biol. Ther. 2004, 3, 1214–1216. [Google Scholar] [CrossRef]
- Albini, A.; Tosetti, F.; Benelli, R.; Noonan, D.M. Tumor inflammatory angiogenesis and its chemoprevention. Cancer Res. 2005, 65, 10637–10641. [Google Scholar] [CrossRef]
- Matsumoto, G.; Namekawa, J.; Muta, M.; Nakamura, T.; Bando, H.; Tohyama, K.; Toi, M.; Umezawa, K. Targeting of nuclear factor kappaB pathways by dehydroxymethylepoxyquinomicin, a novel inhibitor of breast carcinomas: Antitumor and antiangiogenic potential in vivo. Clin. Cancer Res. 2005, 11, 1287–1293. [Google Scholar]
- Mouta, C.; Heroult, M. Inflammatory triggers of lymphangiogenesis. Lymphat. Res. Biol. 2003, 1, 201–218. [Google Scholar] [CrossRef]
- Bharti, A.C.; Aggarwal, B.B. Chemopreventive agents induce suppression of nuclear factor-kappaB leading to chemosensitization. Ann. NY Acad. Sci. 2002, 973, 392–395. [Google Scholar] [CrossRef]
- Huang, S.; Pettaway, C.A.; Uehara, H.; Bucana, C.D.; Fidler, I.J. Blockade of NF-kappaB activity in human prostate cancer cells is associated with suppression of angiogenesis, invasion, and metastasis. Oncogene 2001, 20, 4188–4197. [Google Scholar] [CrossRef]
- Zhang, C.; Chakravarty, D.; Sakabe, I.; Mewani, R.R.; Boudreau, H.E.; Kumar, D.; Ahmad, I.; Kasid, U.N. Role of SCC-S2 in experimental metastasis and modulation of VEGFR-2, MMP-1, and MMP-9 expression. Mol. Ther. 2006, 13, 947–955. [Google Scholar]
- Kurahara, H.; Shinchi, H.; Mataki, Y.; Maemura, K.; Noma, H.; Kubo, F.; Sakoda, M.; Ueno, S.; Natsugoe, S.; Takao, S. Significance of M2-polarized tumor-associated macrophage in pancreatic cancer. J. Surg. Res. 2011, 167, e211–e219. [Google Scholar] [CrossRef]
- Zhang, B.; Yao, G.; Zhang, Y.; Gao, J.; Yang, B.; Rao, Z.; Gao, J. M2-polarized tumor-associated macrophages are associated with poor prognoses resulting from accelerated lymphangiogenesis in lung adenocarcinoma. Clinics (Sao Paulo) 2011, 66, 1879–1886. [Google Scholar] [CrossRef]
- Zhang, B.C.; Gao, J.; Wang, J.; Rao, Z.G.; Wang, B.C.; Gao, J.F. Tumor-associated macrophages infiltration is associated with peritumoral lymphangiogenesis and poor prognosis in lung adenocarcinoma. Med. Oncol. 2011, 28, 1447–1452. [Google Scholar] [CrossRef]
- Bolat, F.; Kayaselcuk, F.; Nursal, T.Z.; Yagmurdur, M.C.; Bal, N.; Demirhan, B. Microvessel density, VEGF expression, and tumor-associated macrophages in breast tumors: Correlations with prognostic parameters. J. Exp. Clin. Cancer Res. 2006, 25, 365–372. [Google Scholar]
- Ohta, M.; Kitadai, Y.; Tanaka, S.; Yoshihara, M.; Yasui, W.; Mukaida, N.; Haruma, K.; Chayama, K. Monocyte chemoattractant protein-1 expression correlates with macrophage infiltration and tumor vascularity in human esophageal squamous cell carcinomas. Int. J. Cancer 2002, 102, 220–224. [Google Scholar] [CrossRef]
- Storr, S.J.; Safuan, S.; Mitra, A.; Elliott, F.; Walker, C.; Vasko, M.J.; Ho, B.; Cook, M.; Mohammed, R.A.; Patel, P.M.; et al. Objective assessment of blood and lymphatic vessel invasion and association with macrophage infiltration in cutaneous melanoma. Mod. Pathol. 2011, 25, 493–504. [Google Scholar]
- Valkovic, T.; Dobrila, F.; Melato, M.; Sasso, F.; Rizzardi, C.; Jonjic, N. Correlation between vascular endothelial growth factor, angiogenesis, and tumor-associated macrophages in invasive ductal breast carcinoma. Virchows Arch. 2002, 440, 583–588. [Google Scholar] [CrossRef]
- Campbell, M.J.; Tonlaar, N.Y.; Garwood, E.R.; Huo, D.; Moore, D.H.; Khramtsov, A.I.; Au, A.; Baehner, F.; Chen, Y.; Malaka, D.O.; et al. Proliferating macrophages associated with high grade, hormone receptor negative breast cancer and poor clinical outcome. Breast Cancer Res. Treat. 2011, 128, 703–711. [Google Scholar] [CrossRef]
- Schoppmann, S.F.; Fenzl, A.; Nagy, K.; Unger, S.; Bayer, G.; Geleff, S.; Gnant, M.; Horvat, R.; Jakesz, R.; Birner, P. VEGF-C expressing tumor-associated macrophages in lymph node positive breast cancer: Impact on lymphangiogenesis and survival. Surgery 2006, 139, 839–846. [Google Scholar] [CrossRef]
- Shi, L.; Lei, D.; Ma, C.; Xu, F.; Li, Y.; Wang, Y.; Cong, N.; Liu, D.; Pan, X.L. Clinicopathological implications of tumour-associated macrophages and vascularization in sinonasal melanoma. J. Int. Med. Res. 2010, 38, 1276–1286. [Google Scholar]
- Kawai, Y.; Hosaka, K.; Kaidoh, M.; Minami, T.; Kodama, T.; Ohhashi, T. Heterogeneity in immunohistochemical, genomic, and biological properties of human lymphatic endothelial cells between initial and collecting lymph vessels. Lymphat. Res. Biol. 2008, 6, 15–27. [Google Scholar] [CrossRef]
- Algars, A.; Irjala, H.; Vaittinen, S.; Huhtinen, H.; Sundstrom, J.; Salmi, M.; Ristamaki, R.; Jalkanen, S. Type and location of tumor-infiltrating macrophages and lymphatic vessels predict survival of colorectal cancer patients. Int. J. Cancer 2012, 131, 864–873. [Google Scholar] [CrossRef]
- Heusinkveld, M.; van der Burg, S.H. Identification and manipulation of tumor associated macrophages in human cancers. J. Transl. Med. 2011, 9, 216. [Google Scholar] [CrossRef]
- Fischer, C.; Jonckx, B.; Mazzone, M.; Zacchigna, S.; Loges, S.; Pattarini, L.; Chorianopoulos, E.; Liesenborghs, L.; Koch, M.; de Mol, M.; et al. Anti-PlGF inhibits growth of VEGF(R)-inhibitor-resistant tumors without affecting healthy vessels. Cell 2007, 131, 463–475. [Google Scholar] [CrossRef]
- Kubota, Y.; Takubo, K.; Shimizu, T.; Ohno, H.; Kishi, K.; Shibuya, M.; Saya, H.; Suda, T. M-CSF inhibition selectively targets pathological angiogenesis and lymphangiogenesis. J. Exp. Med. 2009, 206, 1089–1102. [Google Scholar] [CrossRef]
- Yang, H.; Kim, C.; Kim, M.J.; Schwendener, R.A.; Alitalo, K.; Heston, W.; Kim, I.; Kim, W.J.; Koh, G.Y. Soluble vascular endothelial growth factor receptor-3 suppresses lymphangiogenesis and lymphatic metastasis in bladder cancer. Mol. Cancer 2011, 10, 36. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Schwarz, E.; Davies, M.; Jo, M.; Gates, J.; Wu, J.; Zhang, X.; Lieberman, J.R. Differences in the cytokine profiles associated with prostate cancer cell induced osteoblastic nd osteolytic lesions in bone. J. Orthop. Res. 2003, 21, 62–72. [Google Scholar] [CrossRef]
- Achen, M.G.; Mann, G.B.; Stacker, S.A. Targeting lymphangiogenesis to prevent tumour metastasis. Br. J. Cancer 2006, 94, 1355–1360. [Google Scholar]
- Salven, P.; Mustjoki, S.; Alitalo, R.; Alitalo, K.; Rafii, S. VEGFR-3 and CD133 identify a population of CD34+ lymphatic/vascular endothelial precursor cells. Blood 2003, 101, 168–172. [Google Scholar] [CrossRef]
- Bogos, K.; Renyi-Vamos, F.; Dobos, J.; Kenessey, I.; Tovari, J.; Timar, J.; Strausz, J.; Ostoros, G.; Klepetko, W.; Ankersmit, H.J.; et al. High VEGFR-3-positive circulating lymphatic/vascular endothelial progenitor cell level is associated with poor prognosis in human small cell lung cancer. Clin. Cancer Res. 2009, 15, 1741–1746. [Google Scholar] [CrossRef]
- Jiang, S.; Bailey, A.S.; Goldman, D.C.; Swain, J.R.; Wong, M.H.; Streeter, P.R.; Fleming, W.H. Hematopoietic stem cells contribute to lymphatic endothelium. PLoS ONE 2008, 3, e3812. [Google Scholar]
- Yamashita, M.; Iwama, N.; Date, F.; Shibata, N.; Miki, H.; Yamauchi, K.; Sawai, T.; Sato, S.; Takahashi, T.; Ono, M. Macrophages participate in lymphangiogenesis in idiopathic diffuse alveolar damage through CCL19-CCR7 signal. Hum. Pathol. 2009, 40, 1553–1563. [Google Scholar] [CrossRef]
- Gordon, E.J.; Rao, S.; Pollard, J.W.; Nutt, S.L.; Lang, R.A.; Harvey, N.L. Macrophages define dermal lymphatic vessel calibre during development by regulating lymphatic endothelial cell proliferation. Development 2010, 137, 3899–3910. [Google Scholar]
- Lewis, C.E.; de Palma, M.; Naldini, L. Tie2-expressing monocytes and tumor angiogenesis: Regulation by hypoxia and angiopoietin-2. Cancer Res. 2007, 67, 8429–8432. [Google Scholar] [CrossRef]
- Karikoski, M.; Irjala, H.; Maksimow, M.; Miiluniemi, M.; Granfors, K.; Hernesniemi, S.; Elima, K.; Moldenhauer, G.; Schledzewski, K.; Kzhyshkowska, J.; et al. Clever-1/Stabilin-1 regulates lymphocyte migration within lymphatics and leukocyte entrance to sites of inflammation. Eur. J. Immunol. 2009, 39, 3477–3487. [Google Scholar] [CrossRef]
- Pflicke, H.; Sixt, M. Preformed portals facilitate dendritic cell entry into afferent lymphatic vessels. J. Exp. Med. 2009, 206, 2925–2935. [Google Scholar]
- Randolph, G.J.; Angeli, V.; Swartz, M.A. Dendritic-cell trafficking to lymph nodes through lymphatic vessels. Nat. Rev. Immunol. 2005, 5, 617–628. [Google Scholar] [CrossRef]
- Bellingan, G.J.; Caldwell, H.; Howie, S.E.; Dransfield, I.; Haslett, C. In vivo fate of the inflammatory macrophage during the resolution of inflammation: Inflammatory macrophages do not die locally, but emigrate to the draining lymph nodes. J. Immunol. 1996, 157, 2577–2585. [Google Scholar]
- Weissman, I.L.; Anderson, D.J.; Gage, F. Stem and progenitor cells: Origins, phenotypes, lineage commitments, and transdifferentiations. Annu. Rev. Cell Dev. Biol. 2001, 17, 387–403. [Google Scholar] [CrossRef]
- Staton, C.A.; Stribbling, S.M.; Tazzyman, S.; Hughes, R.; Brown, N.J.; Lewis, C.E. Current methods for assaying angiogenesis in vitro and in vivo. Int. J. Exp. Pathol. 2004, 85, 233–248. [Google Scholar] [CrossRef]
- Verma, S.; Kuliszewski, M.A.; Li, S.H.; Szmitko, P.E.; Zucco, L.; Wang, C.H.; Badiwala, M.V.; Mickle, D.A.; Weisel, R.D.; Fedak, P.W.; et al. C-reactive protein attenuates endothelial progenitor cell survival, differentiation, and function: Further evidence of a mechanistic link between C-reactive protein and cardiovascular disease. Circulation 2004, 109, 2058–2067. [Google Scholar] [CrossRef]
- Ribatti, D. The involvement of endothelial progenitor cells in tumor angiogenesis. J. Cell Mol. Med. 2004, 8, 294–300. [Google Scholar] [CrossRef]
- Bonder, C.S.; Sun, W.Y.; Matthews, T.; Cassano, C.; Li, X.; Ramshaw, H.S.; Pitson, S.M.; Lopez, A.F.; Coates, P.T.; Proia, R.L.; et al. Sphingosine kinase regulates the rate of endothelial progenitor cell differentiation. Blood 2009, 113, 2108–2117. [Google Scholar] [CrossRef]
- El-Chemaly, S.; Malide, D.; Zudaire, E.; Ikeda, Y.; Weinberg, B.A.; Pacheco-Rodriguez, G.; Rosas, I.O.; Aparicio, M.; Ren, P.; MacDonald, S.D.; et al. Abnormal lymphangiogenesis in idiopathic pulmonary fibrosis with insights into cellular and molecular mechanisms. Proc. Natl. Acad. Sci. USA 2009, 106, 3958–3963. [Google Scholar]
- Conrad, C.; Niess, H.; Huss, R.; Huber, S.; von Luettichau, I.; Nelson, P.J.; Ott, H.C.; Jauch, K.W.; Bruns, C.J. Multipotent mesenchymal stem cells acquire a lymphendothelial phenotype and enhance lymphatic regeneration in vivo. Circulation 2009, 119, 281–289. [Google Scholar] [CrossRef]
- Yan, A.; Avraham, T.; Zampell, J.C.; Haviv, Y.S.; Weitman, E.; Mehrara, B.J. Adipose-derived stem cells promote lymphangiogenesis in response to VEGF-C stimulation or TGF-beta1 inhibition. Future Oncol. 2011, 7, 1457–1473. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ran, S.; Montgomery, K.E. Macrophage-Mediated Lymphangiogenesis: The Emerging Role of Macrophages as Lymphatic Endothelial Progenitors. Cancers 2012, 4, 618-657. https://doi.org/10.3390/cancers4030618
Ran S, Montgomery KE. Macrophage-Mediated Lymphangiogenesis: The Emerging Role of Macrophages as Lymphatic Endothelial Progenitors. Cancers. 2012; 4(3):618-657. https://doi.org/10.3390/cancers4030618
Chicago/Turabian StyleRan, Sophia, and Kyle E. Montgomery. 2012. "Macrophage-Mediated Lymphangiogenesis: The Emerging Role of Macrophages as Lymphatic Endothelial Progenitors" Cancers 4, no. 3: 618-657. https://doi.org/10.3390/cancers4030618