Influence of Levamisole and Other Angiogenesis Inhibitors on Angiogenesis and Endothelial Cell Morphology in Vitro

Abstract
:1. Introduction
1.1. Angiogenesis
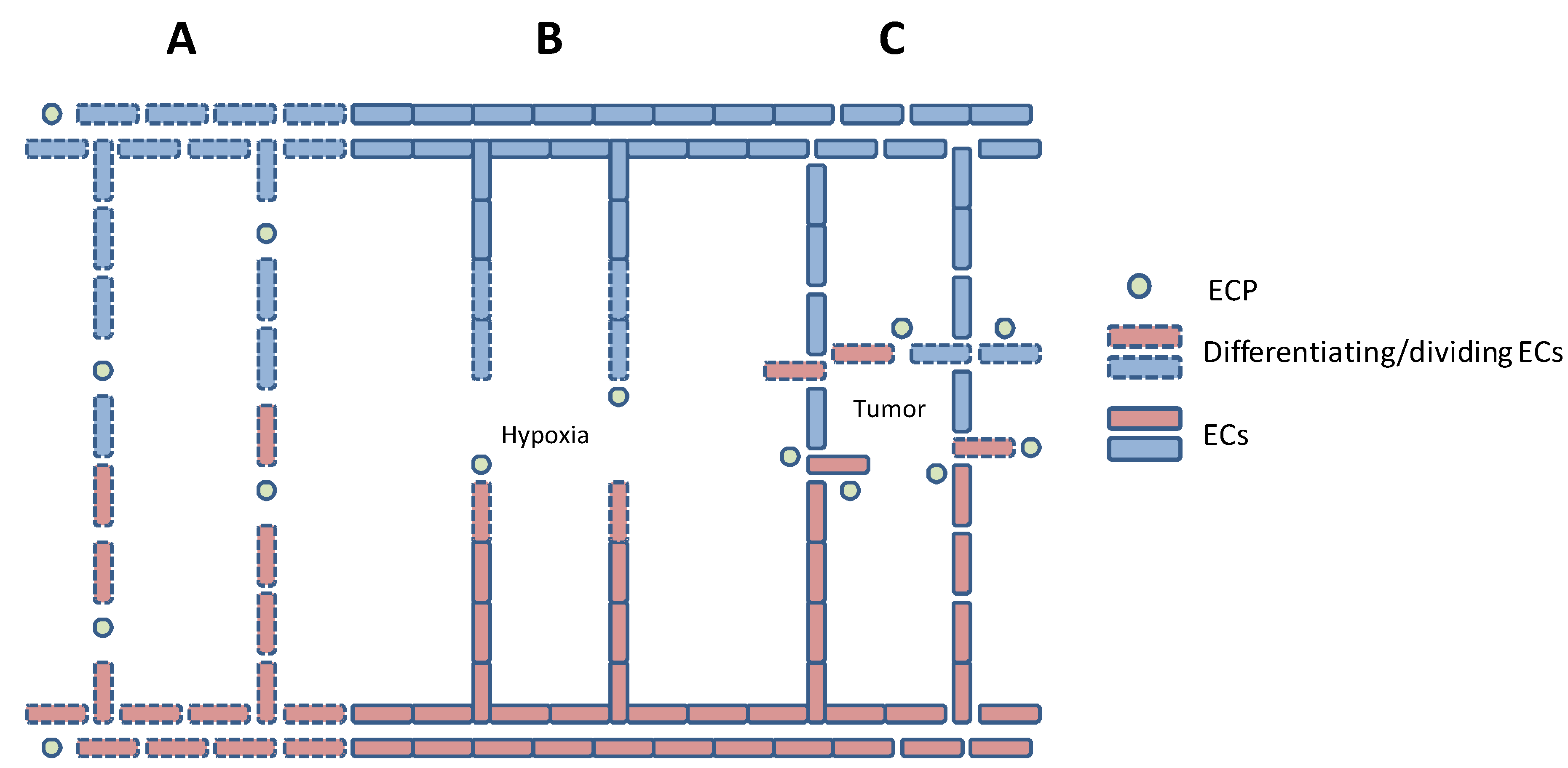
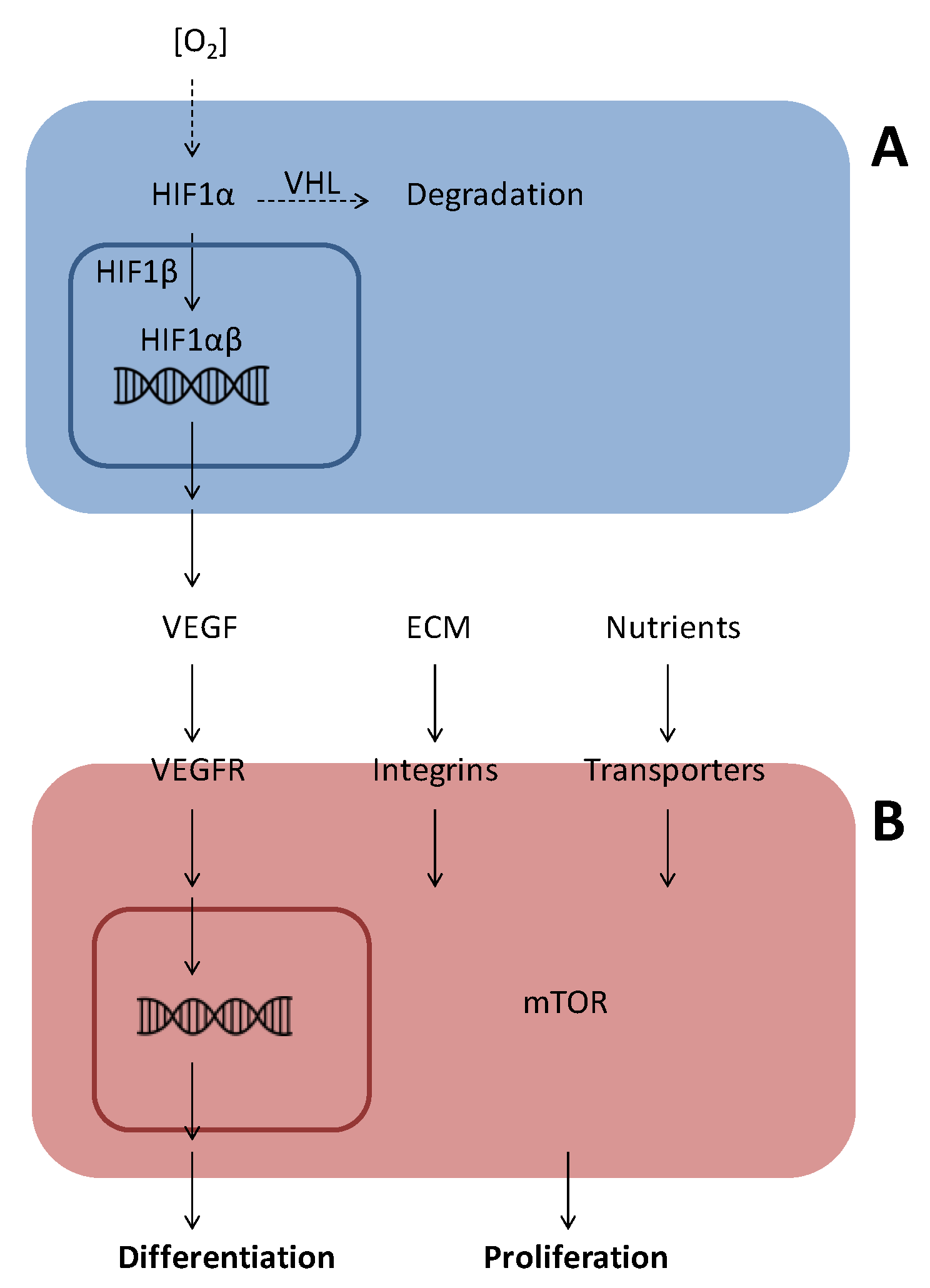
1.2. Angiogenic Factors
1.2.1. Vascular Endothelial Growth Factor (VEGF)
1.2.2. Angiopoietins (Angs)
2. Endothelial Cell Differentiation and Migration
3. Vessel Stabilization
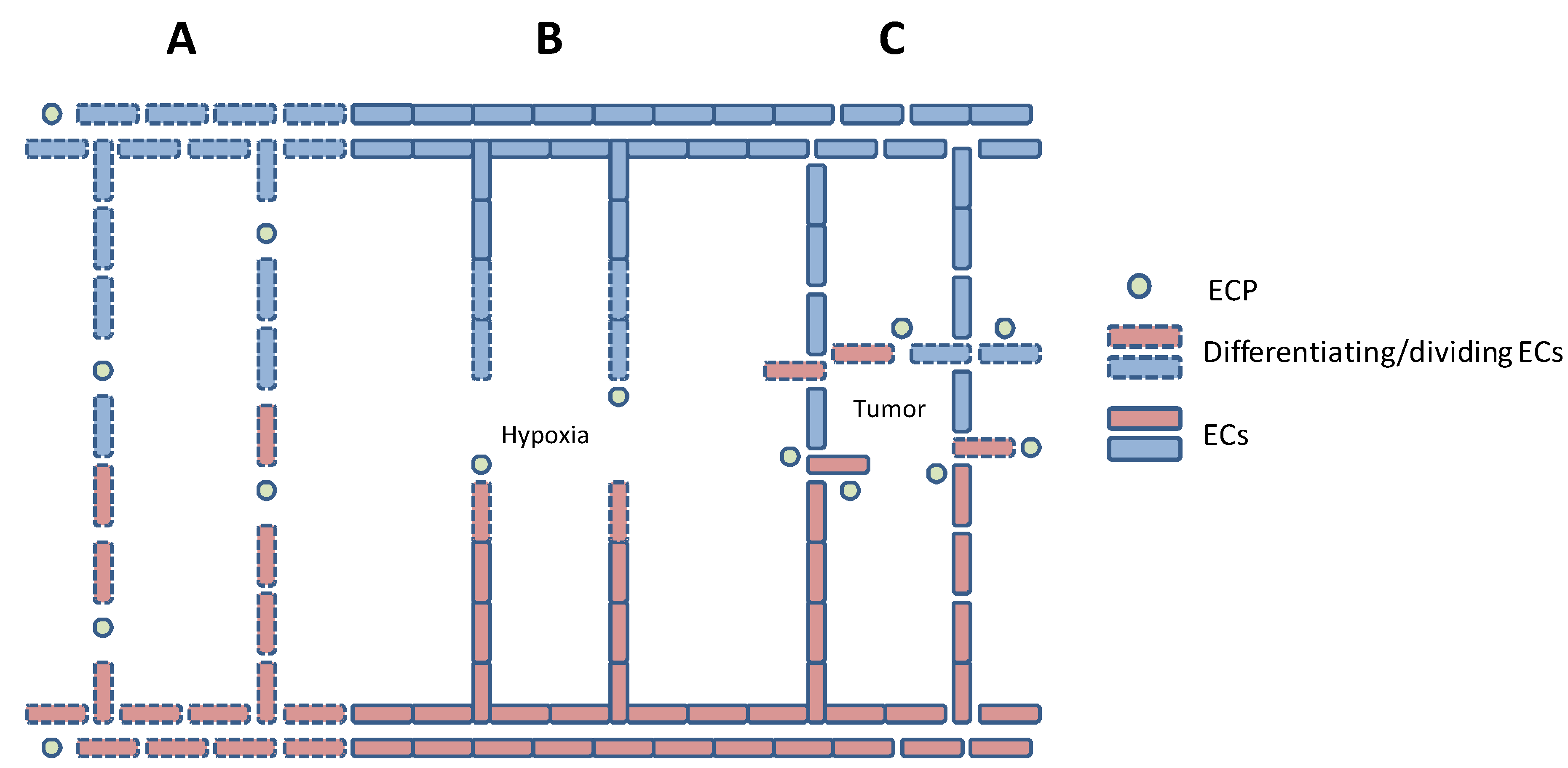
4. Tumor-Induced Angiogenesis
5. In vitro Models of Angiogenesis
5.1. Proliferation Assays
5.2. Migration Assays
5.3. Three-Dimensional Gel Assays
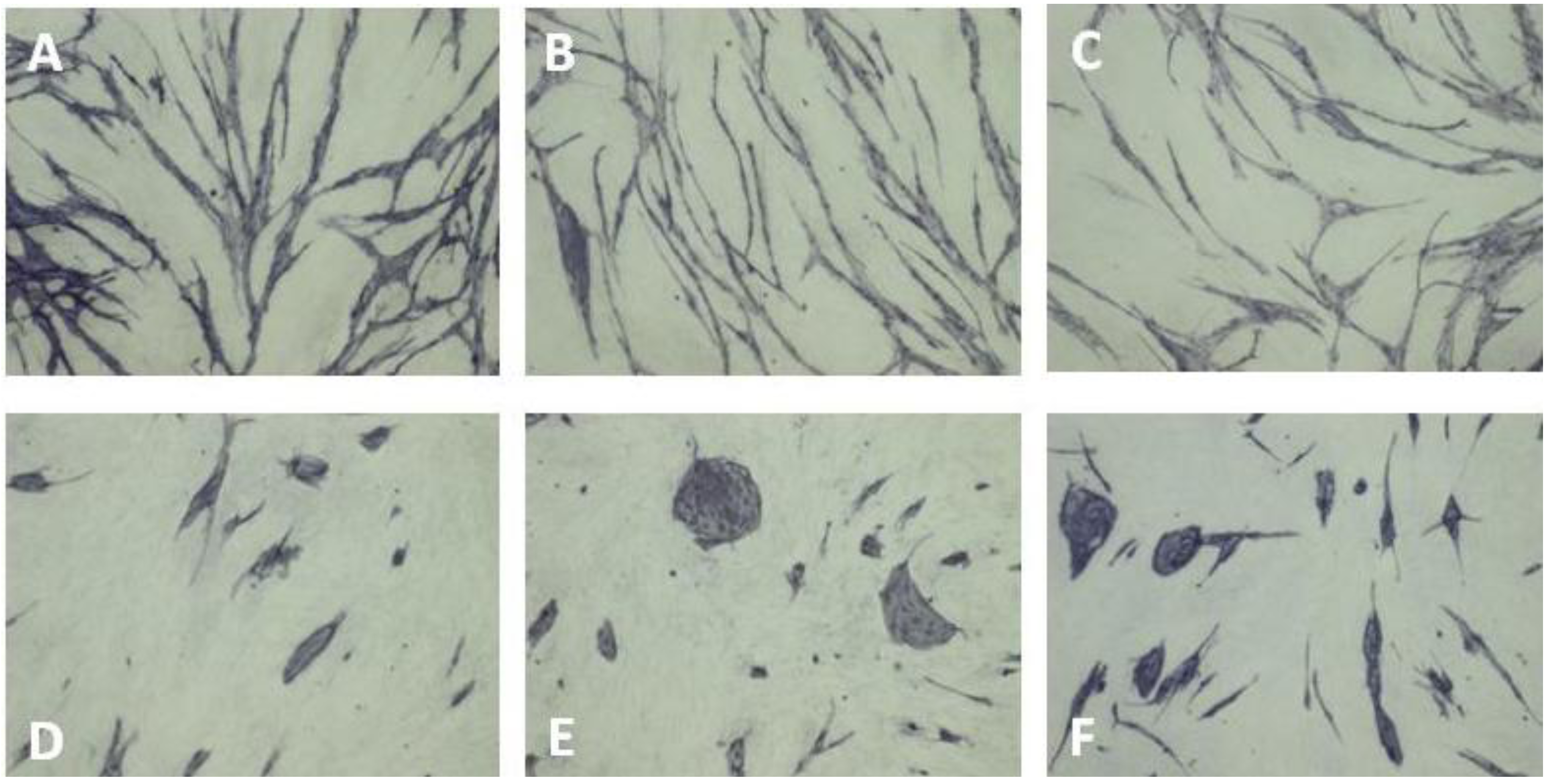
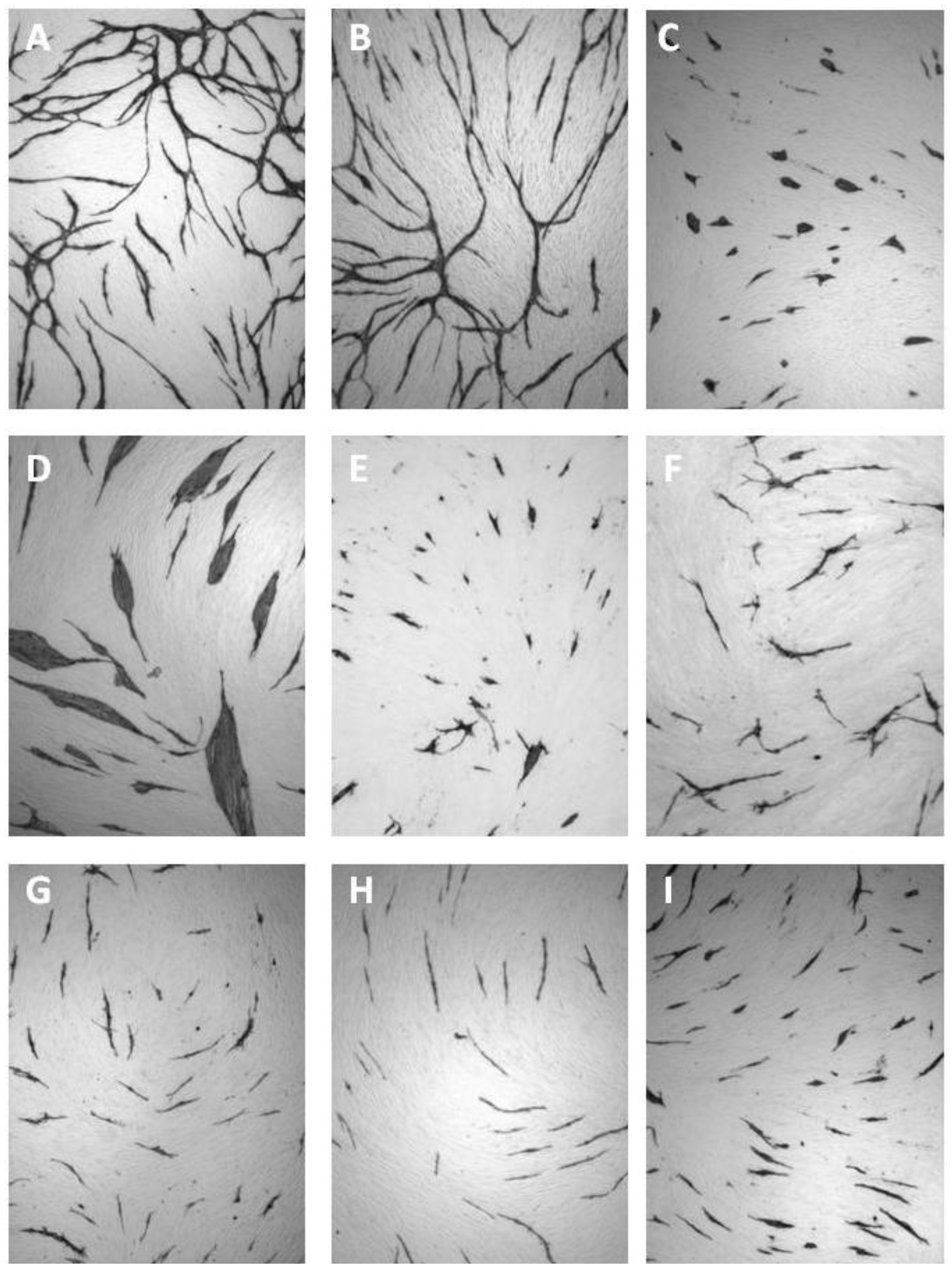
5.4. Coculture Assays with ECs and Fibroblasts
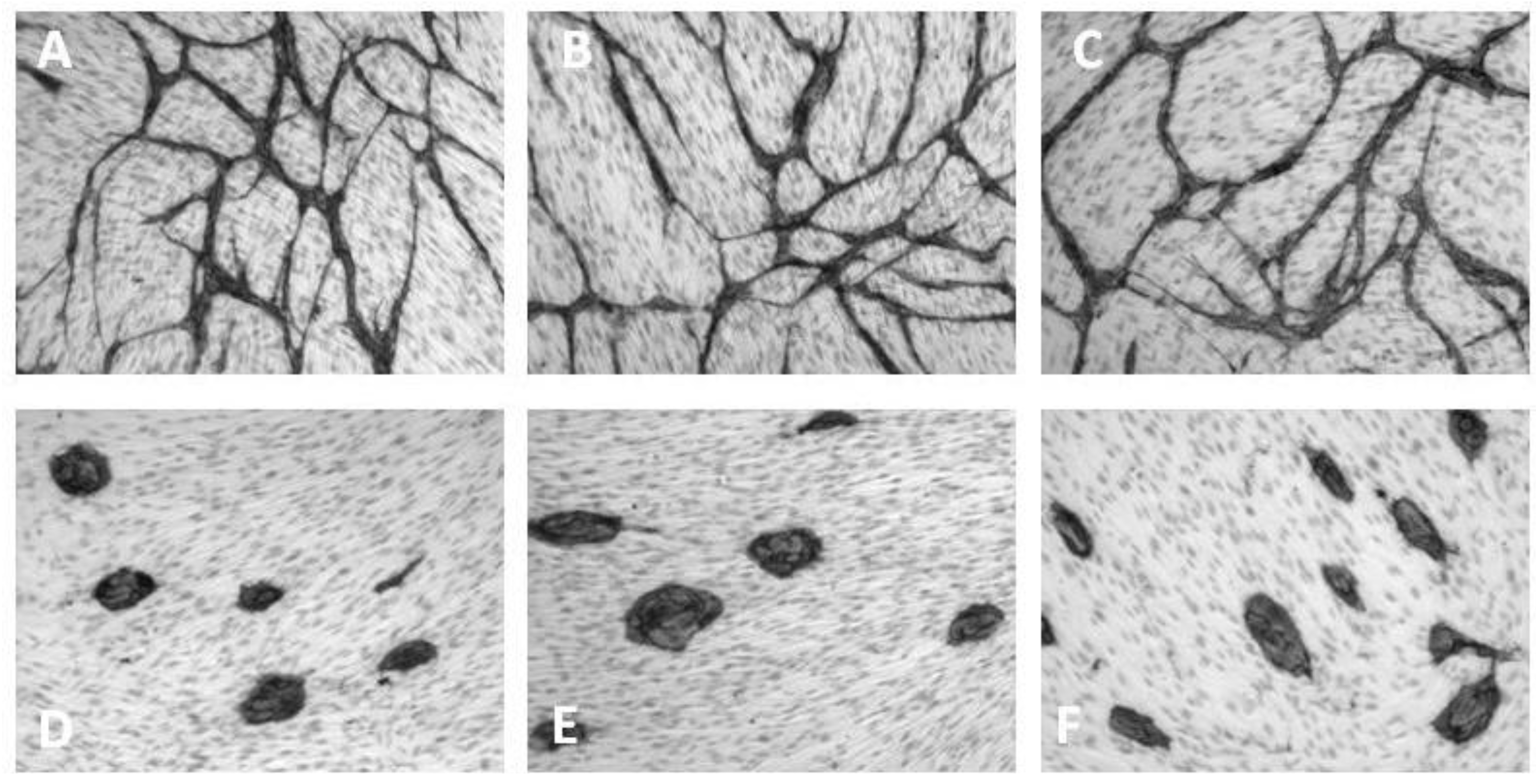
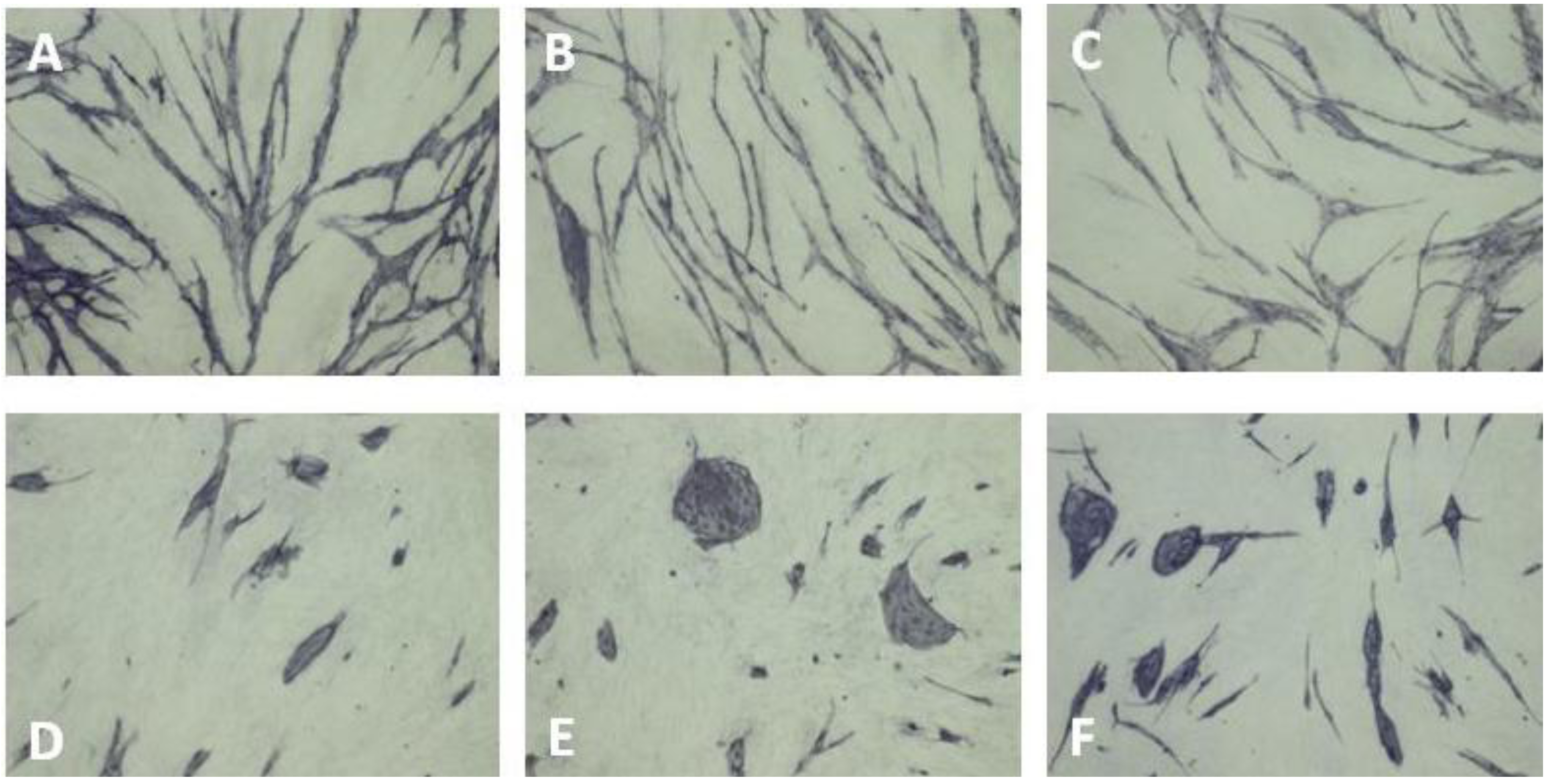
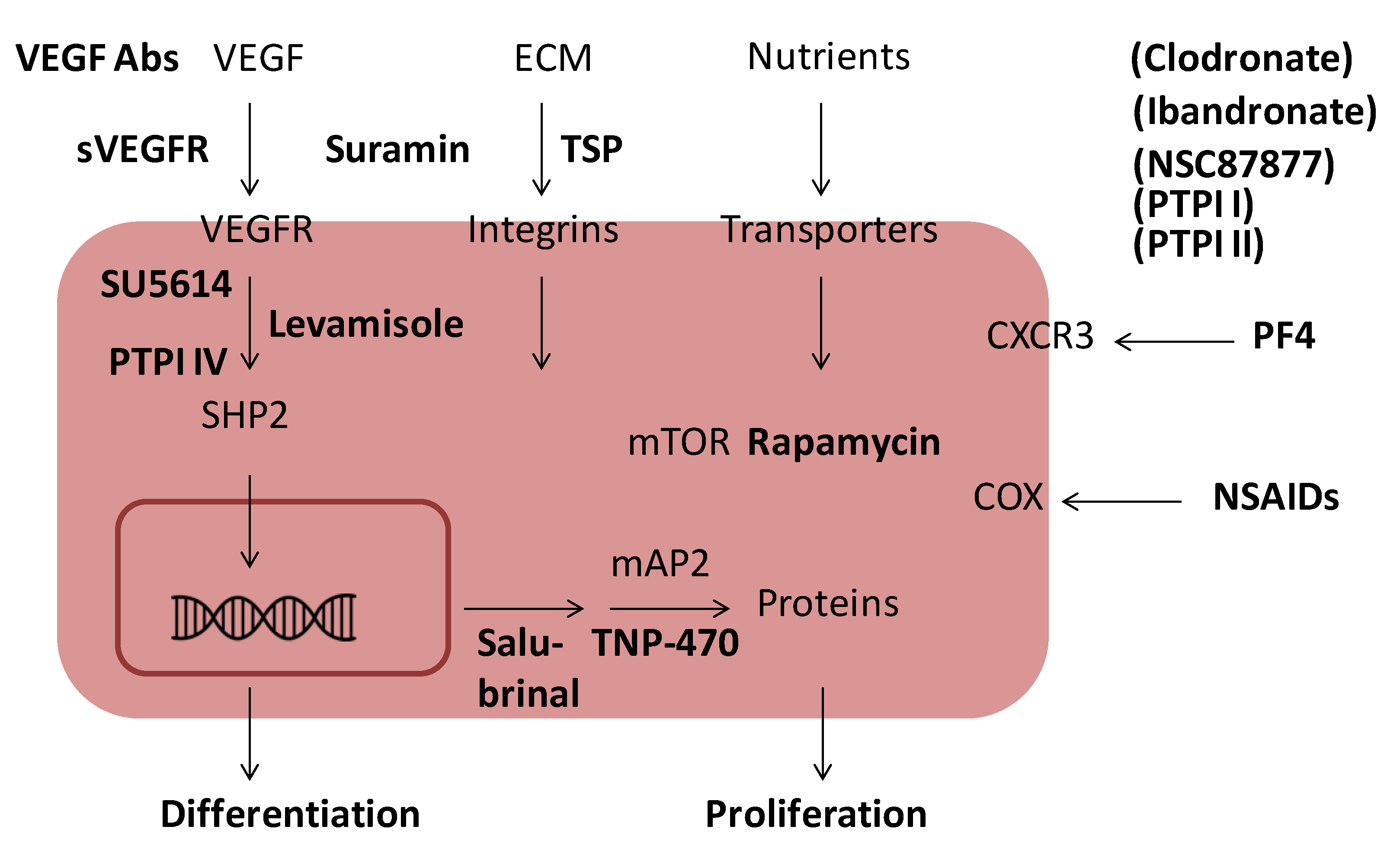
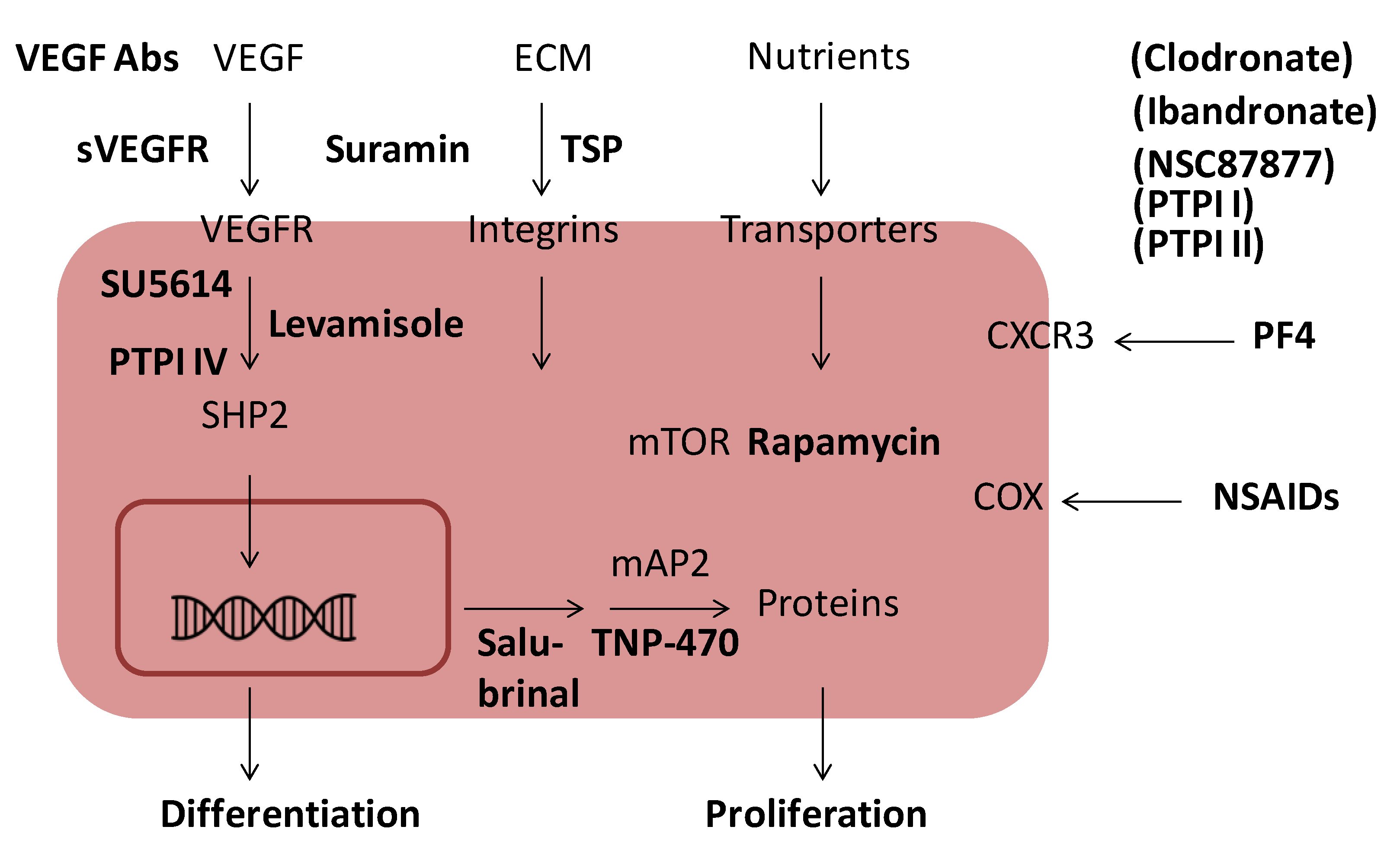
6. Inhibition of Angiogenesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Endogenous angiogenesis inhibitors | Exogenous angiogenesis inhibitors |
|---|---|
| Angiostatin | Fumagillin/TNP-470 |
| Antithrombin | NSAIDs |
| CXCL4 | PTPIs * |
| Endostatin | Rapamycin |
| Thrombospondin | SU5614 |
| Vasostatin | Suramin |
| sVEGFR1 | VEGF antibodies |
7. Endogenous Inhibitors of Angiogenesis
7.1. Angiostatin
7.2. Antithrombin
7.3. Endostatin and Other Globular Collagen Domains
7.4. Platelet Factor 4
7.5. Thrombospondin
7.6. Vasostatin
7.7. VEGF Isoforms and Decoy Receptors
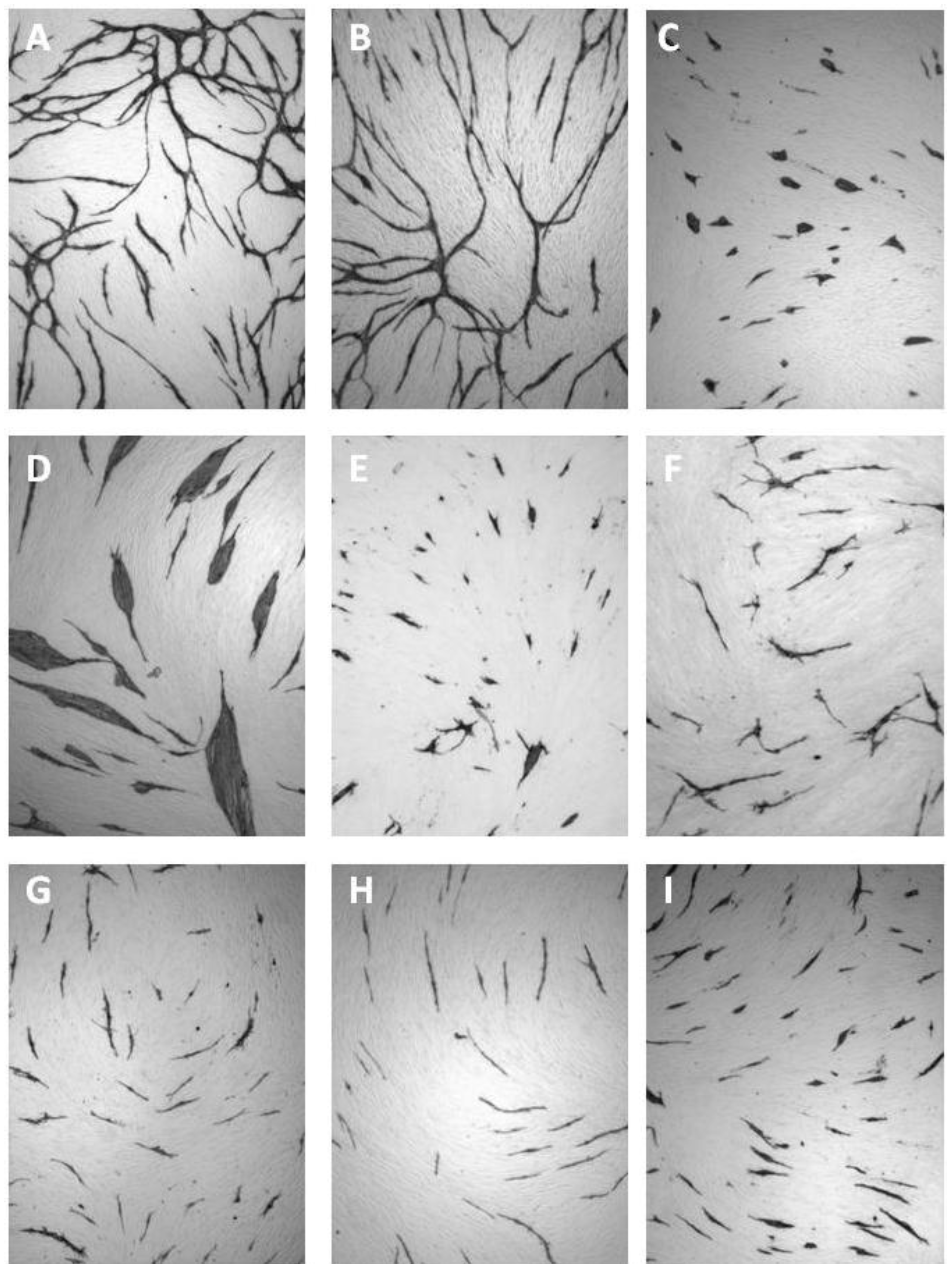
8. Exogenous Inhibitors of Angiogenesis
8.1. Fumagillin and TNP-470
8.2. Non-Steroidal Anti-Inflammatory Drugs (NSAIDs)
8.3. Levamisole and Other Phosphatase Inhibitors
8.4. Rapamycin
8.5. SU5614 and Other Kinase Inhibitors
8.6. Suramin
8.7. VEGF Antibodies
9. In vitro Assays of Angiogenesis
9.1. The Chicken CAM Assay
9.2. Cornea Assays
9.3. Mouse Hindlimb Ischemia Model
9.4. Matrigel Implantation Assay
9.5. Animal Models of Tumor-Angiogenesis
10. Clinical Use of Angiogenesis Inhibitors
11. Discussion
| Cluster morphology | Short cord morphology |
|---|---|
| Levamisole | NSAIDs |
| N-methyllevamisole | Platelet factor 4 |
| p-Bromolevamisole | PTPI I, II |
| PTPI IV | Rapamycin |
| sVEGFR1 | Suramin |
| SU5614 | Thrombospondin |
| VEGF antibodies | TNP-470 |
12. Conclusions
References
- Folkman, J. Angiogenesis: An organizing principle for drug discovery? Nat. Rev. Drug Discov. 2007, 6, 273–286. [Google Scholar] [CrossRef]
- Folkman, J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med. 1995, 1, 27–31. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat. Rev. Drug Discov. 2011, 10, 417–427. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 40, 249–257. [Google Scholar] [CrossRef]
- Patel-Hett, S.; D’Amore, P.A. Signal transduction in vasculogenesis and developmental angiogenesis. Int. J. Dev. Biol. 2011, 55, 353–363. [Google Scholar] [CrossRef]
- De Spiegelaere, W.; Casteleyn, C.; van den Broeck, W.; Plendl, J.; Bahramsoltani, M.; Simoens, P.; Djonov, V.; Cornillie, P. Intussusceptive angiogenesis: A biologically relevant form of angiogenesis. J. Vasc. Res. 2012, 49, 390–404. [Google Scholar] [CrossRef]
- Liu, W.; Shen, S.M.; Zhao, X.Y.; Chen, G.Q. Targeted genes and interacting proteins of hypoxia inducible factor-1. Int. J. Biochem. Mol. Biol. 2012, 3, 165–178. [Google Scholar]
- Greer, S.N.; Metcalf, J.L.; Wang, Y.; Ohh, M. The updated biology of hypoxia-inducible factor. EMBO J. 2012, 31, 2448–2460. [Google Scholar] [CrossRef]
- Tugues, S.; Koch, S.; Gualandi, L.; Li, X.; Claesson-Welsh, L. Vascular endothelial growth factors and receptors: Anti-angiogenic therapy in the treatment of cancer. Mol. Aspects Med. 2011, 32, 88–111. [Google Scholar] [CrossRef]
- Koch, S.; Tugues, S.; Li, X.; Gualandi, L.; Claesson-Welsh, L. Signal transduction by vascular endothelial growth factor receptors. Biochem. J. 2011, 437, 169–183. [Google Scholar] [CrossRef]
- Thomas, M.; Augustin, H.G. The role of the angiopoietins in vascular morphogenesis. Angiogenesis 2009, 12, 125–137. [Google Scholar] [CrossRef]
- Waugh, D.J.; Wilson, C. The interleukin-8 pathway in cancer. Clin. Cancer Res. 2008, 14, 6735–6741. [Google Scholar] [CrossRef]
- Wesche, J.; Haglund, K.; Haugsten, E.M. Fibroblast growth factors and their receptors in cancer. Biochem. J. 2011, 437, 199–213. [Google Scholar]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef]
- Ten Hagen, T.L.; Seynhaeve, A.L.; Eggermont, A.M. Tumor necrosis factor-mediated interactions between inflammatory response and tumor vascular bed. Immunol. Rev. 2008, 222, 299–315. [Google Scholar] [CrossRef]
- Koch, S. Neuropilin signalling in angiogenesis. Biochem. Soc. Trans. 2012, 40, 20–25. [Google Scholar] [CrossRef]
- Tung, J.J.; Tattersall, I.W.; Kitajewski, J. Tips, stalks, tubes: Notch-mediated cell fate determination and mechanisms of tubulogenesis during angiogenesis. Cold Spring Harb. Perspect. Med. 2012, 2, a006601. [Google Scholar] [CrossRef]
- Diermeier-Daucher, S.; Brockhoff, G. Dynamic proliferation assessment in flow cytometry. Curr. Protoc. Cell Biol. 2010. [Google Scholar] [CrossRef]
- Marshall, N.J.; Goodwin, C.J.; Holt, S.J. A critical assessment of the use of microculture tetrazolium assays to measure cell growth and function. Growth Regul. 1995, 5, 69–84. [Google Scholar]
- Cavanagh, B.L.; Walker, T.; Norazit, A.; Meedeniya, A.C. Thymidine analogues for tracking DNA synthesis. Molecules 2011, 16, 7980–7993. [Google Scholar] [CrossRef]
- Vega-Avila, E.; Pugsley, M.K. An overview of colorimetric assay methods used to assess survival or proliferation of mammalian cells. Proc. West. Pharmacol. Soc. 2011, 54, 10–14. [Google Scholar]
- Albrecht-Buehler, G. The phagokinetic tracks of 3T3 cells. Cell 1977, 11, 395–404. [Google Scholar] [CrossRef]
- Cai, G.; Lian, J.; Shapiro, S.S.; Beacham, D.A. Evaluation of endothelial cell migration with a novel in vitro assay system. Methods Cell Sci. 2000, 22, 107–114. [Google Scholar] [CrossRef]
- Hoying, J.B.; Williams, S.K. Measurement of endothelial cell migration using an improved linear migration assay. Microcirculation 1996, 3, 167–174. [Google Scholar] [CrossRef]
- Falasca, M.; Raimondi, C.; Maffucci, T. Boyden chamber. Methods Mol. Biol. 2011, 769, 87–95. [Google Scholar] [CrossRef]
- Maliakal, J.C. Quantitative high throughput endothelial cell migration and invasion assay system. Methods Enzymol. 2002, 352, 175–182. [Google Scholar] [CrossRef]
- Mastyugin, V.; McWhinnie, E.; Labow, M.; Buxton, F. A quantitative high-throughput endothelial cell migration assay. J. Biomol. Screen. 2004, 9, 712–718. [Google Scholar] [CrossRef]
- Riahi, R.; Yang, Y.; Zhang, D.D.; Wong, P.K. Advances in wound-healinh assays for probing collective cell migration. J. Lab. Automat. 2013, 17, 59–65. [Google Scholar]
- Chalupowicz, D.G.; Chowdhury, Z.A.; Bach, T.L.; Barsigian, C.; Martinez, J. Fibrin II induces endothelial cell capillary tube formation. J. Cell Biol. 1995, 130, 207–215. [Google Scholar] [CrossRef]
- Kleinman, H.K.; McGarvey, M.L.; Liotta, L.A.; Robey, P.G.; Tryggvason, K.; Martin, G.R. Isolation and characterization of type IV procollagen, laminin, and heparan sulfate proteoglycan from the EHS sarcoma. Biochemistry 1982, 21, 6188–6193. [Google Scholar] [CrossRef]
- Kubota, Y.; Kleinman, H.K.; Martin, G.R.; Lawley, T.J. Role of laminin and basement membrane in the morphological differentiation of human endothelial cells into capillary-like structures. J. Cell Biol. 1988, 107, 1589–1598. [Google Scholar] [CrossRef]
- Vukicevic, S.; Kleinman, H.K.; Luyten, F.P.; Roberts, A.B.; Roche, N.S.; Reddi, A.H. Identification of multiple active growth factors in basement membrane Matrigel suggests caution in interpretation of cellular activity related to extracellular matrix components. Exp. Cell Res. 1992, 202, 1–8. [Google Scholar] [CrossRef]
- Jeong, G.S.; Kwon, G.H.; Kang, A.R.; Jung, B.Y.; Park, Y.; Chung, S.; Lee, S.H. Microfluidic assay of endothelial cell migration in 3D interpenetrating polymer semi-network HA-Collagen hydrogel. Biomed. Microdevices 2011, 13, 717–723. [Google Scholar]
- Zeitlin, B.D.; Dong, Z.; Nör, J.E. RAIN-Droplet: A novel 3D in vitro angiogenesis model. Lab. Invest. 2012, 92, 988–998. [Google Scholar]
- Bishop, E.T.; Bell, G.T.; Bloor, S.; Broom, I.J.; Hendry, N.F.; Wheatley, D.N. An in vitro model of angiogenesis: Basic features. Angiogenesis 1999, 3, 335–344. [Google Scholar] [CrossRef]
- Friis, T.; Kjaer, S.B.; Engel, A.M.; Rygaard, J.; Houen, G. A quantitative ELISA-based coculture angiogenesis and cell proliferation assay. APMIS 2003, 111, 658–668. [Google Scholar] [CrossRef]
- Friis, T.; Hansen, A.B.; Houen, G.; Engel, A.M. Influence of angiogenesis inhibitors on endothelial cell morphology in vitro. APMIS 2006, 114, 211–224. [Google Scholar] [CrossRef]
- Sarkanen, J.R.; Mannerstrom, M.; Vuorenpaa, H.; Uotila, J.; Ylikomi, T.; Heinonen, T. Intra-laboratory pre-validation of a human cell based in vitro angiogenesis assay for testing angiogenesis modulators. Front. Pharmacol. 2010, 147, 1–13. [Google Scholar]
- Hetheridge, C.; Mavria, G.; Mellor, H. Uses of the in vitro endothelial-fibroblast organotypic coculture assay in angiogenesis research. Biochem. Soc. Trans. 2011, 39, 1597–1600. [Google Scholar] [CrossRef]
- Schulze, A.; Harris, A.L. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature 2012, 491, 364–373. [Google Scholar] [CrossRef]
- Loges, S.; Mazzone, M.; Hohensinner, P.; Carmeliet, P. Silencing or fueling metastasis with VEGF inhibitors: Antiangiogenesis revisited. Cancer Cell 2009, 15, 167–170. [Google Scholar]
- O’Reilly, M.S.; Holmgren, L.; Shing, Y.; Chen, C.; Rosenthal, R.A.; Moses, M.; Lane, W.S.; Cao, Y.; Sage, E.H.; Folkman, J. Angiostatin: A novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell 1994, 79, 315–328. [Google Scholar] [CrossRef]
- O’Reilly, M.S.; Holmgren, L.; Chen, C.; Folkman, J. Angiostatin induces and sustains dormancy of human primary tumors in mice. Nat. Med. 1996, 2, 689–692. [Google Scholar] [CrossRef]
- O’Reilly, M.S.; Pirie-Shepherd, S.; Lane, W.S.; Folkman, J. Antiangiogenic activity of the cleaved conformation of the serpin antithrombin. Science 1999, 285, 1926–1928. [Google Scholar] [CrossRef]
- Mundel, T.M.; Kalluri, R. Type IV collagen-derived angiogenesis inhibitors. Microvasc. Res. 2007, 74, 85–89. [Google Scholar] [CrossRef]
- Ramchandran, R.; Dhanabal, M.; Volk, R.; Waterman, M.J.; Segal, M.; Lu, H.; Knebelmann, B.; Sukhatme, V.P. Antiangiogenic activity of restin, NC10 domain of human collagen XV: Comparison to endostatin. Biochem. Biophys. Res. Commun. 1999, 255, 735–739. [Google Scholar] [CrossRef]
- Folkman, J. Antiangiogenesis in cancer therapy—Endostatin and its mechanisms of action. Exp. Cell Res. 2006, 312, 594–607. [Google Scholar]
- Taddei, L.; Chiarugi, P.; Brogelli, L.; Cirri, P.; Magnelli, L.; Raugei, G.; Ziche, M.; Granger, H.J.; Chiarugi, V.; Ramponi, G. Inhibitory effect of full-length human endostatin on in vitro angiogenesis. Biochem. Biophys. Res. Commun. 1999, 263, 340–345. [Google Scholar]
- Vandercappellen, J.; van Damme, J.; Struyf, S. The role of the CXC chemokines platelet factor-4 (CXCL4/PF-4) and its variant (CXCL4L1/PF-4var) in inflammation, angiogenesis and cancer. Cytokine Growth Factor Rev. 2011, 22, 1–18. [Google Scholar]
- Kasper, B.; Petersen, F. Molecular pathways of platelet factor 4/CXCL4 signaling. Eur. J. Cell Biol. 2011, 90, 521–526. [Google Scholar] [CrossRef]
- Struyf, S.; Salogni, L.; Burdick, M.D.; Vandercappellen, J.; Gouwy, M.; Noppen, S.; Proost, P.; Opdenakker, G.; Parmentier, M.; Gerard, C.; et al. Angiostatic and chemotactic activities of the CXC chemokine CXCL4L1 (platelet factor-4 variant) are mediated by CXCR3. Blood 2011, 117, 480–488. [Google Scholar] [CrossRef]
- Vandercappellen, J.; Liekens, S.; Bronckaers, A.; Noppen, S.; Ronsse, I.; Dillen, C.; Belleri, M.; Mitola, S.; Proost, P.; Presta, M.; et al. The COOH-terminal peptide of platelet factor-4 variant (CXCL4L1/PF-4var47-70) strongly inhibits angiogenesis and suppresses B16 melanoma growth in vivo. Mol. Cancer Res. 2010, 8, 322–334. [Google Scholar] [CrossRef]
- Adams, J.C.; Lawler, J. The thrombospondins. Cold Spring Harb. Perspect. Biol. 2011, 3, a009712. [Google Scholar] [CrossRef]
- Ren, B.; Yee, K.O.; Lawler, J.; Khosravi-Far, R. Regulation of tumor angiogenesis by thrombospondin-1. Biochim. Biophys. Acta 2006, 1765, 178–188. [Google Scholar]
- Jiménez, B.; Volpert, O.V.; Crawford, S.E.; Febbraio, M.; Silverstein, R.L.; Bouck, N. Signals leading to apoptosis-dependent inhibition of neovascularization by thrombospondin-1. Nat. Med. 2000, 6, 41–48. [Google Scholar] [CrossRef]
- Pike, S.E.; Yao, L.; Jones, K.D.; Cherney, B.; Appella, E.; Sakaguchi, K.; Nakhasi, H.; Teruya-Feldstein, J.; Wirth, P.; Gupta, G.; et al. Vasostatin, a calreticulin fragment, inhibits angiogenesis and suppresses tumor growth. J. Exp. Med. 1998, 188, 2349–2356. [Google Scholar] [CrossRef]
- Pike, S.E.; Yao, L.; Setsuda, J.; Jones, K.D.; Cherney, B.; Appella, E.; Sakaguchi, K.; Nakhasi, H.; Atreya, C.D.; Teruya-Feldstein, J.; et al. Calreticulin and calreticulin fragments are endothelial cell inhibitors that suppress tumor growth. Blood 1999, 94, 2461–2468. [Google Scholar]
- Wu, P.C.; Yang, L.C.; Kuo, H.K.; Huang, C.C.; Tsai, C.L.; Lin, P.R.; Wu, P.C.; Shin, S.J.K.; Tai, M.H. Inhibition of corneal angiogenesis by local application of vasostatin. Mol. Vis. 2005, 11, 28–35. [Google Scholar]
- Yao, L.; Pike, S.E.; Tosato, G. Laminin binding to the calreticulin fragment vasostatin regulates endothelial cell function. J. Leukoc. Biol. 2002, 71, 47–53. [Google Scholar]
- Ingber, D.; Fujita, T.; Kishimoto, S.; Sudo, K.; Kanamaru, T.; Brem, H.; Folkman, J. Synthetic analogues of fumagillin that inhibit angiogenesis and suppress tumour growth. Nature 1990, 348, 555–557. [Google Scholar] [CrossRef]
- Kusaka, M.; Sudo, K.; Fujita, T.; Marui, S.; Itoh, F.; Ingber, D.; Folkman, J. Potent anti-angiogenic action of AGM-1470: Comparison to the fumagillin parent. Biochem. Biophys. Res. Commun. 1991, 174, 1070–1076. [Google Scholar]
- Kusaka, M.; Sudo, K.; Matsutani, E.; Kozai, Y.; Marui, S.; Fujita, T.; Ingber, D.; Folkman, J. Cytostatic inhibition of endothelial cell growth by the angiogenesis inhibitor TNP-470 (AGM-1470). Br. J. Cancer 1994, 69, 212–216. [Google Scholar] [CrossRef]
- Satchi-Fainaro, R.; Puder, M.; Davies, J.W.; Tran, H.T.; Sampson, D.A.; Greene, A.K.; Corfas, G.; Folkman, J. Targeting angiogenesis with a conjugate of HPMA copolymer and TNP-470. Nat. Med. 2004, 10, 255–261. [Google Scholar] [CrossRef]
- Satchi-Fainaro, R.; Mamluk, R.; Wang, L.; Short, S.M.; Nagy, J.A.; Feng, D.; Dvorak, A.M.; Dvorak, H.F.; Puder, M.; Mukhopadhyay, D.; Folkman, J. Inhibition of vessel permeability by TNP-470 and its polymer conjugate, caplostatin. Cancer Cell 2005, 7, 251–261. [Google Scholar]
- Griffith, E.C.; Su, Z.; Turk, B.E.; Chen, S.; Chang, Y.H.; Wu, Z.; Biemann, K.; Liu, J.O. Methionine aminopeptidase (type 2) is the common target for angiogenesis inhibitors AGM-1470 and ovalicin. Chem. Biol. 1997, 4, 461–471. [Google Scholar] [CrossRef]
- Sin, N.; Meng, L.; Wang, M.Q.; Wen, J.J.; Bornmann, W.G.; Crews, C.M. The anti-angiogenic agent fumagillin covalently binds and inhibits the methionine aminopeptidase, MetAP-2. Proc. Natl. Acad. Sci. USA 1997, 94, 6099–6103. [Google Scholar] [CrossRef]
- Kragh, M.; Hjarnaa, P.J.; Bramm, E.; Kristjansen, P.E.; Rygaard, J.; Binderup, L. In vivo chamber angiogenesis assay: An optimized Matrigel plug assay for fast assessment of anti-angiogenic activity. Int. J. Oncol. 2003, 22, 305–311. [Google Scholar]
- Lien, W.H.; Chen, C.K.; Lai, L.Y.; Chen, Y.H.; Wu, M.P.; Wu, L.W. Participation of cyclin D1 deregulation in TNP-470-mediated cytostatic effect: Involvement of senescence. Biochem. Pharmacol. 2004, 68, 729–738. [Google Scholar] [CrossRef]
- Yeh, J.R.; Mohan, R.; Crews, C.M. The antiangiogenic agent TNP-470 requires p53 and p21CIP/WAF for endothelial cell growth arrest. Proc. Natl. Acad. Sci. USA 2000, 97, 12782–12787. [Google Scholar] [CrossRef]
- Sawaoka, H.; Tsuji, S.; Tsujii, M.; Gunawan, E.S.; Sasaki, Y.; Kawano, S.; Hori, M. Cyclooxygenase inhibitors suppress angiogenesis and reduce tumor growth in vivo. Lab. Invest. 1999, 79, 1469–1477. [Google Scholar]
- Dormond, O.; Foletti, A.; Paroz, C.; Ruegg, C. NSAIDs inhibit alpha V beta 3 integrin-mediated and Cdc42/Rac-dependent endothelial-cell spreading, migration and angiogenesis. Nat. Med. 2001, 7, 1041–1047. [Google Scholar] [CrossRef]
- Jones, M.K.; Wang, H.; Peskar, B.M.; Levin, E.; Itani, R.M.; Sarfeh, I.J.; Tarnawski, A.S. Inhibition of angiogenesis by nonsteroidal anti-inflammatory drugs: Insight into mechanisms and implications for cancer growth and ulcer healing. Nat. Med. 1999, 5, 1418–1423. [Google Scholar] [CrossRef]
- Miller, M.J. Use of levamisole in parasitic infections. Drugs 1980, 20, 122–130. [Google Scholar] [CrossRef]
- Mutch, R.S.; Hutson, P.R. Levamisole in the adjuvant treatment of colon cancer. Clin. Pharm. 1991, 10, 95–109. [Google Scholar]
- Stevenson, H.C.; Green, I.; Hamilton, J.M.; Calabro, B.A.; Parkinson, D.R. Levamisole: Known effects on the immune system, clinical results, and future applications to the treatment of cancer. J. Clin. Oncol. 1991, 9, 2052–2066. [Google Scholar]
- Friis, T.; Engel, A.M.; Klein, B.M.; Rygaard, J.; Houen, G. Levamisole inhibits angiogenesis in vitro and tumor growth in vivo. Angiogenesis 2005, 8, 25–34. [Google Scholar] [CrossRef]
- Hansen, A.N.; Bendiksen, C.D.; Sylvest, L.; Friis, T.; Staerk, D.; Jørgensen, F.S.; Olsen, C.A.; Houen, G. Synthesis and antiangiogenic activity of N-alkylated levamisole derivatives. PLoS One 2012, 7, e45405. [Google Scholar] [CrossRef]
- Artwohl, M.; Hölzenbein, T.; Wagner, L.; Freudenthaler, A.; Waldhäusl, W.; Baumgartner-Parzer, S.M. Levamisole induced apoptosis in cultured vascular endothelial cells. Br. J. Pharmacol. 2000, 131, 1577–1583. [Google Scholar]
- Ramanadham, M.; Nageshwari, B. Anti-proliferative effect of levamisole on human myeloma cell lines in vitro. J. Immunotoxicol. 2010, 7, 327–332. [Google Scholar] [CrossRef]
- Hegde, M.; Karki, S.S.; Thomas, E.; Kumar, S.; Panjamurthy, K.; Ranganatha, S.R.; Rangappa, K.S.; Choudhary, B.; Raghavan, S.C. Novel levamisole derivative induces extrinsic pathway of apoptosis in cancer cells and inhibits tumor progression in mice. PLoS One 2012, 7, e43632. [Google Scholar]
- MacDonald, M.L.; Lamerdin, J.; Owens, S.; Keon, B.H.; Bilter, G.K.; Shang, Z.; Huang, Z.; Yu, H.; Dias, J.; Minami, T.; et al. Identifying off-target effects and hidden phenotypes of drugs in human cells. Nat. Chem. Biol. 2006, 2, 329–337. [Google Scholar]
- Sylvest, L.; Bendiksen, C.D.; Houen, G. Phosphatase inhibitors with anti-angiogenic effect in vitro. APMIS 2010, 118, 49–59. [Google Scholar] [CrossRef]
- Mannell, H.; Hellwig, N.; Gloe, T.; Plank, C.; Sohn, H.Y.; Groesser, L.; Walzog, B.; Pohl, U.; Krotz, F. Inhibition of the tyrosine phosphatase SHP-2 suppresses angiogenesis in vitro and in vivo. J. Vasc. Res. 2008, 45, 153–163. [Google Scholar] [CrossRef]
- Kwon, Y.S.; Kim, J.C. Inhibition of corneal neovascularization by rapamycin. Exp. Mol. Med. 2006, 38, 173–179. [Google Scholar]
- Guba, M.; von Breitenbuch, P.; Steinbauer, M.; Koehl, G.; Flegel, S.; Hornung, M.; Bruns, C.J.; Zuelke, C.; Farkas, S.; Anthuber, M.; et al. Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: Involvement of vascular endothelial growth factor. Nat. Med. 2002, 8, 128–135. [Google Scholar] [CrossRef]
- Koehl, G.E.; Andrassy, J.; Guba, M.; Richter, S.; Kroemer, A.; Scherer, M.N.; Steinbauer, M.; Graeb, C.; Schlitt, H.J.; Jauch, K.W.; et al. Rapamycin protects allografts from rejection while simultaneously attacking tumors in immunosuppressed mice. Transplantation 2004, 77, 1319–1326. [Google Scholar] [CrossRef]
- Humar, R.; Kiefer, F.N.; Berns, H.; Resink, T.J.; Battegay, E.J. Hypoxia enhances vascular cell proliferation and angiogenesis in vitro via rapamycin (mTOR)-dependent signaling. FASEB J. 2002, 16, 771–780. [Google Scholar] [CrossRef]
- Weichhart, T. Mammalian target of rapamycin: A signaling kinase for every aspect of cellular life. Methods Mol. Biol. 2012, 821, 1–14. [Google Scholar] [CrossRef]
- Gotink, K.J.; Verheul, H.M. Anti-angiogenic tyrosine kinase inhibitors: What is their mechanism of action? Angiogenesis 2010, 13, 1–14. [Google Scholar] [CrossRef]
- Sun, L.; Tran, N.; Tang, F.; App, H.; Hirth, P.; McMahon, G.; Tang, C. Synthesis and biological evaluations of 3-substituted indolin-2-ones: A novel class of tyrosine kinase inhibitors that exhibit selectivity toward particular receptor tyrosine kinases. J. Med. Chem. 1998, 41, 2588–2603. [Google Scholar] [CrossRef]
- Spiekermann, K.; Faber, F.; Voswinckel, R.; Hiddemann, W. The protein tyrosine kinase inhibitor SU5614 inhibits VEGF-induced endothelial cell sprouting and induces growth arrest and apoptosis by inhibition of c-kit in AML cells. Exp. Hematol. 2002, 30, 767–773. [Google Scholar] [CrossRef]
- Jia, H.; Bagherzadeh, A.; Bicknell, R.; Duchen, M.R.; Liu, D.; Zachary, I. Vascular endothelial growth factor (VEGF)-D and VEGF-A differentially regulate KDR-mediated signaling and biological function in vascular endothelial cells. J. Biol. Chem. 2004, 279, 36148–36157. [Google Scholar] [CrossRef]
- McGeary, R.P.; Bennett, A.J.; Tran, Q.B.; Cosgrove, K.L.; Ross, B.P. Suramin: Clinical uses and structure-activity relationships. Mini Rev. Med. Chem. 2008, 8, 1384–1394. [Google Scholar] [CrossRef]
- Waltenberger, J.; Mayr, U.; Frank, H.; Hombach, V. Suramin is a potent inhibitor of vascular endothelial growth factor. A contribution to the molecular basis of its antiangiogenic action. J. Mol. Cell. Cardiol. 1996, 28, 1523–1529. [Google Scholar] [CrossRef]
- Takano, S.; Gately, S.; Neville, M.E.; Herblin, W.F.; Gross, J.L.; Engelhard, H.; Perricone, M.; Eidsvoog, K.; Brem, S. Suramin, an anticancer and angiosuppressive agent, inhibits endothelial cell binding of basic fibroblast growth factor, migration, proliferation, and induction of urokinase-type plasminogen activator. Cancer Res. 1994, 54, 2654–2660. [Google Scholar]
- HoSang, M. Suramin binds to platelet-derived growth factor and inhibits its biological activity. J. Cell. Biochem. 1985, 29, 265–273. [Google Scholar] [CrossRef]
- Hasan, J.; Shnyder, S.D.; Bibby, M.; Double, J.A.; Bicknel, R.; Jayson, G.C. Quantitative angiogenesis assays in vivo—A review. Angiogenesis 2004, 7, 1–16. [Google Scholar]
- Norrby, K. In vivo models of angiogenesis. J. Cell Mol. Med. 2006, 10, 588–612. [Google Scholar] [CrossRef]
- Naumov, G.N.; Akslen, L.A.; Folkman, J. Role of angiogenesis in human tumor dormancy: Animal models of the angiogenic switch. Cell Cycle 2006, 5, 1779–1787. [Google Scholar] [CrossRef]
- Couffinhal, T.; Dufourcq, P.; Barandon, L.; Leroux, L.; Duplaa, C. Mouse models to study angiogenesis in the context of cardiovascular diseases. Front. Biosci. 2009, 14, 3310–3325. [Google Scholar]
- Goldbrunner, R.H.; Wagner, S.; Roosen, K.; Tonn, J.C. Models for assessment of angiogenesis in gliomas. J. Neurooncol. 2000, 50, 53–62. [Google Scholar] [CrossRef]
- Otterness, I.G.; Lachman, L.B.; Bliven, M.L. Effects of levamisole on the proliferation of thymic lymphocyte subpopulations. Immunopharmacology 1981, 3, 61–69. [Google Scholar] [CrossRef]
- Redondo, J.M.; Lopez-Guerrero, J.A.; Fresno, M. Potentiation of interleukin-2 activity by levamisole and imidazole. Immunol. Lett. 1987, 14, 111–116. [Google Scholar] [CrossRef]
- Renoux, G.; Renoux, M. Thymus-like activities of sulphur derivatives on T-cell differentiation. J. Exp. Med. 1977, 145, 466–471. [Google Scholar] [CrossRef]
- Szeto, C.; Gillespie, K.M.; Mathieson, P.W. Levamisole induces interleukin-18 and shifts type 1/type 2 cytokine balance. Immunology 2000, 100, 217–224. [Google Scholar] [CrossRef]
- Gressett, S.M.; Shah, S.R. Intricacies of bevacizumab-induced toxicities and their management. Ann. Pharmacother. 2009, 43, 490–501. [Google Scholar] [CrossRef]
- Shord, S.S.; Bressler, L.R.; Tierney, L.A.; Cuellar, S.; George, A. Understanding and managing the possible adverse effects associated with bevacizumab. Am. J. Health Syst. Pharm. 2009, 66, 999–1013. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Friis, T.; Engel, A.-M.; Bendiksen, C.D.; Larsen, L.S.; Houen, G. Influence of Levamisole and Other Angiogenesis Inhibitors on Angiogenesis and Endothelial Cell Morphology in Vitro. Cancers 2013, 5, 762-785. https://doi.org/10.3390/cancers5030762
Friis T, Engel A-M, Bendiksen CD, Larsen LS, Houen G. Influence of Levamisole and Other Angiogenesis Inhibitors on Angiogenesis and Endothelial Cell Morphology in Vitro. Cancers. 2013; 5(3):762-785. https://doi.org/10.3390/cancers5030762
Chicago/Turabian StyleFriis, Tina, Anne-Marie Engel, Christine D. Bendiksen, Line S. Larsen, and Gunnar Houen. 2013. "Influence of Levamisole and Other Angiogenesis Inhibitors on Angiogenesis and Endothelial Cell Morphology in Vitro" Cancers 5, no. 3: 762-785. https://doi.org/10.3390/cancers5030762