Alterations of 5-Hydroxymethylcytosine in Human Cancers

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
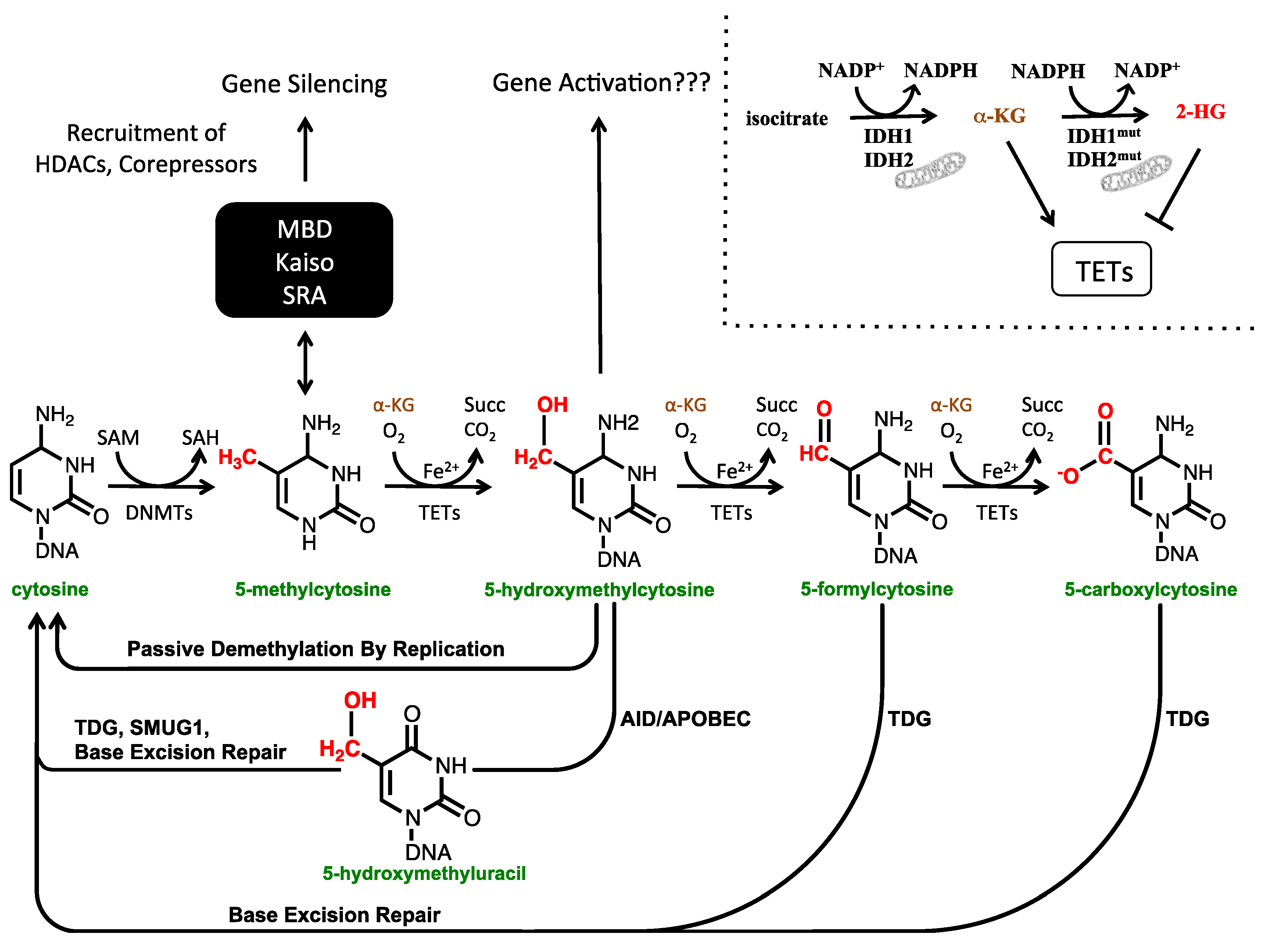
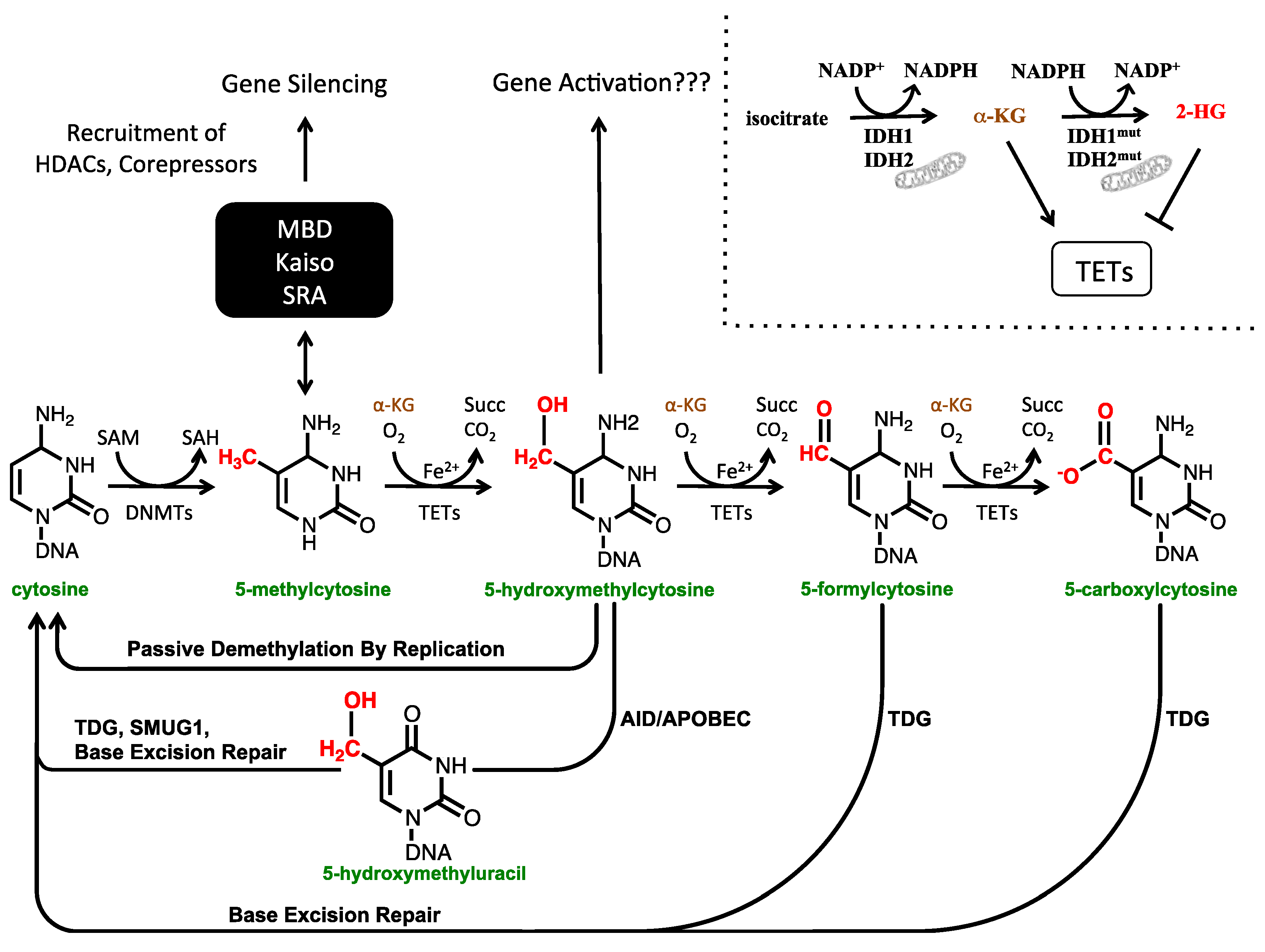
1.1. The Cytosine Modification Pathway
1.2. The Biological Functions of 5-hmC
2. Techniques Used to Study 5-hmC
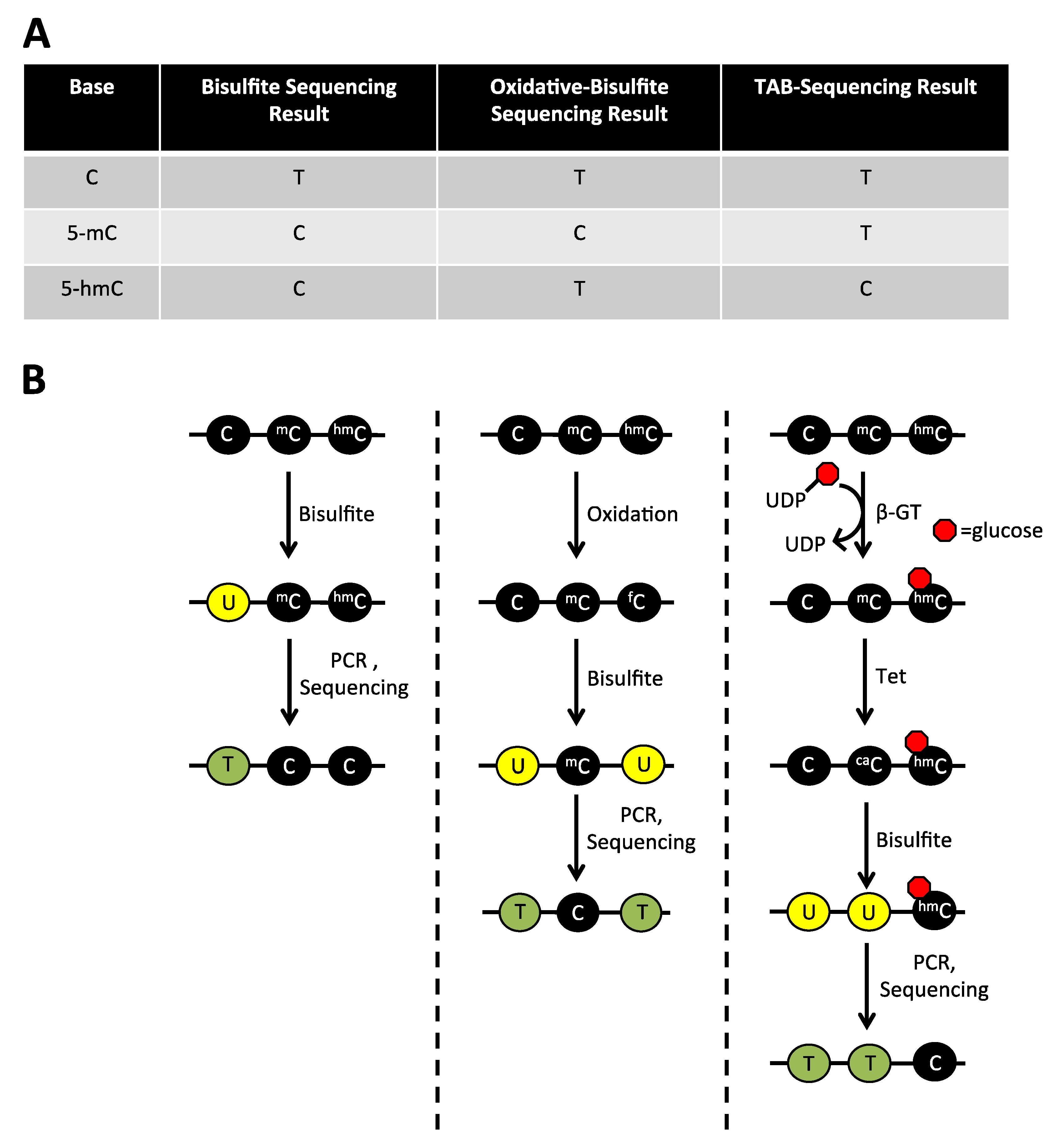
2.1. The Limitations of Sodium Bisulfite Based Technologies
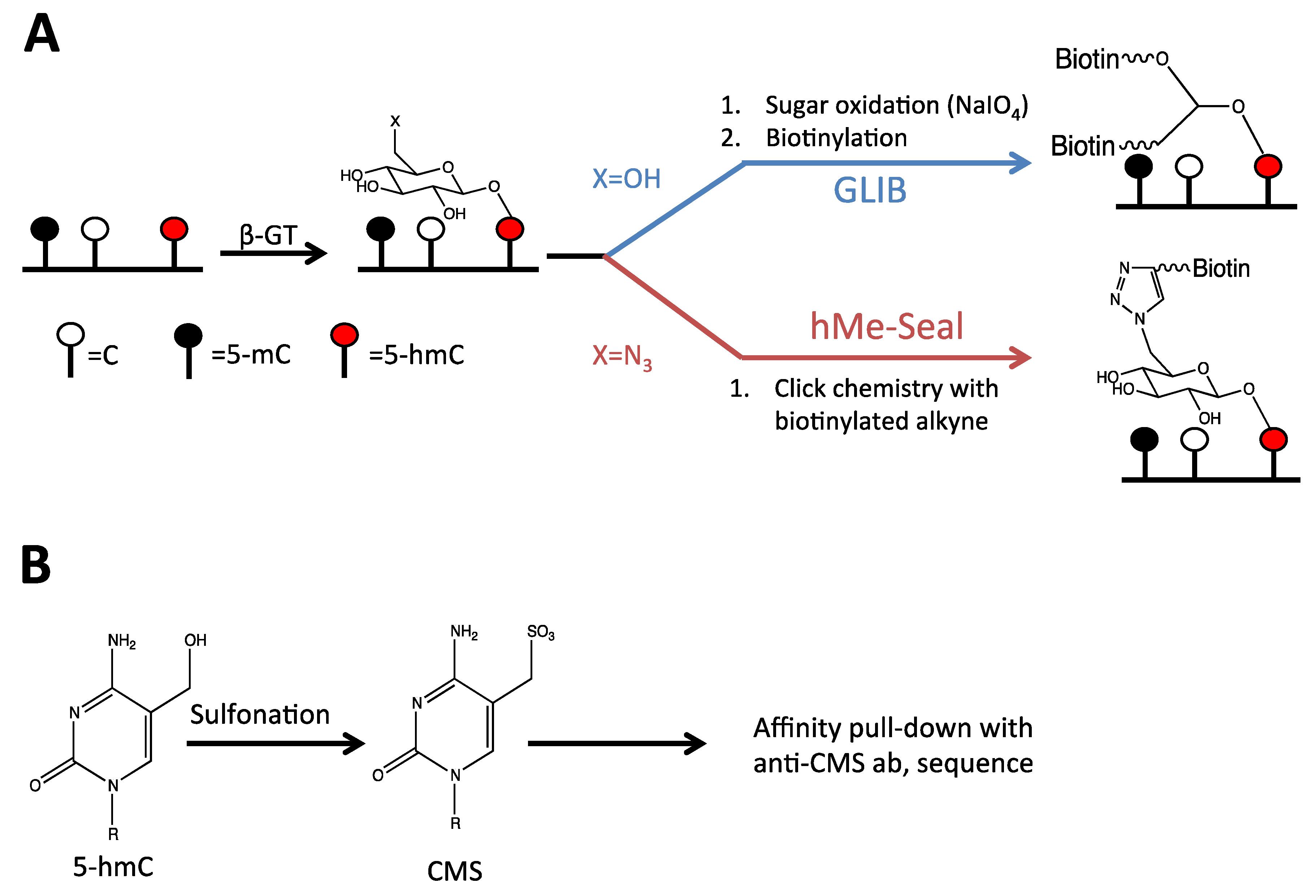
2.2. Detection of 5-hmC at the Global Level

2.3. Detection of 5-hmC at the Regional Level
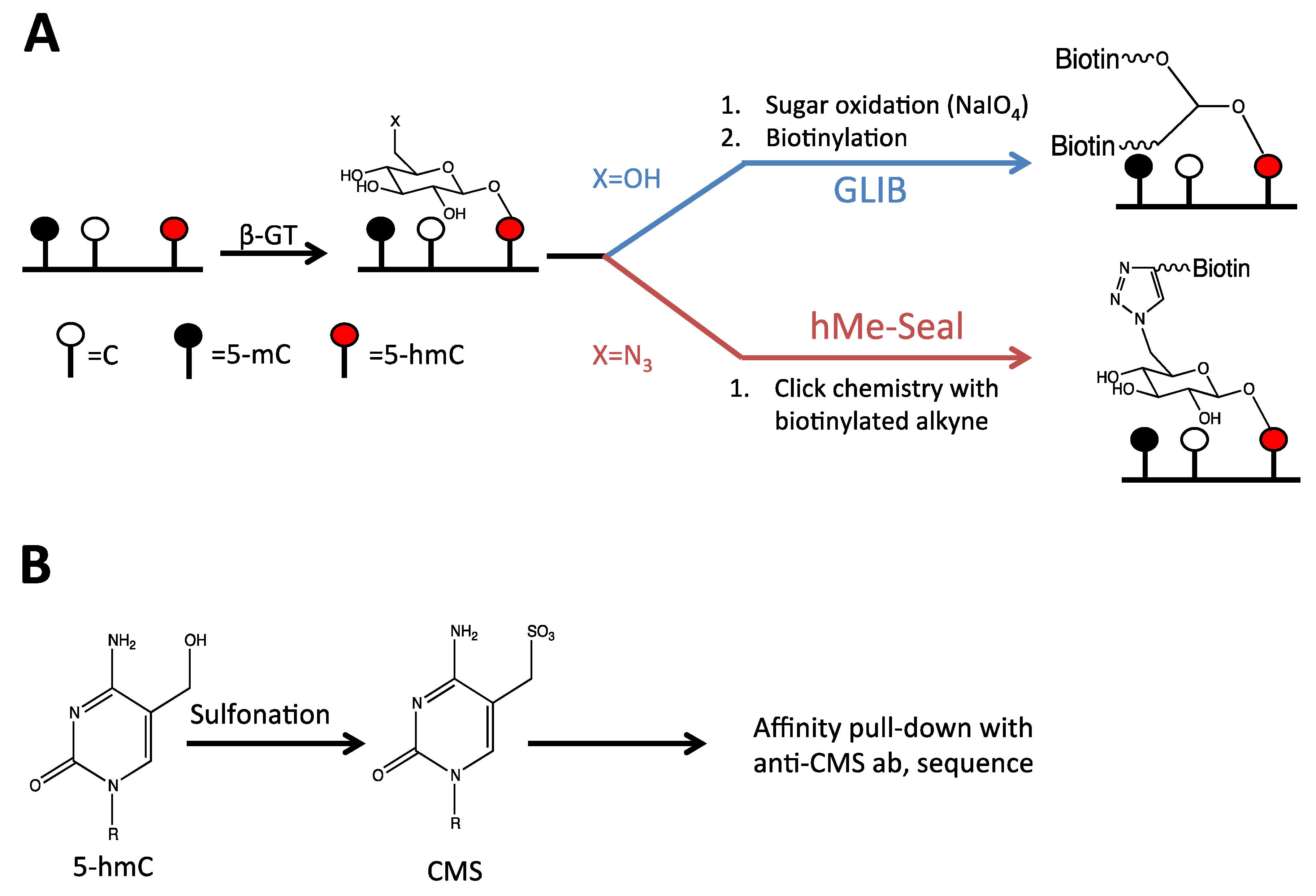
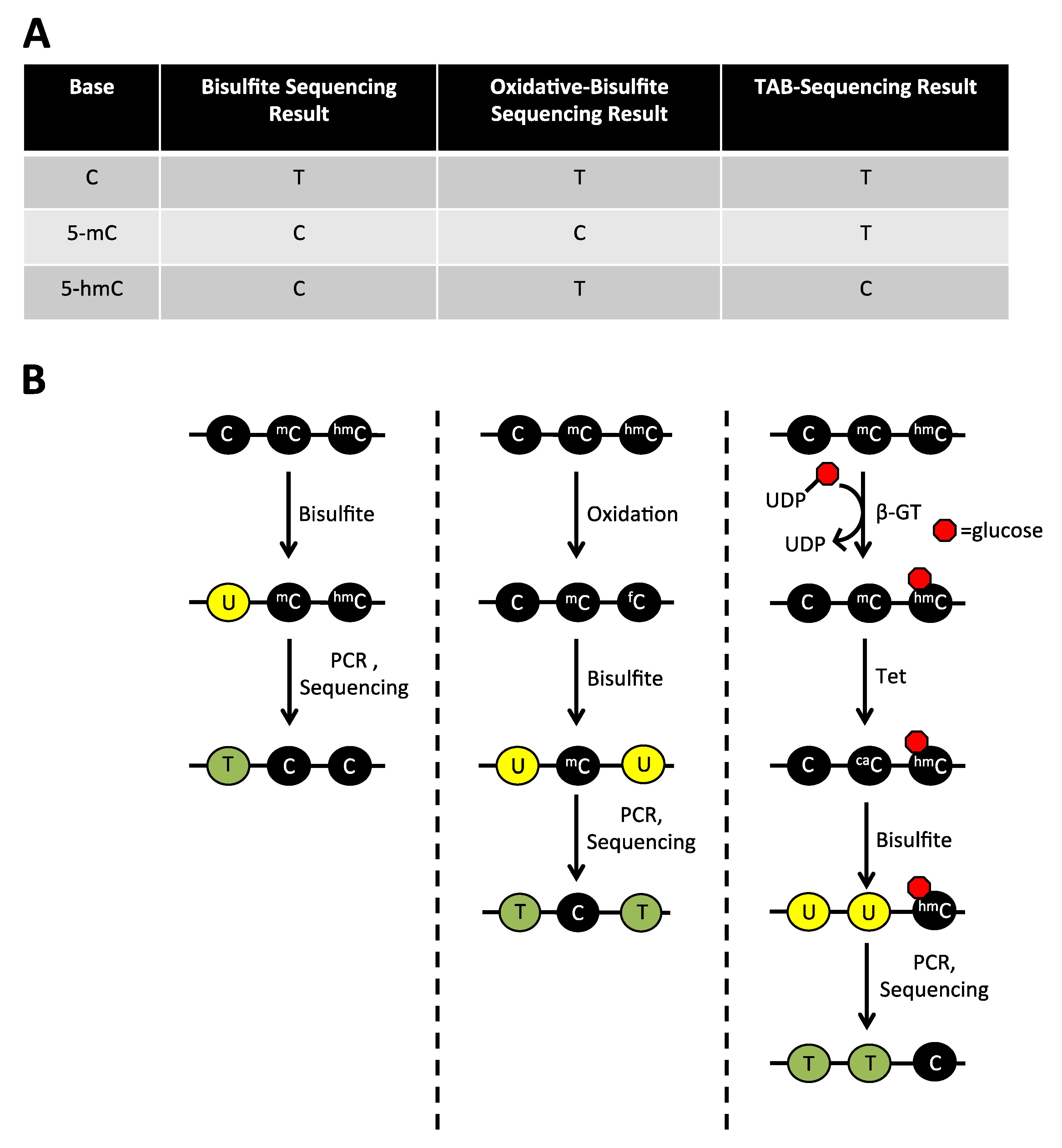
2.4. Detection of 5-hmC at the Single Base Level
3. The Role of Hydroxymethylation in Normal Physiology
3.1. Demethylation of the Paternal Genome after Fertilization
3.2. Embryonic Stem Cells (ESCs)
3.3. Hydroxymethylcytosine in the Brain
4. Glioma
4.1. 5-hmC Levels Are Reduced in Gliomas
4.2. Potential Mechanisms for Loss of 5-hmC in Gliomas
4.3. IDH1/2 Mutations
4.4. TET Silencing or Mislocalization
4.5. Removal of 5-hmC by Base Excision Repair Enzymes
5. Hematological Malignancies
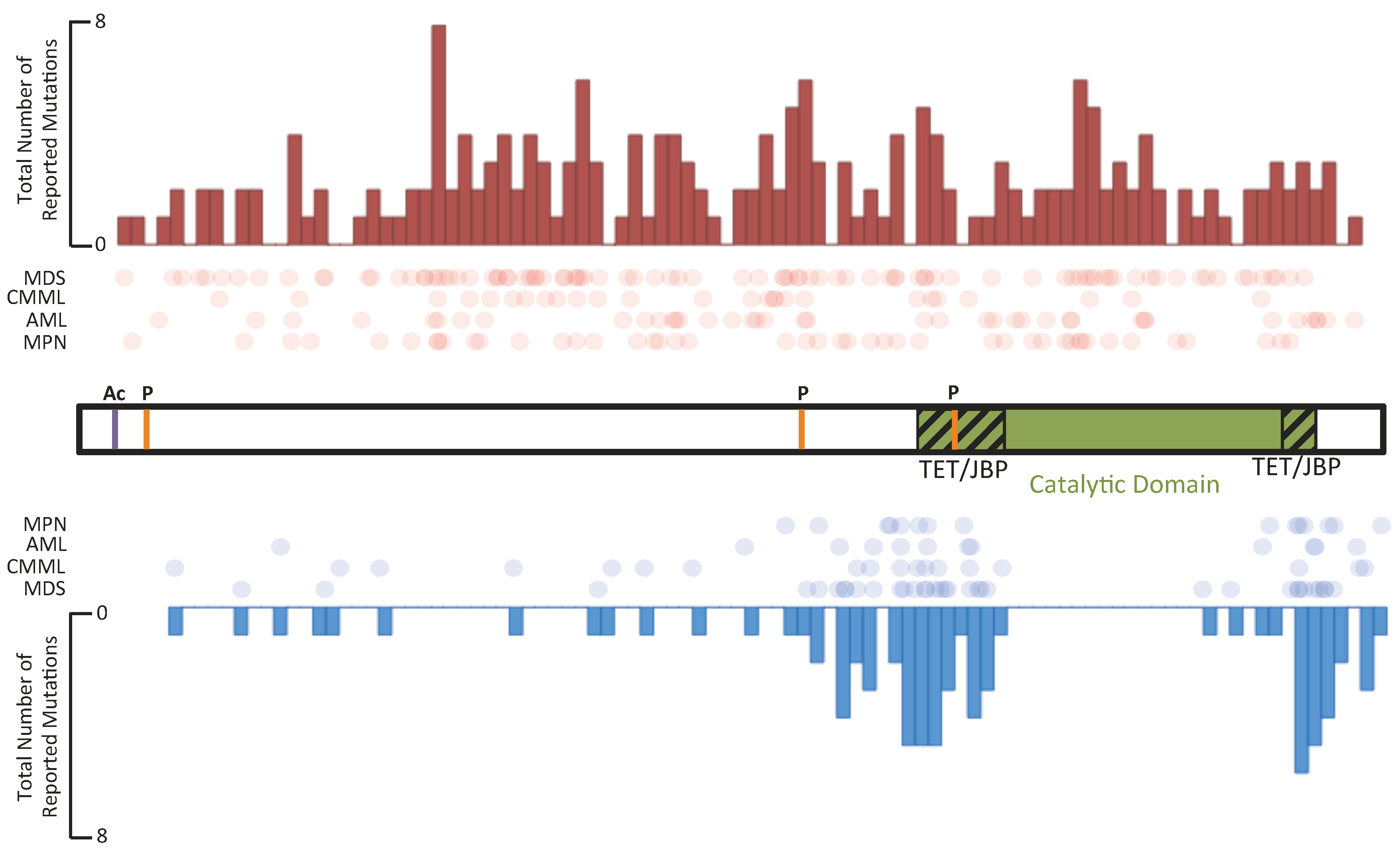
5.1. TET Enzymes in Hematological Malignancies
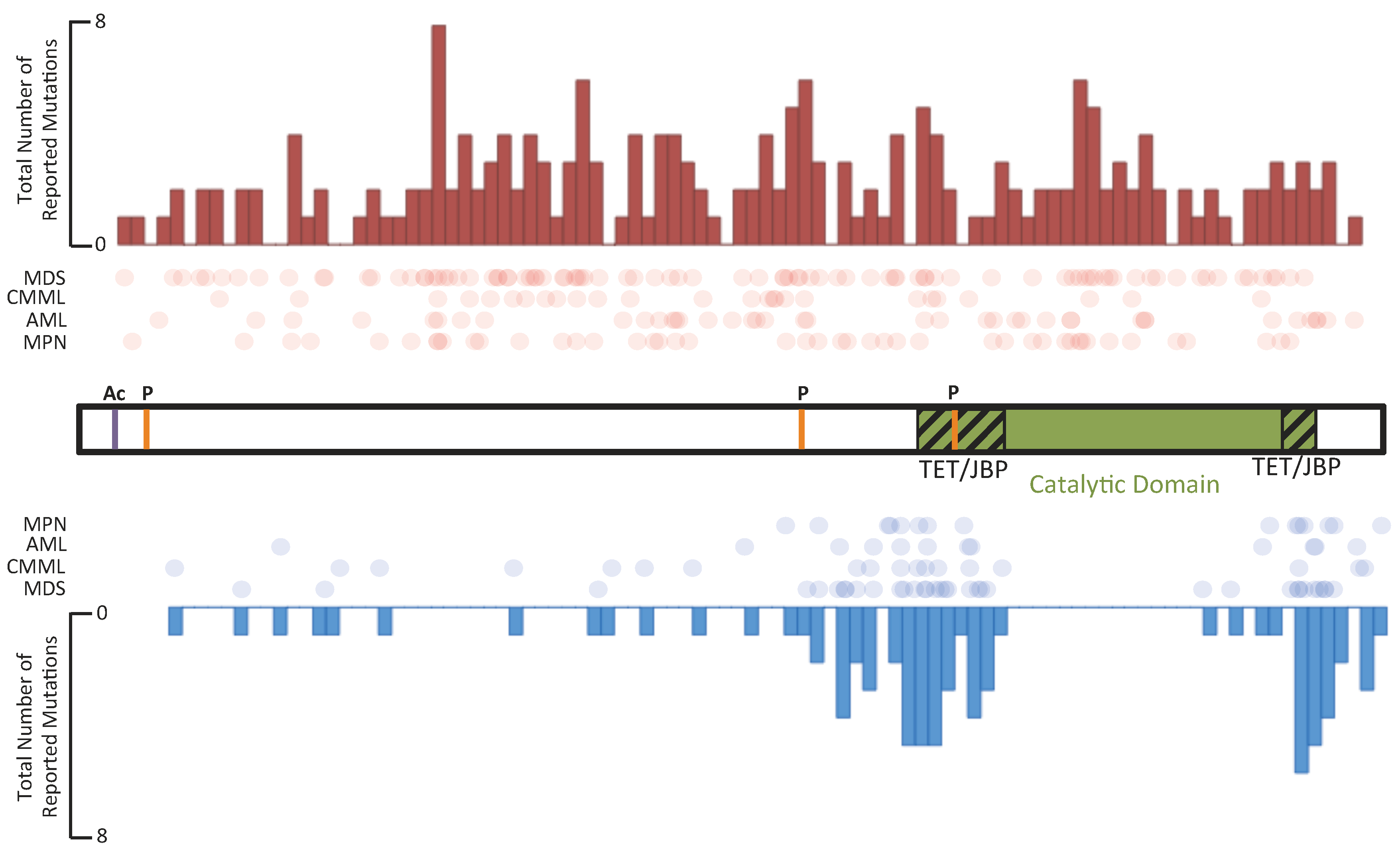
5.2. IDH Enzymes in Hematological Malignancies
6. Hydroxymethylation in Other Solid Tumors
7. Common Themes/Conclusions
References
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Penn, N.W.; Suwalski, R.; O’Riley, C.; Bojanowski, K.; Yura, R. The presence of 5-hydroxymethylcytosine in animal deoxyribonucleic acid. Biochem. J. 1972, 126, 781–790. [Google Scholar]
- Bogdanović, O.; Veenstra, G.J.C. DNA methylation and methyl-CpG binding proteins: developmental requirements and function. Chromosoma 2009, 118, 549–565. [Google Scholar] [CrossRef]
- Ono, R.; Taki, T.; Taketani, T.; Taniwaki, M.; Kobayashi, H.; Hayashi, Y. LCX, leukemia-associated protein with a CXXC domain, is fused to MLL in acute myeloid leukemia with trilineage dysplasia having t (10; 11)(q22; q23). Cancer Res. 2002, 62, 4075–4080. [Google Scholar]
- Lorsbach, R.B.; Moore, J.; Mathew, S.; Raimondi, S.C.; Mukatira, S.T.; Downing, J.R. TET1, a member of a novel protein family, is fused to MLL in acute myeloid leukemia containing the t(10;11)(q22;q23). Leukemia 2003, 17, 637–641. [Google Scholar] [CrossRef]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; Rao, A. Conversion of 5-Methylcytosine to 5-Hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef]
- Kriaucionis, S.; Heintz, N. The Nuclear DNA Base 5-Hydroxymethylcytosine is present in purkinje neurons and the brain. Science 2009, 324, 929–930. [Google Scholar] [CrossRef]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef]
- He, Y.F.; Li, B.Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef]
- Pfaffeneder, T.; Hackner, B.; Truß, M.; Münzel, M.; Müller, M.; Deiml, C.A.; Hagemeier, C.; Carell, T. The discovery of 5-formylcytosine in embryonic stem cell DNA. Angew. Chem. Int. Ed. 2011, 50, 7008–7012. [Google Scholar] [CrossRef]
- Valinluck, V.; Sowers, L.C. Endogenous cytosine damage products alter the site selectivity of human DNA maintenance methyltransferase DNMT1. Cancer Res. 2007, 67, 946–950. [Google Scholar] [CrossRef]
- Rai, K.; Huggins, I.J.; James, S.R.; Karpf, A.R.; Jones, D.A.; Cairns, B.R. DNA demethylation in zebrafish involves the coupling of a deaminase, a glycosylase, and Gadd45. Cell 2008, 135, 1201–1212. [Google Scholar] [CrossRef]
- Cortellino, S.; Xu, J.; Sannai, M.; Moore, R.; Caretti, E.; Cigliano, A.; Le Coz, M.; Devarajan, K.; Wessels, A.; Soprano, D.; et al. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell 2011, 146, 67–79. [Google Scholar] [CrossRef]
- Guo, J.U.; Su, Y.; Zhong, C.; Ming, G.-L.; Song, H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell 2011, 145, 423–434. [Google Scholar] [CrossRef]
- Rangam, G.; Schmitz, K.-M.; Cobb, A.J.A.; Petersen-Mahrt, S.K. AID enzymatic activity is inversely proportional to the size of cytosine C5 orbital cloud. PLoS One 2012, 7, e43279. [Google Scholar]
- Nabel, C.S.; Jia, H.; Ye, Y.; Shen, L.; Goldschmidt, H.L.; Stivers, J.T.; Zhang, Y.; Kohli, R.M. AID/APOBEC deaminases disfavor modified cytosines implicated in DNA demethylation. Nat. Chem. Biol. 2012, 8, 751–758. [Google Scholar] [CrossRef]
- Maiti, A.; Drohat, A.C. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: Potential implications for active demethylation of CpG sites. J. Biol. Chem. 2011, 286, 35334–35338. [Google Scholar] [CrossRef]
- Schiesser, S.; Hackner, B.; Pfaffeneder, T.; Müller, M.; Hagemeier, C.; Truß, M.; Carell, T. Mechanism and stem-cell activity of 5-carboxycytosine decarboxylation determined by isotope tracing. Angew. Chem. Int. Ed. 2012, 51, 6516–6520. [Google Scholar]
- Smiley, J.A.; Kundracik, M.; Landfried, D.A.; Barnes, V.R., Sr.; Axhemi, A.A. Genes of the thymidine salvage pathway: Thymine-7-hydroxylase from a Rhodotorula glutinis cDNA library and iso-orotate decarboxylase from Neurospora crassa. Biochim. Biophys. Acta 2005, 1723, 256–264. [Google Scholar] [CrossRef]
- Branco, M.R.; Ficz, G.; Reik, W. Uncovering the role of 5-hydroxymethylcytosine in the epigenome. Nat. Rev. Genet. 2011, 13, 7–13. [Google Scholar]
- Song, C.-X.; Yi, C.; He, C. Mapping recently identified nucleotide variants in the genome and transcriptome. Nat. Biotechnol. 2012, 30, 1107–1116. [Google Scholar]
- Yildirim, O.; Li, R.; Hung, J.-H.; Chen, P.B.; Dong, X.; Ee, L.-S.; Weng, Z.; Rando, O.J.; Fazzio, T.G. Mbd3/NURD complex regulates expression of 5-hydroxymethylcytosine marked genes in embryonic stem cells. Cell 2011, 147, 1498–1510. [Google Scholar] [CrossRef]
- Frauer, C.; Hoffmann, T.; Bultmann, S.; Casa, V.; Cardoso, M.C.; Antes, I.; Leonhardt, H. Recognition of 5-hydroxymethylcytosine by the Uhrf1 SRA domain. PLoS One 2011, 6, e21306. [Google Scholar]
- Mellén, M.; Ayata, P.; Dewell, S.; Kriaucionis, S.; Heintz, N. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell 2012, 151, 1417–1430. [Google Scholar] [CrossRef]
- Spruijt, C.G.; Gnerlich, F.; Smits, A.H.; Pfaffeneder, T.; Jansen, P.W.T.C.; Bauer, C.; Münzel, M.; Wagner, M.; Müller, M.; Khan, F.; et al. Dynamic Readers for 5-(Hydroxy)Methylcytosine and Its Oxidized Derivatives. Cell 2013, 152, 1146–1159. [Google Scholar] [CrossRef]
- Jin, S.-G.; Kadam, S.; Pfeifer, G.P. Examination of the specificity of DNA methylation profiling techniques towards 5-methylcytosine and 5-hydroxymethylcytosine. Nucleic Acids Res. 2010, 38, e125. [Google Scholar]
- Huang, Y.; Pastor, W.A.; Shen, Y.; Tahiliani, M.; Liu, D.R.; Rao, A. The Behaviour of 5-Hydroxymethylcytosine in Bisulfite Sequencing. PLoS One 2010, 5, e8888. [Google Scholar]
- Hershey, A.D.; Dixon, J.; Chase, M. Nucleic acid economy in bacteria infected with bacteriophage T2. J. Gen. Physiol. 1953, 37, 1–23. [Google Scholar] [CrossRef]
- Wyatt, G.R.; Cohen, S.S. The bases of the nucleic acids of some bacterial and animal viruses: the occurrence of 5-hydroxymethylcytosine. Biochem. J. 1953, 55, 774–782. [Google Scholar]
- Pastor, W.A.; Huang, Y.; Henderson, H.R.; Agarwal, S.; Rao, A. The GLIB technique for genome-wide mapping of 5-hydroxymethylcytosine. Nat. Protoc. 2012, 7, 1909–1917. [Google Scholar] [CrossRef]
- Harris, D.C. Chapter 23: Introduction to Analytical Separations. In Quantitative Chemical Analysis, 7th ed; W.H. Freeman and Company: New York, NY, USA, 2007; pp. 501–528. [Google Scholar]
- Münzel, M.; Globisch, D.; Brückl, T.; Wagner, M.; Welzmiller, V.; Michalakis, S.; Müller, M.; Biel, M.; Carell, T. Quantification of the sixth DNA base hydroxymethylcytosine in the brain. Angew. Chem. Int. Ed. 2010, 49, 5375–5377. [Google Scholar]
- Song, L.; James, S.R.; Kazim, L.; Karpf, A.R. Specific method for the determination of genomic DNA methylation by liquid chromatography-electrospray ionization tandem mass spectrometry. Anal. Chem. 2005, 77, 504–510. [Google Scholar] [CrossRef]
- Kornberg, S.R.; Zimmerman, S.B.; Kornberg, A. Glucosylation of deoxyribonucleic acid by enzymes from bacteriophage-infected Escherichia coli. J. Biol. Chem. 1961, 236, 1487–1493. [Google Scholar]
- Szwagierczak, A.; Bultmann, S.; Schmidt, C.S.; Spada, F.; Leonhardt, H. Sensitive enzymatic quantification of 5-hydroxymethylcytosine in genomic DNA. Nucleic Acids Res. 2010, 38, e181. [Google Scholar]
- Terragni, J.; Bitinaite, J.; Zheng, Y.; Pradhan, S. Biochemical characterization of recombinant β-glucosyltransferase and analysis of global 5-hydroxymethylcytosine in unique genomes. Biochemistry 2012, 51, 1009–1019. [Google Scholar]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.-H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.-T. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar]
- Ko, M.; Huang, Y.; Jankowska, A.M.; Pape, U.J.; Tahiliani, M.; Bandukwala, H.S.; An, J.; Lamperti, E.D.; Koh, K.P.; Ganetzky, R.; et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature 2011, 2011, 839–843. [Google Scholar]
- Song, C.-X.; Szulwach, K.E.; Fu, Y.; Dai, Q.; Yi, C.; Li, X.; Li, Y.; Chen, C.-H.; Zhang, W.; Jian, X.; et al. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat. Biotechnol. 2010, 29, 68–72. [Google Scholar]
- Huang, Y.; Pastor, W.A.; Zepeda-Martínez, J.A.; Rao, A. The anti-CMS technique for genome-wide mapping of 5-hydroxymethylcytosine. Nat. Protoc. 2012, 7, 1897–1908. [Google Scholar] [CrossRef]
- Krueger, F.; Kreck, B.; Franke, A.; Andrews, S.R. DNA methylome analysis using short bisulfite sequencing data. Nat. Meth. 2012, 9, 145–151. [Google Scholar] [CrossRef]
- Lim, J.-Q.; Tennakoon, C.; Li, G.; Wong, E.; Ruan, Y.; Wei, C.-L.; Sung, W.-K. BatMeth: Improved mapper for bisulfite sequencing reads on DNA methylation. Genome Biol. 2012, 13, R82. [Google Scholar]
- Chatterjee, A.; Stockwell, P.A.; Rodger, E.J.; Morison, I.M. Comparison of alignment software for genome-wide bisulphite sequence data. Nucleic Acids Res. 2012, 40, e79. [Google Scholar]
- Waalwijk, C.; Flavell, R.A. MspI, an isoschizomer of hpaII which cleaves both unmethylated and methylated hpaII sites. Nucleic Acids Res. 1978, 5, 3231–3236. [Google Scholar]
- Shen, C.K.; Maniatis, T. Tissue-specific DNA methylation in a cluster of rabbit beta-like globin genes. Proc. Natl. Acad. Sci. USA 1980, 77, 6634–6638. [Google Scholar] [CrossRef]
- Kinney, S.M.; Chin, H.G.; Vaisvila, R.; Bitinaite, J.; Zheng, Y.; Esteve, P.O.; Feng, S.; Stroud, H.; Jacobsen, S.E.; Pradhan, S. Tissue-specific distribution and dynamic changes of 5-hydroxymethylcytosine in mammalian genomes. J. Biol. Chem. 2011, 286, 24685–24693. [Google Scholar] [CrossRef]
- Wang, H.; Guan, S.; Quimby, A.; Cohen-Karni, D.; Pradhan, S.; Wilson, G.; Roberts, R.J.; Zhu, Z.; Zheng, Y. Comparative characterization of the PvuRts1I family of restriction enzymes and their application in mapping genomic 5-hydroxymethylcytosine. Nucleic Acids Res. 2011, 39, 9294–9305. [Google Scholar] [CrossRef]
- Sun, Z.; Jolyon, T.; Borgaro, J.G.; Liu, Y.; Yu, L.; Guan, S.; Wang, H.; Sun, D.; Cheng, X.; Zhu, Z.; et al. High-Resolution Enzymatic Mapping of Genomic 5-Hydroxymethylcytosine in Mouse Embryonic Stem Cells. Cell Rep. 2013, 3, 567–576. [Google Scholar] [CrossRef]
- Booth, M.J.; Branco, M.R.; Ficz, G.; Oxley, D.; Krueger, F.; Reik, W.; Balasubramanian, S. Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science 2012, 336, 934–937. [Google Scholar]
- Yu, M.; Hon, G.C.; Szulwach, K.E.; Song, C.-X.; Zhang, L.; Kim, A.; Li, X.; Dai, Q.; Shen, Y.; Park, B.; et al. Base-Resolution Analysis of 5-Hydroxymethylcytosine in the Mammalian Genome. Cell 2012, 149, 1368–1380. [Google Scholar] [CrossRef]
- Flusberg, B.A.; Webster, D.R.; Lee, J.H.; Travers, K.J.; Olivares, E.C.; Clark, T.A.; Korlach, J.; Turner, S.W. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat. Meth. 2010, 7, 461–465. [Google Scholar] [CrossRef]
- Song, C.-X.; Clark, T.A.; Lu, X.-Y.; Kislyuk, A.; Dai, Q.; Turner, S.W.; He, C.; Korlach, J. Sensitive and specific single-molecule sequencing of 5-hydroxymethylcytosine. Nat. Meth. 2011, 9, 75–77. [Google Scholar]
- Oswald, J.; Engemann, S.; Lane, N.; Mayer, W.; Olek, A.; Fundele, R.; Dean, W.; Reik, W.; Walter, J. Active demethylation of the paternal genome in the mouse zygote. Curr. Biol. 2000, 10, 475–478. [Google Scholar]
- Mayer, W.; Niveleau, A.; Walter, J.; Fundele, R.; Haaf, T. Embryogenesis: demethylation of the zygotic paternal genome. Nature 2000, 403, 501–502. [Google Scholar]
- Wossidlo, M.; Nakamura, T.; Lepikhov, K.; Marques, C.J.; Zakhartchenko, V.; Boiani, M.; Arand, J.; Nakano, T.; Reik, W.; Walter, J.O.R. 5-Hydroxymethylcytosine in the mammalian zygote is linked with epigenetic reprogramming. Nat. Commun. 2011, 2, 241–248. [Google Scholar]
- Iqbal, K.; Jin, S.G.; Pfeifer, G.P.; Szabó, P.E. Reprogramming of the paternal genome upon fertilization involves genome-wide oxidation of 5-methylcytosine. Proc. Natl. Acad. Sci. USA 2011, 108, 3642–3647. [Google Scholar] [CrossRef]
- Inoue, A.; Zhang, Y. Replication-dependent loss of 5-hydroxymethylcytosine in mouse preimplantation embryos. Science 2011, 334, 194. [Google Scholar]
- Gu, T.-P.; Guo, F.; Yang, H.; Wu, H.-P.; Xu, G.-F.; Liu, W.; Xie, Z.-G.; Shi, L.; He, X.; Jin, S.-G.; et al. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature 2011, 477, 606–610. [Google Scholar] [CrossRef]
- Li, G.; Reinberg, D. Chromatin higher-order structures and gene regulation. Curr. Opin. Genet. Dev. 2011, 21, 175–186. [Google Scholar]
- Nakamura, T.; Liu, Y.-J.; Nakashima, H.; Umehara, H.; Inoue, K.; Matoba, S.; Tachibana, M.; Ogura, A.; Shinkai, Y.; Nakano, T. PGC7 binds histone H3K9me2 to protect against conversion of 5mC to 5hmC in early embryos. Nature 2012, 486, 415–419. [Google Scholar]
- Koh, K.P.; Yabuuchi, A.; Rao, S.; Huang, Y.; Cunniff, K.; Nardone, J.; Laiho, A.; Tahiliani, M.; Sommer, C.A.; Mostoslavsky, G.; et al. Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell 2011, 8, 200–213. [Google Scholar] [CrossRef]
- Ficz, G.; Branco, M.R.; Seisenberger, S.; Santos, F.; Krueger, F.; Hore, T.A.; Marques, C.J.; Andrews, S.; Reik, W. Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature 2011, 473, 398–402. [Google Scholar] [CrossRef]
- Ito, S.; D’Alessio, A.C.; Taranova, O.V.; Hong, K.; Sowers, L.C.; Zhang, Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 2010, 466, 1129–1133. [Google Scholar] [CrossRef]
- Costa, Y.; Ding, J.; Theunissen, T.W.; Faiola, F.; Hore, T.A.; Shliaha, P.V.; Fidalgo, M.; Saunders, A.; Lawrence, M.; Dietmann, S.; et al. NANOG-dependent function of TET1 and TET2 in establishment of pluripotency. Nature 2013, 495, 370–374. [Google Scholar] [CrossRef]
- Dawlaty, M.M.; Ganz, K.; Powell, B.E.; Hu, Y.-C.; Markoulaki, S.; Cheng, A.W.; Gao, Q.; Kim, J.; Choi, S.-W.; Page, D.C.; et al. Tet1 Is dispensable for maintaining pluripotency and its loss Is compatible with embryonic and postnatal development. Cell Stem Cell 2011, 9, 166–175. [Google Scholar]
- Dawlaty, M.M.; Breiling, A.; Le, T.; Raddatz, G.; Barrasa, M.I.; Cheng, A.W.; Gao, Q.; Powell, B.E.; Li, Z.; Xu, M.; et al. Combined deficiency of Tet1 and Tet2 causes epigenetic abnormalities but is compatible with postnatal development. Dev. Cell 2013, 3, 310–323. [Google Scholar]
- Xu, Y.; Wu, F.; Tan, L.; Kong, L.; Xiong, L.; Deng, J.; Barbera, A.J.; Zheng, L.; Zhang, H.; Huang, S.; et al. Genome-wide regulation of 5hmC, 5mC, and gene expression by Tet1 hydroxylase in mouse embryonic stem cells. Mol. Cell 2011, 42, 451–464. [Google Scholar] [CrossRef]
- Wu, H.; D’Alessio, A.C.; Ito, S.; Xia, K.; Wang, Z.; Cui, K.; Zhao, K.; Sun, Y.E.; Zhang, Y. Dual functions of Tet1 in transcriptional regulation in mouse embryonic stem cells. Nature 2011, 473, 389–393. [Google Scholar] [CrossRef]
- Globisch, D.; Münzel, M.; Müller, M.; Michalakis, S.; Wagner, M.; Koch, S.; Brückl, T.; Biel, M.; Carell, T. Tissue Distribution of 5-Hydroxymethylcytosine and Search for Active Demethylation Intermediates. PLoS One 2010, 5, e15367. [Google Scholar] [CrossRef] [Green Version]
- Szulwach, K.E.; Li, X.; Li, Y.; Song, C.-X.; Wu, H.; Dai, Q.; Irier, H.; Upadhyay, A.K.; Gearing, M.; Levey, A.I.; et al. 5-hmC–mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat. Neurosci. 2011, 14, 1607–1616. [Google Scholar] [CrossRef]
- Khare, T.; Pai, S.; Koncevicius, K.; Pal, M.; Kriukiene, E.; Liutkeviciute, Z.; Irimia, M.; Jia, P.; Ptak, C.; Xia, M.; et al. 5-hmC in the brain is abundant in synaptic genes and shows differences at the exon-intron boundary. Nat. Struct. Mol. Biol. 2012, 19, 1037–1043. [Google Scholar] [CrossRef]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Fanelli, M.; Caprodossi, S.; Ricci-Vitiani, L.; Porcellini, A.; Tomassoni-Ardori, F.; Amatori, S.; Andreoni, F.; Magnani, M.; de Maria, R.; Santoni, A.; et al. Loss of pericentromeric DNA methylation pattern in human glioblastoma is associated with altered DNA methyltransferases expression and involves the stem cell compartment. Oncogene 2007, 27, 358–365. [Google Scholar]
- Cadieux, B.; Ching, T.-T.; VandenBerg, S.R.; Costello, J.F. Genome-wide hypomethylation in human glioblastomas associated with specific copy number alteration, methylenetetrahydrofolate reductase allele status, and increased proliferation. Cancer Res. 2006, 66, 8469–8476. [Google Scholar] [CrossRef]
- Orr, B.A.; Haffner, M.C.; Nelson, W.G.; Yegnasubramanian, S.; Eberhart, C.G. Decreased 5-hydroxymethylcytosine is associated with neural progenitor phenotype in normal brain and shorter survival in malignant glioma. PLoS One 2012, 7, e41036. [Google Scholar]
- Kraus, T.F.J.; Globisch, D.; Wagner, M.; Eigenbrod, S.; Widmann, D.; Münzel, M.; Müller, M.; Pfaffeneder, T.; Hackner, B.; Feiden, W.; et al. Low values of 5-hydroxymethylcytosine (5hmC), the “sixth base,” are associated with anaplasia in human brain tumors. Int. J. Cancer 2012, 131, 1577–1590. [Google Scholar] [CrossRef]
- Reitman, Z.J.; Yan, H. Isocitrate dehydrogenase 1 and 2 mutations in cancer: alterations at a crossroads of cellular metabolism. J. Natl. Cancer Inst. 2010, 102, 932–941. [Google Scholar] [CrossRef]
- Balss, J.; Meyer, J.; Mueller, W.; Korshunov, A.; Hartmann, C.; Deimling, A. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol. 2008, 116, 597–602. [Google Scholar] [CrossRef]
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 2010, 17, 510–522. [Google Scholar] [CrossRef]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.-M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef]
- Christensen, B.C.; Smith, A.A.; Zheng, S.; Koestler, D.C.; Houseman, E.A.; Marsit, C.J.; Wiemels, J.L.; Nelson, H.H.; Karagas, M.R.; Wrensch, M.R.; et al. DNA Methylation, Isocitrate Dehydrogenase Mutation, and Survival in Glioma. J. Natl. Cancer Inst. 2011, 103, 143–153. [Google Scholar] [CrossRef]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.M.; Lu, C.; Ward, P.S.; et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef]
- Müller, T.; Gessi, M.; Waha, A.; Isselstein, L.J.; Luxen, D.; Freihoff, D.; Freihoff, J.; Becker, A.; Simon, M.; Hammes, J.; et al. Nuclear exclusion of TET1 is associated with loss of 5-hydroxymethylcytosine in IDH1 wild-type gliomas. Am. J. Pathol. 2012, 181, 675–683. [Google Scholar] [CrossRef]
- Liu, Y.; Jiang, W.; Liu, J.; Zhao, S.; Xiong, J.; Mao, Y.; Wang, Y. IDH1 mutations inhibit multiple α-ketoglutarate-dependent dioxygenase activities in astroglioma. J. Neurooncol. 2012, 109, 253–260. [Google Scholar] [CrossRef]
- Kim, Y.H.; Pierscianek, D.; Mittelbronn, M.; Vital, A.; Mariani, L.; Hasselblatt, M.; Ohgaki, H. TET2 promoter methylation in low-grade diffuse gliomas lacking IDH1/2 mutations. J. Clin. Pathol. 2011, 64, 850–852. [Google Scholar] [CrossRef]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- McLendon, R.; Friedman, A.; Bigner, D.; van Meir, E.G.; Brat, D.J.; M Mastrogianakis, G.; Olson, J.J.; Mikkelsen, T.; Lehman, N.; et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Ko, M.; Bandukwala, H.S.; An, J.; Lamperti, E.D.; Thompson, E.C.; Hastie, R.; Tsangaratou, A.; Rajewsky, K.; Koralov, S.B.; Rao, A. Ten-Eleven-Translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc. Natl. Acad. Sci. USA 2011, 108, 14566–14571. [Google Scholar] [CrossRef]
- Moran-Crusio, K.; Reavie, L.; Shih, A.; Abdel-Wahab, O.; Ndiaye-Lobry, D.; Lobry, C.; Figueroa, M.E.; Vasanthakumar, A.; Patel, J.; Zhao, X.; et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 2011, 20, 11–24. [Google Scholar] [CrossRef]
- Sasaki, M.; Knobbe, C.B.; Munger, J.C.; Lind, E.F.; Brenner, D.; Brüstle, A.; Harris, I.S.; Holmes, R.; Wakeham, A.; Haight, J.; et al. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature 2012, 488, 656–659. [Google Scholar] [CrossRef]
- Delhommeau, F.; Dupont, S.; Valle Della, V.; James, C.; Trannoy, S.; Massé, A.; Kosmider, O.; le Couedic, J.-P.; Robert, F.; Alberdi, A.; et al. Mutation in TET2 in myeloid cancers. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef]
- Langemeijer, S.M.C.; Kuiper, R.P.; Berends, M.; Knops, R.; Aslanyan, M.G.; Massop, M.; Stevens-Linders, E.; van Hoogen, P.; van Kessel, A.G.; Raymakers, R.A.P.; et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat. Genet. 2009, 41, 838–842. [Google Scholar] [CrossRef]
- Abdel-Wahab, O.; Mullally, A.; Hedvat, C.; Garcia-Manero, G.; Patel, J.; Wadleigh, M.; Malinge, S.; Yao, J.; Kilpivaara, O.; Bhat, R.; et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood 2009, 114, 144–147. [Google Scholar] [CrossRef]
- Beer, P.A.; Delhommeau, F.; LeCouedic, J.P.; Dawson, M.A.; Chen, E.; Bareford, D.; Kusec, R.; McMullin, M.F.; Harrison, C.N.; Vannucchi, A.M.; et al. Two routes to leukemic transformation after a JAK2 mutation-positive myeloproliferative neoplasm. Blood 2010, 115, 2891–2900. [Google Scholar] [CrossRef]
- Colaizzo, D.; Tiscia, G.L.; Pisanelli, D.; Bafunno, V.; Amitrano, L.; Grandone, E.; Guardascione, M.A.; Margaglione, M. New TET2 gene mutations in patients with myeloproliferative neoplasms and splanchnic vein thrombosis. J. Thromb. Haemost. 2010, 5, 1142–1144. [Google Scholar]
- Couronné, L.; Lippert, E.; Andrieux, J.; Kosmider, O.; Radford-Weiss, I.; Penther, D.; Dastugue, N.; Mugneret, F.; Lafage, M.; Gachard, N.; et al. Analyses of TET2 mutations in post-myeloproliferative neoplasm acute myeloid leukemias. Leukemia 2010, 24, 201–203. [Google Scholar] [CrossRef]
- Flach, J.; Dicker, F.; Schnittger, S.; Kohlmann, A.; Haferlach, T.; Haferlach, C. Mutations of JAK2 and TET2, but not CBL are detectable in a high portion of patients with refractory anemia with ring sideroblasts and thrombocytosis. Haematologica 2010, 95, 518–519. [Google Scholar] [CrossRef]
- Gelsi-Boyer, V.; Trouplin, V.; Adélaïde, J.; Bonansea, J.; Cervera, N.; Carbuccia, N.; Lagarde, A.; Prebet, T.; Nezri, M.; Sainty, D.; et al. Mutations of polycomb-associated gene ASXL1in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br. J. Haematol. 2009, 145, 788–800. [Google Scholar] [CrossRef]
- Jankowska, A.M.; Szpurka, H.; Tiu, R.V.; Makishima, H.; Afable, M.; Huh, J.; O’Keefe, C.L.; Ganetzky, R.; McDevitt, M.A.; Maciejewski, J.P. Loss of heterozygosity 4q24 and TET2 mutations associated with myelodysplastic/myeloproliferative neoplasms. Blood 2009, 113, 6403–6410. [Google Scholar] [CrossRef]
- Kosmider, O.; Gelsi-Boyer, V.; Ciudad, M.; Racoeur, C.; Jooste, V.; Vey, N.; Quesnel, B.; Fenaux, P.; Bastie, J.N.; Beyne-Rauzy, O.; et al. TET2 gene mutation is a frequent and adverse event in chronic myelomonocytic leukemia. Haematologica 2009, 94, 1676–1681. [Google Scholar] [CrossRef]
- Mohamedali, A.M.; Smith, A.E.; Gaken, J.; Lea, N.C.; Mian, S.A.; Westwood, N.B.; Strupp, C.; Gattermann, N.; Germing, U.; Mufti, G.J. Novel TET2 mutations associated with UPD4q24 in myelodysplastic syndrome. J. Clin. Orthod. 2009, 27, 4002–4006. [Google Scholar]
- Nibourel, O.; Kosmider, O.; Cheok, M.; Boissel, N.; Renneville, A.; Philippe, N.; Dombret, H.; Dreyfus, F.; Quesnel, B.; Geffroy, S.; et al. Incidence and prognostic value of TET2 alterations in de novo acute myeloid leukemia achieving complete remission. Blood 2010, 116, 1132–1135. [Google Scholar] [CrossRef]
- Schaub, F.X.; Looser, R.; Li, S.; Hao-Shen, H.; Lehmann, T.; Tichelli, A.; Skoda, R.C. Clonal analysis of TET2 and JAK2 mutations suggests that TET2 can be a late event in the progression of myeloproliferative neoplasms. Blood 2010, 115, 2003–2007. [Google Scholar] [CrossRef]
- Szpurka, H.; Jankowska, A.M.; Makishima, H.; Bodo, J.; Bejanyan, N.; Hsi, E.D.; Sekeres, M.A.; Maciejewski, J.P. Spectrum of mutations in RARS-T patients includes TET2 and ASXL1 mutations. Leuk. Res. 2010, 34, 969–973. [Google Scholar] [CrossRef]
- Yamazaki, J.; Taby, R.; Vasanthakumar, A.; Macrae, T.; Ostler, K.R.; Shen, L.; Kantarjian, H.M.; Estecio, M.R.; Jelinek, J.; Godley, L.A.; et al. Effects of TET2 mutations on DNA methylation in chronic myelomonocytic leukemia. Epigenetics 2012, 7, 201–207. [Google Scholar] [CrossRef]
- Quivoron, C.; Couronné, L.; Valle, D.V.; Lopez, C.K.; Plo, I.; Wagner-Ballon, O.; do Cruzeiro, M.; Delhommeau, F.; Arnulf, B.; et al. TET2 Inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell 2011, 20, 25–38. [Google Scholar] [CrossRef]
- Kunimoto, H.; Fukuchi, Y.; Sakurai, M.; Sadahira, K.; Ikeda, Y.; Okamoto, S.; Nakajima, H. Tet2 disruption leads to enhanced self-renewal and altered differentiation of fetal liver hematopoietic stem cells. Sci. Rep. 2012, 2, 273. [Google Scholar]
- Li, Z.; Cai, X.; Cai, C.L.; Wang, J.; Zhang, W.; Petersen, B.E.; Yang, F.C.; Xu, M. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood 2011, 118, 4509–4518. [Google Scholar] [CrossRef]
- Pronier, E.; Almire, C.; Mokrani, H.; Vasanthakumar, A.; Simon, A.; da Costa, R.M.B.; Masse, A.; Le Couedic, J.P.; Pendino, F.; Carbonne, B.; et al. Inhibition of TET2-mediated conversion of 5-methylcytosine to 5-hydroxymethylcytosine disturbs erythroid and granulomonocytic differentiation of human hematopoietic progenitors. Blood 2011, 118, 2551–2555. [Google Scholar] [CrossRef]
- Abdel-Wahab, O.; Manshouri, T.; Patel, J.; Harris, K.; Yao, J.; Hedvat, C.; Heguy, A.; Bueso-Ramos, C.; Kantarjian, H.; Levine, R.L.; et al. Genetic analysis of transforming events that convert chronic myeloproliferative neoplasms to leukemias. Cancer Res. 2010, 70, 447–452. [Google Scholar] [CrossRef]
- Metzeler, K.H.; Maharry, K.; Radmacher, M.D.; Mrozek, K.; Margeson, D.; Becker, H.; Curfman, J.; Holland, K.B.; Schwind, S.; Whitman, S.P.; et al. TET2 mutations improve the new european leukemianet risk classification of acute myeloid leukemia: A cancer and leukemia group B study. J. Clin. Orhtop. 2011, 29, 1373–1381. [Google Scholar]
- Bejar, R.; Stevenson, K.; Abdel-Wahab, O.; Galili, N.; Nilsson, B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.L.; Neuberg, D. Clinical effect of point mutations in myelodysplastic syndromes. N. Engl. J. Med. 2011, 364, 2496–2506. [Google Scholar] [CrossRef]
- Liu, X.; Guangsen, Z.; Yi, Y.; Xiao, L.; Pei, M.; Liu, S.; Luo, Y.; Zhong, H.; Xu, Y.; Zheng, W.; Shen, J. Decreased 5-hydroxymethylcytosine levels are associated with TET2 mutation and unfavorable overall survival in myelodysplastic syndromes. Leuk. Lymphoma 2013. [Google Scholar] [CrossRef]
- Sjoblom, T.; Jones, S.; Wood, L.D.; Parsons, D.W.; Lin, J.; Barber, T.D.; Mandelker, D.; Leary, R.J.; Ptak, J.; Silliman, N.; Szabo, S.; et al. The Consensus Coding Sequences of Human Breast and Colorectal Cancers. Science 2006, 314, 268–274. [Google Scholar] [CrossRef]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Patel, J.P.; Gonen, M.; Figueroa, M.E.; Fernandez, H.; Sun, Z.; Racevskis, J.; van Vlierberghe, P.; Dolgalev, I.; Thomas, S.; Aminova, O.; et al. Prognostic relevance of integrated genetic profiling in acute myeloid Leukemia. N. Engl. J. Med. 2012, 366, 1079–1089. [Google Scholar] [CrossRef]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef]
- Gross, S.; Cairns, R.A.; Minden, M.D.; Driggers, E.M.; Bittinger, M.A.; Jang, H.G.; Sasaki, M.; Jin, S.; Schenkein, D.P.; Su, S.M.; et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J. Exp. Med. 2010, 207, 339–344. [Google Scholar] [CrossRef]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef]
- Chowdhury, R.; Yeoh, K.K.; Tian, Y.-M.; Hillringhaus, L.; Bagg, E.A.; Rose, N.R.; Leung, I.K.H.; Li, X.S.; Woon, E.C.Y.; Yang, M.; et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011, 12, 463–469. [Google Scholar] [CrossRef]
- Koivunen, P.; Lee, S.; Duncan, C.G.; Lopez, G.; Lu, G.; Ramkissoon, S.; Losman, J.A.; Joensuu, P.; Bergmann, U.; Gross, S.; et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 2012, 483, 484–488. [Google Scholar] [CrossRef]
- Losman, J.-A.; Looper, R.; Koivunen, P.; Lee, S.; Schneider, R.K.; McMahon, C.; Cowley, G.; Root, D.; Ebert, B.L.; Kaelin, W.G. (R)-2-Hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 2013, 339, 1621–1625. [Google Scholar] [CrossRef]
- Pollyea, D.A.; Kohrt, H.E.; Zhang, B.; Zehnder, J.; Schenkein, D.; Fantin, V.; Straley, K.; Vasanthakumar, A.; Abdel-Wahab, O.; Levine, R.; et al. 2-Hydroxyglutarate in IDH mutant acute myeloid leukemia: predicting patient responses, minimal residual disease and correlations with methylcytosine and hydroxymethylcytosine levels. Leuk. Lymphoma 2013, 54, 408–410. [Google Scholar] [CrossRef]
- Popovici-Muller, J.; Saunders, J.O.; Salituro, F.G.; Travins, J.M.; Yan, S.; Zhao, F.; Gross, S.; Dang, L.; Yen, K.E.; Yang, H.; et al. Discovery of the first potent inhibitors of mutant IDH1 that lower tumor 2-HG in vivo. ACS Med. Chem. Lett. 2012, 3, 850–855. [Google Scholar] [CrossRef]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar]
- Green, C.L.; Evans, C.M.; Hills, R.K.; Burnett, A.K.; Linch, D.C.; Gale, R.E. The prognostic significance of IDH1 mutations in younger adult patients with acute myeloid leukemia is dependent on FLT3/ITD status. Blood 2010, 116, 2779–2782. [Google Scholar] [CrossRef]
- Green, C.L.; Evans, C.M.; Zhao, L.; Hills, R.K.; Burnett, A.K.; Linch, D.C.; Gale, R.E. The prognostic significance of IDH2 mutations in AML depends on the location of the mutation. Blood 2011, 118, 409–412. [Google Scholar] [CrossRef]
- Döhner, K.; Schlenk, R.F.; Habdank, M.; Scholl, C.; Rücker, F.G.; Corbacioglu, A.; Bullinger, L.; Fröhling, S.; Döhner, H. Mutant nucleophosmin (NPM1) predicts favorable prognosis in younger adults with acute myeloid leukemia and normal cytogenetics: interaction with other gene mutations. Blood 2005, 106, 3740–3746. [Google Scholar] [CrossRef]
- Chotirat, S.; Thongnoppakhun, W.; Promsuwicha, O.; Boonthimat, C.; Auewarakul, C.U. Molecular alterations of isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2) metabolic genes and additional genetic mutations in newly diagnosed acute myeloid leukemia patients. J. Hematol. Oncol. 2012, 5, 5. [Google Scholar] [CrossRef]
- Marcucci, G.; Maharry, K.; Wu, Y.Z.; Radmacher, M.D.; Mrozek, K.; Margeson, D.; Holland, K.B.; Whitman, S.P.; Becker, H.; Schwind, S.; et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: A cancer and leukemia group B study. J. Clin. Orthod. 2010, 28, 2348–2355. [Google Scholar]
- Paschka, P.; Schlenk, R.F.; Gaidzik, V.I.; Habdank, M.; Kronke, J.; Bullinger, L.; Spath, D.; Kayser, S.; Zucknick, M.; Gotze, K.; et al. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J. Clin. Orhtod. 2010, 28, 3636–3643. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Lasho, T.L.; Guglielmelli, P.; Biamonte, F.; Pardanani, A.; Pereira, A.; Finke, C.; Score, J.; Gangat, N.; Mannarelli, C.; et al. Mutations and prognosis in primary myelofibrosis. Leukemia 2013, 1–33. [Google Scholar]
- Haffner, M.C.; Chaux, A.; Meeker, A.K.; Esopi, D.M.; Gerber, J.; Pellakuru, L.G.; Toubaji, A.; Argani, P.; Iacobuzio-Donahue, C.; Nelson, W.G.; et al. Global 5-hydroxymethylcytosine content is significantly reduced in tissue stem/progenitor cell compartments and in human cancers. Oncotarget 2011, 2, 627–637. [Google Scholar]
- Jin, S.G.; Jiang, Y.; Qiu, R.; Rauch, T.A.; Wang, Y.; Schackert, G.; Krex, D.; Lu, Q.; Pfeifer, G.P. 5-Hydroxymethylcytosine is strongly depleted in human cancers but its levels do not correlate with IDH1 mutations. Cancer Res. 2011, 71, 7360–7365. [Google Scholar] [CrossRef]
- Yang, H.; Liu, Y.; Bai, F.; Zhang, J.-Y.; Ma, S.-H.; Liu, J.; Xu, Z.-D.; Zhu, H.-G.; Ling, Z.-Q.; Ye, D.; et al. Tumor development is associated with decrease of TET gene expression and 5-methylcytosine hydroxylation. Oncogene 2013, 32, 663–669. [Google Scholar] [CrossRef]
- Chen, M.L.; Shen, F.; Huang, W.; Qi, J.H.; Wang, Y.; Feng, Y.Q.; Liu, S.M.; Yuan, B.F. Quantification of 5-methylcytosine and 5-hydroxymethylcytosine in genomic DNA from hepatocellular carcinoma tissues by capillary hydrophilic-Interaction liquid chromatography/quadrupole TOF mass spectrometry. Clin. Chem. 2013, 59, 824–832. [Google Scholar] [CrossRef]
- Kudo, Y.; Tateishi, K.; Yamamoto, K.; Yamamoto, S.; Asaoka, Y.; Ijichi, H.; Nagae, G.; Yoshida, H.; Aburatani, H.; Koike, K. Loss of 5-hydroxymethylcytosine is accompanied with malignant cellular transformation. Cancer Sci. 2012, 103, 670–676. [Google Scholar] [CrossRef]
- Lian, C.G.; Xu, Y.; Ceol, C.; Wu, F.; Larson, A.; Dresser, K.; Xu, W.; Tan, L.; Hu, Y.; Zhan, Q.; et al. Loss of 5-Hydroxymethylcytosine Is an Epigenetic Hallmark of Melanoma. Cell 2012, 150, 1135–1146. [Google Scholar] [CrossRef]
- Gambichler, T.; Sand, M.; Skrygan, M. Loss of 5-hydroxymethylcytosine and ten-eleven translocation 2 protein expression in malignant melanoma. Melanoma Res. 2013, 23, 218–220. [Google Scholar] [CrossRef]
- Shibata, T.; Kokubu, A.; Miyamoto, M.; Sasajima, Y.; Yamazaki, N. Mutant IDH1 confers an in vivo growth in a melanoma cell line with BRAF mutation. Am. J. Pathol. 2011, 178, 1395–1402. [Google Scholar] [CrossRef]
- Hsu, C.-H.; Peng, K.-L.; Kang, M.-L.; Chen, Y.-R.; Yang, Y.-C.; Tsai, C.-H.; Chu, C.-S.; Jeng, Y.-M.; Chen, Y.-T.; Lin, F.-M.; et al. TET1 suppresses cancer invasion by activating the tissue Inhibitors of metalloproteinases. Cell Rep. 2012, 2, 568–579. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mariani, C.J.; Madzo, J.; Moen, E.L.; Yesilkanal, A.; Godley, L.A. Alterations of 5-Hydroxymethylcytosine in Human Cancers. Cancers 2013, 5, 786-814. https://doi.org/10.3390/cancers5030786
Mariani CJ, Madzo J, Moen EL, Yesilkanal A, Godley LA. Alterations of 5-Hydroxymethylcytosine in Human Cancers. Cancers. 2013; 5(3):786-814. https://doi.org/10.3390/cancers5030786
Chicago/Turabian StyleMariani, Christopher J., Jozef Madzo, Erika L. Moen, Ali Yesilkanal, and Lucy A. Godley. 2013. "Alterations of 5-Hydroxymethylcytosine in Human Cancers" Cancers 5, no. 3: 786-814. https://doi.org/10.3390/cancers5030786