Gene Regulatory Scenarios of Primary 1,25-Dihydroxyvitamin D3 Target Genes in a Human Myeloid Leukemia Cell Line


Abstract
:1. Introduction
2. Experimental
2.1. Cell Culture
2.2. RNA Extraction, cDNA Synthesis and qPCR
2.3. ChIP
3. Results
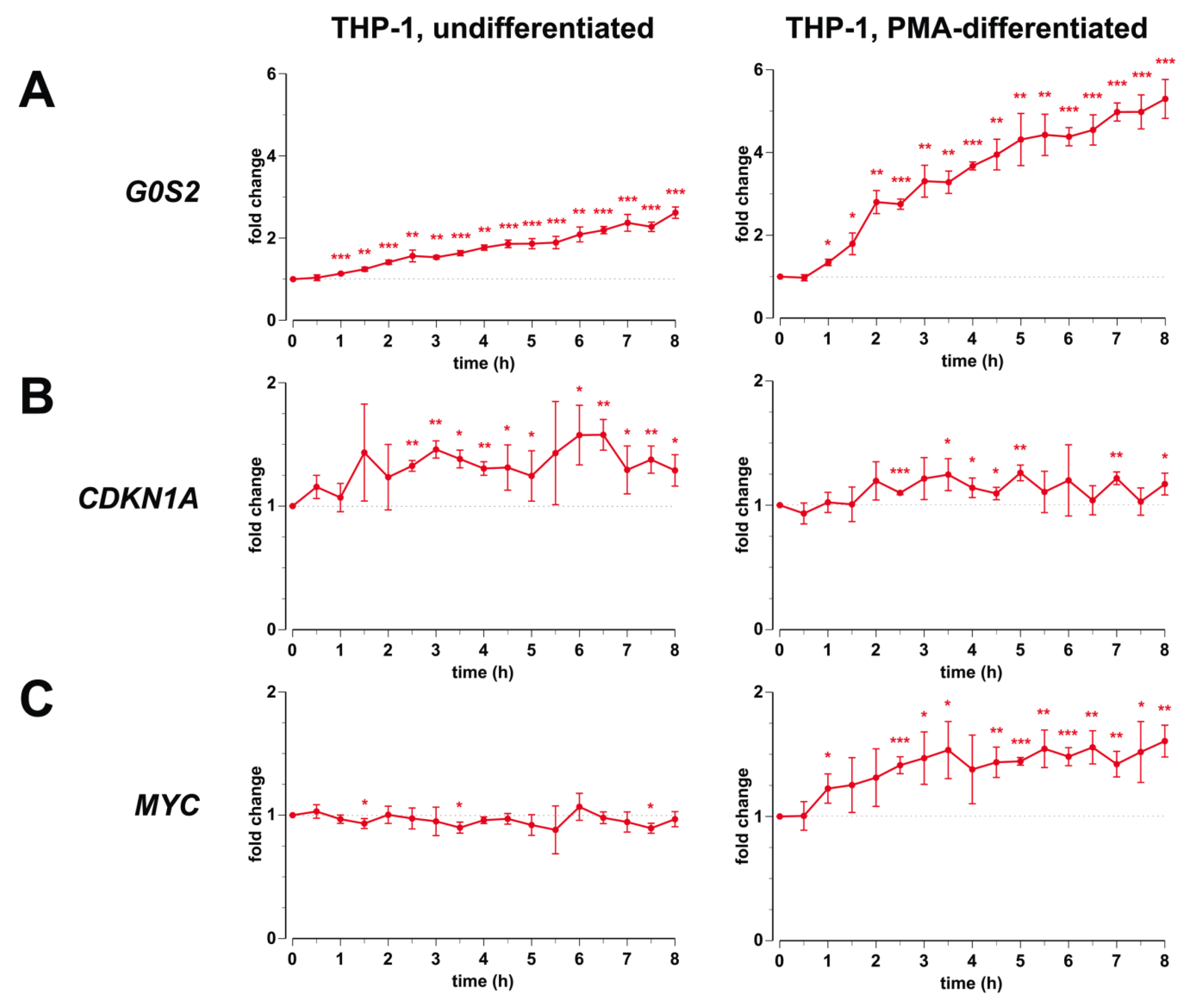
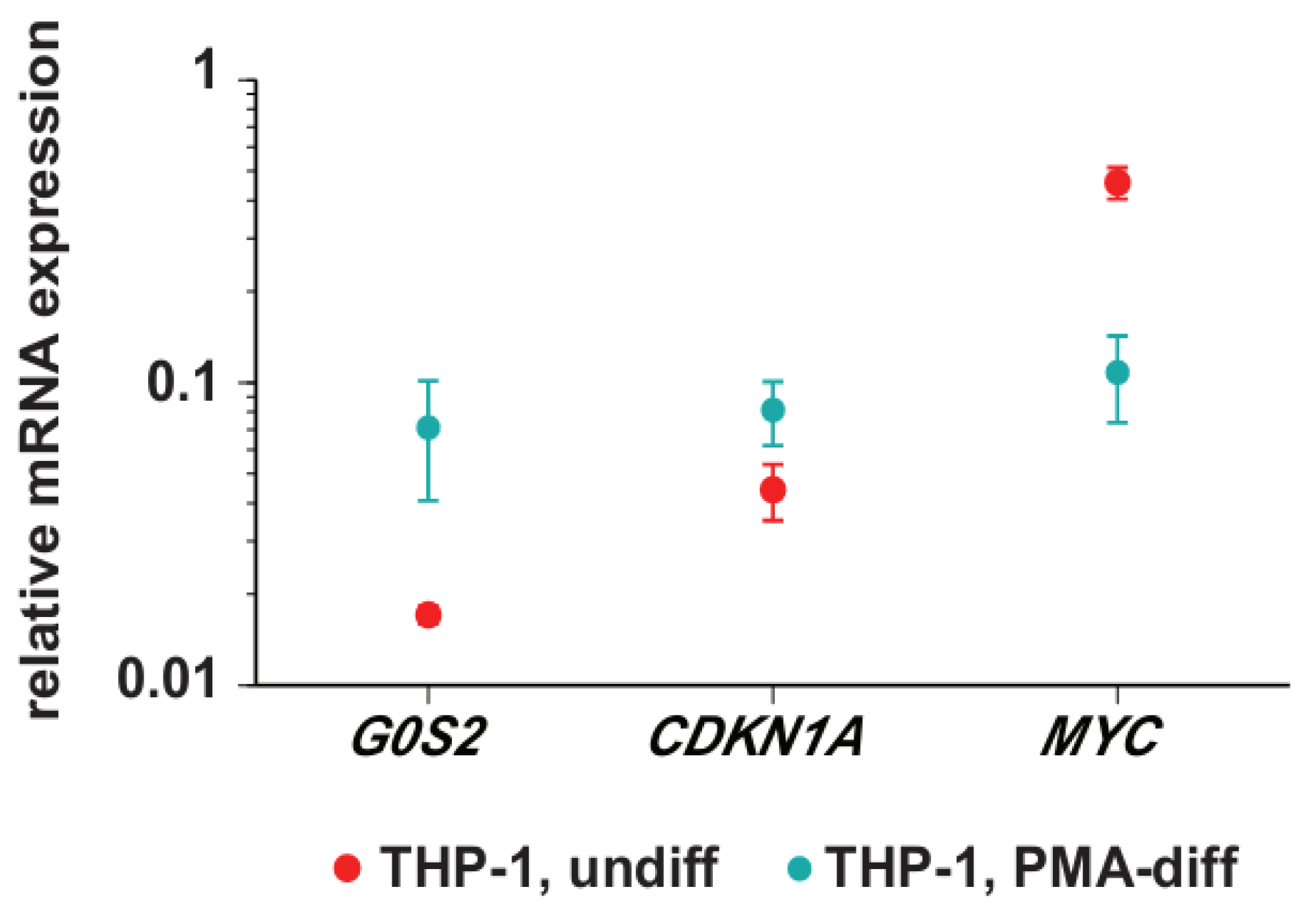
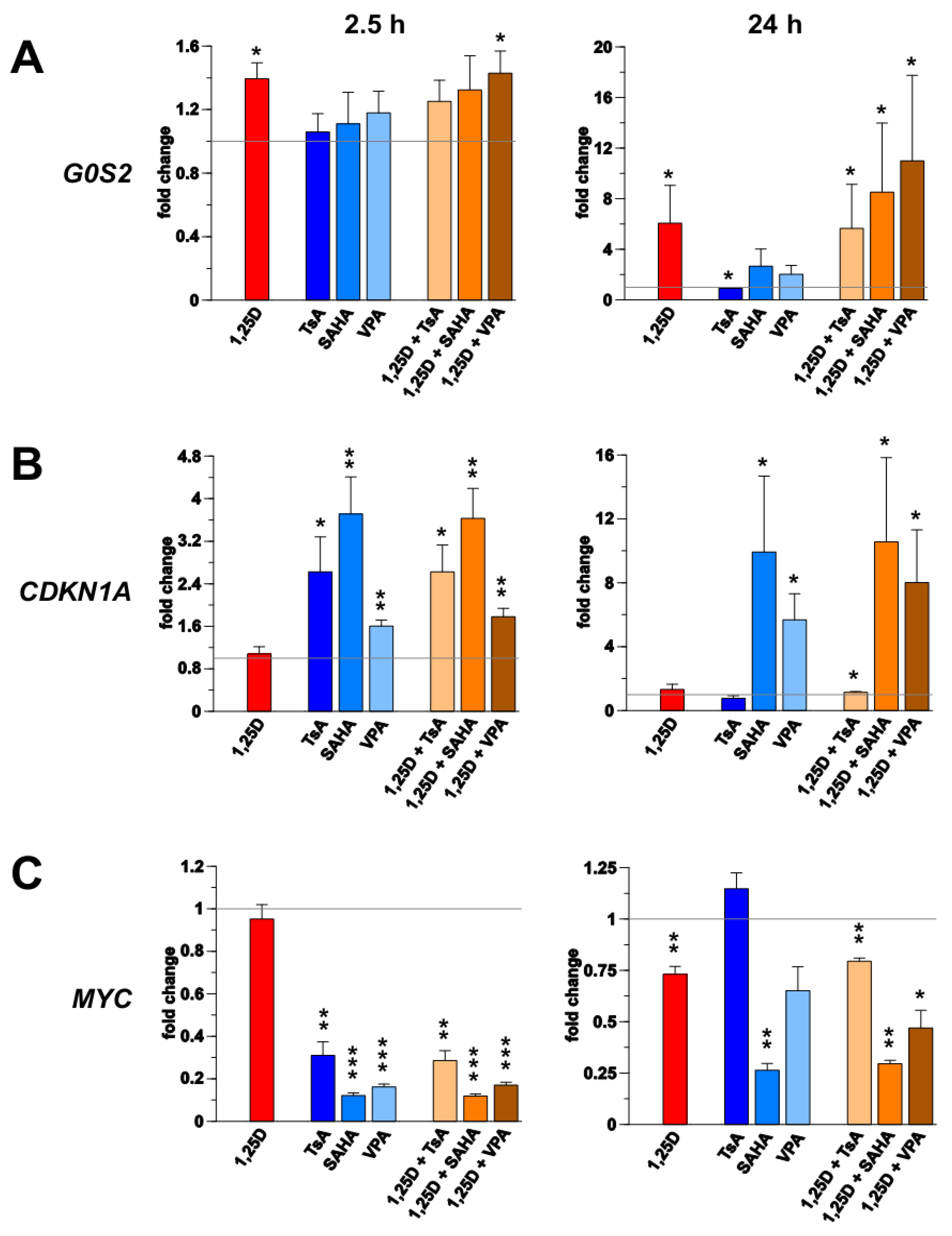
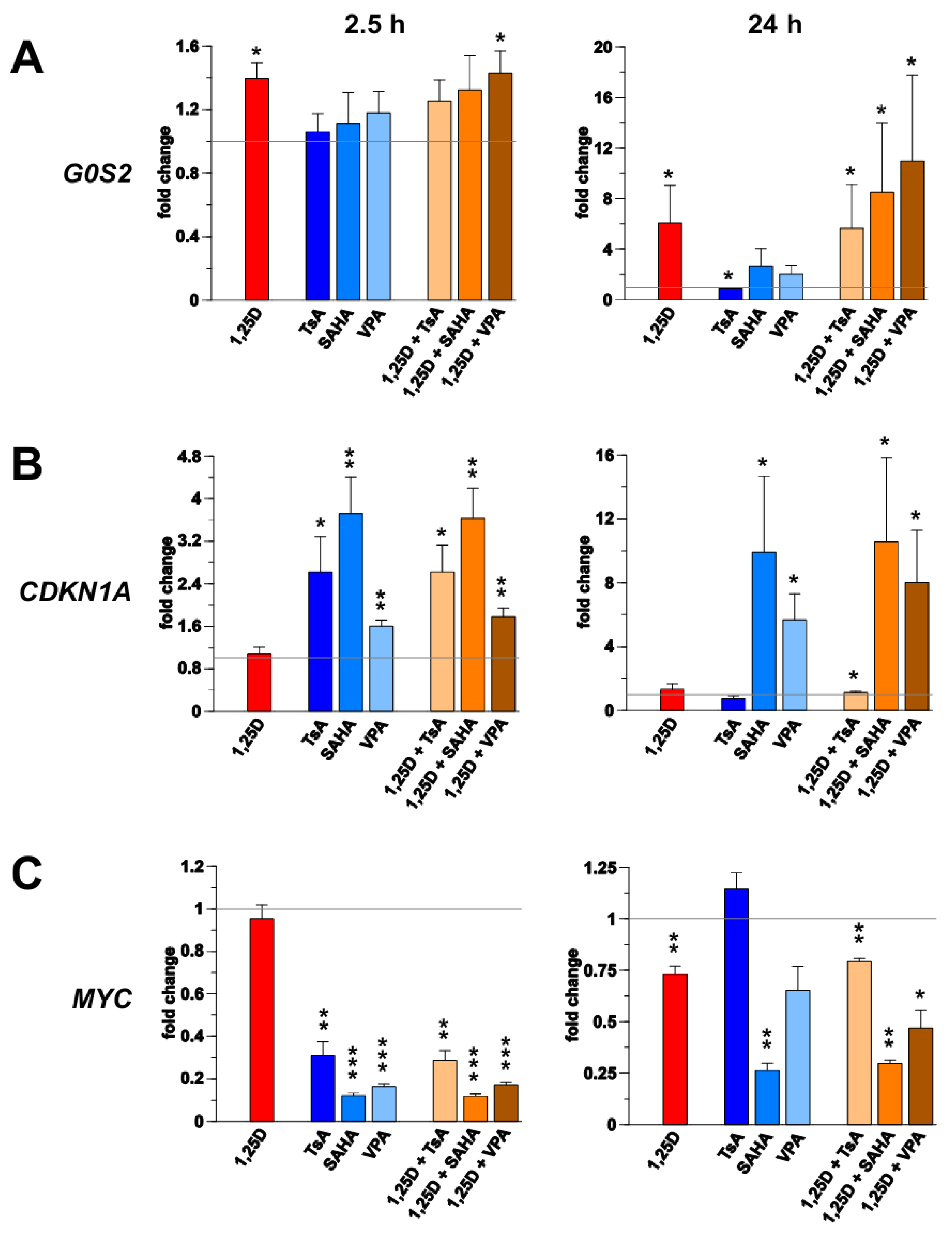
3.1. Transcription of G0S2, CDKN1A and MYC in Monocyte- and Macrophage-Like Cells

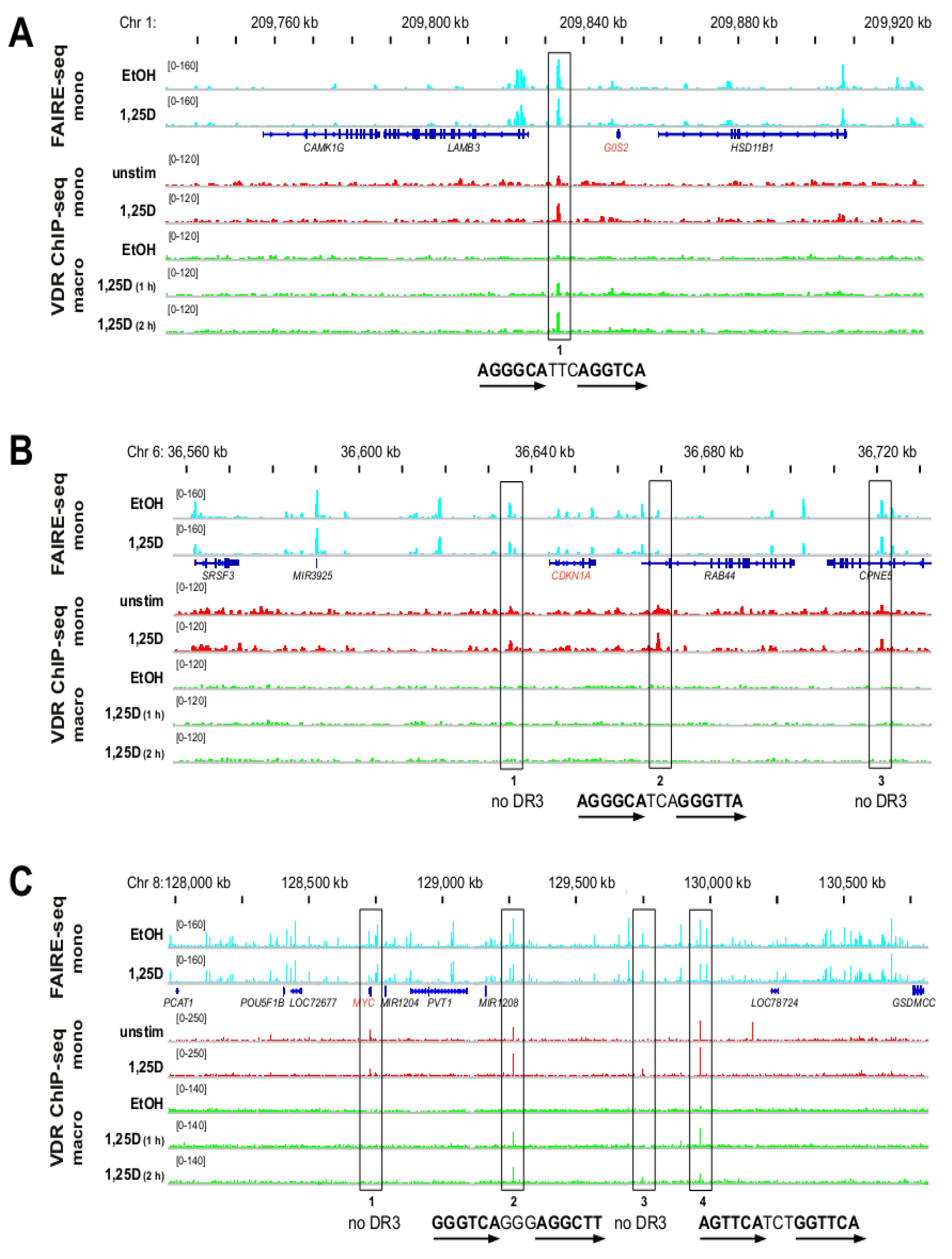
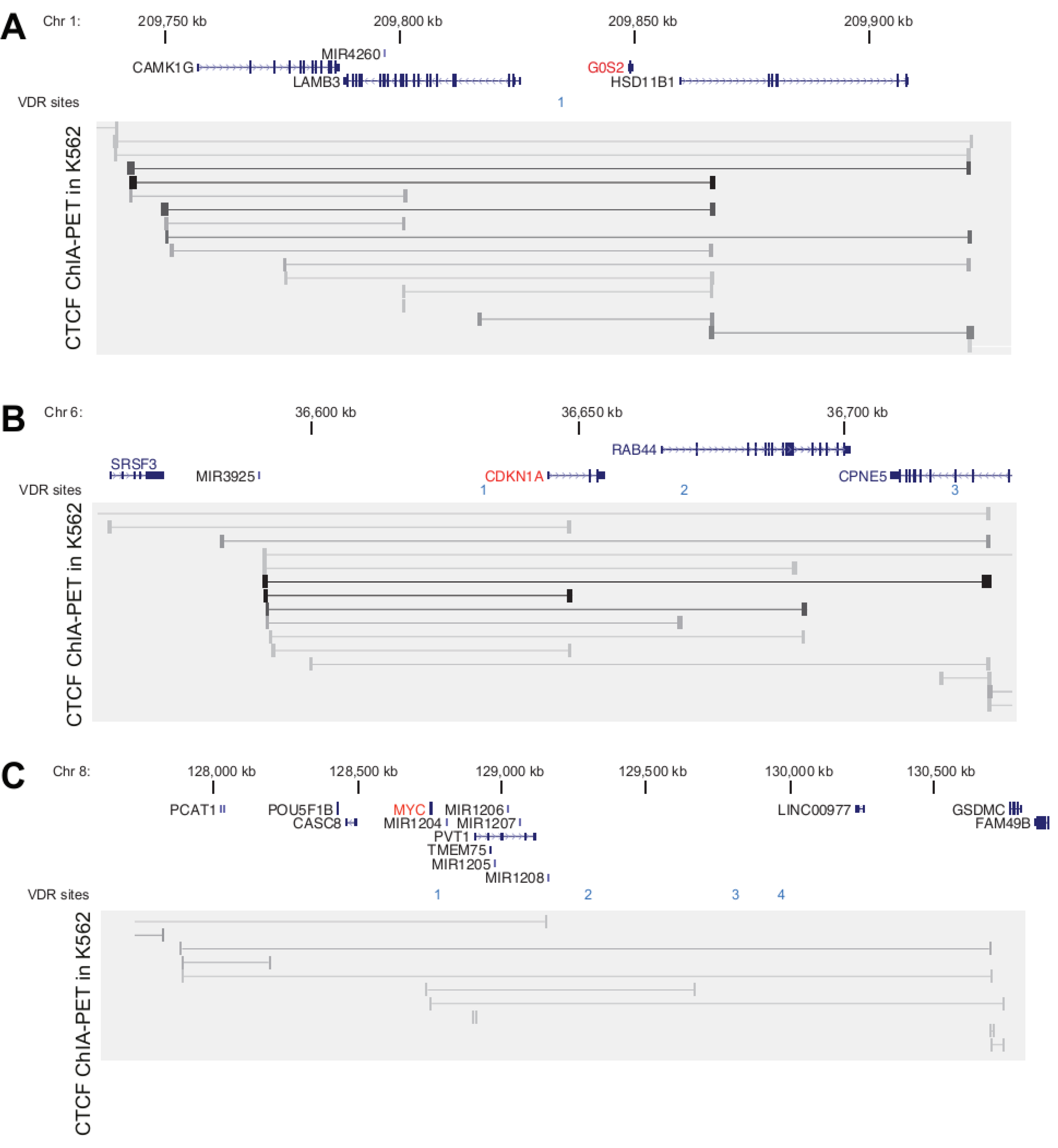
3.2. Genomic Profile of G0S2, CDKN1A and MYC in Monocyte- and Macrophage-like Cells

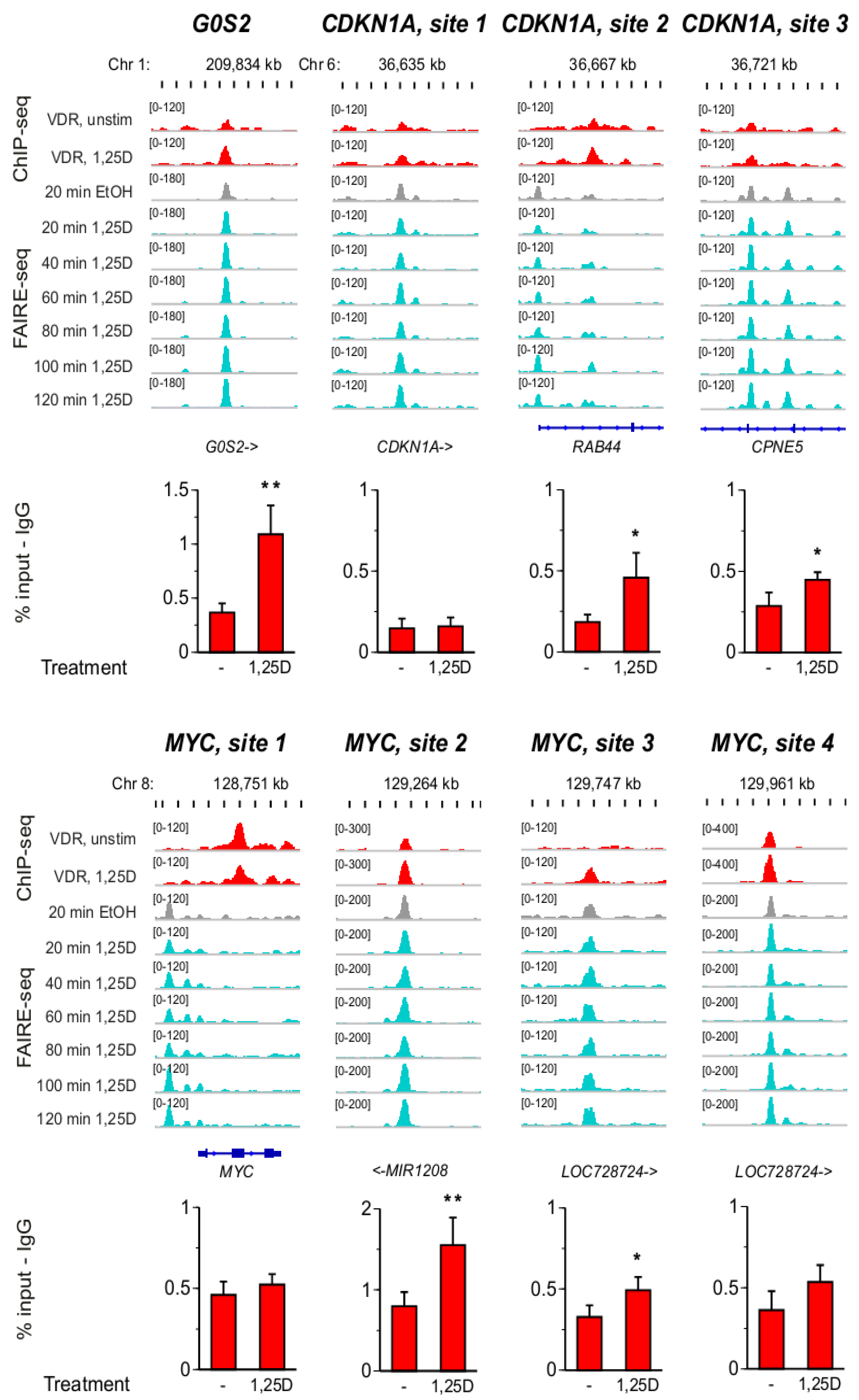
3.3. VDR Binding Sites of G0S2, CDKN1A and MYC

4. Discussion
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Carlberg, C.; Molnar, F. Current status of vitamin D signaling and its therapeutic applications. Curr. Top. Med. Chem. 2012, 12, 528–547. [Google Scholar]
- Furey, T.S. ChIP-seq and beyond: New and improved methodologies to detect and characterize protein-DNA interactions. Nat. Rev. Genet. 2012, 13, 840–852. [Google Scholar] [CrossRef]
- Ramagopalan, S.V.; Heger, A.; Berlanga, A.J.; Maugeri, N.J.; Lincoln, M.R.; Burrell, A.; Handunnetthi, L.; Handel, A.E.; Disanto, G.; Orton, S.M.; et al. A ChIP-seq defined genome-wide map of vitamin D receptor binding: associations with disease and evolution. Genome Res. 2010, 20, 1352–1360. [Google Scholar] [CrossRef]
- Heikkinen, S.; Väisänen, S.; Pehkonen, P.; Seuter, S.; Benes, V.; Carlberg, C. Nuclear hormone 1α,25-dihydroxyvitamin D3 elicits a genome-wide shift in the locations of VDR chromatin occupancy. Nucleic Acids Res. 2011, 39, 9181–9193. [Google Scholar] [CrossRef]
- Meyer, M.B.; Goetsch, P.D.; Pike, J.W. VDR/RXR and TCF4/beta-catenin cistromes in colonic cells of colorectal tumor origin: Impact on c-FOS and c-MYC gene expression. Mol. Endocrinol. 2012, 26, 37–51. [Google Scholar] [CrossRef]
- Ding, N.; Yu, R.T.; Subramaniam, N.; Sherman, M.H.; Wilson, C.; Rao, R.; Leblanc, M.; Coulter, S.; He, M.; Scott, C.; et al. A vitamin D receptor/SMAD genomic circuit gates hepatic fibrotic response. Cell 2013, 153, 601–613. [Google Scholar] [CrossRef]
- Handel, A.E.; Sandve, G.K.; Disanto, G.; Berlanga-Taylor, A.J.; Gallone, G.; Hanwell, H.; Drablos, F.; Giovannoni, G.; Ebers, G.C.; Ramagopalan, S.V. Vitamin D receptor ChIP-seq in primary CD4+ cells: Relationship to serum 25-hydroxyvitamin D levels and autoimmune disease. BMC Med. 2013. [Google Scholar] [CrossRef] [Green Version]
- Carlberg, C.; Seuter, S.; Heikkinen, S. The first genome-wide view of vitamin D receptor locations and their mechanistic implications. Anticancer Res. 2012, 32, 271–282. [Google Scholar]
- Umesono, K.; Murakami, K.K.; Thompson, C.C.; Evans, R.M. Direct repeats as selective response elements for the thyroid hormone, retinoic acid, and vitamin D3 receptors. Cell 1991, 65, 1255–1266. [Google Scholar] [CrossRef]
- Carlberg, C.; Bendik, I.; Wyss, A.; Meier, E.; Sturzenbecker, L.J.; Grippo, J.F.; Hunziker, W. Two nuclear signalling pathways for vitamin D. Nature 1993, 361, 657–660. [Google Scholar] [CrossRef]
- Carlberg, C.; Campbell, M.J. Vitamin D receptor signaling mechanisms: Integrated actions of a well-defined transcription factor. Steroids 2013, 78, 127–136. [Google Scholar] [CrossRef]
- Stambolsky, P.; Tabach, Y.; Fontemaggi, G.; Weisz, L.; Maor-Aloni, R.; Siegfried, Z.; Shiff, I.; Kogan, I.; Shay, M.; Kalo, E.; et al. Modulation of the vitamin D3 response by cancer-associated mutant p53. Cancer Cell 2010, 17, 273–285. [Google Scholar] [CrossRef]
- Razin, A. CpG methylation, chromatin structure and gene silencing-a three-way connection. EMBO J. 1998, 17, 4905–4908. [Google Scholar] [CrossRef]
- Eberharter, A.; Becker, P.B. Histone acetylation: A switch between repressive and permissive chromatin. Second in review series on chromatin dynamics. EMBO Rep. 2002, 3, 224–229. [Google Scholar] [CrossRef]
- Polly, P.; Herdick, M.; Moehren, U.; Baniahmad, A.; Heinzel, T.; Carlberg, C. VDR-Alien: A novel, DNA-selective vitamin D3 receptor-corepressor partnership. FASEB J. 2000, 14, 1455–1463. [Google Scholar] [CrossRef]
- Khanim, F.L.; Gommersall, L.M.; Wood, V.H.; Smith, K.L.; Montalvo, L.; O’Neill, L.P.; Xu, Y.; Peehl, D.M.; Stewart, P.M.; Turner, B.M.; et al. Altered SMRT levels disrupt vitamin D3 receptor signalling in prostate cancer cells. Oncogene 2004, 23, 6712–6725. [Google Scholar] [CrossRef] [Green Version]
- Malinen, M.; Saramäki, A.; Ropponen, A.; Degenhardt, T.; Väisänen, S.; Carlberg, C. Distinct HDACs regulate the transcriptional response of human cyclin-dependent kinase inhibitor genes to trichostatin A and 1α,25-dihydroxyvitamin D3. Nucleic Acids Res. 2008, 36, 121–132. [Google Scholar] [CrossRef]
- Banwell, C.M.; O’Neill, L.P.; Uskokovic, M.R.; Campbell, M.J. Targeting 1α,25-dihydroxyvitamin D3 antiproliferative insensitivity in breast cancer cells by co-treatment with histone deacetylation inhibitors. J. Steroid Biochem. Mol. Biol. 2004, 89, 245–249. [Google Scholar]
- Gronemeyer, H.; Gustafsson, J.A.; Laudet, V. Principles for modulation of the nuclear receptor superfamily. Nat. Rev. Drug Discov. 2004, 3, 950–964. [Google Scholar] [CrossRef]
- Carlberg, C.; Seuter, S. Dynamics of nuclear receptor target gene regulation. Chromosoma 2010, 119, 479–484. [Google Scholar] [CrossRef]
- Tan, J.; Cang, S.; Ma, Y.; Petrillo, R.L.; Liu, D. Novel histone deacetylase inhibitors in clinical trials as anti-cancer agents. J. Hematol. Oncol. 2010. [Google Scholar] [CrossRef]
- Seuter, S.; Heikkinen, S.; Carlberg, C. Chromatin acetylation at transcription start sites and vitamin D receptor binding regions relates to effects of 1α,25-dihydroxyvitamin D3 and histone deacetylase inhibitors on gene expression. Nucleic Acids Res. 2013, 41, 110–124. [Google Scholar] [CrossRef]
- Giresi, P.G.; Kim, J.; McDaniell, R.M.; Iyer, V.R.; Lieb, J.D. FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res. 2007, 17, 877–885. [Google Scholar] [CrossRef]
- Song, L.; Zhang, Z.; Grasfeder, L.L.; Boyle, A.P.; Giresi, P.G.; Lee, B.K.; Sheffield, N.C.; Graf, S.; Huss, M.; Keefe, D.; et al. Open chromatin defined by DNaseI and FAIRE identifies regulatory elements that shape cell-type identity. Genome Res. 2011, 21, 1757–1767. [Google Scholar] [CrossRef]
- Tsuchiya, S.; Yamabe, M.; Yamaguchi, Y.; Kobayashi, Y.; Konno, T.; Tada, K. Establishment and characterization of a human acute monocytic leukemia cell line (THP-1). Int. J. Cancer 1980, 26, 171–176. [Google Scholar] [CrossRef]
- Tsuchiya, S.; Kobayashi, Y.; Goto, Y.; Okumura, S.; Nakae, S.; Konno, T.; Tada, K. Induction of maturation in culture human monocytic leukemia cells by phorbol diester. Cancer Res. 1982, 42, 1530–1536. [Google Scholar]
- Vandesompele, J.; de Preter, K.; Pattyn, F.; Poppe, B.; van Roy, N.; de Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002. [Google Scholar] [CrossRef] [Green Version]
- Toropainen, S.; Väisänen, S.; Heikkinen, S.; Carlberg, C. The down-regulation of the human MYC gene by the nuclear hormone 1α,25-dihydroxyvitamin D3 is associated with cycling of corepressors and histone deacetylases. J. Mol. Biol. 2010, 400, 284–294. [Google Scholar] [CrossRef]
- Phillips, J.E.; Corces, V.G. CTCF: Master weaver of the genome. Cell 2009, 137, 1194–1211. [Google Scholar] [CrossRef]
- Schmidt, D.; Schwalie, P.C.; Wilson, M.D.; Ballester, B.; Gonçalves, A.; Kutter, C.; Brown, G.D.; Marshall, A.; Flicek, P.; Odom, D.T. Waves of retrotransposon expansion remodel genome organization and CTCF binding in multiple mammalian lineages. Cell 2012, 148, 335–348. [Google Scholar] [CrossRef]
- Klein, E.; Ben-Bassat, H.; Neumann, H.; Ralph, P.; Zeuthen, J.; Polliack, A.; Vánky, F. Properties of the K562 cell line, derived from a patient with chronic myeloid leukemia. Int. J. Cancer 1976, 18, 421–431. [Google Scholar] [CrossRef]
- Handoko, L.; Xu, H.; Li, G.; Ngan, C.Y.; Chew, E.; Schnapp, M.; Lee, C.W.; Ye, C.; Ping, J.L.; Mulawadi, F.; et al. CTCF-mediated functional chromatin interactome in pluripotent cells. Nat. Genet. 2011, 43, 630–638. [Google Scholar] [CrossRef]
- Seuter, S.; Pehkonen, P.; Heikkinen, S.; Carlberg, C. The gene for the transcription factor BHLHE40/DEC1/stra13 is a dynamically regulated primary target of the vitamin D receptor. J. Steroid Biochem. Mol. Biol. 2013, 136, 62–67. [Google Scholar] [CrossRef]
- Seuter, S.; Pehkonen, P.; Heikkinen, S.; Carlberg, C. Dynamics of 1α,25-dihydroxyvitamin D3-dependent chromatin accessibility of early vitamin D receptor target genes. 2013; submitted for publication. [Google Scholar]
- ENCODE-Project-Consortium; Bernstein, B.E.; Birney, E.; Dunham, I.; Green, E.D.; Gunter, C.; Snyder, M. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Reitsma, P.H.; Rothberg, P.G.; Astrin, S.M.; Trial, J.; Bar-Shavit, Z.; Hall, A.; Teitelbaum, S.L.; Kahn, A.J. Regulation of myc gene expression in HL-60 leukaemia cells by a vitamin D metabolite. Nature 1983, 306, 492–494. [Google Scholar] [CrossRef]
- Gordan, J.D.; Thompson, C.B.; Simon, M.C. HIF and c-Myc: Sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell 2007, 12, 108–113. [Google Scholar] [CrossRef]
- Evan, G.I.; Wyllie, A.H.; Gilbert, C.S.; Littlewood, T.D.; Land, H.; Brooks, M.E.; Waters, C.M.; Penn, L.Z.; Hancock, D.C. Induction of apoptosis in fibroblasts by c-Myc protein. Cell 1992, 69, 119–128. [Google Scholar] [CrossRef]
- Thompson, E.B. The many roles of c-Myc in apoptosis. Annu. Rev. Physiol. 1998, 60, 575–600. [Google Scholar] [CrossRef]
- Hansen, C.M.; Binderup, L.; Hamberg, K.J.; Carlberg, C. Vitamin D and cancer: Effects of 1,25(OH)2D3 and its analogs on growth control and tumorigenesis. Front. Biosci. 2001, 6, D820–D848. [Google Scholar] [CrossRef]
- Danielsson, C.; Mathiasen, I.S.; James, S.Y.; Nayeri, S.; Bretting, C.; Hansen, C.M.; Colston, K.W.; Carlberg, C. Sensitive induction of apoptosis in breast cancer cells by a novel 1,25-dihydroxyvitamin D3 analogue shows relation to promoter selectivity. J. Cell. Biochem. 1997, 66, 552–562. [Google Scholar] [CrossRef]
- Warfel, N.A.; El-Deiry, W.S. p21WAF1 and tumourigenesis: 20 years after. Curr. Opin. Oncol. 2013, 25, 52–58. [Google Scholar] [CrossRef]
- Liu, M.; Lee, M.-H.; Cohen, M.; Bommakanti, M.; Freedman, L.P. Transcriptional activation of the Cdk inhibitor p21 by vitamin D3 leads to the induced differentiation of the myelomonocytic cell line U937. Genes Dev. 1996, 10, 142–153. [Google Scholar] [CrossRef]
- Schwaller, J.; Koeffler, H.P.; Niklaus, G.; Loetscher, P.; Nagel, S.; Fey, M.F.; Tobler, A. Posttranscriptional stabilization underlies p53-independent induction of p21WAF1/CIP1/SDI1 in differentiating human leukemic cells. J. Clin. Invest. 1995, 95, 973–979. [Google Scholar] [CrossRef]
- Heckmann, B.L.; Zhang, X.; Xie, X.; Liu, J. The G0/G1 switch gene 2 (G0S2): Regulating metabolism and beyond. Biochim. Biophys. Acta 2013, 1831, 276–281. [Google Scholar]
- Zandbergen, F.; Mandard, S.; Escher, P.; Tan, N.S.; Patsouris, D.; Jatkoe, T.; Rojas-Caro, S.; Madore, S.; Wahli, W.; Tafuri, S.; et al. The G0/G1 switch gene 2 is a novel PPAR target gene. Biochem. J. 2005, 392, 313–324. [Google Scholar] [CrossRef]
- Kitareewan, S.; Blumen, S.; Sekula, D.; Bissonnette, R.P.; Lamph, W.W.; Cui, Q.; Gallagher, R.; Dmitrovsky, E. G0S2 is an all-trans-retinoic acid target gene. Int. J. Oncol. 2008, 33, 397–404. [Google Scholar]
- Tuupanen, S.; Turunen, M.; Lehtonen, R.; Hallikas, O.; Vanharanta, S.; Kivioja, T.; Bjorklund, M.; Wei, G.; Yan, J.; Niittymäki, I.; et al. The common colorectal cancer predisposition SNP rs6983267 at chromosome 8q24 confers potential to enhanced Wnt signaling. Nat. Genet. 2009, 41, 885–890. [Google Scholar] [CrossRef]
- Saramäki, A.; Banwell, C.M.; Campbell, M.J.; Carlberg, C. Regulation of the human p21waf1/cip1 gene promoter via multiple binding sites for p53 and the vitamin D3 receptor. Nucleic Acids Res. 2006, 34, 543–554. [Google Scholar] [CrossRef]
- Thorne, J.L.; Maguire, O.; Doig, C.L.; Battaglia, S.; Fehr, L.; Sucheston, L.E.; Heinäniemi, M.; O’Neill, L.P.; McCabe, C.J.; Turner, B.M.; et al. Epigenetic control of a VDR-governed feed-forward loop that regulates p21(waf1/cip1) expression and function in non-malignant prostate cells. Nucleic Acids Res. 2011, 39, 2045–2056. [Google Scholar] [CrossRef]
Supplementary Materials

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Fragment size (bp) | Annealing temperature (°C) | Primer sequences (5'-3') |
|---|---|---|---|
| B2M 1,2 | 246 | 60 | GGCTATCCAGCGTACTCCAAA
CGGCAGGCATACTCATCTTTTT |
| CDKN1A 3 | 99 | 60 | GCCACTAAGGTCATTCCCGCCT
CCTTGCGCTTCTGGGCCATCAT |
| G0S2 4 | 102 | 60 | GCCACTAAGGTCATTCCCGCCT
CCTTGCGCTTCTGGGCCATCAT |
| GAPDH 1,2 | 113 | 60 | CATGAGAAGTATGACAACAGCCTAGTC
CTTCCACGATACCAAAGT |
| HPRTI 1,5 | 94 | 60 | TGACACTGGCAAAACAATGCA
GGTCCTTTTCACCAGCAAGCT |
| MYC 3 | 147 | 60 | CCAGCAGCGACTCTGAGG
GGACCAGTGGGCTGTGAG |
| Genomic region | Fragment size (bp) | Annealing temperature (°C) | Primer sequences (5'-3') |
|---|---|---|---|
| G0S2 1 | 119 | 62 | GCCTGAGAGTATGCTGTGTACGTTT CTAAGTGCTCTCTTGGCGTAACAAA |
| CDKN1A 2, peak 1 | 129 | 58 | TGTGGGGAGGGTGTTTCAG GAGGGAAGGAAGGAGTGAG |
| CDKN1A 2, peak 2 | 91 | 58 | CAGAGGAAGTGGGTTGAG AGCAGGGCAGGAGAGATTATAC |
| CDKN1A 2, peak 3 | 69 | 62 | GCACTCTTGACCTTGACGGA CTAACACCCTTGGCTTGGAC |
| CDKN1A 2, control | 180 | 62 | ATCACAGGGGTCAGCACATC CGCAGCATTTGGGTTCACAC |
| MYC 2, peak 1 | 181 | 62 | GTCACACCCTTCTCCCTTC CGCTCCACATACAGTCCTG |
| MYC 2, peak 2 | 93 | 58 | CCACACTAACCTCTCAGTTC GTAATGATACTCCCAGCAAAG |
| MYC 2, peak 3 | 137 | 62 | GGATGTCAGCAGGGTTTCTC GGAAGTGATTTCGGGAGTAG |
| MYC 2, peak 4 | 96 | 58 | GCTCTGTTGGTGTGGACTG GATTAGGGTGCCATAGAATAC |



© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ryynänen, J.; Seuter, S.; Campbell, M.J.; Carlberg, C. Gene Regulatory Scenarios of Primary 1,25-Dihydroxyvitamin D3 Target Genes in a Human Myeloid Leukemia Cell Line. Cancers 2013, 5, 1221-1241. https://doi.org/10.3390/cancers5041221
Ryynänen J, Seuter S, Campbell MJ, Carlberg C. Gene Regulatory Scenarios of Primary 1,25-Dihydroxyvitamin D3 Target Genes in a Human Myeloid Leukemia Cell Line. Cancers. 2013; 5(4):1221-1241. https://doi.org/10.3390/cancers5041221
Chicago/Turabian StyleRyynänen, Jussi, Sabine Seuter, Moray J. Campbell, and Carsten Carlberg. 2013. "Gene Regulatory Scenarios of Primary 1,25-Dihydroxyvitamin D3 Target Genes in a Human Myeloid Leukemia Cell Line" Cancers 5, no. 4: 1221-1241. https://doi.org/10.3390/cancers5041221