HspB1, HspB5 and HspB4 in Human Cancers: Potent Oncogenic Role of Some of Their Client Proteins

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Small Hsps in Human Cancer Pathologies
2.1. HspB1
2.2. HspB5 and HspB4
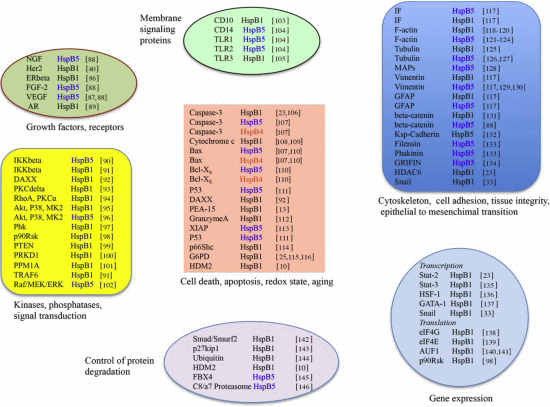
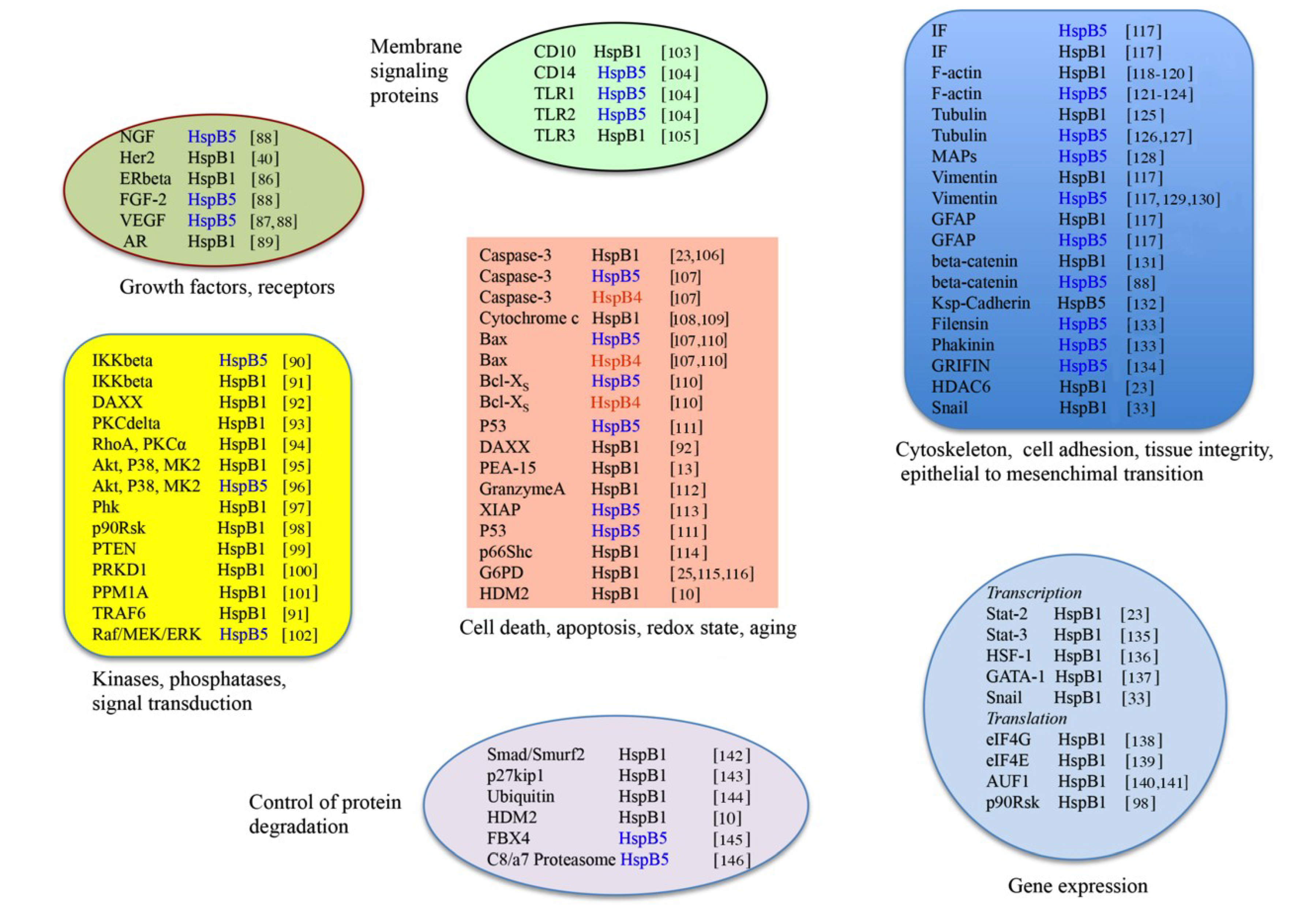
3. HspB1, HspB5 and HspB4 Clients and Their Potential Role in Cancer Pathologies
3.1. Clients as Receptors and Growth Factors Stimulating Cell Growth
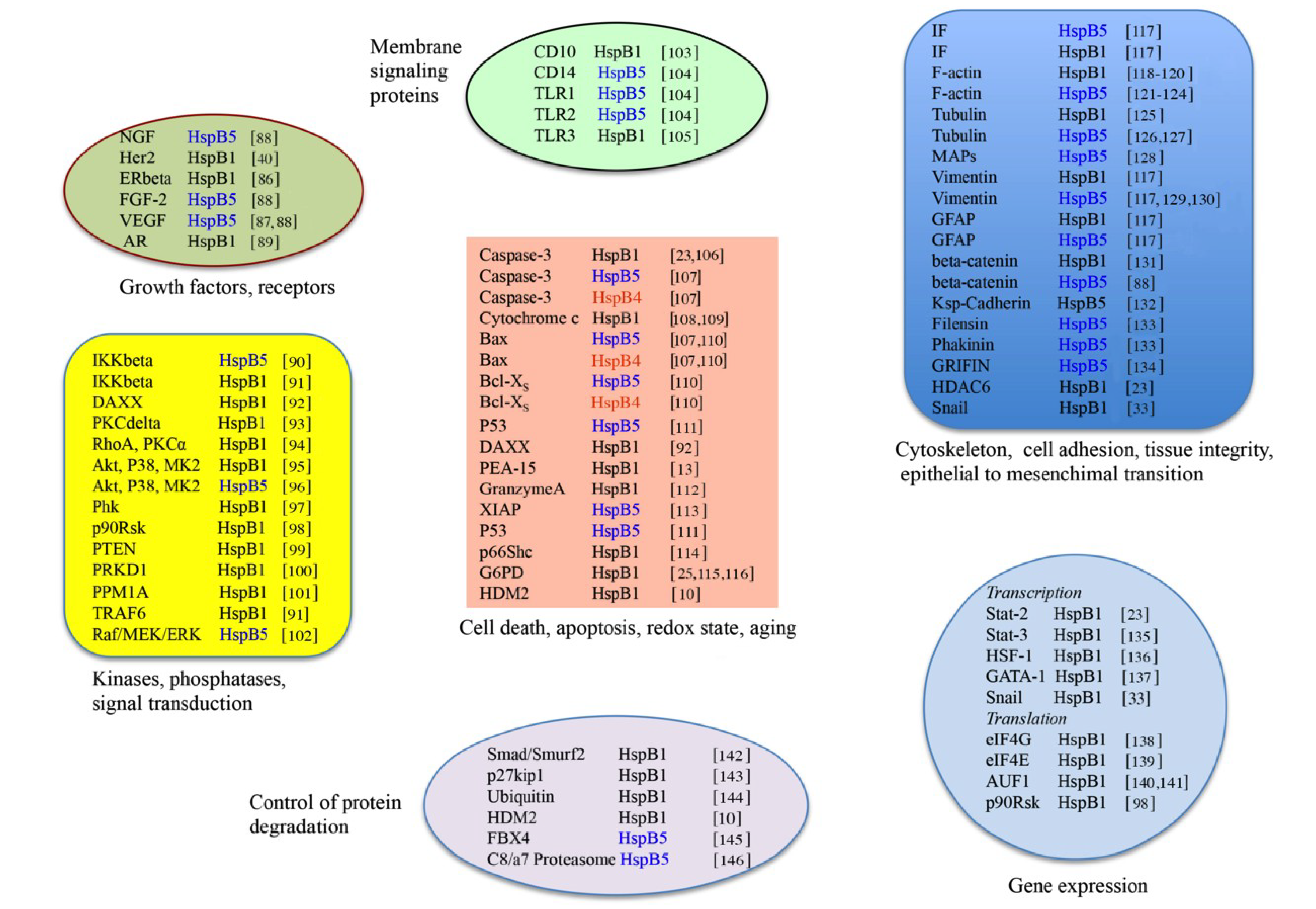
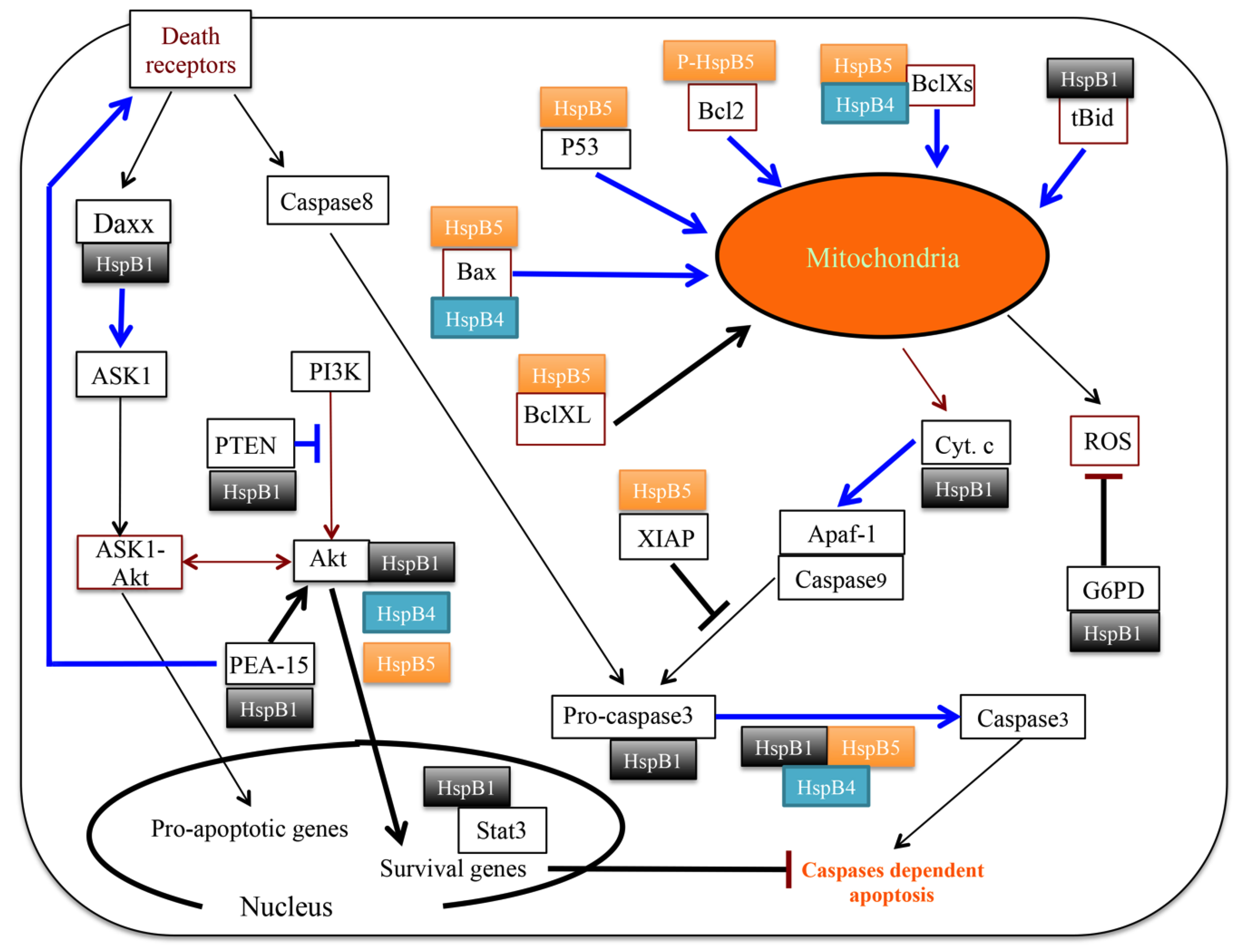
3.2. Clients Modulating Survival Signaling Pathways and Apoptosis
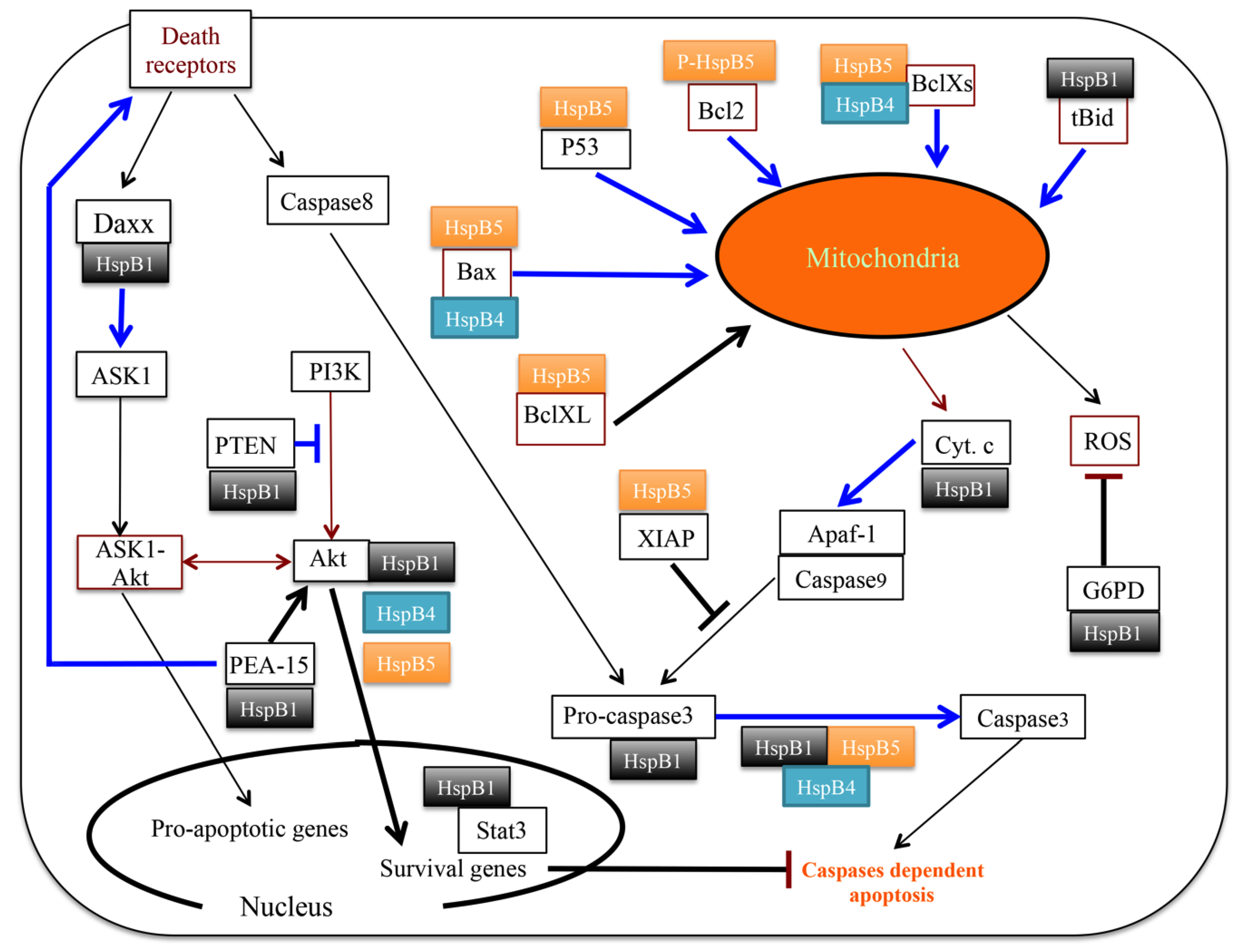
3.3. Senescence
3.4. Clients Involved in Tumor Progression, Epithelial-to-Mesenchymal Transition, Metastasis
3.5. Clients on Cell Surface and Outside of Cells, Immunosurveillance, Angiogenesis and Autoantibodies
3.6. Clients Involved in Inflammation and in Controling Intracellular Redox State
3.7. Clients Regulating Gene Expression
3.7.1. Transcription
3.7.2. Translation
4. Peptides, Natural Molecules and Drugs that Inhibit the Proliferation of Human Cancer Cells through Their Ability to Interact with Small Hsps, Therapeutic Approaches
- (1)
- Biphenyl isooxazoles such as 5-(5-ethyl-2-hydroxy-4-methoxyphenyl)-4-(4-methoxyphenyl) isoxazole (KRIBB3) inhibit tumor cell migration and invasion by blocking protein kinase C-dependent phosphorylation of HspB1 through their direct binding to HspB1 [228].
- (2)
- RP101 (E)-5-(2-bromovinyl)-deoxyuridine, BVDU, brivudine), a 2'-deoxyuridine derivative (thymidine analogue). This well-known anti-viral drug, particularly efficient againt Herpes simplex virus (HSV-1) and varicella zoster virus (VZV), was recently reported to drastically enhance the efficiency of human pancreatic cancer chemotherapy. RP101 acts by inhibiting HspB1 interaction with pro-cancerous binding partners, such as cytochrome c, pro-caspase-3, and Akt1 [229]. This results in a stimulation of the activity of caspase-9 and apoptosis efficiency. In addition of being incorporated into viral DNAs and inhibiting the action of DNA polymerase and viral replication, RP101 can bind to a specific site of the N-terminal domain of HspB1 characterized by two phenylalanine residues (Phe29 and Phe33). This probably interferes with the dynamic oligomerization of this chaperone and its ability to recognize client proteins. In addition of being approved as an anti-viral drug, RP101 efficiency towards pancreatic cancer patients is actually in clinical phase II/III. Of interest, an increased efficiency has recently been reported for novel HspB1-targetting derivatives of RP101 [230].
- (3)
- Clerodane diterpenoids form a class of terpene derivatives biosynthesized in plants, particularly those of the Lamiaceae family. Among them, neo-clerodane diterpenoids from Salvia spp. were recently described as compounds inhibiting the proliferation of several human cancer cell lines. Chemical proteomics approach analyzing the cellular interactome of a representative clerodane diterpenoid molecule, hardwickiic acid (HAA), revealed HspB1 as a major HAA interacting protein partner in U937 leukemic cells [231]. The interaction was confirmed by several other approaches, including surface plasmon resonance measurements, limited proteolysis, and biochemical assays. Hence, the antitumor potential of clerodane diterpenoids may be linked to the inhibition HspB1 oncogenic properties. Moreover, this observation may be crucial for the future discovery of potent HspB1 diterpenoid-based chemical inhibitors.
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sherman, M.; Multhoff, G. Heat shock proteins in cancer. Ann. NY Acad. Sci. 2007, 1113, 192–201. [Google Scholar]
- Ciocca, D.R.; Arrigo, A.P.; Calderwood, S.K. Heat shock proteins and heat shock factor 1 in carcinogenesis and tumor development: An update. Arch. Toxicol. 2013, 87, 19–48. [Google Scholar]
- Trepel, J.; Mollapour, M.; Giaccone, G.; Neckers, L. Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 2010, 10, 537–549. [Google Scholar] [CrossRef]
- Calderwood, S.K. HSF1, a versatile factor in tumorogenesis. Curr. Mol. Med. 2012, 12, 1102–1107. [Google Scholar] [CrossRef]
- Arrigo, A.P.; Simon, S.; Gibert, B.; Kretz-Remy, C.; Nivon, M.; Czekalla, A.; Guillet, D.; Moulin, M.; Diaz-Latoud, C.; Vicart, P. Hsp27 (HspB1) and alphaB-crystallin (HspB5) as therapeutic targets. FEBS Lett. 2007, 581, 3665–3674. [Google Scholar] [CrossRef]
- Shiota, M.; Bishop, J.L.; Nip, K.M.; Zardan, A.; Takeuchi, A.; Cordonnier, T.; Beraldi, E.; Bazov, J.; Fazli, L.; Chi, K.; et al. Hsp27 regulates epithelial mesenchymal transition, metastasis, and circulating tumor cells in prostate cancer. Cancer Res. 2013, 73, 3109–3119. [Google Scholar] [CrossRef]
- Mehlen, P.; Schulze-Osthoff, K.; Arrigo, A.P. Small stress proteins as novel regulators of apoptosis. Heat shock protein 27 blocks Fas/APO-1- and staurosporine-induced cell death. J. Biol. Chem. 1996, 271, 16510–16514. [Google Scholar] [CrossRef]
- Kamradt, M.C.; Chen, F.; Cryns, V.L. The small heat shock protein alpha B-crystallin negatively regulates cytochrome c- and caspase-8-dependent activation of caspase-3 by inhibiting its autoproteolytic maturation. J. Biol. Chem. 2001, 276, 16059–16063. [Google Scholar] [CrossRef]
- Garrido, C.; Brunet, M.; Didelot, C.; Zermati, Y.; Schmitt, E.; Kroemer, G. Heat shock proteins 27 and 70: Anti-apoptotic proteins with tumorigenic properties. Cell Cycle 2006, 5, 22. [Google Scholar]
- O’Callaghan-Sunol, C.; Gabai, V.L.; Sherman, M.Y. Hsp27 modulates p53 signaling and suppresses cellular senescence. Cancer Res. 2007, 67, 11779–11788. [Google Scholar] [CrossRef]
- Zhuang, H.; Jiang, W.; Cheng, W.; Qian, K.; Dong, W.; Cao, L.; Huang, Q.; Li, S.; Dou, F.; Chiu, J.F.; et al. Down-regulation of HSP27 sensitizes TRAIL-resistant tumor cell to TRAIL-induced apoptosis. Lung Cancer 2009, 68, 27–38. [Google Scholar]
- Gibert, B.; Eckel, B.; Gonin, V.; Goldschneider, D.; Fombonne, J.; Deux, B.; Mehlen, P.; Arrigo, A.P.; Clezardin, P.; Diaz-Latoud, C. Targeting heat shock protein 27 (HspB1) interferes with bone metastasis and tumour formation in vivo. Br. J. Cancer 2012, 107, 63–70. [Google Scholar] [CrossRef]
- Hayashi, N.; Peacock, J.W.; Beraldi, E.; Zoubeidi, A.; Gleave, M.E.; Ong, C.J. Hsp27 silencing coordinately inhibits proliferation and promotes Fas-induced apoptosis by regulating the PEA-15 molecular switch. Cell Death Differ. 2012, 19, 990–1002. [Google Scholar] [CrossRef]
- Garrido, C.; Fromentin, A.; Bonnotte, B.; Favre, N.; Moutet, M.; Arrigo, A.P.; Mehlen, P.; Solary, E. Heat shock protein 27 enhances the tumorigenicity of immunogenic rat colon carcinoma cell clones. Cancer Res. 1998, 58, 5495–5499. [Google Scholar]
- Bruey, J.M.; Paul, C.; Fromentin, A.; Hilpert, S.; Arrigo, A.P.; Solary, E.; Garrido, C. Differential regulation of HSP27 oligomerization in tumor cells grown in vitro and in vivo. Oncogene 2000, 19, 4855–4863. [Google Scholar] [CrossRef]
- Gibert, B.; Hadchity, E.; Czekalla, A.; Aloy, M.T.; Colas, P.; Rodriguez-Lafrasse, C.; Arrigo, A.P.; Diaz-Latoud, C. Inhibition of heat shock protein 27 (HspB1) tumorigenic functions by peptide aptamers. Oncogene 2011, 34, 3672–3681. [Google Scholar]
- Lemieux, P.; Oesterreich, S.; Lawrence, J.A.; Steeg, P.S.; Hilsenbeck, S.G.; Harvey, J.M.; Fuqua, S.A. The small heat shock protein hsp27 increases invasiveness but decreases motility of breast cancer cells. Invasion Metastasis 1997, 17, 113–123. [Google Scholar]
- Bausero, M.A.; Page, D.T.; Osinaga, E.; Asea, A. Surface expression of Hsp25 and Hsp72 differentially regulates tumor growth and metastasis. Tumour Biol. 2004, 25, 243–251. [Google Scholar] [CrossRef]
- Nagaraja, G.N.; Kaur, P.; Asea, A. Role of human and mouse HspB1 in metastasis. Curr. Mol. Med. 2012, 12, 1142–1150. [Google Scholar] [CrossRef]
- Calderwood, S.K.; Khaleque, M.A.; Sawyer, D.B.; Ciocca, D.R. Heat shock proteins in cancer: Chaperones of tumorigenesis. Trends Biochem. Sci. 2006, 31, 164–172. [Google Scholar] [CrossRef]
- Arrigo, A.P.; Gibert, B. HspB1 dynamic phospho-oligomeric structure dependent interactome as cancer therapeutic target. Curr. Mol. Med. 2012, 12, 1151–1163. [Google Scholar] [CrossRef]
- Arrigo, A.; Simon, S. Dual, Beneficial and Deleterous, Roles of Small Stress Proteins in Human Diseases: Implications for Therapeutic Strategies. In Small Stress Proteins in Human Diseases; Book Serie: Protein Science Engineering; Arrigo, A.P., Simon, S., Eds.; Nova Sciences: New York, NY, USA, 2010; pp. 457–476. [Google Scholar]
- Gibert, B.; Eckel, B.; Fasquelle, L.; Moulin, M.; Bouhallier, F.; Gonin, V.; Mellier, G.; Simon, S.; Kretz-Remy, C.; Arrigo, A.P.; et al. Knock down of heat shock protein 27 (HspB1) induces degradation of several putative client proteins. PLoS One 2012, 7, e29719. [Google Scholar] [CrossRef]
- Arrigo, A.P.; Gibert, B. Protein interactomes of three stress inducible small heat shock proteins: HspB1, HspB5 and HspB8. Int. J. Hyperth. 2013, 29, 409–422. [Google Scholar] [CrossRef]
- Arrigo, A.P. Human small heat shock proteins: Protein interactomes of homo- and hetero-oligomeric complexes: An update. FEBS Lett. 2013, 587, 1959–1969. [Google Scholar] [CrossRef]
- Ciocca, D.R.; Calderwood, S.K. Heat shock proteins in cancer: Diagnostic, prognostic, predictive, and treatment implications. Cell Stress Chaperones 2005, 10, 86–103. [Google Scholar] [CrossRef]
- Arrigo, A.P. Pathology-dependent effects linked to small heat shock proteins expression. Scientifica 2012, 2012, 19. [Google Scholar]
- Blackburn, R.V.; Galoforo, S.S.; Berns, C.M.; Armour, E.P.; McEachern, D.; Corry, P.M.; Lee, Y.J. Comparison of tumor growth between Hsp25- and Hsp27- transfected murine L929 cells in nude mice. Int. J. Cancer 1997, 72, 871–877. [Google Scholar] [CrossRef]
- Katoh, M.; Koninkx, J.; Schumacher, U. Heat shock protein expression in human tumours grown in severe combined immunodeficient mice. Cancer Lett. 2000, 161, 113–120. [Google Scholar] [CrossRef]
- Hsu, H.S.; Lin, J.H.; Huang, W.C.; Hsu, T.W.; Su, K.; Chiou, S.H.; Tsai, Y.T.; Hung, S.C. Chemoresistance of lung cancer stemlike cells depends on activation of Hsp27. Cancer 2011, 117, 1516–1528. [Google Scholar] [CrossRef]
- Wei, L.; Liu, T.T.; Wang, H.H.; Hong, H.M.; Yu, A.L.; Feng, H.P.; Chang, W.W. Hsp27 participates in the maintenance of breast cancer stem cells through regulation of epithelial-mesenchymal transition and nuclear factor-kappaB. Breast Cancer Res. 2011, 13, R101. [Google Scholar]
- Straume, O.; Shimamura, T.; Lampa, M.J.; Carretero, J.; Oyan, A.M.; Jia, D.; Borgman, C.L.; Soucheray, M.; Downing, S.R.; Short, S.M.; et al. Suppression of heat shock protein 27 induces long-term dormancy in human breast cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 8699–8704. [Google Scholar]
- Wettstein, G.; Bellaye, P.S.; Kolb, M.; Hammann, A.; Crestani, B.; Soler, P.; Marchal-Somme, J.; Hazoume, A.; Gauldie, J.; Gunther, A.; et al. Inhibition of HSP27 blocks fibrosis development and EMT features by promoting Snail degradation. FASEB J. 2013, 27, 4169–4183. [Google Scholar] [CrossRef]
- Castro, G.N.; Cayado-Gutierrez, N.; Moncalero, V.L.; Lima, P.; de Angelis, R.L.; Chavez, V.; Cuello-Carrion, F.D.; Ciocca, D.R. Hsp27 (HSPB1): A possible surrogate molecular marker for loss of heterozygosity (LOH) of chromosome 1p in oligodendrogliomas but not in astrocytomas. Cell Stress Chaperones 2012, 17, 779–790. [Google Scholar] [CrossRef]
- Huot, J.; Roy, G.; Lambert, H.; Chretien, P.; Landry, J. Increased survival after treatments with anticancer agents of Chinese hamster cells expressing the human Mr 27,000 heat shock protein. Cancer Res. 1991, 51, 5245–5252. [Google Scholar]
- Richards, E.H.; Hickey, E.; Weber, L.A.; Master, J.R. Effect of overexpression of the small heat shock protein HSP27 on the heat and drug sensitivities of human testis tumor cells. Cancer Res. 1996, 56, 2446–2451. [Google Scholar]
- Arrigo, A.P. sHsp as novel regulators of programmed cell death and tumorigenicity. Pathol. Biol. Paris 2000, 48, 280–288. [Google Scholar]
- Garrido, C.; Gurbuxani, S.; Ravagnan, L.; Kroemer, G. Heat shock proteins: Endogenous modulators of apoptotic cell death. Biochem. Biophys. Res. Commun. 2001, 286, 433–442. [Google Scholar] [CrossRef]
- Kamada, M.; So, A.; Muramaki, M.; Rocchi, P.; Beraldi, E.; Gleave, M. Hsp27 knockdown using nucleotide-based therapies inhibit tumor growth and enhance chemotherapy in human bladder cancer cells. Mol. Cancer Ther. 2007, 6, 299–308. [Google Scholar] [CrossRef]
- Kang, S.H.; Kang, K.W.; Kim, K.H.; Kwon, B.; Kim, S.K.; Lee, H.Y.; Kong, S.Y.; Lee, E.S.; Jang, S.G.; Yoo, B.C. Upregulated HSP27 in human breast cancer cells reduces Herceptin susceptibility by increasing Her2 protein stability. BMC Cancer 2008, 8, 286. [Google Scholar] [CrossRef]
- Kase, S.; Parikh, J.G.; Rao, N.A. Expression of heat shock protein 27 and alpha-crystallins in human retinoblastoma after chemoreduction. Br. J. Ophthalmol. 2009, 93, 541–544. [Google Scholar] [CrossRef]
- Zhang, Y.; Shen, X. Heat shock protein 27 protects L929 cells from cisplatin-induced apoptosis by enhancing Akt activation and abating suppression of thioredoxin reductase activity. Clin. Cancer Res. 2007, 13, 2855–2864. [Google Scholar] [CrossRef]
- Chen, R.; Dai, R.Y.; Duan, C.Y.; Liu, Y.P.; Chen, S.K.; Yan, D.M.; Chen, C.N.; Wei, M.; Li, H. Unfolded protein response suppresses cisplatin-induced apoptosis via autophagy regulation in human hepatocellular carcinoma cells. Folia Biol. Praha 2011, 57, 87–95. [Google Scholar]
- Fujita, R.; Ounzain, S.; Wang, A.C.; Heads, R.J.; Budhram-Mahadeo, V.S. Hsp-27 induction requires POU4F2/Brn-3b TF in doxorubicin-treated breast cancer cells, whereas phosphorylation alters its cellular localisation following drug treatment. Cell Stress Chaperones 2011, 16, 427–439. [Google Scholar] [CrossRef]
- Ounzain, S.; Bowen, S.; Patel, C.; Fujita, R.; Heads, R.J.; Budhram-Mahadeo, V.S. Proliferation-associated POU4F2/Brn-3b transcription factor expression is regulated by oestrogen through ERalpha and growth factors via MAPK pathway. Breast Cancer Res. 2011, 13, R5. [Google Scholar] [CrossRef]
- Oesterreich, S.; Schunck, H.; Benndorf, R.; Bielka, H. Cisplatin induces the small heat shock protein hsp25 and thermotolerance in Ehrlich ascites tumor cells. Biochem. Biophys. Res. Commun. 1991, 180, 243–248. [Google Scholar] [CrossRef]
- Schafer, C.; Seeliger, H.; Bader, D.C.; Assmann, G.; Buchner, D.; Guo, Y.; Ziesch, A.; Palagyi, A.; Ochs, S.; Laubender, R.P.; et al. Heat shock protein 27 as a prognostic and predictive biomarker in pancreatic ductal adenocarcinoma. J. Cell Mol. Med. 2012, 16, 1776–1791. [Google Scholar]
- Mori-Iwamoto, S.; Kuramitsu, Y.; Ryozawa, S.; Mikuria, K.; Fujimoto, M.; Maehara, S.; Maehara, Y.; Okita, K.; Nakamura, K.; Sakaida, I. Proteomics finding heat shock protein 27 as a biomarker for resistance of pancreatic cancer cells to gemcitabine. Int. J. Oncol. 2007, 31, 1345–1350. [Google Scholar]
- Nakashima, M.; Adachi, S.; Yasuda, I.; Yamauchi, T.; Kawaguchi, J.; Itani, M.; Yoshioka, T.; Matsushima-Nishiwaki, R.; Hirose, Y.; Kozawa, O.; et al. Phosphorylation status of heat shock protein 27 plays a key role in gemcitabine-induced apoptosis of pancreatic cancer cells. Cancer Lett. 2011, 313, 218–225. [Google Scholar] [CrossRef]
- Zhang, D.; Wong, L.L.; Koay, E.S. Phosphorylation of Ser78 of Hsp27 correlated with HER-2/neu status and lymph node positivity in breast cancer. Mol. Cancer 2007, 6, 52. [Google Scholar] [CrossRef] [Green Version]
- Paul, C.; Simon, S.; Gibert, B.; Virot, S.; Manero, F.; Arrigo, A.P. Dynamic processes that reflect anti-apoptotic strategies set up by HspB1 (Hsp27). Exp. Cell Res. 2010, 316, 1535–1552. [Google Scholar] [CrossRef]
- Tweedle, E.M.; Khattak, I.; Ang, C.W.; Nedjadi, T.; Jenkins, R.; Park, B.K.; Kalirai, H.; Dodson, A.; Azadeh, B.; Terlizzo, M.; et al. Low molecular weight heat shock protein HSP27 is a prognostic indicator in rectal cancer but not colon cancer. Gut 2010, 59, 1501–1510. [Google Scholar] [CrossRef]
- Moyano, J.V.; Evans, J.R.; Chen, F.; Lu, M.; Werner, M.E.; Yehiely, F.; Diaz, L.K.; Turbin, D.; Karaca, G.; Wiley, E.; et al. AlphaB-crystallin is a novel oncoprotein that predicts poor clinical outcome in breast cancer. J. Clin. Invest. 2006, 116, 261–270. [Google Scholar]
- Chen, P.; Ji, W.; Liu, F.Y.; Tang, H.Z.; Fu, S.; Zhang, X.; Liu, M.; Gong, L.; Deng, M.; Hu, W.F.; et al. Alpha-crystallins and tumorigenesis. Curr. Mol. Med. 2012, 12, 1164–1173. [Google Scholar]
- Chelouche-Lev, D.; Kluger, H.M.; Berger, A.J.; Rimm, D.L.; Price, J.E. alphaB-crystallin as a marker of lymph node involvement in breast carcinoma. Cancer 2004, 100, 2543–2548. [Google Scholar] [CrossRef]
- Kim, H.S.; Lee, Y.; Lim, Y.A.; Kang, H.J.; Kim, L.S. AlphaB-crystallin is a novel oncoprotein associated with poor prognosis in breast cancer. J. Breast Cancer 2011, 14, 14–19. [Google Scholar]
- Sitterding, S.M.; Wiseman, W.R.; Schiller, C.L.; Luan, C.; Chen, F.; Moyano, J.V.; Watkin, W.G.; Wiley, E.L.; Cryns, V.L.; Diaz, L.K. AlphaB-crystallin: A novel marker of invasive basal-like and metaplastic breast carcinomas. Ann. Diagn. Pathol. 2008, 12, 33–40. [Google Scholar] [CrossRef]
- Ivanov, O.; Chen, F.; Wiley, E.L.; Keswani, A.; Diaz, L.K.; Memmel, H.C.; Rademaker, A.; Gradishar, W.J.; Morrow, M.; Khan, S.A.; et al. alphaB-crystallin is a novel predictor of resistance to neoadjuvant chemotherapy in breast cancer. Breast Cancer Res. Treat. 2008, 111, 411–417. [Google Scholar] [CrossRef]
- Aoyama, A.; Steiger, R.; Frohli, E.; Schafer, R.; von Deimling, A.; Wiestler, O.; Klemenz, R. Expression of alpha B-crystallin in human brain tumors. Int. J. Cancer 1993, 55, 760–764. [Google Scholar]
- Khalil, A.A. Biomarker discovery: A proteomic approach for brain cancer profiling. Cancer Sci. 2007, 98, 201–213. [Google Scholar] [CrossRef]
- Tang, Q.; Liu, Y.F.; Zhu, X.J.; Li, Y.H.; Zhu, J.; Zhang, J.P.; Feng, Z.Q.; Guan, X.H. Expression and prognostic significance of the alpha B-crystallin gene in human hepatocellular carcinoma. Hum. Pathol. 2009, 40, 300–305. [Google Scholar] [CrossRef]
- Liu, X.L.; Guo, K.P.; Ma, F.; Xie, G.Y.; He, Y.; Hu, C.H. Expression profile of heat shock proteins in tissues and cells of lung adenocarcinoma. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2007, 32, 660–664. [Google Scholar]
- Takashi, M.; Katsuno, S.; Sakata, T.; Ohshima, S.; Kato, K. Different concentrations of two small stress proteins, alphaB crystallin and HSP27 in human urological tumor tissues. Urol. Res. 1998, 26, 395–399. [Google Scholar] [CrossRef]
- Bai, F.; Xi, J.H.; Wawrousek, E.F.; Fleming, T.P.; Andley, U.P. Hyperproliferation and p53 status of lens epithelial cells derived from alphaB-crystallin knockout mice. J. Biol. Chem. 2003, 278, 36876–36886. [Google Scholar] [CrossRef]
- Shi, T.; Dong, F.; Liou, L.S.; Duan, Z.H.; Novick, A.C.; DiDonato, J.A. Differential protein profiling in renal-cell carcinoma. Mol. Carcinog. 2004, 40, 47–61. [Google Scholar] [CrossRef]
- Mineva, I.; Gartner, W.; Hauser, P.; Kainz, A.; Loffler, M.; Wolf, G.; Oberbauer, R.; Weissel, M.; Wagner, L. Differential expression of alphaB-crystallin and Hsp27–1 in anaplastic thyroid carcinomas because of tumor-specific alphaB-crystallin gene (CRYAB) silencing. Cell Stress Chaperones 2005, 10, 171–184. [Google Scholar] [CrossRef]
- Lung, H.L.; Lo, C.C.; Wong, C.C.; Cheung, A.K.; Cheong, K.F.; Wong, N.; Kwong, F.M.; Chan, K.C.; Law, E.W.; Tsao, S.W.; et al. Identification of tumor suppressive activity by irradiation microcell-mediated chromosome transfer and involvement of alpha B-crystallin in nasopharyngeal carcinoma. Int. J. Cancer 2008, 122, 1288–1296. [Google Scholar]
- Solares, C.A.; Boyle, G.M.; Brown, I.; Parsons, P.G.; Panizza, B. Reduced alphaB-crystallin staining in perineural invasion of head and neck cutaneous squamous cell carcinoma. Otolaryngol. Head. Neck. Surg. 2010, 142, S15–S19. [Google Scholar] [CrossRef]
- Chen, J.; He, Q.Y.; Yuen, A.P.; Chiu, J.F. Proteomics of buccal squamous cell carcinoma: The involvement of multiple pathways in tumorigenesis. Proteomics 2004, 4, 2465–2475. [Google Scholar] [CrossRef] [Green Version]
- He, Q.Y.; Chen, J.; Kung, H.F.; Yuen, A.P.; Chiu, J.F. Identification of tumor-associated proteins in oral tongue squamous cell carcinoma by proteomics. Proteomics 2004, 4, 271–278. [Google Scholar] [CrossRef] [Green Version]
- Stronach, E.A.; Sellar, G.C.; Blenkiron, C.; Rabiasz, G.J.; Taylor, K.J.; Miller, E.P.; Massie, C.E.; Al-Nafussi, A.; Smyth, J.F.; Porteous, D.J.; et al. Identification of clinically relevant genes on chromosome 11 in a functional model of ovarian cancer tumor suppression. Cancer Res. 2003, 63, 8648–8655. [Google Scholar]
- Bosman, J.D.; Yehiely, F.; Evans, J.R.; Cryns, V.L. Regulation of alphaB-crystallin gene expression by the transcription factor Ets1 in breast cancer. Breast Cancer Res Treat 2010, 119, 63–70. [Google Scholar] [CrossRef]
- Deng, M.; Chen, P.C.; Xie, S.; Zhao, J.; Gong, L.; Liu, J.; Zhang, L.; Sun, S.; Liu, J.; Ma, H.; et al. The small heat shock protein alphaA-crystallin is expressed in pancreas and acts as a negative regulator of carcinogenesis. Biochim. Biophys. Acta 2010, 1802, 621–631. [Google Scholar]
- Mahon, K.A.; Chepelinsky, A.B.; Khillan, J.S.; Overbeek, P.A.; Piatigorsky, J.; Westphal, H. Oncogenesis of the lens in transgenic mice. Science 1987, 235, 1622–1628. [Google Scholar]
- Rigas, P.K.; Kase, S.; Rao, N.A. Expression of alpha-crystallins in human sebaceous carcinoma of the eyelid. Eur. J. Ophthalmol. 2009, 19, 702–707. [Google Scholar]
- Basha, E.; O’Neill, H.; Vierling, E. Small heat shock proteins and alpha-crystallins: Dynamic proteins with flexible functions. Trends Biochem. Sci. 2011, 37, 106–117. [Google Scholar]
- Arrigo, A.P. Structure-functions of HspB1 (Hsp27). Methods Mol. Biol. 2011, 787, 105–119. [Google Scholar] [CrossRef]
- Simon, S.; Dimitrova, V.; Gibert, B.; Virot, S.; Mounier, N.; Nivon, M.; Kretz-Remy, C.; Corset, V.; Mehlen, P.; Arrigo, A.P. Analysis of the dominant effects mediated by wild type or R120G mutant of alphaB-crystallin (HspB5) towards Hsp27 (HspB1). PLoS One 2013, 8, e70545. [Google Scholar]
- Arrigo, A.-P.; Suhan, J.P.; Welch, W.J. Dynamic changes in the structure and intracellular locale of the mammalian low-molecular-weight heat shock protein. Mol. Cell. Biol. 1988, 8, 5059–5071. [Google Scholar]
- Mehlen, P.; Arrigo, A.-P. The serum-induced phosphorylation of mammalian hsp27 correlates with changes in its intracellular localization and levels of oligomerization. Eur. J. Biochem. 1994, 221, 327–334. [Google Scholar] [CrossRef]
- Mehlen, P.; Hickey, E.; Weber, L.; Arrigo, A.-P. Large unphosphorylated aggregates as the active form of hsp27 which controls intracellular reactive oxygen species and glutathione levels and generates a protection against TNFα in NIH-3T3-ras cells. Biochem. Biophys. Res. Comm. 1997, 241, 187–192. [Google Scholar] [CrossRef]
- Garrido, C. Size matters: Of the small HSP27 and its large oligomers. Cell Death Differ. 2002, 9, 483–485. [Google Scholar] [CrossRef]
- Arrigo, A.P. The cellular “networking” of mammalian Hsp27 and its functions in the control of protein folding, redox state and apoptosis. Adv. Exp. Med. Biol. 2007, 594, 14–26. [Google Scholar]
- Stengel, F.; Baldwin, A.J.; Painter, A.J.; Jaya, N.; Basha, E.; Kay, L.E.; Vierling, E.; Robinson, C.V.; Benesch, J.L. Quaternary dynamics and plasticity underlie small heat shock protein chaperone function. Proc. Natl. Acad. Sci. USA 2010, 107, 2007–2012. [Google Scholar] [CrossRef]
- Ciocca, D.R.; Luque, E.H. Immunological evidence for the identity between the hsp27 estrogen- regulated heat shock protein and the p29 estrogen receptor-associated protein in breast and endometrial cancer. Breast. Cancer Res. Treat. 1991, 20, 33–42. [Google Scholar] [CrossRef]
- Al-Madhoun, A.S.; Chen, Y.X.; Haidari, L.; Rayner, K.; Gerthoffer, W.; McBride, H.; O’Brien, E.R. The interaction and cellular localization of HSP27 and ERbeta are modulated by 17beta-estradiol and HSP27 phosphorylation. Mol. Cell Endocrinol. 2007, 270, 33–42. [Google Scholar]
- Kerr, B.A.; Byzova, T.V. alphaB-crystallin: A novel VEGF chaperone. Blood 2010, 115, 3181–3183. [Google Scholar] [CrossRef]
- Ghosh, J.G.; Shenoy, A.K., Jr.; Clark, J.I. Interactions between important regulatory proteins and human alphaB crystallin. Biochemistry 2007, 46, 6308–6317. [Google Scholar]
- Zoubeidi, A.; Zardan, A.; Beraldi, E.; Fazli, L.; Sowery, R.; Rennie, P.; Nelson, C.; Gleave, M. Cooperative interactions between androgen receptor (AR) and heat-shock protein 27 facilitate AR transcriptional activity. Cancer Res. 2007, 67, 10455–10465. [Google Scholar] [CrossRef]
- Adhikari, A.S.; Singh, B.N.; Rao, K.S.; Rao Ch, M. alphaB-crystallin, a small heat shock protein, modulates NF-kappaB activity in a phosphorylation-dependent manner and protects muscle myoblasts from TNF-alpha induced cytotoxicity. Biochim. Biophys. Acta 2011, 1813, 1532–1542. [Google Scholar] [CrossRef]
- Wu, Y.; Liu, J.; Zhang, Z.; Huang, H.; Shen, J.; Zhang, S.; Jiang, Y.; Luo, L.; Yin, Z. HSP27 regulates IL-1 stimulated IKK activation through interacting with TRAF6 and affecting its ubiquitination. Cell. Signal. 2009, 21, 143–150. [Google Scholar]
- Charette, S.J.; Landry, J. The interaction of HSP27 with Daxx identifies a potential regulatory role of HSP27 in Fas-induced apoptosis. Ann. NY Acad. Sci. 2000, 926, 126–131. [Google Scholar] [CrossRef]
- Lee, H.J.; Lee, Y.S. Repeated-dose toxicity of HSP27-binding heptapeptide in mice. Drug Chem. Toxicol. 2010, 33, 284–290. [Google Scholar] [CrossRef]
- Patil, S.B.; Pawar, M.D.; Bitar, K.N. Direct association and translocation of PKC-alpha with calponin. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, G954–G963. [Google Scholar] [CrossRef]
- Wu, R.; Kausar, H.; Johnson, P.; Montoya-Durango, D.E.; Merchant, M.; Rane, M.J. Hsp27 regulates Akt activation and polymorphonuclear leukocyte apoptosis by scaffolding MK2 to Akt signal complex. J. Biol. Chem. 2007, 282, 21598–21608. [Google Scholar]
- Liu, J.P.; Schlosser, R.; Ma, W.Y.; Dong, Z.; Feng, H.; Lui, L.; Huang, X.Q.; Liu, Y.; Li, D.W. Human alphaA- and alphaB-crystallins prevent UVA-induced apoptosis through regulation of PKCalpha, RAF/MEK/ERK and AKT signaling pathways. Exp. Eye Res. 2004, 79, 393–403. [Google Scholar] [CrossRef]
- Chebotareva, N.A.; Makeeva, V.F.; Bazhina, S.G.; Eronina, T.B.; Gusev, N.B.; Kurganov, B.I. Interaction of Hsp27 with native phosphorylase kinase under crowding conditions. Macromol. Biosci. 2010, 10, 783–789. [Google Scholar] [CrossRef]
- Zoubeidi, A.; Zardan, A.; Wiedmann, R.M.; Locke, J.; Beraldi, E.; Fazli, L.; Gleave, M.E. Hsp27 promotes insulin-like growth factor-I survival signaling in prostate cancer via p90Rsk-dependent phosphorylation and inactivation of BAD. Cancer Res. 2010, 70, 2307–2317. [Google Scholar] [CrossRef]
- Cayado-Gutierrez, N.; Moncalero, V.L.; Rosales, E.M.; Beron, W.; Salvatierra, E.E.; Alvarez-Olmedo, D.; Radrizzani, M.; Ciocca, D.R. Downregulation of Hsp27 (HSPB1) in MCF-7 human breast cancer cells induces upregulation of PTEN. Cell Stress Chaperones 2012, 18, 243–249. [Google Scholar]
- Doppler, H.; Storz, P.; Li, J.; Comb, M.J.; Toker, A. A phosphorylation state-specific antibody recognizes Hsp27, a novel substrate of protein kinase D. J. Biol. Chem. 2005, 280, 15013–15019. [Google Scholar] [CrossRef]
- Wang, J.; Huo, K.; Ma, L.; Tang, L.; Li, D.; Huang, X.; Yuan, Y.; Li, C.; Wang, W.; Guan, W.; et al. Toward an understanding of the protein interaction network of the human liver. Mol. Syst. Biol. 2011, 7, 536. [Google Scholar]
- Li, D.W.; Liu, J.P.; Mao, Y.W.; Xiang, H.; Wang, J.; Ma, W.Y.; Dong, Z.; Pike, H.M.; Brown, R.E.; Reed, J.C. Calcium-activated RAF/MEK/ERK signaling pathway mediates p53-dependent apoptosis and is abrogated by alpha B-crystallin through inhibition of RAS activation. Mol. Biol. Cell. 2005, 16, 4437–4453. [Google Scholar] [CrossRef]
- Dall’Era, M.A.; Oudes, A.; Martin, D.B.; Liu, A.Y. HSP27 and HSP70 interact with CD10 in C4–2 prostate cancer cells. Prostate 2007, 67, 714–721. [Google Scholar] [CrossRef]
- Van Noort, J.M.; Bsibsi, M.; Nacken, P.J.; Gerritsen, W.H.; Amor, S.; Holtman, I.R.; Boddeke, E.; van Ark, I.; Leusink-Muis, T.; Folkerts, G.; et al. Activation of an immune-regulatory macrophage response and inhibition of lung inflammation in a mouse model of COPD using heat-shock protein alpha B-crystallin-loaded PLGA microparticles. Biomaterials 2013, 34, 831–840. [Google Scholar]
- Thuringer, D.; Jego, G.; Wettstein, G.; Terrier, O.; Cronier, L.; Yousfi, N.; Hebrard, S.; Bouchot, A.; Hazoume, A.; Joly, A.L.; et al. Extracellular HSP27 mediates angiogenesis through Toll-like receptor 3. FASEB J. 2013, 27, 4169–4183. [Google Scholar]
- Pandey, P.; Farber, R.; Nakazawa, A.; Kumar, S.; Bharti, A.; Nalin, C.; Weichselbaum, R.; Kufe, D.; Kharbanda, S. Hsp27 functions as a negative regulator of cytochrome c-dependent activation of procaspase-3. Oncogene 2000, 19, 1975–1981. [Google Scholar]
- Hu, W.F.; Gong, L.; Cao, Z.; Ma, H.; Ji, W.; Deng, M.; Liu, M.; Hu, X.H.; Chen, P.; Yan, Q.; et al. alphaA- and alphaB-crystallins interact with caspase-3 and Bax to guard mouse lens development. Curr. Mol. Med. 2012, 12, 177–187. [Google Scholar] [CrossRef]
- Bruey, J.M.; Ducasse, C.; Bonniaud, P.; Ravagnan, L.; Susin, S.A.; Diaz-Latoud, C.; Gurbuxani, S.; Arrigo, A.P.; Kroemer, G.; Solary, E.; et al. Hsp27 negatively regulates cell death by interacting with cytochrome c. Nat. Cell. Biol. 2000, 2, 645–652. [Google Scholar] [CrossRef]
- Padival, A.K.; Crabb, J.W.; Nagaraj, R.H. Methylglyoxal modifies heat shock protein 27 in glomerular mesangial cells. FEBS Lett. 2003, 551, 113–118. [Google Scholar] [CrossRef]
- Mao, Y.W.; Liu, J.P.; Xiang, H.; Li, D.W. Human alphaA- and alphaB-crystallins bind to Bax and Bcl-X(S) to sequester their translocation during staurosporine-induced apoptosis. Cell Death Differ. 2004, 11, 512–526. [Google Scholar] [CrossRef]
- Liu, S.; Li, J.; Tao, Y.; Xiao, X. Small heat shock protein alphaB-crystallin binds to p53 to sequester its translocation to mitochondria during hydrogen peroxide-induced apoptosis. Biochem. Biophys. Res. Commun. 2007, 354, 109–114. [Google Scholar] [CrossRef]
- Beresford, P.J.; Jaju, M.; Friedman, R.S.; Yoon, M.J.; Lieberman, J. A role for heat shock protein 27 in CTL-mediated cell death. J. Immunol. 1998, 161, 161–167. [Google Scholar]
- Lee, J.S.; Kim, H.Y.; Jeong, N.Y.; Lee, S.Y.; Yoon, Y.G.; Choi, Y.H.; Yan, C.; Chu, I.S.; Koh, H.; Park, H.T.; et al. Expression of alphaB-crystallin overrides the anti-apoptotic activity of XIAP. Neuro. Oncol. 2012, 14, 1332–1345. [Google Scholar] [CrossRef]
- Arany, I.; Clark, J.S.; Reed, D.K.; Ember, I.; Juncos, L.A. Cisplatin enhances interaction between p66Shc and HSP27: Its role in reorganization of the actin cytoskeleton in renal proximal tubule cells. Anticancer Res. 2012, 32, 4759–4763. [Google Scholar]
- Preville, X.; Salvemini, F.; Giraud, S.; Chaufour, S.; Paul, C.; Stepien, G.; Ursini, M.V.; Arrigo, A.P. Mammalian small stress proteins protect against oxidative stress through their ability to increase glucose-6-phosphate dehydrogenase activity and by maintaining optimal cellular detoxifying machinery. Exp. Cell Res. 1999, 247, 61–78. [Google Scholar] [CrossRef]
- Cosentino, C.; Grieco, D.; Costanzo, V. ATM activates the pentose phosphate pathway promoting anti-oxidant defence and DNA repair. EMBO J. 2011, 30, 546–555. [Google Scholar] [CrossRef]
- Perng, M.D.; Cairns, L.; van den, I.P.; Prescott, A.; Hutcheson, A.M.; Quinlan, R.A. Intermediate filament interactions can be altered by HSP27 and alphaB-crystallin. J. Cell Sci. 1999, 112, 2099–2112. [Google Scholar]
- Mounier, N.; Arrigo, A.P. Actin cytoskeleton and small heat shock proteins: How do they interact? Cell Stress Chaperones 2002, 7, 167–176. [Google Scholar] [CrossRef]
- Ke, L.; Meijering, R.A.; Hoogstra-Berends, F.; Mackovicova, K.; Vos, M.J.; van Gelder, I.C.; Henning, R.H.; Kampinga, H.H.; Brundel, B.J. HSPB1, HSPB6, HSPB7 and HSPB8 protect against RhoA GTPase-induced remodeling in tachypaced atrial myocytes. PLoS One 2011, 6, e20395. [Google Scholar] [CrossRef]
- Seit-Nebi, A.S.; Datskevich, P.; Gusev, N.B. Commentary on paper: Small heat shock proteins and the cytoskeleton: An essential interplay for cell integrity? (Wettstein et al.). Int. J. Biochem. Cell Biol. 2013, 45, 344–346. [Google Scholar] [CrossRef]
- Del Vecchio, P.J.; MacElroy, K.S.; Rosser, M.P.; Church, R.L. Association of alpha-crystallin with actin in cultured lens cells. Curr. Eye Res. 1984, 3, 1213–1219. [Google Scholar] [CrossRef]
- Wang, K.; Spector, A. alpha-Crystallin stabilizes actin filaments and prevents cytochalasin- induced depolymerization in a phosphorylation-dependent manner. Eur. J. Biochem. 1996, 242, 56–66. [Google Scholar]
- Singh, B.N.; Rao, K.S.; Ramakrishna, T.; Rangaraj, N.; Rao Ch, M. Association of alphab-crystallin, a small heat shock protein, with actin: Role in modulating actin filament dynamics in vivo. J. Mol. Biol. 2007, 366, 756–767. [Google Scholar] [CrossRef]
- Ghosh, J.G.; Houck, S.A.; Clark, J.I. Interactive sequences in the stress protein and molecular chaperone human alphaB crystallin recognize and modulate the assembly of filaments. Int. J. Biochem. Cell Biol. 2007, 39, 1804–1815. [Google Scholar] [CrossRef]
- Hino, M.; Kurogi, K.; Okubo, M.A.; Murata-Hori, M.; Hosoya, H. Small heat shock protein 27 (HSP27) associates with tubulin/microtubules in HeLa cells. Biochem. Biophys. Res. Commun. 2000, 271, 164–169. [Google Scholar] [CrossRef]
- Ohto-Fujita, E.; Fujita, Y.; Atomi, Y. Analysis of the alphaB-crystallin domain responsible for inhibiting tubulin aggregation. Cell Stress Chaperones 2007, 12, 163–171. [Google Scholar] [CrossRef]
- Ghosh, J.G.; Houck, S.A.; Clark, J.I. Interactive domains in the molecular chaperone human alphaB crystallin modulate microtubule assembly and disassembly. PLoS One 2007, 2, e498. [Google Scholar] [CrossRef]
- Xi, J.H.; Bai, F.; McGaha, R.; Andley, U.P. Alpha-crystallin expression affects microtubule assembly and prevents their aggregation. FASEB J. 2006, 20, 846–857. [Google Scholar] [CrossRef]
- Djabali, K.; de Nechaud, B.; Landon, F.; Portier, M.M. AlphaB-crystallin interacts with intermediate filaments in response to stress. J. Cell Sci. 1997, 110, 2759–2769. [Google Scholar]
- Djabali, K.; Piron, G.; de Nechaud, B.; Portier, M.M. alphaB-crystallin interacts with cytoplasmic intermediate filament bundles during mitosis. Exp. Cell Res. 1999, 253, 649–662. [Google Scholar] [CrossRef]
- Fanelli, M.A.; Montt-Guevara, M.; Diblasi, A.M.; Gago, F.E.; Tello, O.; Cuello-Carrion, F.D.; Callegari, E.; Bausero, M.A.; Ciocca, D.R. P-cadherin and beta-catenin are useful prognostic markers in breast cancer patients; beta-catenin interacts with heat shock protein Hsp27. Cell Stress Chaperones 2008, 13, 207–220. [Google Scholar] [CrossRef]
- Thedieck, C.; Kalbacher, H.; Kratzer, U.; Lammers, R.; Stevanovic, S.; Klein, G. alpha B-crystallin is a cytoplasmic interaction partner of the kidney-specific cadherin-16. J. Mol. Biol. 2008, 378, 145–153. [Google Scholar] [CrossRef]
- Muchowski, P.J.; Valdez, M.M.; Clark, J.I. AlphaB-crystallin selectively targets intermediate filament proteins during thermal stress. Invest. Ophthalmol. Vis. Sci. 1999, 40, 951–958. [Google Scholar]
- Barton, K.A.; Hsu, C.D.; Petrash, J.M. Interactions between small heat shock protein alpha-crystallin and galectin-related interfiber protein (GRIFIN) in the ocular lens. Biochemistry 2009, 48, 3956–3966. [Google Scholar] [CrossRef]
- Rocchi, P.; Beraldi, E.; Ettinger, S.; Fazli, L.; Vessella, R.L.; Nelson, C.; Gleave, M. Increased Hsp27 after androgen ablation facilitates androgen-independent progression in prostate cancer via signal transducers and activators of transcription 3-mediated suppression of apoptosis. Cancer Res. 2005, 65, 11083–11093. [Google Scholar] [CrossRef]
- Brunet Simioni, M.; de Thonel, A.; Hammann, A.; Joly, A.L.; Bossis, G.; Fourmaux, E.; Bouchot, A.; Landry, J.; Piechaczyk, M.; Garrido, C. Heat shock protein 27 is involved in SUMO-2/3 modification of heat shock factor 1 and thereby modulates the transcription factor activity. Oncogene 2009, 28, 3332–3344. [Google Scholar] [CrossRef]
- De Thonel, A.; Vandekerckhove, J.; Lanneau, D.; Selvakumar, S.; Courtois, G.; Hazoume, A.; Brunet, M.; Maurel, S.; Hammann, A.; Ribeil, J.A.; et al. HSP27 controls GATA-1 protein level during erythroid cell differentiation. Blood 2010, 116, 85–96. [Google Scholar] [CrossRef]
- Cuesta, R.; Laroia, G.; Schneider, R.J. Chaperone Hsp27 inhibits translation during heat shock by binding eIF4G and facilitating dissociation of cap-initiation complexes. Genes Dev. 2000, 14, 1460–1470. [Google Scholar]
- Andrieu, C.; Taieb, D.; Baylot, V.; Ettinger, S.; Soubeyran, P.; De-Thonel, A.; Nelson, C.; Garrido, C.; So, A.; Fazli, L.; et al. Heat shock protein 27 confers resistance to androgen ablation and chemotherapy in prostate cancer cells through eIF4E. Oncogene 2010, 29, 1883–1896. [Google Scholar] [CrossRef]
- Sinsimer, K.S.; Gratacos, F.M.; Knapinska, A.M.; Lu, J.; Krause, C.D.; Wierzbowski, A.V.; Maher, L.R.; Scrudato, S.; Rivera, Y.M.; Gupta, S.; et al. Chaperone Hsp27, a novel subunit of AUF1 protein complexes, functions in AU-rich element-mediated mRNA decay. Mol. Cell. Biol. 2008, 28, 5223–5237. [Google Scholar] [CrossRef]
- Knapinska, A.M.; Gratacos, F.M.; Krause, C.D.; Hernandez, K.; Jensen, A.G.; Bradley, J.J.; Wu, X.; Pestka, S.; Brewer, G. Chaperone Hsp27 modulates AUF1 proteolysis and AU-rich element-mediated mRNA degradation. Mol. Cell. Biol. 2011, 31, 1419–1431. [Google Scholar] [CrossRef]
- Sun, Y.; Zhou, M.; Fu, D.; Xu, B.; Fang, T.; Ma, Y.; Chen, J.; Zhang, J. Ubiquitination of heat shock protein 27 is mediated by its interaction with Smad ubiquitination regulatory factor 2 in A549 cells. Exp. Lung Res. 2011, 37, 568–573. [Google Scholar] [CrossRef]
- Parcellier, A.; Brunet, M.; Schmitt, E.; Col, E.; Didelot, C.; Hammann, A.; Nakayama, K.; Nakayama, K.I.; Khochbin, S.; Solary, E.; et al. HSP27 favors ubiquitination and proteasomal degradation of p27Kip1 and helps S-phase re-entry in stressed cells. FASEB J. 2006, 20, 1179–1181. [Google Scholar] [CrossRef]
- Parcellier, A.; Schmitt, E.; Gurbuxani, S.; Seigneurin-Berny, D.; Pance, A.; Chantome, A.; Plenchette, S.; Khochbin, S.; Solary, E.; Garrido, C. HSP27 is a ubiquitin-binding protein involved in I-kappaBalpha proteasomal degradation. Mol. Cell. Biol. 2003, 23, 5790–5802. [Google Scholar] [CrossRef]
- Lin, D.I.; Barbash, O.; Kumar, K.G.; Weber, J.D.; Harper, J.W.; Klein-Szanto, A.J.; Rustgi, A.; Fuchs, S.Y.; Diehl, J.A. Phosphorylation-dependent ubiquitination of cyclin D1 by the SCF(FBX4-alphaB crystallin) complex. Mol. Cell. 2006, 24, 355–366. [Google Scholar] [CrossRef]
- Boelens, W.C.; Croes, Y.; de Jong, W.W. Interaction between alphaB-crystallin and the human 20S proteasomal subunit C8/alpha7. Biochim. Biophys. Acta 2001, 1544, 311–319. [Google Scholar]
- Mehlen, P.; Mehlen, A.; Godet, J.; Arrigo, A.-P. Hsp27 as a switch between differentiation and apoptosis in murine embryonic stem cells. J. Biol. Chem. 1997, 272, 31657–31665. [Google Scholar]
- Arrigo, A.P. Editorial: Heat shock proteins in cancer. Curr. Mol. Med. 2012, 12, 1099–1101. [Google Scholar] [CrossRef]
- Paul, C.; Manero, F.; Gonin, S.; Kretz-Remy, C.; Virot, S.; Arrigo, A.P. Hsp27 as a negative regulator of cytochrome C release. Mol. Cell Biol. 2002, 22, 816–834. [Google Scholar] [CrossRef]
- Chauhan, D.; Li, G.; Hideshima, T.; Podar, K.; Mitsiades, C.; Mitsiades, N.; Catley, L.; Tai, Y.T.; Hayashi, T.; Shringarpure, R.; et al. Hsp27 inhibits release of mitochondrial protein Smac in multiple myeloma cells and confers dexamethasone resistance. Blood 2003, 102, 3379–3386. [Google Scholar]
- Garrido, C.; Bruey, J.M.; Fromentin, A.; Hammann, A.; Arrigo, A.P.; Solary, E. HSP27 inhibits cytochrome c-dependent activation of procaspase-9. FASEB J. 1999, 13, 2061–2070. [Google Scholar]
- Mehlen, P.; Préville, X.; Chareyron, P.; Briolay, J.; Klemenz, R.; Arrigo, A.-P. Constitutive expression of human hsp27, Drosophila hsp27, or human alpha B-crystallin confers resistance to TNF- and oxidative stress-induced cytotoxicity in stably transfected murine L929 fibroblasts. J. Immunol. 1995, 154, 363–374. [Google Scholar]
- Charette, S.J.; Lavoie, J.N.; Lambert, H.; Landry, J. Inhibition of daxx-mediated apoptosis by heat shock protein 27. Mol. Cell. Biol. 2000, 20, 7602–7612. [Google Scholar] [CrossRef]
- Rane, M.J.; Pan, Y.; Singh, S.; Powell, D.W.; Wu, R.; Cummins, T.; Chen, Q.; McLeish, K.R.; Klein, J.B. Heat shock protein 27 controls apoptosis by regulating Akt activation. J. Biol. Chem. 2003, 278, 27828–27835. [Google Scholar] [CrossRef]
- Golembieski, W.A.; Thomas, S.L.; Schultz, C.R.; Yunker, C.K.; McClung, H.M.; Lemke, N.; Cazacu, S.; Barker, T.; Sage, E.H.; Brodie, C.; et al. HSP27 mediates SPARC-induced changes in glioma morphology, migration, and invasion. Glia 2008, 56, 1061–1075. [Google Scholar] [CrossRef]
- McClung, H.M.; Golembieski, W.A.; Schultz, C.R.; Jankowski, M.; Schultz, L.R.; Rempel, S.A. Deletion of the SPARC acidic domain or EGF-like module reduces SPARC-induced migration and signaling through p38 MAPK/HSP27 in glioma. Carcinogenesis 2012, 33, 275–284. [Google Scholar] [CrossRef]
- Andley, U.P.; Song, Z.; Wawrousek, E.F.; Fleming, T.P.; Bassnett, S. Differential protective activity of {alpha}A- and {alpha}B-crystallin in lens epithelial cells. J. Biol. Chem. 2000, 275, 36823–36831. [Google Scholar]
- Launay, N.; Tarze, A.; Vicart, P.; Lilienbaum, A. Serine 59 phosphorylation of {alpha}B-crystallin down-regulates its anti-apoptotic function by binding and sequestering Bcl-2 in breast cancer cells. J. Biol. Chem. 2010, 285, 37324–37332. [Google Scholar]
- Lee, Y.S.; Lim, K.H.; Guo, X.; Kawaguchi, Y.; Gao, Y.; Barrientos, T.; Ordentlich, P.; Wang, X.F.; Counter, C.M.; Yao, T.P. The cytoplasmic deacetylase HDAC6 is required for efficient oncogenic tumorigenesis. Cancer Res. 2008, 68, 7561–7569. [Google Scholar] [CrossRef]
- Ghosh, A.; Lai, C.; McDonald, S.; Suraweera, N.; Sengupta, N.; Propper, D.; Dorudi, S.; Silver, A. HSP27 expression in primary colorectal cancers is dependent on mutation of KRAS and PI3K/AKT activation status and is independent of TP53. Exp. Mol. Pathol. 2013, 94, 103–108. [Google Scholar] [CrossRef]
- Yang, Y.; Ludwig, R.L.; Jensen, J.P.; Pierre, S.A.; Medaglia, M.V.; Davydov, I.V.; Safiran, Y.J.; Oberoi, P.; Kenten, J.H.; Phillips, A.C.; et al. Small molecule inhibitors of HDM2 ubiquitin ligase activity stabilize and activate p53 in cells. Cancer Cell 2005, 7, 547–559. [Google Scholar] [CrossRef]
- Bausero, M.A.; Bharti, A.; Page, D.T.; Perez, K.D.; Eng, J.W.; Ordonez, S.L.; Asea, E.E.; Jantschitsch, C.; Kindas-Muegge, I.; Ciocca, D.; et al. Silencing the hsp25 gene eliminates migration capability of the highly metastatic murine 4T1 breast adenocarcinoma cell. Tumour Biol. 2006, 27, 17–26. [Google Scholar] [CrossRef]
- Ciocca, D.R.; Fanelli, M.A.; Cuello-Carrion, F.D.; Castro, G.N. Heat shock proteins in prostate cancer: From tumorigenesis to the clinic. Int. J. Hyperth. 2010, 26, 737–747. [Google Scholar] [CrossRef]
- Lavoie, J.N.; Hickey, E.; Weber, L.A.; Landry, J. Modulation of actin microfilament dynamics and fluid phase pinocytosis by phosphorylation of Heat Shock Protein 27. J. Biol. Chem. 1993, 268, 24210–24214. [Google Scholar]
- Huot, J.; Houle, F.; Spitz, D.R.; Landry, J. HSP27 phosphorylation-mediated resistance against actin fragmentation and cell death induced by oxidative stress. Cancer Res. 1996, 56, 273–279. [Google Scholar]
- Dalle-Donne, I.; Rossi, R.; Milzani, A.; di Simplicio, P.; Colombo, R. The actin cytoskeleton response to oxidants: From small heat shock protein phosphorylation to changes in the redox state of actin itself. Free Radic. Biol. Med. 2001, 31, 1624–1632. [Google Scholar] [CrossRef]
- Wettstein, G.; Bellaye, P.S.; Micheau, O.; Bonniaud, P. Small heat shock proteins and the cytoskeleton: An essential interplay for cell integrity? Int. J. Biochem. Cell Biol. 2012, 44, 1680–1686. [Google Scholar]
- Xu, L.; Chen, S.; Bergan, R.C. MAPKAPK2 and HSP27 are downstream effectors of p38 MAP kinase-mediated matrix metalloproteinase type 2 activation and cell invasion in human prostate cancer. Oncogene 2006, 25, 2987–2998. [Google Scholar] [CrossRef]
- Yoshimura, T.; Nagahara, M.; Kuo, C.; Turner, R.R.; Soon-Shiong, P.; Hoon, D.S. Lymphovascular invasion of colorectal cancer is correlated to SPARC expression in the tumor stromal microenvironment. Epigenetics 2011, 6, 1001–1011. [Google Scholar] [CrossRef]
- Schultz, C.R.; Golembieski, W.A.; King, D.A.; Brown, S.L.; Brodie, C.; Rempel, S.A. Inhibition of HSP27 alone or in combination with pAKT inhibition as therapeutic approaches to target SPARC-induced glioma cell survival. Mol. Cancer 2012, 11, 20. [Google Scholar] [CrossRef]
- Nagaraja, G.M.; Kaur, P.; Neumann, W.; Asea, E.E.; Bausero, M.A.; Multhoff, G.; Asea, A. Silencing Hsp25/Hsp27 gene expression augments proteasome activity and increases CD8+ T-cell-mediated tumor killing and memory responses. Cancer Prev. Res. Phila 2012, 5, 122–137. [Google Scholar] [CrossRef]
- Gruvberger-Saal, S.K.; Parsons, R. Is the small heat shock protein alphaB-crystallin an oncogene? J. Clin. Invest. 2006, 116, 30–32. [Google Scholar] [CrossRef]
- De Maio, A. Extracellular heat shock proteins, cellular export vesicles, and the Stress Observation System: A form of communication during injury, infection, and cell damage. It is never known how far a controversial finding will go! Dedicated to Ferruccio Ritossa. Cell Stress Chaperones 2011, 16, 235–249. [Google Scholar] [CrossRef]
- Tsvetkova, N.M.; Horvath, I.; Torok, Z.; Wolkers, W.F.; Balogi, Z.; Shigapova, N.; Crowe, L.M.; Tablin, F.; Vierling, E.; Crowe, J.H.; et al. Small heat-shock proteins regulate membrane lipid polymorphism. Proc. Natl. Acad. Sci. USA 2002, 99, 13504–13509. [Google Scholar] [CrossRef]
- Chowdary, T.K.; Bakthisaran, R.; Tangirala, R.; Rao, M.C. Interaction of mammalian Hsp22 with lipid membranes. Biochem. J. 2006, 401, 437–445. [Google Scholar]
- Rayner, K.; Chen, Y.X.; McNulty, M.; Simard, T.; Zhao, X.; Wells, D.J.; de Belleroche, J.; O’Brien, E.R. Extracellular release of the atheroprotective heat shock protein 27 is mediated by estrogen and competitively inhibits acLDL binding to scavenger receptor-A. Circ. Res. 2008, 103, 133–141. [Google Scholar] [CrossRef]
- Delneste, Y.; Magistrelli, G.; Gauchat, J.; Haeuw, J.; Aubry, J.; Nakamura, K.; Kawakami-Honda, N.; Goetsch, L.; Sawamura, T.; Bonnefoy, J.; et al. Involvement of LOX-1 in dendritic cell-mediated antigen cross-presentation. Immunity 2002, 17, 353–362. [Google Scholar] [CrossRef]
- Chalmin, F.; Ladoire, S.; Mignot, G.; Vincent, J.; Bruchard, M.; Remy-Martin, J.P.; Boireau, W.; Rouleau, A.; Simon, B.; Lanneau, D.; et al. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J. Clin. Invest. 2010, 120, 457–471. [Google Scholar] [Green Version]
- Salari, S.; Seibert, T.; Chen, Y.X.; Hu, T.; Shi, C.; Zhao, X.; Cuerrier, C.M.; Raizman, J.E.; O’Brien, E.R. Extracellular HSP27 acts as a signaling molecule to activate NF-kappaB in macrophages. Cell Stress Chaperones 2012, 18, 53–63. [Google Scholar]
- Gruden, G.; Bruno, G.; Chaturvedi, N.; Burt, D.; Schalkwijk, C.; Pinach, S.; Stehouwer, C.D.; Witte, D.R.; Fuller, J.H.; Perin, P.C. Serum heat shock protein 27 and diabetes complications in the EURODIAB prospective complications study: A novel circulating marker for diabetic neuropathy. Diabetes 2008, 57, 1966–1970. [Google Scholar] [CrossRef]
- Joachim, S.C.; Bruns, K.; Lackner, K.J.; Pfeiffer, N.; Grus, F.H. Antibodies to alpha B-crystallin, vimentin, and heat shock protein 70 in aqueous humor of patients with normal tension glaucoma and IgG antibody patterns against retinal antigen in aqueous humor. Curr. Eye Res. 2007, 32, 501–509. [Google Scholar] [CrossRef]
- Tezel, G.; Wax, M.B. The mechanisms of hsp27 antibody-mediated apoptosis in retinal neuronal cells. J. Neurosci. 2000, 20, 3552–3562. [Google Scholar]
- Lu, H.; Ouyang, W.; Huang, C. Inflammation, a key event in cancer development. Mol. Cancer Res. 2006, 4, 221–233. [Google Scholar] [CrossRef]
- Alford, K.A.; Glennie, S.; Turrell, B.R.; Rawlinson, L.; Saklatvala, J.; Dean, J.L. HSP27 functions in inflammatory gene expression and TAK1-mediated signalling. J. Biol. Chem. 2007, 282, 6232–6241. [Google Scholar]
- Rothbard, J.B.; Kurnellas, M.P.; Brownell, S.; Adams, C.M.; Su, L.; Axtell, R.C.; Chen, R.; Fathman, C.G.; Robinson, W.H.; Steinman, L. Therapeutic effects of systemic administration of chaperone alphaB-crystallin associated with binding proinflammatory plasma proteins. J. Biol. Chem. 2012, 287, 9708–9721. [Google Scholar] [CrossRef]
- Mehlen, P.; Préville, X.; Kretz-Remy, C.; Arrigo, A.-P. Human hsp27, Drosophila hsp27 and human αB-crystallin expression-mediated increase in glutathione is essential for the protective activity of these protein against TNFα-induced cell death. EMBO J. 1996, 15, 2695–2706. [Google Scholar]
- Arrigo, A.P. Small stress proteins: Chaperones that act as regulators of intracellular redox state and programmed cell death. Biol. Chem. 1998, 379, 19–26. [Google Scholar]
- Park, Y.M.; Han, M.Y.; Blackburn, R.V.; Lee, Y.J. Overexpression of HSP25 reduces the level of TNF alpha-induced oxidative DNA damage biomarker, 8-hydroxy-2’-deoxyguanosine, in L929 cells. J. Cell. Physiol. 1998, 174, 27–34. [Google Scholar] [CrossRef]
- Rogalla, T.; Ehrnsperger, M.; Preville, X.; Kotlyarov, A.; Lutsch, G.; Ducasse, C.; Paul, C.; Wieske, M.; Arrigo, A.P.; Buchner, J.; et al. Regulation of Hsp27 oligomerization, chaperone function, and protective activity against oxidative stress/tumor necrosis factor alpha by phosphorylation. J. Biol. Chem. 1999, 274, 18947–18956. [Google Scholar] [CrossRef]
- Arrigo, A.P. Hsp27: Novel regulator of intracellular redox state. IUBMB Life 2001, 52, 303–307. [Google Scholar] [CrossRef]
- Merendino, A.M.; Paul, C.; Vignola, A.M.; Costa, M.A.; Melis, M.; Chiappara, G.; Izzo, V.; Bousquet, J.; Arrigo, A.P. Heat shock protein-27 protects human bronchial epithelial cells against oxidative stress-mediated apoptosis: Possible implication in asthma. Cell Stress Chaperones 2002, 7, 269–280. [Google Scholar] [CrossRef]
- Wyttenbach, A.; Sauvageot, O.; Carmichael, J.; Diaz-Latoud, C.; Arrigo, A.P.; Rubinsztein, D.C. Heat shock protein 27 prevents cellular polyglutamine toxicity and suppresses the increase of reactive oxygen species caused by huntingtin. Hum. Mol. Genet. 2002, 11, 1137–1151. [Google Scholar] [CrossRef]
- Yan, L.J.; Christians, E.S.; Liu, L.; Xiao, X.; Sohal, R.S.; Benjamin, I.J. Mouse heat shock transcription factor 1 deficiency alters cardiac redox homeostasis and increases mitochondrial oxidative damage. EMBO J. 2002, 21, 5164–5172. [Google Scholar] [CrossRef]
- Arrigo, A.P.; Virot, S.; Chaufour, S.; Firdaus, W.; Kretz-Remy, C.; Diaz-Latoud, C. Hsp27 consolidates intracellular redox homeostasis by upholding glutathione in its reduced form and by decreasing iron intracellular levels. Antioxid. Redox Signal. 2005, 7, 414–422. [Google Scholar] [CrossRef]
- Firdaus, W.J.; Wyttenbach, A.; Diaz-Latoud, C.; Currie, R.W.; Arrigo, A.P. Analysis of oxidative events induced by expanded polyglutamine huntingtin exon 1 that are differentially restored by expression of heat shock proteins or treatment with an antioxidant. FEBS J. 2006, 273, 3076–3093. [Google Scholar] [CrossRef]
- Aloy, M.T.; Hadchity, E.; Bionda, C.; Diaz-Latoud, C.; Claude, L.; Rousson, R.; Arrigo, A.P.; Rodriguez-Lafrasse, C. Protective role of Hsp27 protein against gamma radiation-induced apoptosis and radiosensitization effects of Hsp27 gene silencing in different human tumor cells. Int. J. Radiat. Oncol. Biol. Phys. 2008, 70, 543–553. [Google Scholar] [CrossRef]
- Christians, E.S.; Ishiwata, T.; Benjamin, I.J. Small heat shock proteins in redox metabolism: Implications for cardiovascular diseases. Int. J. Biochem. Cell Biol. 2012, 44, 1632–1645. [Google Scholar] [CrossRef]
- Chen, H.; Zheng, C.; Zhang, Y.; Chang, Y.Z.; Qian, Z.M.; Shen, X. Heat shock protein 27 downregulates the transferrin receptor 1-mediated iron uptake. Int. J. Biochem. Cell Biol. 2006, 38, 1402–1416. [Google Scholar] [CrossRef]
- Preville, X.; Gaestel, M.; Arrigo, A.P. Phosphorylation is not essential for protection of L929 cells by Hsp25 against H2O2-mediated disruption actin cytoskeleton, a protection which appears related to the redox change mediated by Hsp25. Cell Stress Chaperones 1998, 3, 177–187. [Google Scholar] [CrossRef]
- Kim, E.H.; Lee, H.J.; Lee, D.H.; Bae, S.; Soh, J.W.; Jeoung, D.; Kim, J.; Cho, C.K.; Lee, Y.J.; Lee, Y.S. Inhibition of heat shock protein 27-mediated resistance to DNA damaging agents by a novel PKC delta-V5 heptapeptide. Cancer Res. 2007, 67, 6333–6341. [Google Scholar] [CrossRef]
- Kuo, W.Y.; Tang, T.K. Effects of G6PD overexpression in NIH3T3 cells treated with tert-butyl hydroperoxide or paraquat. Free Radic. Biol. Med. 1998, 24, 1130–1138. [Google Scholar] [CrossRef]
- Salvemini, F.; Franze, A.; Iervolino, A.; Filosa, S.; Salzano, S.; Ursini, M.V. Enhanced glutathione levels and oxidoresistance mediated by increased glucose-6-phosphate dehydrogenase expression. J. Biol. Chem. 1999, 274, 2750–2757. [Google Scholar]
- McCollum, A.K.; Teneyck, C.J.; Sauer, B.M.; Toft, D.O.; Erlichman, C. Up-regulation of heat shock protein 27 induces resistance to 17-allylamino-demethoxygeldanamycin through a glutathione-mediated mechanism. Cancer Res. 2006, 66, 10967–10975. [Google Scholar]
- Aoki, Y.; Feldman, G.M.; Tosato, G. Inhibition of STAT3 signaling induces apoptosis and decreases survivin expression in primary effusion lymphoma. Blood 2003, 101, 1535–1542. [Google Scholar] [CrossRef]
- Brierley, M.M.; Fish, E.N. Stats: Multifaceted regulators of transcription. J. Interferon Cytokine Res. 2005, 25, 733–744. [Google Scholar] [CrossRef]
- Gritsko, T.; Williams, A.; Turkson, J.; Kaneko, S.; Bowman, T.; Huang, M.; Nam, S.; Eweis, I.; Diaz, N.; Sullivan, D.; et al. Persistent activation of stat3 signaling induces survivin gene expression and confers resistance to apoptosis in human breast cancer cells. Clin. Cancer Res. 2006, 12, 11–19. [Google Scholar] [CrossRef]
- Diaz, N.; Minton, S.; Cox, C.; Bowman, T.; Gritsko, T.; Garcia, R.; Eweis, I.; Wloch, M.; Livingston, S.; Seijo, E.; et al. Activation of stat3 in primary tumors from high-risk breast cancer patients is associated with elevated levels of activated SRC and survivin expression. Clin. Cancer Res. 2006, 12, 20–28. [Google Scholar] [CrossRef]
- Song, H.; Ethier, S.P.; Dziubinski, M.L.; Lin, J. Stat3 modulates heat shock 27 kDa protein expression in breast epithelial cells. Biochem. Biophys. Res. Commun. 2004, 314, 143–150. [Google Scholar]
- Silva, C.M. Role of STATs as downstream signal transducers in Src family kinase-mediated tumorigenesis. Oncogene 2004, 23, 8017–8023. [Google Scholar] [CrossRef]
- Yuan, Z.L.; Guan, Y.J.; Wang, L.; Wei, W.; Kane, A.B.; Chin, Y.E. Central role of the threonine residue within the p+1 loop of receptor tyrosine kinase in STAT3 constitutive phosphorylation in metastatic cancer cells. Mol. Cell Biol. 2004, 24, 9390–9400. [Google Scholar] [CrossRef]
- De la Iglesia, N.; Konopka, G.; Puram, S.V.; Chan, J.A.; Bachoo, R.M.; You, M.J.; Levy, D.E.; Depinho, R.A.; Bonni, A. Identification of a PTEN-regulated STAT3 brain tumor suppressor pathway. Genes Dev. 2008, 22, 449–462. [Google Scholar] [CrossRef]
- Lee, J.; Kim, J.C.; Lee, S.E.; Quinley, C.; Kim, H.; Herdman, S.; Corr, M.; Raz, E. Signal transducer and activator of transcription 3 (STAT3) protein suppresses adenoma-to-carcinoma transition in Apcmin/+ mice via regulation of Snail-1 (SNAI) protein stability. J. Biol. Chem. 2012, 287, 18182–18189. [Google Scholar]
- Zheng, R.; Blobel, G.A. GATA transcription factors and cancer. Genes Cancer 2010, 1, 1178–1188. [Google Scholar] [CrossRef]
- Diaz-Latoud, C.; Buache, E.; Javouhey, E.; Arrigo, A.P. Substitution of the unique cysteine residue of murine hsp25 interferes with the protective activity of this stress protein through inhibition of dimer formation. Antioxid. Redox. Signal 2005, 7, 436–445. [Google Scholar] [CrossRef]
- Ghosh, J.G.; Houck, S.A.; Clark, J.I. Interactive sequences in the molecular chaperone, human alphaB crystallin modulate the fibrillation of amyloidogenic proteins. Int. J. Biochem. Cell Biol. 2008, 40, 954–967. [Google Scholar] [CrossRef]
- Colas, P.; Cohen, B.; Jessen, T.; Grishina, I.; McCoy, J.; Brent, R. Genetic selection of peptide aptamers that recognize and inhibit cyclin-dependent kinase 2. Nature 1996, 380, 548–550. [Google Scholar] [CrossRef]
- De Chassey, B.; Mikaelian, I.; Mathieu, A.L.; Bickle, M.B.; Olivier, D.; Negre, D.; Cosset, F.L.; Rudkin, B.B.; Colas, P. An antiproliferative genetic screening identifies a peptide aptamer that targets calcineurin and upregulates its activity. Mol. Cell. Proteomics 2006, 6, 551–549. [Google Scholar]
- Bickle, M.B.; Dusserre, E.; Moncorge, O.; Bottin, H.; Colas, P. Selection and characterization of large collections of peptide aptamers through optimized yeast two-hybrid procedures. Nat. Protoc. 2006, 1, 1066–1091. [Google Scholar] [CrossRef]
- Nouvion, A.L.; Thibaut, J.; Lohez, O.D.; Venet, S.; Colas, P.; Gillet, G.; Lalle, P. Modulation of Nr-13 antideath activity by peptide aptamers. Oncogene 2007, 26, 701–710. [Google Scholar] [CrossRef]
- Rerole, A.L.; Gobbo, J.; de Thonel, A.; Schmitt, E.; Pais de Barros, J.P.; Hammann, A.; Lanneau, D.; Fourmaux, E.; Deminov, O.; Micheau, O.; et al. Peptides and aptamers targeting HSP70: A novel approach for anticancer chemotherapy. Cancer Res. 2011, 71, 484–495. [Google Scholar]
- Gibert, B.; Simon, S.; Dimitrova, V.; Diaz-Latoud, C.; Arrigo, A.P. Peptide aptamers: Tools to negatively or positively modulate HSPB1(27) function. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368, 20120075. [Google Scholar] [CrossRef]
- Rocchi, P.; Jugpal, P.; So, A.; Sinneman, S.; Ettinger, S.; Fazli, L.; Nelson, C.; Gleave, M. Small interference RNA targeting heat-shock protein 27 inhibits the growth of prostatic cell lines and induces apoptosis via caspase-3 activation in vitro. BJU Int. 2006, 28, 28. [Google Scholar]
- Morino, M.; Tsuzuki, T.; Ishikawa, Y.; Shirakami, T.; Yoshimura, M.; Kiyosuke, Y.; Matsunaga, K.; Yoshikumi, C.; Saijo, N. Specific regulation of HSPs in human tumor cell lines by flavonoids. In Vivo 1997, 11, 265–270. [Google Scholar]
- Tanaka, Y.; Fujiwara, K.; Tanaka, H.; Maehata, K.; Kohno, I. Paclitaxel inhibits expression of heat shock protein 27 in ovarian and uterine cancer cells. Int. J. Gynecol. Cancer 2004, 14, 616–620. [Google Scholar] [CrossRef]
- Taba, K.; Kuramitsu, Y.; Ryozawa, S.; Yoshida, K.; Tanaka, T.; Mori-Iwamoto, S.; Maehara, S.; Maehara, Y.; Sakaida, I.; Nakamura, K. KNK437 downregulates heat shock protein 27 of pancreatic cancer cells and enhances the cytotoxic effect of gemcitabine. Chemotherapy 2011, 57, 12–16. [Google Scholar]
- Oba, M.; Yano, S.; Shuto, T.; Suico, M.A.; Eguma, A.; Kai, H. IFN-gamma down-regulates Hsp27 and enhances hyperthermia-induced tumor cell death in vitro and tumor suppression in vivo. Int. J. Oncol. 2008, 32, 1317–1324. [Google Scholar]
- Kuramitsu, Y.; Wang, Y.; Taba, K.; Suenaga, S.; Ryozawa, S.; Kaino, S.; Sakaida, I.; Nakamura, K. Heat-shock protein 27 plays the key role in gemcitabine-resistance of pancreatic cancer cells. Anticancer Res. 2012, 32, 2295–2299. [Google Scholar]
- Shin, K.D.; Lee, M.Y.; Shin, D.S.; Lee, S.; Son, K.H.; Koh, S.; Paik, Y.K.; Kwon, B.M.; Han, D.C. Blocking tumor cell migration and invasion with biphenyl isoxazole derivative KRIBB3, a synthetic molecule that inhibits Hsp27 phosphorylation. J. Biol. Chem. 2005, 280, 41439–41448. [Google Scholar]
- Heinrich, J.C.; Tuukkanen, A.; Schroeder, M.; Fahrig, T.; Fahrig, R. RP101 (brivudine) binds to heat shock protein HSP27 (HSPB1) and enhances survival in animals and pancreatic cancer patients. J. Cancer Res. Clin. Oncol. 2011, 137, 1349–1361. [Google Scholar]
- Resprotect—Prevention of Chemoresistance. Available online: http://www.resprotect.de/Pipeline-Summary/Pipeline-Summary.html/ (Accessed on 16 November 2013).
- Faiella, L.; Piaz, F.D.; Bisio, A.; Tosco, A.; de Tommasi, N. A chemical proteomics approach reveals Hsp27 as a target for proapoptotic clerodane diterpenes. Mol. Biosyst. 2012, 8, 2637–2644. [Google Scholar] [CrossRef]
- Simon, S.; Michiel, M.; Skouri-Panet, F.; Lechaire, J.P.; Vicart, P.; Tardieu, A. Residue R120 is essential for the quaternary structure and functional integrity of human alphaB-crystallin. Biochemistry 2007, 46, 9605–9614. [Google Scholar] [CrossRef]
- Bagneris, C.; Bateman, O.A.; Naylor, C.E.; Cronin, N.; Boelens, W.C.; Keep, N.H.; Slingsby, C. Crystal structures of alpha-crystallin domain dimers of alphab-crystallin and hsp20. J. Mol. Biol. 2009, 392, 1242–1252. [Google Scholar] [CrossRef]
- Mymrikov, E.V.; Seit-Nebi, A.S.; Gusev, N.B. Large potentials of small heat shock proteins. Physiol. Rev. 2011, 91, 1123–1159. [Google Scholar] [CrossRef]
- Fragoso, M.A.; Patel, A.K.; Nakamura, R.E.; Yi, H.; Surapaneni, K.; Hackam, A.S. The Wnt/beta-catenin pathway cross-talks with STAT3 signaling to regulate survival of retinal pigment epithelium cells. PLoS One 2012, 7, e46892. [Google Scholar]
- Zantema, A.; Vries, M.V.-D.; Maasdam, D.; Bol, S.; Eb, A.V.D. Heat shock protein 27 and αB-cristallin can form a complex, which dissociates by heat shock. J. Biol. Chem. 1992, 267, 12936–12941. [Google Scholar]
- Mymrikov, E.V.; Seit-Nebi, A.S.; Gusev, N.B. Heterooligomeric complexes of human small heat shock proteins. Cell Stress Chaperones 2012, 17, 157–169. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Arrigo, A.-P.; Gibert, B. HspB1, HspB5 and HspB4 in Human Cancers: Potent Oncogenic Role of Some of Their Client Proteins. Cancers 2014, 6, 333-365. https://doi.org/10.3390/cancers6010333
Arrigo A-P, Gibert B. HspB1, HspB5 and HspB4 in Human Cancers: Potent Oncogenic Role of Some of Their Client Proteins. Cancers. 2014; 6(1):333-365. https://doi.org/10.3390/cancers6010333
Chicago/Turabian StyleArrigo, André-Patrick, and Benjamin Gibert. 2014. "HspB1, HspB5 and HspB4 in Human Cancers: Potent Oncogenic Role of Some of Their Client Proteins" Cancers 6, no. 1: 333-365. https://doi.org/10.3390/cancers6010333