STAT3 Activation in Glioblastoma: Biochemical and Therapeutic Implications

Abstract
:1. Introduction
2. STAT3 Signaling Pathways

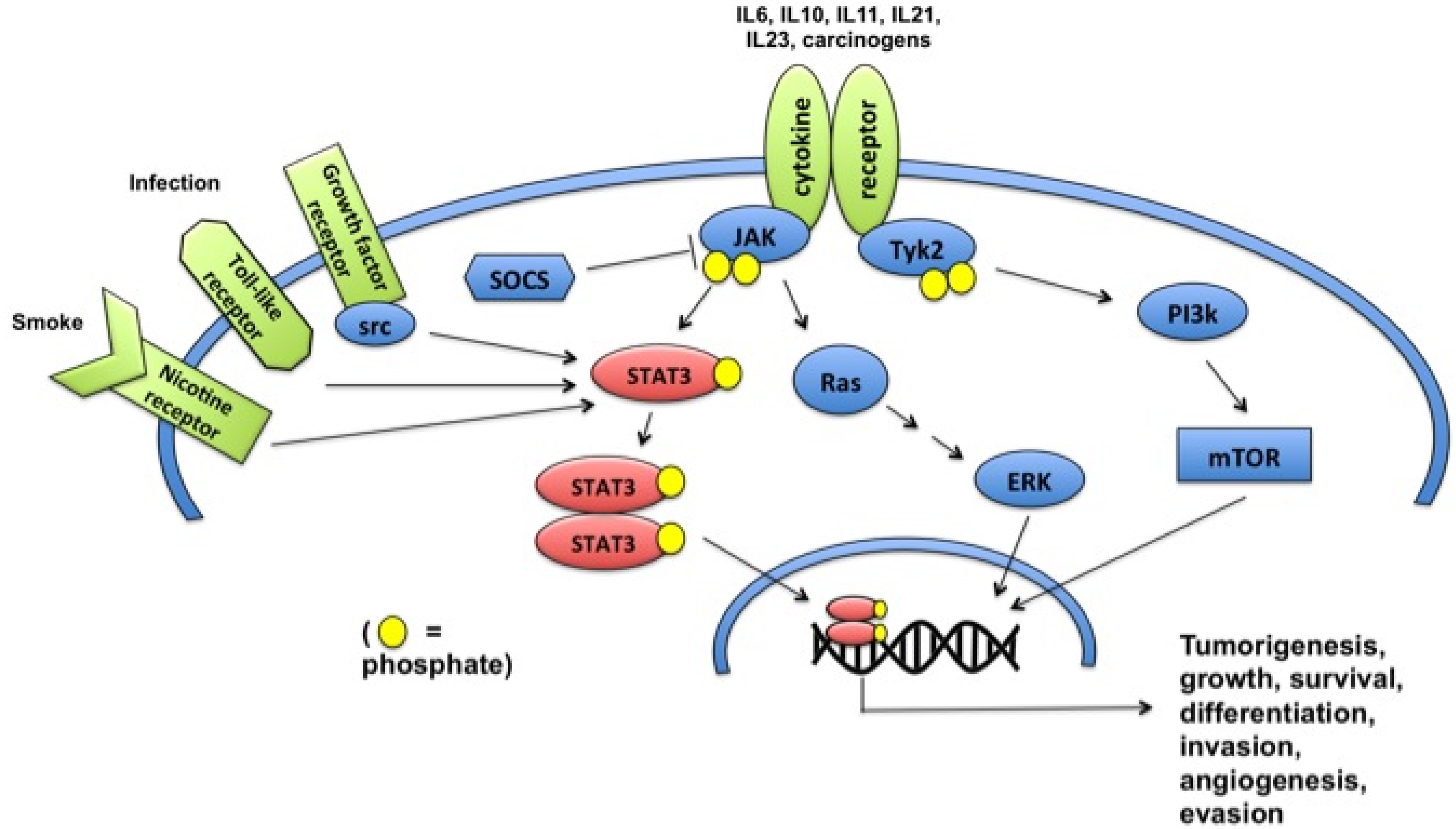{kind=link}
| Site of disruption | Potential mediators of disruption |
|---|---|
| Cell surface receptor-ligand interaction | Ligand/receptor antagonists, i.e., anti-EGF-R antibodies [5,40] |
| Tyrosine or serine kinase activity | TRK, EGF-R, FBGF-R, JAK, Src, PSK inhibitors [15,20,21,28] |
| Endogenous STAT3 activity or function | Biological protein inhibitors of STAT3 activity, i.e., SOCS, PIAS3, StIP1 modulators [36,37,41,42] |
| De-phosphorylation of phospho-STATs | Protein tyrosine or serine phosphatases [25,43] |
| STAT3 dimerization | Small molecule inhibitors of dimerization [43] |
| STAT3 nuclear translocation | Small molecule inhibitors of dimerization, inhibitors of nuclear endocytosis [43,44] |
| STAT3 transcription activation | Antisense or STAT3 decoy oligonucleotide sequences, dominant negative mutants [45,46,47] |
3. Constitutive Activation

4. Role of STAT3 in Tumorigenesis
4.1. Survival
4.2. Invasion
4.3. Angiogenesis
4.4. Immune Suppression and Evasion
5. STAT3 as a Tumor Suppressor
6. STAT3 as a Prognostic Indicator
7. STAT3 as a Therapeutic Target
| Drug | Mechanism of Action |
|---|---|
| Oleanolic acid | Suppresses IL-10 secretion which suppresses M2 polarization of tumor-associated macrophages [130] |
| LLL12 | Suppress phosphorylation of STAT3; inhibit STAT3 DNA binding [26,131] |
| LLL3 | |
| WP1193 | Inhibitor of JAK2/STAT3 pathway in glioma-like stem cells resultin gin G1 arrest [132] |
| RNAi | Downregulation of cyclin D1 in glioma cells [133] |
| Oligodeoxynucleotides | Induce cell cycle arrest and apoptosis by mimicking STAT3 specific cis elements [36] |
| AG490 | Inhibits JAK2, resulting in decreased activation of STAT3 and downstream decreased expression of MMP-2 and MMP-9 and inhibition of tumor cell invasiveness [84] |
8. Conclusions
Conflicts of Interest
References
- Ostrom, Q.T.; Gittleman, H.; Farah, P.; Ondracek, A.; Chen, Y.; Wolinsky, Y.; Stroup, N.E.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the united states in 2006–2010. Neuro-oncology 2013, 15, ii1–ii56. [Google Scholar]
- Stewart, L.A. Chemotherapy in adult high-grade glioma: A systematic review and meta-analysis of individual patient data from 12 randomised trials. Lancet 2002, 359, 1011–1018. [Google Scholar] [CrossRef]
- Rich, J.N.; Reardon, D.A.; Peery, T.; Dowell, J.M.; Quinn, J.A.; Penne, K.L.; Wikstrand, C.J.; van Duyn, L.B.; Dancey, J.E.; McLendon, R.E.; et al. Phase II trial of gefitinib in recurrent glioblastoma. J. Clin. Oncol. 2004, 22, 133–142. [Google Scholar]
- Taylor, T.E.; Furnari, F.B.; Cavenee, W.K. Targeting EGFR for treatment of glioblastoma: Molecular basis to overcome resistance. Curr. Cancer Drug Targets 2012, 12, 197–209. [Google Scholar] [CrossRef]
- Mellinghoff, I.K.; Wang, M.Y.; Vivanco, I.; Haas-Kogan, D.A.; Zhu, S.; Dia, E.Q.; Lu, K.V.; Yoshimoto, K.; Huang, J.H.; Chute, D.J.; et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N. Engl. J. Med. 2005, 353, 2012–2024. [Google Scholar] [CrossRef]
- Inda, M.M.; Bonavia, R.; Mukasa, A.; Narita, Y.; Sah, D.W.; Vandenberg, S.; Brennan, C.; Johns, T.G.; Bachoo, R.; Hadwiger, P.; et al. Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Genes Dev. 2010, 24, 1731–1745. [Google Scholar] [CrossRef]
- Fan, Q.W.; Knight, Z.A.; Goldenberg, D.D.; Yu, W.; Mostov, K.E.; Stokoe, D.; Shokat, K.M.; Weiss, W.A. A dual PI3 kinase/mTOR inhibitor reveals emergent efficacy in glioma. Cancer Cell 2006, 9, 341–349. [Google Scholar] [CrossRef]
- Hu, X.; Pandolfi, P.P.; Li, Y.; Koutcher, J.A.; Rosenblum, M.; Holland, E.C. mTOR promotes survival and astrocytic characteristics induced by pten/AKT signaling in glioblastoma. Neoplasia 2005, 7, 356–368. [Google Scholar] [CrossRef]
- Rubenstein, J.L.; Kim, J.; Ozawa, T.; Zhang, M.; Westphal, M.; Deen, D.F.; Shuman, M.A. Anti-VEGF antibody treatment of glioblastoma prolongs survival but results in increased vascular cooption. Neoplasia 2000, 2, 306–134. [Google Scholar]
- Zagzag, D.; Lukyanov, Y.; Lan, L.; Ali, M.A.; Esencay, M.; Mendez, O.; Yee, H.; Voura, E.B.; Newcomb, E.W. Hypoxia-inducible factor 1 and VEGF upregulate CXCR4 in glioblastoma: Implications for angiogenesis and glioma cell invasion. Lab. Investig. 2006, 86, 1221–1232. [Google Scholar] [CrossRef]
- Batchelor, T.T.; Sorensen, A.G.; di Tomaso, E.; Zhang, W.T.; Duda, D.G.; Cohen, K.S.; Kozak, K.R.; Cahill, D.P.; Chen, P.J.; Zhu, M.; et al. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell 2007, 11, 83–95. [Google Scholar] [CrossRef]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Wu, C.Y.; Wang, T.N.; Chen, Y.T.; Lin, K.C.; Chen, Y.A.; Li, H.T.; Tsai, P.L. Effects of constraint-induced therapy combined with eye patching on functional outcomes and movement kinematics in poststroke neglect. Am. J. Occup. Ther. 2013, 67, 236–245. [Google Scholar] [CrossRef]
- Kohsaka, S.; Wang, L.; Yachi, K.; Mahabir, R.; Narita, T.; Itoh, T.; Tanino, M.; Kimura, T.; Nishihara, H.; Tanaka, S. STAT3 inhibition overcomes temozolomide resistance in glioblastoma by downregulating MGMT expression. Mol. Cancer Ther. 2012, 11, 1289–1299. [Google Scholar] [CrossRef]
- Carro, M.S.; Lim, W.K.; Alvarez, M.J.; Bollo, R.J.; Zhao, X.; Snyder, E.Y.; Sulman, E.P.; Anne, S.L.; Doestch, F.; Colman, H.; et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature 2010, 463, 318–325. [Google Scholar] [CrossRef]
- Kortylewski, M.; Yu, H. Stat3 as a potential target for cancer immunotherapy. J. Immunother. 2007, 30, 131–139. [Google Scholar] [CrossRef]
- See, A.P.; Han, J.E.; Phallen, J.; Binder, Z.; Gallia, G.; Pan, F.; Jinasena, D.; Jackson, C.; Belcaid, Z.; Jeong, S.J.; et al. The role of STAT3 activation in modulating the immune microenvironment of GBM. J. Neurooncol. 2012, 110, 359–368. [Google Scholar] [CrossRef]
- Nefedova, Y.; Nagaraj, S.; Rosenbauer, A.; Muro-Cacho, C.; Sebti, S.M.; Gabrilovich, D.I. Regulation of dendritic cell differentiation and antitumor immune response in cancer by pharmacologic-selective inhibition of the janus-activated kinase 2/signal transducers and activators of transcription 3 pathway. Cancer Res. 2005, 65, 9525–9535. [Google Scholar] [CrossRef]
- Cheng, F.; Wang, H.W.; Cuenca, A.; Huang, M.; Ghansah, T.; Brayer, J.; Kerr, W.G.; Takeda, K.; Akira, S.; Schoenberger, S.P.; et al. A critical role for Stat3 signaling in immune tolerance. Immunity 2003, 19, 425–436. [Google Scholar]
- Bromberg, J.F.; Horvath, C.M.; Besser, D.; Lathem, W.W.; Darnell, J.E., Jr. Stat3 activation is required for cellular transformation by v-src. Mol. Cell. Biol. 1998, 18, 2553–2558. [Google Scholar]
- Yu, C.L.; Meyer, D.J.; Campbell, G.S.; Larner, A.C.; Carter-Su, C.; Schwartz, J.; Jove, R. Enhanced DNA-binding activity of a Stat3-related protein in cells transformed by the src oncoprotein. Science 1995, 269, 81–83. [Google Scholar]
- Bowman, T.; Broome, M.A.; Sinibaldi, D.; Wharton, W.; Pledger, W.J.; Sedivy, J.M.; Irby, R.; Yeatman, T.; Courtneidge, S.A.; Jove, R. Stat3-mediated myc expression is required for src transformation and PDGF-induced mitogenesis. Proc. Natl. Acad. Sci. USA 2001, 98, 7319–7324. [Google Scholar]
- Turkson, J.; Bowman, T.; Garcia, R.; Caldenhoven, E.; de Groot, R.P.; Jove, R. Stat3 activation by src induces specific gene regulation and is required for cell transformation. Mol. Cell. Biol. 1998, 18, 2545–2552. [Google Scholar]
- Wei, J.; Wu, A.; Kong, L.Y.; Wang, Y.; Fuller, G.; Fokt, I.; Melillo, G.; Priebe, W.; Heimberger, A.B. Hypoxia potentiates glioma-mediated immunosuppression. PLoS One 2011, 6, e16195. [Google Scholar]
- Ashizawa, T.; Miyata, H.; Iizuka, A.; Komiyama, M.; Oshita, C.; Kume, A.; Nogami, M.; Yagoto, M.; Ito, I.; Oishi, T.; et al. Effect of the STAT3 inhibitor STX-0119 on the proliferation of cancer stem-like cells derived from recurrent glioblastoma. Int. J. Oncol. 2013, 43, 219–227. [Google Scholar]
- Fuh, B.; Sobo, M.; Cen, L.; Josiah, D.; Hutzen, B.; Cisek, K.; Bhasin, D.; Regan, N.; Lin, L.; Chan, C.; et al. LLL-3 inhibits STAT3 activity, suppresses glioblastoma cell growth and prolongs survival in a mouse glioblastoma model. Br. J. Cancer 2009, 100, 106–112. [Google Scholar] [CrossRef]
- Gao, L.; Li, F.; Dong, B.; Zhang, J.; Rao, Y.; Cong, Y.; Mao, B.; Chen, X. Inhibition of STAT3 and ErbB2 suppresses tumor growth, enhances radiosensitivity, and induces mitochondria-dependent apoptosis in glioma cells. Int. J. Radiat. Oncol. Biol. Phys. 2010, 77, 1223–1231. [Google Scholar] [CrossRef]
- Li, G.H.; Wei, H.; Lv, S.Q.; Ji, H.; Wang, D.L. Knockdown of STAT3 expression by RNAi suppresses growth and induces apoptosis and differentiation in glioblastoma stem cells. Int. J. Oncol. 2010, 37, 103–110. [Google Scholar]
- Zhong, Z.; Wen, Z.; Darnell, J.E., Jr. Stat3: A STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 1994, 264, 95–98. [Google Scholar]
- Bromberg, J.; Darnell, J.E., Jr. The role of STATs in transcriptional control and their impact on cellular function. Oncogene 2000, 19, 2468–2473. [Google Scholar] [CrossRef]
- Levy, D.E.; Darnell, J.E., Jr. Stats: Transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 2002, 3, 651–662. [Google Scholar] [CrossRef]
- Takeda, K.; Clausen, B.E.; Kaisho, T.; Tsujimura, T.; Terada, N.; Forster, I.; Akira, S. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity 1999, 10, 39–49. [Google Scholar] [CrossRef]
- Akira, S. Roles of STAT3 defined by tissue-specific gene targeting. Oncogene 2000, 19, 2607–2611. [Google Scholar]
- Yamamoto, T.; Sekine, Y.; Kashima, K.; Kubota, A.; Sato, N.; Aoki, N.; Matsuda, T. The nuclear isoform of protein-tyrosine phosphatase TC-PTP regulates interleukin-6-mediated signaling pathway through STAT3 dephosphorylation. Biochem. Biophys. Res. Commun. 2002, 297, 811–817. [Google Scholar] [CrossRef]
- Zhang, X.; Guo, A.; Yu, J.; Possemato, A.; Chen, Y.; Zheng, W.; Polakiewicz, R.D.; Kinzler, K.W.; Vogelstein, B.; Velculescu, V.E.; et al. Identification of STAT3 as a substrate of receptor protein tyrosine phosphatase T. Proc. Natl. Acad. Sci. USA 2007, 104, 4060–4064. [Google Scholar] [CrossRef]
- Yoshikawa, H.; Matsubara, K.; Qian, G.S.; Jackson, P.; Groopman, J.D.; Manning, J.E.; Harris, C.C.; Herman, J.G. SOCS-1, a negative regulator of the JAK/STAT pathway, is silenced by methylation in human hepatocellular carcinoma and shows growth-suppression activity. Nat. Genet. 2001, 28, 29–35. [Google Scholar]
- Shuai, K. Modulation of STAT signaling by STAT-interacting proteins. Oncogene 2000, 19, 2638–2644. [Google Scholar] [CrossRef]
- Collum, R.G.; Brutsaert, S.; Lee, G.; Schindler, C. A Stat3-interacting protein (StIP1) regulates cytokine signal transduction. Proc. Natl. Acad. Sci. USA 2000, 97, 10120–10125. [Google Scholar] [CrossRef]
- Jackson, C.; Ruzevick, J.; Amin, A.G.; Lim, M. Potential role for STAT3 inhibitors in glioblastoma. Neurosurg. Clin. N. Am. 2012, 23, 379–389. [Google Scholar] [CrossRef]
- Lindemann, C.; Hackmann, O.; Delic, S.; Schmidt, N.; Reifenberger, G.; Riemenschneider, M.J. SOCS3 promoter methylation is mutually exclusive to EGFR amplification in gliomas and promotes glioma cell invasion through STAT3 and FAK activation. Acta Neuropathol. 2011, 122, 241–251. [Google Scholar] [CrossRef]
- Lo, H.W.; Cao, X.; Zhu, H.; Ali-Osman, F. Constitutively activated STAT3 frequently coexpresses with epidermal growth factor receptor in high-grade gliomas and targeting STAT3 sensitizes them to iressa and alkylators. Clin. Cancer Res. 2008, 14, 6042–6054. [Google Scholar]
- Liu, J.; Xu, X.; Feng, X.; Zhang, B.; Wang, J. Adenovirus-mediated delivery of bFGF small interfering RNA reduces STAT3 phosphorylation and induces the depolarization of mitochondria and apoptosis in glioma cells U251. J. Exp. Clin. Cancer Res. 2011, 30, 80. [Google Scholar] [CrossRef]
- Schust, J.; Sperl, B.; Hollis, A.; Mayer, T.U.; Berg, T. Stattic: A small-molecule inhibitor of STAT3 activation and dimerization. Chem. Biol. 2006, 13, 1235–1242. [Google Scholar]
- Bild, A.H.; Turkson, J.; Jove, R. Cytoplasmic transport of STAT3 by receptor mediated endocytosis. EMBO J. 2002, 21, 3255–3273. [Google Scholar] [CrossRef]
- Gu, J.; Li, G.; Sun, T.; Su, Y.; Zhang, X.; Shen, J.; Tian, Z.; Zhang, J. Blockage of the STAT3 signaling pathway with a decoy oligonucleotide suppresses growth of human malignant glioma cells. J. Neurooncol. 2008, 89, 9–17. [Google Scholar] [CrossRef]
- Leong, P.L.; Andrews, G.A.; Johnson, D.E.; Dyer, K.F.; Xi, S.; Mai, J.C.; Robbins, P.D.; Gadiparthi, S.; Burke, N.A.; Watkins, S.F.; et al. Targeted inhibition of Stat3 with a decoy oligonucleotide abrogates head and neck cancer cell growth. Proc. Natl. Acad. Sci. USA 2003, 100, 4138–4143. [Google Scholar] [CrossRef]
- Sen, M.; Grandis, J.R. Nucleic acid-based approaches to STAT inhibition. JAKSTAT 2012, 1, 285–291. [Google Scholar]
- Buettner, R.; Mora, L.B.; Jove, R. Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clin. Cancer Res. 2002, 8, 945–954. [Google Scholar]
- Yu, H.; Jove, R. The STATs of cancer—New molecular targets come of age. Nat. Rev. Cancer 2004, 4, 97–105. [Google Scholar]
- Hodge, D.R.; Hurt, E.M.; Farrar, W.L. The role of IL-6 and STAT3 in inflammation and cancer. Eur. J. Cancer 2005, 41, 2502–2512. [Google Scholar] [CrossRef]
- He, B.; You, L.; Uematsu, K.; Zang, K.; Xu, Z.; Lee, A.Y.; Costello, J.F.; McCormick, F.; Jablons, D.M. SOCS-3 is frequently silenced by hypermethylation and suppresses cell growth in human lung cancer. Proc. Natl. Acad. Sci. USA 2003, 100, 14133–14138. [Google Scholar] [CrossRef]
- Catlett-Falcone, R.; Landowski, T.H.; Oshiro, M.M.; Turkson, J.; Levitzki, A.; Savino, R.; Ciliberto, G.; Moscinski, L.; Fernandez-Luna, J.L.; Nunez, G.; et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity 1999, 10, 105–115. [Google Scholar] [CrossRef]
- Grandis, J.R.; Drenning, S.D.; Zeng, Q.; Watkins, S.C.; Melhem, M.F.; Endo, S.; Johnson, D.E.; Huang, L.; He, Y.; Kim, J.D. Constitutive activation of Stat3 signaling abrogates apoptosis in squamous cell carcinogenesis in vivo. Proc. Natl. Acad. Sci. USA 2000, 97, 4227–4232. [Google Scholar] [CrossRef]
- Luwor, R.B.; Stylli, S.S.; Kaye, A.H. The role of Stat3 in glioblastoma multiforme. J. Clin. Neurosci. 2013, 20, 907–911. [Google Scholar] [CrossRef]
- Mizoguchi, M.; Betensky, R.A.; Batchelor, T.T.; Bernay, D.C.; Louis, D.N.; Nutt, C.L. Activation of STAT3, MAPK, and AKT in malignant astrocytic gliomas: Correlation with EGFR status, tumor grade, and survival. J. Neuropathol. Exp. Neurol. 2006, 65, 1181–1188. [Google Scholar] [CrossRef]
- Abou-Ghazal, M.; Yang, D.S.; Qiao, W.; Reina-Ortiz, C.; Wei, J.; Kong, L.Y.; Fuller, G.N.; Hiraoka, N.; Priebe, W.; Sawaya, R.; et al. The incidence, correlation with tumor-infiltrating inflammation, and prognosis of phosphorylated STAT3 expression in human gliomas. Clin. Cancer Res. 2008, 14, 8228–8235. [Google Scholar] [CrossRef]
- Brantley, E.C.; Nabors, L.B.; Gillespie, G.Y.; Choi, Y.H.; Palmer, C.A.; Harrison, K.; Roarty, K.; Benveniste, E.N. Loss of protein inhibitors of activated STAT-3 expression in glioblastoma multiforme tumors: Implications for STAT-3 activation and gene expression. Clin. Cancer Res. 2008, 14, 4694–4704. [Google Scholar] [CrossRef]
- Schaefer, L.K.; Ren, Z.; Fuller, G.N.; Schaefer, T.S. Constitutive activation of Stat3alpha in brain tumors: Localization to tumor endothelial cells and activation by the endothelial tyrosine kinase receptor (VEGFR-2). Oncogene 2002, 21, 2058–2065. [Google Scholar] [CrossRef]
- Weissenberger, J.; Loeffler, S.; Kappeler, A.; Kopf, M.; Lukes, A.; Afanasieva, T.A.; Aguzzi, A.; Weis, J. IL-6 is required for glioma development in a mouse model. Oncogene 2004, 23, 3308–3316. [Google Scholar] [CrossRef]
- Wang, H.; Wang, H.; Zhang, W.; Huang, H.J.; Liao, W.S.; Fuller, G.N. Analysis of the activation status of akt, NFkappaB, and Stat3 in human diffuse gliomas. Lab. Investig. 2004, 84, 941–951. [Google Scholar] [CrossRef]
- Rahaman, S.O.; Harbor, P.C.; Chernova, O.; Barnett, G.H.; Vogelbaum, M.A.; Haque, S.J. Inhibition of constitutively active Stat3 suppresses proliferation and induces apoptosis in glioblastoma multiforme cells. Oncogene 2002, 21, 8404–8413. [Google Scholar] [CrossRef]
- Rolhion, C.; Penault-Llorca, F.; Kemeny, J.L.; Lemaire, J.J.; Jullien, C.; Labit-Bouvier, C.; Finat-Duclos, F.; Verrelle, P. Interleukin-6 overexpression as a marker of malignancy in human gliomas. J. Neurosurg. 2001, 94, 97–101. [Google Scholar] [CrossRef]
- Chang, C.Y.; Li, M.C.; Liao, S.L.; Huang, Y.L.; Shen, C.C.; Pan, H.C. Prognostic and clinical implication of IL-6 expression in glioblastoma multiforme. J. Clin. Neurosci. 2005, 12, 930–933. [Google Scholar] [CrossRef]
- Sasaki, A.; Ishiuchi, S.; Kanda, T.; Hasegawa, M.; Nakazato, Y. Analysis of interleukin-6 gene expression in primary human gliomas, glioblastoma xenografts, and glioblastoma cell lines. Brain Tumor Pathol. 2001, 18, 13–21. [Google Scholar] [CrossRef]
- Tchirkov, A.; Rolhion, C.; Bertrand, S.; Dore, J.F.; Dubost, J.J.; Verrelle, P. IL-6 gene amplification and expression in human glioblastomas. Br. J. Cancer 2001, 85, 518–522. [Google Scholar] [CrossRef]
- Lee, J.C.; Vivanco, I.; Beroukhim, R.; Huang, J.H.; Feng, W.L.; DeBiasi, R.M.; Yoshimoto, K.; King, J.C.; Nghiemphu, P.; Yuza, Y.; et al. Epidermal growth factor receptor activation in glioblastoma through novel missense mutations in the extracellular domain. PLoS Med. 2006, 3, e485. [Google Scholar]
- Ghosh, M.K.; Sharma, P.; Harbor, P.C.; Rahaman, S.O.; Haque, S.J. PI3K-AKT pathway negatively controls EGFR-dependent DNA-binding activity of Stat3 in glioblastoma multiforme cells. Oncogene 2005, 24, 7290–7300. [Google Scholar] [CrossRef]
- Hatanpaa, K.J.; Burma, S.; Zhao, D.; Habib, A.A. Epidermal growth factor receptor in glioma: Signal transduction, neuropathology, imaging, and radioresistance. Neoplasia 2010, 12, 675–684. [Google Scholar]
- Guryanova, O.A.; Wu, Q.; Cheng, L.; Lathia, J.D.; Huang, Z.; Yang, J.; MacSwords, J.; Eyler, C.E.; McLendon, R.E.; Heddleston, J.M.; et al. Nonreceptor tyrosine kinase BMX maintains self-renewal and tumorigenic potential of glioblastoma stem cells by activating STAT3. Cancer Cell 2011, 19, 498–511. [Google Scholar] [CrossRef]
- Solomon, D.A.; Kim, J.S.; Cronin, J.C.; Sibenaller, Z.; Ryken, T.; Rosenberg, S.A.; Ressom, H.; Jean, W.; Bigner, D.; Yan, H.; et al. Mutational inactivation of PTPRD in glioblastoma multiforme and malignant melanoma. Cancer Res. 2008, 68, 10300–10306. [Google Scholar] [CrossRef]
- Veeriah, S.; Brennan, C.; Meng, S.; Singh, B.; Fagin, J.A.; Solit, D.B.; Paty, P.B.; Rohle, D.; Vivanco, I.; Chmielecki, J.; et al. The tyrosine phosphatase PTPRD is a tumor suppressor that is frequently inactivated and mutated in glioblastoma and other human cancers. Proc. Natl. Acad. Sci. USA 2009, 106, 9435–9440. [Google Scholar] [CrossRef]
- Yuan, Z.L.; Guan, Y.J.; Chatterjee, D.; Chin, Y.E. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science 2005, 307, 269–273. [Google Scholar] [CrossRef]
- Hirano, T.; Ishihara, K.; Hibi, M. Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene 2000, 19, 2548–2556. [Google Scholar] [CrossRef]
- Chen, F.; Xu, Y.; Luo, Y.; Zheng, D.; Song, Y.; Yu, K.; Li, H.; Zhang, L.; Zhong, W.; Ji, Y. Down-regulation of Stat3 decreases invasion activity and induces apoptosis of human glioma cells. J. Mol. Neurosci. 2010, 40, 353–359. [Google Scholar] [CrossRef]
- Epling-Burnette, P.K.; Liu, J.H.; Catlett-Falcone, R.; Turkson, J.; Oshiro, M.; Kothapalli, R.; Li, Y.; Wang, J.M.; Yang-Yen, H.F.; Karras, J.; et al. Inhibition of STAT3 signaling leads to apoptosis of leukemic large granular lymphocytes and decreased mcl-1 expression. J. Clin. Investig. 2001, 107, 351–362. [Google Scholar] [CrossRef]
- Aoki, Y.; Feldman, G.M.; Tosato, G. Inhibition of STAT3 signaling induces apoptosis and decreases survivin expression in primary effusion lymphoma. Blood 2003, 101, 1535–1542. [Google Scholar] [CrossRef]
- Iwamaru, A.; Szymanski, S.; Iwado, E.; Aoki, H.; Yokoyama, T.; Fokt, I.; Hess, K.; Conrad, C.; Madden, T.; Sawaya, R.; et al. A novel inhibitor of the STAT3 pathway induces apoptosis in malignant glioma cells both in vitro and in vivo. Oncogene 2007, 26, 2435–2444. [Google Scholar] [CrossRef]
- Ren, Y.; Zhou, X.; Mei, M.; Yuan, X.B.; Han, L.; Wang, G.X.; Jia, Z.F.; Xu, P.; Pu, P.Y.; Kang, C.S. MicroRNA-21 inhibitor sensitizes human glioblastoma cells U251 (PTEN-mutant) and LN229 (PTEN-wild type) to taxol. BMC Cancer 2010, 10, 27. [Google Scholar] [CrossRef]
- Yang, I.; Han, S.J.; Kaur, G.; Crane, C.; Parsa, A.T. The role of microglia in central nervous system immunity and glioma immunology. J. Clin. Neurosci. 2010, 17, 6–10. [Google Scholar] [CrossRef]
- Stechishin, O.D.; Luchman, H.A.; Ruan, Y.; Blough, M.D.; Nguyen, S.A.; Kelly, J.J.; Cairncross, J.G.; Weiss, S. On-target JAK2/STAT3 inhibition slows disease progression in orthotopic xenografts of human glioblastoma brain tumor stem cells. Neuro-oncology 2013, 15, 198–207. [Google Scholar] [CrossRef]
- Sherry, M.M.; Reeves, A.; Wu, J.K.; Cochran, B.H. STAT3 is required for proliferation and maintenance of multipotency in glioblastoma stem cells. Stem Cells 2009, 27, 2383–2392. [Google Scholar] [CrossRef]
- Birner, P.; Toumangelova-Uzeir, K.; Natchev, S.; Guentchev, M. STAT3 tyrosine phosphorylation influences survival in glioblastoma. J. Neurooncol. 2010, 100, 339–343. [Google Scholar] [CrossRef]
- Claes, A.; Idema, A.J.; Wesseling, P. Diffuse glioma growth: A guerilla war. Acta Neuropathol. 2007, 114, 443–458. [Google Scholar] [CrossRef]
- Senft, C.; Priester, M.; Polacin, M.; Schroderm, K.; Seifertm, V.; Kogelm, D.; Weissenbergerm, J. Inhibition of the JAK-2/STAT3 signaling pathway impedes the migratory and invasive potential of human glioblastoma cells. J. Neurooncol. 2011, 101, 393–403. [Google Scholar] [CrossRef]
- Liu, Q.; Li, G.; Li, R.; Shen, J.; He, Q.; Deng, L.; Zhang, C.; Zhang, J. IL-6 promotion of glioblastoma cell invasion and angiogenesis in U251 and T98G cell lines. J. Neurooncol. 2010, 100, 165–176. [Google Scholar] [CrossRef]
- Tabernero, J. The role of VEGF and EGFR inhibition: Implications for combining anti-VEGF and anti-EGFR agents. Mol. Cancer Res. 2007, 5, 203–220. [Google Scholar] [CrossRef]
- Eliceiri, B.P.; Paul, R.; Schwartzberg, P.L.; Hood, J.D.; Leng, J.; Cheresh, D.A. Selective requirement for src kinases during VEGF-induced angiogenesis and vascular permeability. Mol. Cell 1999, 4, 915–924. [Google Scholar] [CrossRef]
- Dankbar, B.; Padro, T.; Leo, R.; Feldmann, B.; Kropff, M.; Mesters, R.M.; Serve, H.; Berdel, W.E.; Kienast, J. Vascular endothelial growth factor and interleukin-6 in paracrine tumor-stromal cell interactions in multiple myeloma. Blood 2000, 95, 2630–2636. [Google Scholar]
- Niu, G.; Wright, K.L.; Huang, M.; Song, L.; Haura, E.; Turkson, J.; Zhang, S.; Wang, T.; Sinibaldi, D.; Coppola, D.; et al. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene 2002, 21, 2000–2008. [Google Scholar] [CrossRef]
- Maussang, D.; Verzijl, D.; van Walsum, M.; Leurs, R.; Holl, J.; Pleskoff, O.; Michel, D.; van Dongen, G.A.; Smit, M.J. Human cytomegalovirus-encoded chemokine receptor US28 promotes tumorigenesis. Proc. Natl. Acad. Sci. 2006, 103, 13068–13073. [Google Scholar] [CrossRef]
- Soroceanu, L.; Matlaf, L.; Bezrookove, V.; Harkins, L.; Martinez, R.; Greene, M.; Soteropoulos, P.; Cobbs, C.S. Human cytomegalovirus US28 found in glioblastoma promotes an invasive and angiogenic phenotype. Cancer Res. 2011, 71, 6643–6653. [Google Scholar] [CrossRef]
- Plate, K.H. Mechanisms of angiogenesis in the brain. J. Neuropathol. Exp. Neurol. 1999, 58, 313–320. [Google Scholar] [CrossRef]
- Keunen, O.; Johansson, M.; Oudin, A.; Sanzey, M.; Rahim, S.A.; Fack, F.; Thorsen, F.; Taxt, T.; Bartos, M.; Jirik, R.; et al. Anti-VEGF treatment reduces blood supply and increases tumor cell invasion in glioblastoma. Proc. Natl. Acad. Sci. USA 2011, 108, 3749–3754. [Google Scholar] [CrossRef]
- De Groot, J.; Liang, J.; Kong, L.Y.; Wei, J.; Piao, Y.; Fuller, G.; Qiao, W.; Heimberger, A.B. Modulating antiangiogenic resistance by inhibiting the signal transducer and activator of transcription 3 pathway in glioblastoma. Oncotarget 2012, 3, 1036–1048. [Google Scholar]
- Albesiano, E.; Han, J.E.; Lim, M. Mechanisms of local immunoresistance in glioma. Neurosurg. Clin. N. Am. 2010, 21, 17–29. [Google Scholar] [CrossRef]
- Avril, T.; Saikali, S.; Vauleon, E.; Jary, A.; Hamlat, A.; de Tayrac, M.; Mosser, J.; Quillien, V. Distinct effects of human glioblastoma immunoregulatory molecules programmed cell death ligand-1 (PDL-1) and indoleamine 2,3-dioxygenase (IDO) on tumour-specific T cell functions. J. Neuroimmunol. 2010, 225, 22–33. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I.; Chen, H.L.; Girgis, K.R.; Cunningham, H.T.; Meny, G.M.; Nadaf, S.; Kavanaugh, D.; Carbone, D.P. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat. Med. 1996, 2, 1096–1103. [Google Scholar] [CrossRef]
- Wang, T.; Niu, G.; Kortylewski, M.; Burdelya, L.; Shain, K.; Zhang, S.; Bhattacharya, R.; Gabrilovich, D.; Heller, R.; Coppola, D.; et al. Regulation of the innate and adaptive immune responses by stat-3 signaling in tumor cells. Nat. Med. 2004, 10, 48–54. [Google Scholar] [CrossRef]
- Zhang, L.; Alizadeh, D.; van Handel, M.; Kortylewski, M.; Yu, H.; Badie, B. Stat3 inhibition activates tumor macrophages and abrogates glioma growth in mice. Glia 2009, 57, 1458–1467. [Google Scholar] [CrossRef]
- Kostianovsky, A.M.; Maier, L.M.; Anderson, R.C.; Bruce, J.N.; Anderson, D.E. Astrocytic regulation of human monocytic/microglial activation. J. Immunol. 2008, 181, 5425–5432. [Google Scholar]
- Hussain, S.F.; Yang, D.; Suki, D.; Aldape, K.; Grimm, E.; Heimberger, A.B. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro-oncology 2006, 8, 261–279. [Google Scholar] [CrossRef]
- Komohara, Y.; Horlad, H.; Ohnishi, K.; Fujiwara, Y.; Bai, B.; Nakagawa, T.; Suzu, S.; Nakamura, H.; Kuratsu, J.; Takeya, M. Importance of direct macrophage-tumor cell interaction on progression of human glioma. Cancer Sci. 2012, 103, 2165–2172. [Google Scholar] [CrossRef]
- Zou, W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat. Rev. Cancer 2005, 5, 263–274. [Google Scholar] [CrossRef]
- Nishikawa, H.; Kato, T.; Tawara, I.; Ikeda, H.; Kuribayashi, K.; Allen, P.M.; Schreiber, R.D.; Old, L.J.; Shiku, H. IFN-gamma controls the generation/activation of CD4+ CD25+ regulatory T cells in antitumor immune response. J. Immunol. 2005, 175, 4433–4440. [Google Scholar]
- Jacobs, J.F.; Idema, A.J.; Bol, K.F.; Grotenhuis, J.A.; de Vries, I.J.; Wesseling, P.; Adema, G.J. Prognostic significance and mechanism of treg infiltration in human brain tumors. J. Neuroimmunol. 2010, 225, 195–199. [Google Scholar] [CrossRef]
- Wei, J.; Barr, J.; Kong, LY.; Wang, Y.; Wu, A.; Sharma, A.K.; Gumin, J.; Henry, V.; Colman, H.; Priebe, W.; et al. Glioblastoma cancer-initiating cells inhibit T-cell proliferation and effector responses by the signal transducers and activators of transcription 3 pathway. Mol. Cancer Ther. 2010, 9, 67–78. [Google Scholar]
- Kortylewski, M.; Kujawski, M.; Wang, T.; Wei, S.; Zhang, S.; Pilon-Thomas, S.; Niu, G.; Kay, H.; Mule, J.; Kerr, W.G.; et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat. Med. 2005, 11, 1314–1321. [Google Scholar] [CrossRef]
- Spiotto, M.T.; Chung, T.D. STAT3 mediates IL-6-induced neuroendocrine differentiation in prostate cancer cells. Prostate 2000, 42, 186–195. [Google Scholar] [CrossRef]
- Spiotto, M.T.; Chung, T.D. STAT3 mediates IL-6-induced growth inhibition in the human prostate cancer cell line LNCaP. Prostate 2000, 42, 88–98. [Google Scholar] [CrossRef]
- Kortylewski, M.; Heinrich, P.C.; Mackiewicz, A.; Schniertshauer, U.; Klingmuller, U.; Nakajima, K.; Hirano, T.; Horn, F.; Behrmann, I. Interleukin-6 and oncostatin M-induced growth inhibition of human A375 melanoma cells is STAT-dependent and involves upregulation of the cyclin-dependent kinase inhibitor p27/Kip1. Oncogene 1999, 18, 3742–3753. [Google Scholar] [CrossRef]
- McLemore, M.L.; Grewal, S.; Liu, F.; Archambault, A.; Poursine-Laurent, J.; Haug, J.; Link, D.C. STAT-3 activation is required for normal G-CSF-dependent proliferation and granulocytic differentiation. Immunity 2001, 14, 193–204. [Google Scholar] [CrossRef]
- Tian, S.S.; Lamb, P.; Seidel, H.M.; Stein, R.B.; Rosen, J. Rapid activation of the STAT3 transcription factor by granulocyte colony-stimulating factor. Blood 1994, 84, 1760–1764. [Google Scholar]
- De Koning, J.P.; Soede-Bobok, A.A.; Ward, A.C.; Schelen, A.M.; Antonissen, C.; van Leeuwen, D.; Lowenberg, B.; Touw, I.P. STAT3-mediated differentiation and survival and of myeloid cells in response to granulocyte colony-stimulating factor: Role for the cyclin-dependent kinase inhibitor p27(Kip1). Oncogene 2000, 19, 3290–3298. [Google Scholar] [CrossRef]
- Wu, R.; Sun, S.; Steinberg, B.M. Requirement of STAT3 activation for differentiation of mucosal stratified squamous epithelium. Mol. Med. 2003, 9, 77–84. [Google Scholar] [CrossRef]
- Philp, J.A.; Burdon, T.G.; Watson, C.J. Differential activation of STATs 3 and 5 during mammary gland development. FEBS Lett. 1996, 396, 77–80. [Google Scholar] [CrossRef]
- Hauser, P.J.; Agrawal, D.; Hackney, J.; Pledger, W.J. STAT3 activation accompanies keratinocyte differentiation. Cell Growth Differ. 1998, 9, 847–855. [Google Scholar]
- Boccaccio, C.; Ando, M.; Tamagnone, L.; Bardelli, A.; Michieli, P.; Battistini, C.; Comoglio, P.M. Induction of epithelial tubules by growth factor HGF depends on the STAT pathway. Nature 1998, 391, 285–288. [Google Scholar] [CrossRef]
- De la Iglesia, N.; Konopka, G.; Lim, K.L.; Nutt, C.L.; Bromberg, J.F.; Frank, D.A.; Mischel, P.S.; Louis, D.N.; Bonni, A. Deregulation of a STAT3-interleukin 8 signaling pathway promotes human glioblastoma cell proliferation and invasiveness. J. Neurosci. 2008, 28, 5870–5878. [Google Scholar] [CrossRef]
- De la Iglesia, N.; Puram, S.V.; Bonni, A. STAT3 regulation of glioblastoma pathogenesis. Curr. Mol. Med. 2009, 9, 580–590. [Google Scholar] [CrossRef]
- Bonni, A.; Sun, Y.; Nadal-Vicens, M.; Bhatt, A.; Frank, D.A.; Rozovsky, I.; Stahl, N.; Yancopoulos, G.D.; Greenberg, M.E. Regulation of gliogenesis in the central nervous system by the JAK-STAT signaling pathway. Science 1997, 278, 477–483. [Google Scholar] [CrossRef]
- Rajan, P.; McKay, R.D. Multiple routes to astrocytic differentiation in the CNS. J. Neurosci. 1999, 18, 3620–3629. [Google Scholar]
- Louis, D.N. Molecular pathology of malignant gliomas. Annu. Rev. Pathol. 2006, 1, 97–117. [Google Scholar] [CrossRef]
- Bachoo, R.M.; Maher, E.A.; Ligon, K.L.; Sharpless, N.E.; Chan, S.S.; You, M.J.; Tang, Y.; DeFrances, J.; Stover, E.; Weissleder, R.; et al. Epidermal growth factor receptor and Ink4a/arf: Convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell 2002, 1, 269–277. [Google Scholar] [CrossRef]
- Uhrbom, L.; Dai, C.; Celestino, J.C.; Rosenblum, M.K.; Fuller, G.N.; Holland, E.C. Ink4a-arf loss cooperates with KRas activation in astrocytes and neural progenitors to generate glioblastomas of various morphologies depending on activated akt. Cancer Res. 2002, 62, 5551–5558. [Google Scholar]
- Jin, C.; Wang, A.; Chen, J.; Liu, X.; Wang, G. Relationship between expression and prognostic ability of PTEN, STAT3 and VEGF-C in colorectal cancer. Exp. Ther. Med. 2012, 4, 633–639. [Google Scholar]
- Gordziel, C.; Bratsch, J.; Moriggl, R.; Knosel, T.; Friedrich, K. Both STAT1 and STAT3 are favourable prognostic determinants in colorectal carcinoma. Br. J. Cancer 2013, 109, 138–146. [Google Scholar] [CrossRef]
- Chakravarti, A.; Noll, E.; Black, P.M.; Finkelstein, D.F.; Finkelstein, D.M.; Dyson, N.J.; Loeffler, J.S. Quantitatively determined survivin expression levels are of prognostic value in human gliomas. J. Clin. Oncol. 2002, 20, 1063–1068. [Google Scholar] [CrossRef]
- Kajiwara, Y.; Yamasaki, F.; Hama, S.; Yahara, K.; Yoshioka, H.; Sugiyama, K.; Arita, K.; Kurisu, K. Expression of survivin in astrocytic tumors: Correlation with malignant grade and prognosis. Cancer 2003, 97, 1077–1083. [Google Scholar] [CrossRef]
- Tu, Y.; Zhong, Y.; Fu, J.; Cao, Y.; Fu, G.; Tian, X.; Wang, B. Activation of JAK/STAT signal pathway predicts poor prognosis of patients with gliomas. Med. Oncol. 2011, 28, 15–23. [Google Scholar]
- Fujiwara, Y.; Komohara, Y.; Kudo, R.; Tsurushima, K.; Ohnishi, K.; Ikeda, T.; Takeya, M. Oleanolic acid inhibits macrophage differentiation into the M2 phenotype and glioblastoma cell proliferation by suppressing the activation of STAT3. Oncol. Rep. 2011, 26, 1533–1537. [Google Scholar]
- Ball, S.; Li, C.; Li, P.K.; Lin, J. The small molecule, LLL12, inhibits STAT3 phosphorylation and induces apoptosis in medulloblastoma and glioblastoma cells. PLoS One 2011, 6, e18820. [Google Scholar] [CrossRef]
- Sai, K.; Wang, S.; Balasubramaniyan, V.; Conrad, C.; Lang, F.F.; Aldape, K.; Szymanski, S.; Fokt, I.; Dasgupta, A.; Madden, T.; et al. Induction of cell-cycle arrest and apoptosis in glioblastoma stem-like cells by WP1193, a novel small molecule inhibitor of the JAK2/STAT3 pathway. J. Neurooncol. 2012, 107, 487–501. [Google Scholar] [CrossRef]
- Li, G.H.; Wei, H.; Chen, Z.T.; Lv, S.Q.; Yin, C.L.; Wang, D.L. STAT3 silencing with lentivirus inhibits growth and induces apoptosis and differentiation of U251 cells. J. Neurooncol. 2009, 91, 165–174. [Google Scholar] [CrossRef]
- Harris, T.J.; Grosso, J.F.; Yen, H.R.; Xin, H.; Kortylewski, M.; Albesiano, E.; Hipkiss, E.L.; Getnet, D.; Goldberg, M.V.; Maris, C.H.; et al. Cutting edge: An in vivo requirement for STAT3 signaling in TH17 development and TH17-dependent autoimmunity. J. Immunol. 2007, 179, 4313–4317. [Google Scholar]
- Vehlow, A.; Cordes, N. Invasion as target for therapy of glioblastoma multiforme. Biochim. Biophys. Acta 2013, 1836, 236–244. [Google Scholar]
- Yang, F.; Brown, C.; Buettner, R.; Hedvat, M.; Starr, R.; Scuto, A.; Schroeder, A.; Jensen, M.; Jove, R. Sorafenib induces growth arrest and apoptosis of human glioblastoma cells through the dephosphorylation of signal transducers and activators of transcription 3. Mol. Cancer Ther. 2010, 9, 953–962. [Google Scholar] [CrossRef]
- Sutmuller, R.P.; van Duivenvoorde, L.M.; van Elsas, A.; Schumacher, T.N.; Wildenberg, M.E.; Allison, J.P.; Toes, R.E.; Offringa, R.; Melief, C.J. Synergism of cytotoxic T lymphocyte-associated antigen 4 blockade and depletion of CD25(+) regulatory T cells in antitumor therapy reveals alternative pathways for suppression of autoreactive cytotoxic T lymphocyte responses. J. Exp. Med. 2001, 194, 823–832. [Google Scholar] [CrossRef]
- Phan, G.Q.; Yang, J.C.; Sherry, R.M.; Hwu, P.; Topalian, S.L.; Schwartzentruber, D.J.; Restifo, N.P.; Haworth, L.R.; Seipp, C.A.; Freezer, L.J.; et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc. Natl. Acad. Sci. USA 2003, 100, 8372–8377. [Google Scholar] [CrossRef]
- Hodi, F.S.; Mihm, M.C.; Soiffer, R.J.; Haluska, F.G.; Butler, M.; Seiden, M.V.; Davis, T.; Henry-Spires, R.; MacRae, S.; Willman, A.; et al. Biologic activity of cytotoxic T lymphocyte-associated antigen 4 antibody blockade in previously vaccinated metastatic melanoma and ovarian carcinoma patients. Proc. Natl. Acad. Sci. USA 2003, 100, 4712–4717. [Google Scholar] [CrossRef]
- Manuel, E.R.; Blache, C.A.; Paquette, R.; Kaltcheva, T.I.; Ishizaki, H.; Ellenhorn, J.D.; Hensel, M.; Metelitsa, L.; Diamond, D.J. Enhancement of cancer vaccine therapy by systemic delivery of a tumor-targeting salmonella-based STAT3 shRNA suppresses the growth of established melanoma tumors. Cancer Res. 2011, 71, 4183–4191. [Google Scholar] [CrossRef]
- Van Elsas, A.; Hurwitz, A.A.; Allison, J.P. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/ macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J. Exp. Med. 1999, 190, 355–366. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kim, J.E.; Patel, M.; Ruzevick, J.; Jackson, C.M.; Lim, M. STAT3 Activation in Glioblastoma: Biochemical and Therapeutic Implications. Cancers 2014, 6, 376-395. https://doi.org/10.3390/cancers6010376
Kim JE, Patel M, Ruzevick J, Jackson CM, Lim M. STAT3 Activation in Glioblastoma: Biochemical and Therapeutic Implications. Cancers. 2014; 6(1):376-395. https://doi.org/10.3390/cancers6010376
Chicago/Turabian StyleKim, Jennifer E., Mira Patel, Jacob Ruzevick, Christopher M. Jackson, and Michael Lim. 2014. "STAT3 Activation in Glioblastoma: Biochemical and Therapeutic Implications" Cancers 6, no. 1: 376-395. https://doi.org/10.3390/cancers6010376