The Role of STAT3 in Non-Small Cell Lung Cancer

1
Department of Thoracic Oncology, NHO Shikoku Cancer Center, 160 Minami-Umemoto-cho, Matsuyama 791-0280, Japan
2
Department of General Internal Medicine 4, Kawasaki Medical School, 2-1-80 Nakasange, Kita-ku, Okayama 700-8505, Japan
3
Department of Allergy and Respiratory Medicine, Okayama University Hospital, 2-5-1 Shikata-cho, Kita-ku, Okayama 700-8558, Japan
*
Author to whom correspondence should be addressed.
Cancers 2014, 6(2), 708-722; https://doi.org/10.3390/cancers6020708
Submission received: 5 December 2013
/
Revised: 23 February 2014
/
Accepted: 7 March 2014
/
Published: 26 March 2014
(This article belongs to the Special Issue STAT3 Signalling in Cancer: Friend or Foe)
Abstract
:Persistent phosphorylation of signal transducer and activator of transcription 3 (STAT3) has been demonstrated in 22%~65% of non-small cell lung cancers (NSCLC). STAT3 activation is mediated by receptor tyrosine kinases, such as epidermal growth factor receptor (EGFR) and MET, cytokine receptors, such as IL-6, and non-receptor kinases, such as Src. Overexpression of total or phosphorylated STAT3 in resected NSCLC leads to poor prognosis. In a preclinical study, overexpression of STAT3 was correlated with chemoresistance and radioresistance in NSCLC cells. Here, we review the role of STAT3 and the mechanisms of treatment resistance in malignant diseases, especially NSCLC. As STAT3 is a critical mediator of the oncogenic effects of EGFR mutations, we discuss STAT3 pathways in EGFR-mutated NSCLC, referring to mechanisms of EGFR tyrosine kinase inhibitor resistance.
1. Introduction
Signal transducer and activator of transcription 3 (STAT3) is an important signaling mediator in malignant diseases and is persistently activated in 22%~65% of non-small cell lung cancers (NSCLC) [1,2,3]. STAT3 activation by cytokines, such as interleukin-6 (IL-6), is mediated through the Janus family kinases (JAK) or Src [4]. STAT3 is also involved in one of the epidermal growth factor receptor (EGFR) downstream pathways [5]. Several JAK/STAT3 inhibitors suppressed activation of STAT3 and showed anticancer and antiangiogenic effects in vitro and in vivo. Some have been introduced into clinical trials for solid tumors [6,7,8]. Here, we discuss the role of STAT3 in malignant diseases, especially NSCLC. In addition, we discuss the STAT3 pathways in EGFR-mutated NSCLC. Although STATs have numerous functions in immunoregulation, they have been well reviewed and summarized [9,10]. Therefore, in this review we focus on the oncogenic effects of STAT3.
2. Characteristics of STATs
A variety of signal pathways concerning carcinogenesis, malignant transformation, tumor progression and metastasis have been discovered. The RAS/mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K)/AKT pathways have been investigated extensively in oncogenic signaling [11,12,13,14]. One of the more recently recognized oncogenic signaling pathways involves the STAT, which are cytoplasmic proteins with Src homology 2 (SH2) domains. They function as transcription factors responding to cytokines and growth factors in normal cells. This family of proteins consists of seven members: STAT1–4, STAT6, and the closely related STAT5a and STAT5b proteins. The STATs are activated by phosphorylation of a conserved tyrosine residue. Subsequently, they dimerize, translocate to nucleus, bind to DNA and activate the target genes [15,16]. Cytokines and growth factors which phosphorylate the tyrosine residue include IL-6, IL-2, IL-7, epidermal growth factor (EGF), fibroblast growth factor, and platelet-derived growth factor. Soon after growth factors or cytokines bind to their corresponding receptors, intrinsic receptor tyrosine kinases or receptor-associated kinases including JAK or Src tyrosine kinase are activated [17,18]. Subsequently, the tyrosine kinases phosphorylate the receptor to provide docking sites for the recruitment of monomeric STATs. Once they have been adopted, STATs themselves become substrates for tyrosine phosphorylation. Non-receptor tyrosine kinases, such as Src and Bcr-Abl, can phosphorylate STATs independently of receptor activation. Phosphorylated (p) STATs dimerize and translocate to the nucleus, and the dimers regulate gene expression [18]. STAT1, STAT3, and STAT5 are also phosphorylated on a serine residue within their COOH terminus; this phosphorylation is dispensable for dimerization, nuclear translocation, and DNA binding, but is required for maximal transcriptional activity of certain genes [19]. These proteins have dual roles as cytoplasmic signaling proteins and as nuclear transcription factors that activate a diverse set of genes implicated in malignant progression. Since cell cycle is also regulated by the target genes of STATs, the activation promotes cell growth and induces transformation to malignant phenotype.
3. STAT3 in Tumor Cell Proliferation
STAT3 has been reported to be associated with oncogenesis and resistance to chemotherapy. While STAT activation is strictly controlled in normal cells and plays a key role in multiple cellular functions, such as cell growth and differentiation, metabolism, hematopoiesis, host defense and immunoregulation, the durable activation of tyrosine kinases in malignant tumor causes constitutive activation of STATs, particularly STAT3 and STAT5 [10,18,20]. Following the activation of STATs, expressions of genes which regulate cancer progression processes including uncontrolled proliferation, apoptosis resistance, sustained angiogenesis, and evasion of immune surveillance would change [18,20,21,22]. Recent evidence suggests a crucial role for STAT family proteins, especially STAT3, in selectively inducing and maintaining a pro-carcinogenic inflammatory microenvironment, both at the initiation of malignant transformation and during cancer progression [23,24,25,26,27,28,29,30,31,32]. Several reports showed that inhibition of STAT3 suppressed the growth of cancer cells and enhanced the sensitivity to anticancer agents in multiple types of cancer [33,34,35,36,37,38,39]. Therefore, STAT3 has been considered a potential target for anticancer therapy.
4. Prognostic Impact of STAT3 Activation in NSCLC
To explore the relationship between STAT3 or pSTAT3 expression and its prognostic function on NSCLC, several studies evaluated the clinical significance in patients to determine their effects on tumor angiogenesis and proliferation [3,40]. Jiang et al. showed the positive rate of pSTAT3 expression in patients with lymph node metastasis was 78.8% (41/52), which was significantly higher than that in NSCLC patients with non-lymph node metastasis (54.7%, 41/75) using immunohistochemical staining among the 127 cases of p-STAT3 immunoreactivity, was significantly correlated with sex (p = 0.004), smoking history (p = 0.006), EGFR mutation status (p = 0.003), clinical stage (p = 0.034), and lymph node metastasis (p = 0.009) [3]. Xu et al. performed a meta-analysis including a total of 17 retrospective trials and found high STAT3 or pSTAT3 expression was a strong predictor of poor prognosis among patients with NSCLC [40]. Zhao et al. reported that NSCLC patients with high pJAK2 expression who underwent surgery had a significantly poorer overall survival rate compared with those with low pJAK2 expression [41]. They also found that microvessel density (MVD) was higher in NSCLC samples with high pJAK2 and pSTAT3 expression, and that patients with high MVD had poor survival rates. These data suggests that high pSTAT3 expression is a strong predictor of poor prognosis in patients with NSCLC. However, it should be confirmed by a large prospective study with long-term follow-up.
5. STAT3 in Primary Resistance to Treatment
Primary resistance to treatment includes (1) cytotoxic chemotherapy (2) radiotherapy, and (3) targeted therapy. First, Barre et al. reviewed that the STAT3 oncogenic pathway was associated with intrinsic resistance to chemotherapeutic agents in several malignancies [42,43]. In NSCLC cells, overexpression of STAT3 mRNA was related with cisplatin-resistance [42,43]. Silencing STAT3 by siRNA led the resistant cells more sensitive to cytotoxic agents such as paclitaxel, doxorubicin and cisplatin [44,45,46]. Secondary, several studies have suggested that the upregulation of STAT3 directly confers a drug-resistant phenotype, such as resistance to ultraviolet radiation-induced apoptosis [32,47]. You et al. showed that ionizing radiation induces phosphorylation of JAK2 and STAT3 and higher levels of STAT3 were found to be accumulated in the nucleus of radioresistant NSCLC cells. They showed niclosamide, a potent STAT3 inhibitor that disrupted STAT3 transcriptional activity by blocking its phosphorylation and nuclear translocation, alone or in combination with radiotherapy overcame radioresistance in xenograft models [48]. Sun et al. reported that a small molecule inhibitor of JAK2 (TG101209) was able to inhibit survivin, reduce pSTAT3, and sensitize lung cancer cells to radiation in vitro [49]. Thirdly, Looyenga et al. demonstrated that STAT3 activity was unaffected by the TKIs such as sunitinib or crizotinib in spite of PDGFR or MET amplified NSCLC cells, respectively [2]. They also showed the treatment of NSCLC cell lines with ruxolitinib, a JAK1/2 selective inhibitor which has shown promising results in phase I/II study for patients with myelofibrosis, inhibited the cell growth in soft agar and in xenograft assays [2]. Inhibition of STAT3 was an effective strategy for overcoming resistance to a dual inhibitor of PI3K and mTOR [50]. These suggest that persistent STAT3 may be leading to primary resistance to targeted therapies. Thus, the JAK2/STAT3 pathway seemed to be associated with primary resistance to treatments, including cytotoxic chemotherapy, radiotherapy and targeted therapy.
6. Activation of STAT3 Independent of EGFR Downstream Pathways
There are a variety of pro-inflammatory cytokines and growth factors causing EGFR-independent STAT3 activation. Some tumor cell lines need constitutive activation of STAT3 in order to survive [20,51]. STAT3 was activated by IL-6 in myeloma and prostate cancer cells [24,52,53], hepatocyte growth factor (HGF) and its receptor (c-MET) in leiomyosarcoma and breast cancer cells [54,55], and Src in breast cancer and melanoma cells [54,56].
In NSCLC, STAT3 is an oncogene that is expressed in alveolar type II epithelial cells [21,57]. Li et al. showed that persistent STAT3 activation induced pulmonary tumorigenesis in STAT3 transgenic mice [58]. The activation of STAT3 by receptor tyrosine kinases, such as EGFR and MET, cytokine receptors, such as IL-6 receptor, and non-receptor kinases, such as Src, regulates survival pathways in certain NSCLC cells (Figure 1) [59]. Zimmer et al. suggested that STAT3 activity contributes to the carcinogenic potential of NSCLC independently of EGFR mutations [1]. Looyenga et al. demonstrated that STAT3 is constitutively activated in human NSCLC samples and in a variety of NSCLC lines independent of activating KRAS, EGFR and PDGFR mutations or MET amplification. They further showed that STAT3 is activated via autocrine signaling by IL-6 family ligands and genetic or pharmacologic inhibition of the gp130/JAK2 signaling pathway disrupts activation of STAT3 [2]. These data suggest that autocrine inflammatory cytokine signaling via JAK2 is the primary driver of STAT3 activation in NSCLC cell lines and in patient tissues.
7. STAT3 in EGFR Signaling Pathways
Somatic mutations have been discovered in the EGFR tyrosine kinase in a subset of NSCLC patients [60,61,62,63,64]. These mutations strongly sensitize the cancer cells to the growth suppressive effects of EGFR-TKI, leading to clinical responses [60,61,63,64]. PI3K/AKT, RAS/MAPK, and STAT3 are three major downstream pathways activated by EGFR phosphorylation [65].
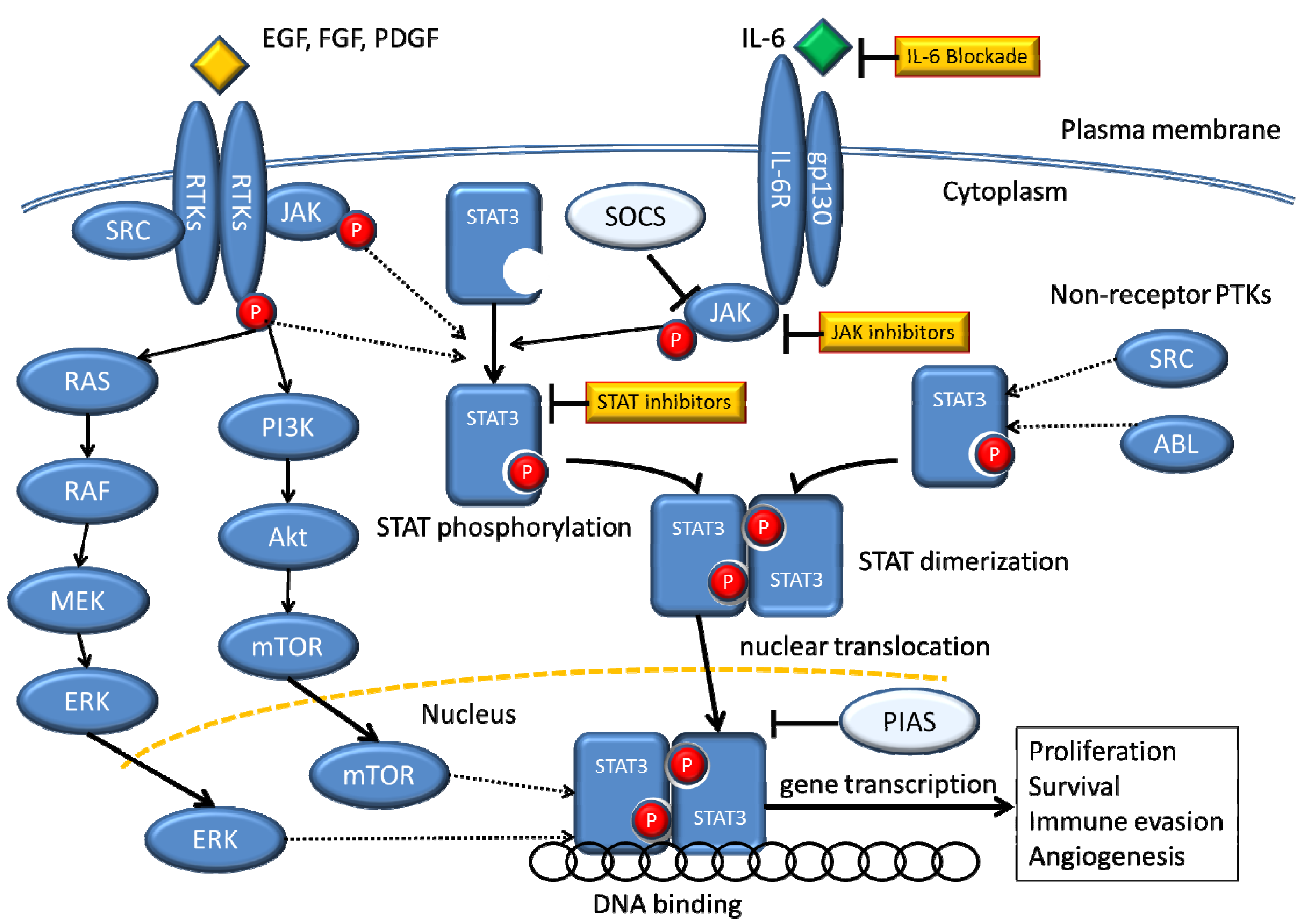
Figure 1.
Signaling pathways that converge on STAT3.
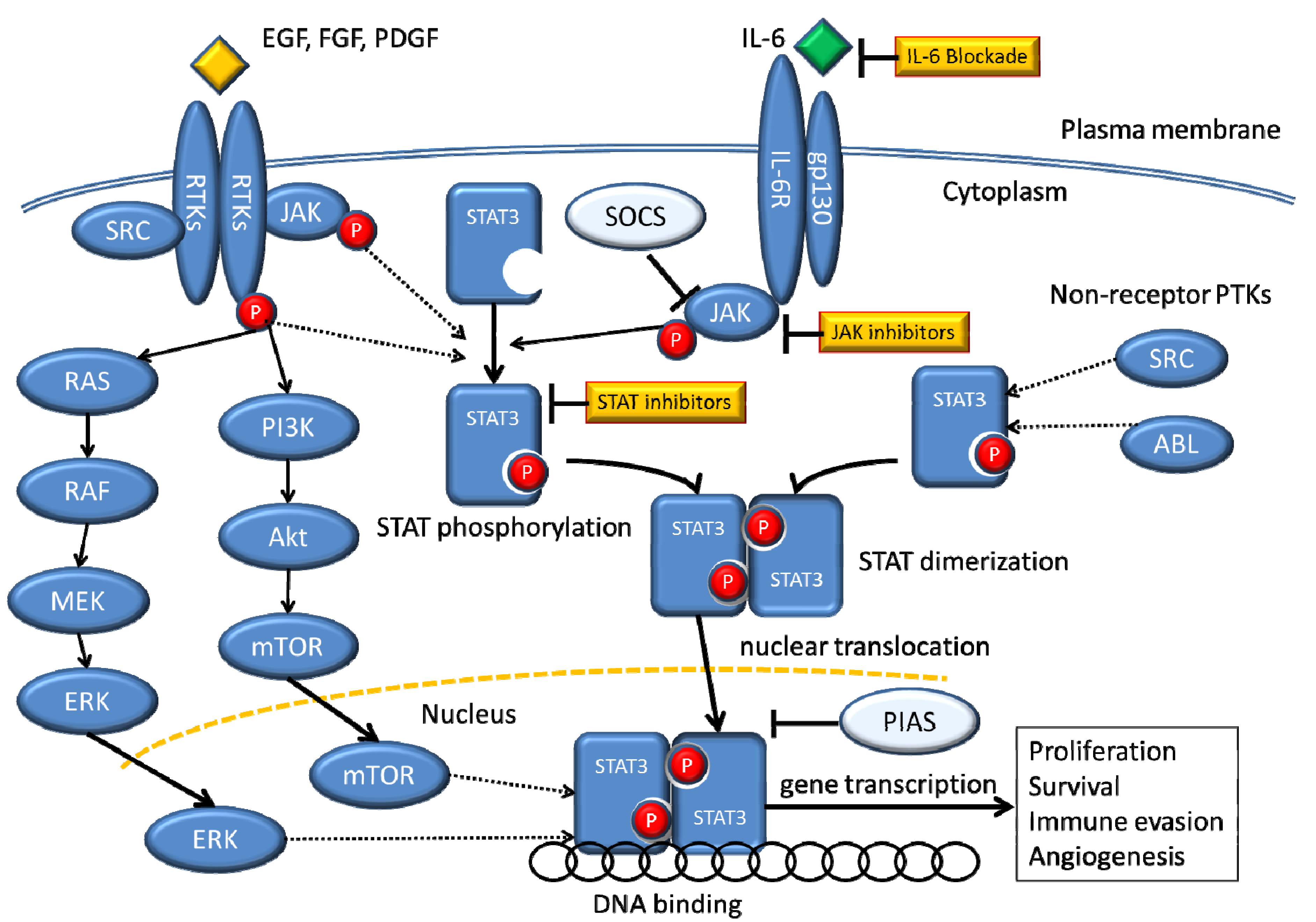
STATs are activated by receptor and non-receptor tyrosine kinases through several mechanisms. The receptor-associated Janus family tyrosine kinases (JAKs) are activated by cytokine-receptor binding through cross-phosphorylation, and then the activated JAKs phosphorylate tyrosine residues within the receptor molecules, which become docking elements for cytoplasmic STATs. STATs are subsequently phosphorylated on a single tyrosine residue and form dimers through reciprocal interaction between the phosphotyrosine of one STAT molecule and the Src homology-2 (SH2) domain of another. Dimerized STATs translocate to the nucleus to induce target gene transcription by binding to specific regulatory elements. Receptors with intrinsic tyrosine kinase activity (RTKs), including the platelet-derived growth factor receptor (PDGFR) and the epidermal growth factor receptor (EGFR), may directly activate STATs without activation of JAKs. STATs can be phosphorylated by constitutively active non-receptor protein tyrosine kinases (PTKs), such as Src and Bcr-Abl. Abbreviation: PTK, protein tyrosine kinase; IL-6, interleukin-6; P, phosphorus; SOCS, suppressor of cytokine signaling; PIAS, protein inhibitor of activated STAT.
STAT3 has also been reported as a critical mediator of the oncogenic effects caused by EGFR mutations [66]. Several NSCLC cell lines contain constitutively active STAT3 [59]. Non-transformed epithelial cells engineered to express various NSCLC-associated EGFR mutations had elevated levels of activated STAT5 and STAT3 [63]. Greulich et al. reported that STAT3 was activated by various EGFR mutations, including the exon 19 in-frame deletion or the exon 21 L858R point mutation, and may contribute to the oncogenic effects of these mutations in fibroblasts and human lung cancer cells [67]. The mechanism whereby mutant EGFR drives STAT3 activation is indirect, as it requires upregulation of the cytokine IL-6, which activated the gp130/JAK pathway [68] (Figure 1). There has been no strong data suggesting that tyrosine receptor kinases such as EGFR, MET or PDGFR directly drive constitutive STAT3 activation in NSCLC, so far.
8. Relationship between STAT3 and Acquired Resistance to EGFR-TKI
First-generation EGFR-TKIs (gefitinib and erlotinib) inhibit EGFR signaling cascade by reversible binding to the adenosine triphosphate (ATP) binding site of the EGFR [69]. Although initial responses to reversible EGFR-TKIs in most patients with EGFR activating mutations were observed, almost all patients acquired resistance to these agents via diverse mechanisms [70]. A secondary T790M mutation (leading to acquired resistance) in exon 20 of EGFR occurred in approximately half of the patients [71].
Kim et al. demonstrated a role for the IL-6R/JAK1/STAT3 signaling pathway, leading to STAT3 activation, in de novo resistance to irreversible EGFR-TKIs, such as afatinib, in NSCLC cells with EGFR T790M [72]. Because STAT3 is not only a downstream effector molecule of IL-6 but also a transcription factor for IL-6 activation [73], autocrine IL-6 production by afatinib could be explained by the activation of a positive feedback in the IL-6/STAT3 axis [72]. It was also suggested that activation of NF-κB stimulated production of IL-6, and that the IL-6-stimulated activation of STAT3 is a mechanism of resistance to EGFR-TKI [74]. As paracrine effect of IL-6 secreted by fibroblasts, IL-6 receptor signal was activated and resistance to afatinib in cancer cells was shown [72]. The tumor microenvironment producing soluble factors such as cytokines and growth factors would help tumor cells to escape from anticancer therapy-induced apoptosis [72,75]. The IL-6 in the tumor microenvironment is known to induce resistance to anticancer agents in malignant diseases [24,76,77]. Because IL-6 is mainly derived from fibroblasts, macrophages and endothelial cells [78,79], high IL-6 levels in the tumor microenvironment might reduce the tumor response to afatinib [72].
Furthermore, serum IL-6 levels were elevated in patients with lung cancer in comparison to healthy individuals [80,81]. Under such circumstance, the combination of an IL-6R/JAK1/STAT3 pathway inhibitor with EGFR-TKI may be effective in NSCLC patients with acquired resistance to EGFR-TKI [72]. We established an erlotinib-resistant lung cancer cell line, PC-9/ER3, by continuous exposure of PC-9 cells to erlotinib. The PC-9/ER3 cells did not carry the T790M mutation or MET gene amplification. Although STAT3 was activated in the resistant cell line, inhibition of JAK2 rather than STAT3 restored sensitivity of PC-9/ER3 cells to erlotinib. Thus, activation of STAT3 in this cell line did not directly cause the resistance to EGFR-TKI in NSCLC with activating EGFR mutations. As IL-6 levels in PC-9/ER3 cells were similar to those in PC-9 cells, activation of STAT3 did not seem to be dependent on the IL-6R/JAK1/STAT3 axis [82].
Li et al. reported that inhibition of EGFR by erlotinib induced STAT3 phosphorylation on Tyr705 together with increased Bcl2/Bcl-XL at both the mRNA and protein levels in human lung cancer cell lines [83]. They suggested that erlotinib-enhanced phosphorylation of STAT3 may occur from suppression of PTPMeg2 expression, as PTPMeg2 is a physiological STAT3 phosphatase that can directly dephosphorylate STAT3 on Tyr705. Niclosamide, a STAT3 inhibitor, was able to restore sensitivity to erlotinib in vitro and in vivo [83]. These findings suggested that blocking the PTPMeg2/STAT3/Bcl2/Bcl-XL survival signaling pathway in human lung cancer may provide a novel strategy to overcome acquired resistance to EGFR-TKI.
9. STAT3 as a Target for Cancer Therapy
One approach to inhibiting STAT signaling is to block the upstream tyrosine kinases that are responsible for their activation. For example, small molecule inhibitors of JAK, Src, Bcr-Abl, and EGFR have all been shown to block STAT3 signaling and induce tumor cell apoptosis (Figure 1) [24,56,84,85,86]. A STAT3 decoy which comprises a 15 bp double-stranded oligonucleotide competes between the endogenous cis-elements within the regulatory regions of target genes and the exogenously added molecules mimicking the specific cis-elements [87]. The decoy which corresponds to the STAT3 response element in the c-fos promoter and binds competitively to STAT3 has been shown to inhibit the growth of human lung cancer [88,89]. In addition, a human phase 0 study of a STAT3 decoy oligonucleotide suggested that it might yield effective therapeutic agent for reducing STAT3 target genes and inhibiting tumor growth [90]. Although studies regarding JAK inhibitors in malignant diseases began in the mid-1990s, more potent JAK inhibitors have been developed since the discovery of the JAK2 mutation in myeloproliferative disorders. These inhibitors were extremely effective for treatment of myeloproliferative preclinical models, leading to clinical trials [7]. For solid tumors, the role of JAK inhibition was examined in models of IL-6-driven breast, ovarian, and prostate cancers using the JAK1/2 inhibitor AZD1480, which led to the suppression of tumor growth [91]. There have been clinical trials testing STAT3 inhibitors, including upstream TKIs for solid tumors, such as lung cancers (Table 1).

{kind=link}
| Drug | Target | Indication | Status |
|---|---|---|---|
| Siltuximab (CNTO-328) | IL-6 (monoclonal antibody) | Ovarian, pancreatic, colorectal, head and neck, and lung cancer | Phase I Phase II |
| Ruxolitinib (INCB018424) | JAK1/2 | Chronic myeloproliferative disorders, leukemia, myelodysplastic syndrome, myeloproliferative neoplasms, unspecified childhood solid tumor | Phase I |
| AZD1480 | JAK1/2 | Advanced solid tumor, hepatocellular carcinomaEGFR and/or ROS mutant lung cancer, metastatic lung cancer, gastric cancer | Phase I |
| INCB047986 | JAK | Advanced solid tumor, Hodgkin's lymphoma, non-Hodgkin's lymphoma | Phase I |
| OPB-31121 | JAK2 and gp130 | Advanced solid tumor | Phase I |
| WP1066 | JAK2 and gp130 | Glioblastoma multiforme, melanoma | Phase I |
| OPB-51602 | STAT3 phosphorylation | Advanced cancer | Phase I |
| SAR302503 | JAK | Solid malignancy | Phase I |
Several agents targeting the IL-6R/JAK/STAT3 signaling pathway, such as siltuximab (IL-6-neutralizing monoclonal antibody [92]), OPB-31121 (small molecule inhibitor JAK2 [93]), and AZD1480 (small molecule inhibitor of JAK [8,94]), have been clinically developed and may be suitable candidates for future combination therapy with EGFR-TKIs. At present ruxolitinib [2] is only JAK1/2 selective inhibitors that has been approved for use by the FDA to treat JAK2-dependent malignancies such as myelofibrosis. Recently, microRNAs were reported to regulate the activity state of STAT3 or to be regulated by STAT3 activation in tumors [95,96,97,98]. The microRNA upregulated by STAT3 activation might be associated with the mechanisms of cancer invasion, migration and resistance to anti-cancer agents including cytotoxic or targeted therapies. In particular, miR-337-3p directly targeted to STAT3 and sensitized NSCLC cells to paclitaxel [99]. The treatment of anticancer agents with microRNA down-regulating STAT3 might be investigated.
10. Conclusions
STAT3 signaling is an important pathway in a variety of malignant diseases, including NSCLC. JAK/STAT3 inhibitors may be effective for treatment of NSCLC irrespective of the EGFR mutation status. We need to elucidate the mechanism(s) of STAT3 activation from autocrine, paracrine or stromal sources of ligand. STAT3 activation by stromal cells may contribute to the oncogenic effects of the NSCLC cells. Subsequently, the unknown role(s) of STAT3 in therapeutic resistance should be resolved. Finally, the most promising avenues for use of JAK or STAT inhibitors in a clinical context, for example, whether they would work as monotherapy or only as combination therapy should be explored. More translational and clinical trials are required to clarify the potential roles of STAT3 inhibitors in the treatment of NSCLC.
Acknowledgments
Grant support: Ministry of Education, Culture, Sports, Science, and Technology, Japan grants 24591182 (N. Takigawa) and 23390221 (K. Kiura).
Conflicts of Interest
N. Takigawa and K. Kiura received honoraria from the speaker’s bureau, AstraZeneca, Chugai Pharmaceutical Company and Boehringer-Ingelheim in Japan. The other author disclosed no conflict of interest.
References
- Zimmer, S.; Kahl, P.; Buhl, T.M.; Steiner, S.; Wardelmann, E.; Merkelbach-Bruse, S.; Buettner, R.; Heukamp, L.C. Epidermal growth factor receptor mutations in non-small cell lung cancer influence downstream Akt, MAPK and Stat3 signaling. J. Cancer Res. Clin. Oncol. 2009, 135, 723–730. [Google Scholar] [CrossRef]
- Looyenga, B.D.; Hutchings, D.; Cherni, I.; Kingsley, C.; Weiss, G.J.; Mackeigan, J.P. STAT3 is activated by JAK2 independent of key oncogenic driver mutations in non-small cell lung carcinoma. PLoS One 2012, 7, e30820. [Google Scholar]
- Jiang, R.; Jin, Z.; Liu, Z.; Sun, L.; Wang, L.; Li, K. Correlation of activated STAT3 expression with clinicopathologic features in lung adenocarcinoma and squamous cell carcinoma. Mol. Diagn. Ther. 2011, 15, 347–352. [Google Scholar] [CrossRef]
- Levy, D.E.; Darnell, J.E., Jr. Stats: Transcriptional control and biological impact. Nat. Rev. Mol. Cell. Biol. 2002, 3, 651–662. [Google Scholar] [CrossRef]
- Zhong, Z.; Wen, Z.; Darnell, J.E., Jr. Stat3: A STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 1994, 264, 95–98. [Google Scholar]
- Lesina, M.; Kurkowski, M.U.; Ludes, K.; Rose-John, S.; Treiber, M.; Kloppel, G.; Yoshimura, A.; Reindl, W.; Sipos, B.; Akira, S.; et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell. 2011, 19, 456–469. [Google Scholar] [CrossRef]
- Verstovsek, S. Therapeutic potential of JAK2 inhibitors. Hematol. Am. Soc. Hematol. Educ. Program. 2009. [Google Scholar] [CrossRef]
- Murakami, T.; Takigawa, N.; Ninomiya, T.; Ochi, N.; Yasugi, M.; Honda, Y.; Kubo, T.; Ichihara, E.; Hotta, K.; Tanimoto, M.; et al. Effect of AZD1480 in an epidermal growth factor receptor-driven lung cancer model. Lung Cancer 2014, 83, 30–36. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Plenge, R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity 2012, 36, 542–550. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Holland, S.M.; Staudt, L.M. JAKs and STATs in immunity, immunodeficiency, and cancer. N. Engl. J. Med. 2013, 368, 161–170. [Google Scholar] [CrossRef]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- Egan, S.E.; Weinberg, R.A. The pathway to signal achievement. Nature 1993, 365, 781–783. [Google Scholar] [CrossRef]
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Barre, B.; Avril, S.; Coqueret, O. Opposite regulation of myc and p21waf1 transcription by STAT3 proteins. J. Biol. Chem. 2003, 278, 2990–2996. [Google Scholar] [CrossRef]
- Darnell, J.E., Jr. STATs and gene regulation. Science 1997, 277, 1630–1635. [Google Scholar] [CrossRef]
- Blaskovich, M.A.; Sun, J.; Cantor, A.; Turkson, J.; Jove, R.; Sebti, S.M. Discovery of JSI-124 (cucurbitacin I), a selective Janus kinase/signal transducer and activator of transcription 3 signaling pathway inhibitor with potent antitumor activity against human and murine cancer cells in mice. Cancer Res. 2003, 63, 1270–1279. [Google Scholar]
- Yu, H.; Jove, R. The STATs of cancer—New molecular targets come of age. Nat. Rev. Cancer 2004, 4, 97–105. [Google Scholar] [CrossRef]
- Decker, T.; Kovarik, P. Serine phosphorylation of STATs. Oncogene 2000, 19, 2628–2637. [Google Scholar] [CrossRef]
- Bowman, T.; Garcia, R.; Turkson, J.; Jove, R. Stats in oncogenesis. Oncogene 2000, 19, 2474–2488. [Google Scholar] [CrossRef]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.E., Jr. Stat3 as an oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef]
- Haura, E.B.; Zheng, Z.; Song, L.; Cantor, A.; Bepler, G. Activated epidermal growth factor receptor-Stat-3 signaling promotes tumor survival in vivo in non-small cell lung cancer. Clin. Cancer Res. 2005, 11, 8288–8294. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Catlett-Falcone, R.; Landowski, T.H.; Oshiro, M.M.; Turkson, J.; Levitzki, A.; Savino, R.; Ciliberto, G.; Moscinski, L.; Fernandez-Luna, J.L.; Nunez, G.; et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity 1999, 10, 105–115. [Google Scholar] [CrossRef]
- Bollrath, J.; Phesse, T.J.; von Burstin, V.A.; Putoczki, T.; Bennecke, M.; Bateman, T.; Nebelsiek, T.; Lundgren-May, T.; Canli, O.; Schwitalla, S.; et al. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell 2009, 15, 91–102. [Google Scholar] [CrossRef]
- Kujawski, M.; Kortylewski, M.; Lee, H.; Herrmann, A.; Kay, H.; Yu, H. Stat3 mediates myeloid cell-dependent tumor angiogenesis in mice. J. Clin. Invest. 2008, 118, 3367–3377. [Google Scholar] [CrossRef]
- Kortylewski, M.; Xin, H.; Kujawski, M.; Lee, H.; Liu, Y.; Harris, T.; Drake, C.; Pardoll, D.; Yu, H. Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironment. Cancer Cell 2009, 15, 114–123. [Google Scholar] [CrossRef]
- Wang, L.; Yi, T.; Kortylewski, M.; Pardoll, D.M.; Zeng, D.; Yu, H. Il-17 can promote tumor growth through an IL-6-STAT3 signaling pathway. J. Exp. Med. 2009, 206, 1457–1464. [Google Scholar] [CrossRef]
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; et al. IL-6 and STAT3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 2009, 15, 103–113. [Google Scholar] [CrossRef]
- Shain, K.H.; Yarde, D.N.; Meads, M.B.; Huang, M.; Jove, R.; Hazlehurst, L.A.; Dalton, W.S. Beta1 integrin adhesion enhances IL-6-mediated STAT3 signaling in myeloma cells: Implications for microenvironment influence on tumor survival and proliferation. Cancer Res. 2009, 69, 1009–1015. [Google Scholar] [CrossRef]
- Ara, T.; Song, L.; Shimada, H.; Keshelava, N.; Russell, H.V.; Metelitsa, L.S.; Groshen, S.G.; Seeger, R.C.; DeClerck, Y.A. Interleukin-6 in the bone marrow microenvironment promotes the growth and survival of neuroblastoma cells. Cancer Res. 2009, 69, 329–337. [Google Scholar] [CrossRef]
- Shen, Y.; Devgan, G.; Darnell, J.E., Jr.; Bromberg, J.F. Constitutively activated Stat3 protects fibroblasts from serum withdrawal and UV-induced apoptosis and antagonizes the proapoptotic effects of activated Stat1. Proc. Natl. Acad. Sci. USA 2001, 98, 1543–1548. [Google Scholar] [CrossRef]
- Cai, L.; Zhang, G.; Tong, X.; You, Q.; An, Y.; Wang, Y.; Guo, L.; Wang, T.; Zhu, D.; Zheng, J. Growth inhibition of human ovarian cancer cells by blocking STAT3 activation with small interfering RNA. Eur. J. Obstet. Gynecol. Reprod. Biol. 2010, 148, 73–80. [Google Scholar] [CrossRef]
- Burke, W.M.; Jin, X.; Lin, H.J.; Huang, M.; Liu, R.; Reynolds, R.K.; Lin, J. Inhibition of constitutively active Stat3 suppresses growth of human ovarian and breast cancer cells. Oncogene 2001, 20, 7925–7934. [Google Scholar] [CrossRef]
- Horiguchi, A.; Asano, T.; Kuroda, K.; Sato, A.; Asakuma, J.; Ito, K.; Hayakawa, M.; Sumitomo, M.; Asano, T. STAT3 inhibitor WP1066 as a novel therapeutic agent for renal cell carcinoma. Br. J. Cancer 2010, 102, 1592–1599. [Google Scholar] [CrossRef]
- Byers, L.A.; Sen, B.; Saigal, B.; Diao, L.; Wang, J.; Nanjundan, M.; Cascone, T.; Mills, G.B.; Heymach, J.V.; Johnson, F.M. Reciprocal regulation of c-Src and STAT3 in non-small cell lung cancer. Clin. Cancer Res. 2009, 15, 6852–6861. [Google Scholar] [CrossRef]
- Chiu, H.C.; Chou, D.L.; Huang, C.T.; Lin, W.H.; Lien, T.W.; Yen, K.J.; Hsu, J.T. Suppression of STAT3 activity sensitizes gefitinib-resistant non small cell lung cancer cells. Biochem. Pharmacol. 2011, 81, 1263–1270. [Google Scholar] [CrossRef]
- Song, L.; Rawal, B.; Nemeth, J.A.; Haura, E.B. JAK1 activates STAT3 activity in non-small-cell lung cancer cells and IL-6 neutralizing antibodies can suppress JAK1-STAT3 signaling. Mol. Cancer Ther. 2011, 10, 481–494. [Google Scholar] [CrossRef]
- Sen, B.; Saigal, B.; Parikh, N.; Gallick, G.; Johnson, F.M. Sustained Src inhibition results in signal transducer and activator of transcription 3 (STAT3) activation and cancer cell survival via altered Janus-activated kinase-STAT3 binding. Cancer Res. 2009, 69, 1958–1965. [Google Scholar] [CrossRef]
- Xu, Y.H.; Lu, S. A meta-analysis of STAT3 and phospho-STAT3 expression and survival of patients with non-small-cell lung cancer. Eur. J. Surg. Oncol. 2014, 40, 311–317. [Google Scholar] [CrossRef]
- Zhao, M.; Gao, F.H.; Wang, J.Y.; Liu, F.; Yuan, H.H.; Zhang, W.Y.; Jiang, B. JAK2/STAT3 signaling pathway activation mediates tumor angiogenesis by upregulation of VEGF and bFGF in non-small-cell lung cancer. Lung Cancer 2011, 73, 366–374. [Google Scholar] [CrossRef]
- Barre, B.; Vigneron, A.; Perkins, N.; Roninson, I.B.; Gamelin, E.; Coqueret, O. The STAT3 oncogene as a predictive marker of drug resistance. Trends Mol. Med. 2007, 13, 4–11. [Google Scholar] [CrossRef]
- Ikuta, K.; Takemura, K.; Kihara, M.; Nishimura, M.; Ueda, N.; Naito, S.; Lee, E.; Shimizu, E.; Yamauchi, A. Overexpression of constitutive signal transducer and activator of transcription 3 mRNA in cisplatin-resistant human non-small cell lung cancer cells. Oncol. Rep. 2005, 13, 217–222. [Google Scholar]
- Su, W.P.; Cheng, F.Y.; Shieh, D.B.; Yeh, C.S.; Su, W.C. PLGA nanoparticles codeliver paclitaxel and Stat3 siRNA to overcome cellular resistance in lung cancer cells. Int. J. Nanomed. 2012, 7, 4269–4283. [Google Scholar]
- Kulesza, D.W.; Carre, T.; Chouaib, S.; Kaminska, B. Silencing of the transcription factor STAT3 sensitizes lung cancer cells to DNA damaging drugs, but not to TNFalpha- and NK cytotoxicity. Exp. Cell Res. 2013, 319, 506–516. [Google Scholar] [CrossRef]
- Zhang, W.; Wan, M.; Ma, L.; Liu, X.; He, J. Protective effects of ADAM8 against cisplatin-mediated apoptosis in non-small-cell lung cancer. Cell Biol. Int. 2013, 37, 47–53. [Google Scholar] [CrossRef]
- Sano, S.; Chan, K.S.; Kira, M.; Kataoka, K.; Takagi, S.; Tarutani, M.; Itami, S.; Kiguchi, K.; Yokoi, M.; Sugasawa, K.; et al. Signal transducer and activator of transcription 3 is a key regulator of keratinocyte survival and proliferation following UV irradiation. Cancer Res. 2005, 65, 5720–5729. [Google Scholar] [CrossRef]
- You, S.; Li, R.; Park, D.; Xie, M.; Sica, G.L.; Cao, Y.; Xiao, Z.Q.; Deng, X. Disruption of STAT3 by niclosamide reverses radioresistance of human lung cancer. Mol. Cancer Ther. 2014, 13, 606–616. [Google Scholar] [CrossRef]
- Sun, Y.; Moretti, L.; Giacalone, N.J.; Schleicher, S.; Speirs, C.K.; Carbone, D.P.; Lu, B. Inhibition of JAK2 signaling by TG101209 enhances radiotherapy in lung cancer models. J. Thorac. Oncol. 2011, 6, 699–706. [Google Scholar] [CrossRef]
- Jin, H.O.; Lee, Y.H.; Park, J.A.; Kim, J.H.; Hong, S.E.; Kim, H.A.; Kim, E.K.; Noh, W.C.; Kim, B.H.; Ye, S.K.; et al. Blockage of STAT3 enhances the sensitivity of NSCLC cells to PI3K/mTOR inhibition. Biochem. Biophys. Res. Commun. 2014, 444, 502–508. [Google Scholar] [CrossRef]
- Frank, D.A. STAT signaling in the pathogenesis and treatment of cancer. Mol. Med. 1999, 5, 432–456. [Google Scholar]
- Lou, W.; Ni, Z.; Dyer, K.; Tweardy, D.J.; Gao, A.C. Interleukin-6 induces prostate cancer cell growth accompanied by activation of stat3 signaling pathway. Prostate 2000, 42, 239–242. [Google Scholar] [CrossRef]
- Mora, L.B.; Buettner, R.; Seigne, J.; Diaz, J.; Ahmad, N.; Garcia, R.; Bowman, T.; Falcone, R.; Fairclough, R.; Cantor, A.; et al. Constitutive activation of Stat3 in human prostate tumors and cell lines: Direct inhibition of Stat3 signaling induces apoptosis of prostate cancer cells. Cancer Res. 2002, 62, 6659–6666. [Google Scholar]
- Hung, W.; Elliott, B. Co-operative effect of c-Src tyrosine kinase and Stat3 in activation of hepatocyte growth factor expression in mammary carcinoma cells. J. Biol. Chem. 2001, 276, 12395–12403. [Google Scholar] [CrossRef]
- Zhang, Y.W.; Wang, L.M.; Jove, R.; vande Woude, G.F. Requirement of Stat3 signaling for HGF/SF-Met mediated tumorigenesis. Oncogene 2002, 21, 217–226. [Google Scholar] [CrossRef]
- Niu, G.; Bowman, T.; Huang, M.; Shivers, S.; Reintgen, D.; Daud, A.; Chang, A.; Kraker, A.; Jove, R.; Yu, H. Roles of activated Src and Stat3 signaling in melanoma tumor cell growth. Oncogene 2002, 21, 7001–7010. [Google Scholar] [CrossRef]
- Yan, C.; Naltner, A.; Martin, M.; Naltner, M.; Fangman, J.M.; Gurel, O. Transcriptional stimulation of the surfactant protein B gene by STAT3 in respiratory epithelial cells. J. Biol. Chem. 2002, 277, 10967–10972. [Google Scholar]
- Li, Y.; Du, H.; Qin, Y.; Roberts, J.; Cummings, O.W.; Yan, C. Activation of the signal transducers and activators of the transcription 3 pathway in alveolar epithelial cells induces inflammation and adenocarcinomas in mouse lung. Cancer Res. 2007, 67, 8494–8503. [Google Scholar]
- Song, L.; Turkson, J.; Karras, J.G.; Jove, R.; Haura, E.B. Activation of Stat3 by receptor tyrosine kinases and cytokines regulates survival in human non-small cell carcinoma cells. Oncogene 2003, 22, 4150–4165. [Google Scholar] [CrossRef]
- Paez, J.G.; Janne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef]
- Pao, W.; Miller, V.; Zakowski, M.; Doherty, J.; Politi, K.; Sarkaria, I.; Singh, B.; Heelan, R.; Rusch, V.; Fulton, L.; et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl. Acad. Sci. USA 2004, 101, 13306–13311. [Google Scholar] [CrossRef]
- Sordella, R. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science 2004, 305, 1163–1167. [Google Scholar] [CrossRef]
- Tsao, M.S.; Sakurada, A.; Cutz, J.C.; Zhu, C.Q.; Kamel-Reid, S.; Squire, J.; Lorimer, I.; Zhang, T.; Liu, N.; Daneshmand, M.; et al. Erlotinib in lung cancer—Molecular and clinical predictors of outcome. N. Engl. J. Med. 2005, 353, 133–144. [Google Scholar] [CrossRef]
- Mitsudomi, T.; Yatabe, Y. Mutations of the epidermal growth factor receptor gene and related genes as determinants of epidermal growth factor receptor tyrosine kinase inhibitors sensitivity in lung cancer. Cancer Sci. 2007, 98, 1817–1824. [Google Scholar] [CrossRef]
- Alvarez, J.V.; Greulich, H.; Sellers, W.R.; Meyerson, M.; Frank, D.A. Signal transducer and activator of transcription 3 is required for the oncogenic effects of non-small-cell lung cancer-associated mutations of the epidermal growth factor receptor. Cancer Res. 2006, 66, 3162–3168. [Google Scholar] [CrossRef]
- Greulich, H.; Chen, T.H.; Feng, W.; Janne, P.A.; Alvarez, J.V.; Zappaterra, M.; Bulmer, S.E.; Frank, D.A.; Hahn, W.C.; Sellers, W.R.; et al. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med. 2005, 2, e313. [Google Scholar] [CrossRef]
- Gao, S.P.; Mark, K.G.; Leslie, K.; Pao, W.; Motoi, N.; Gerald, W.L.; Travis, W.D.; Bornmann, W.; Veach, D.; Clarkson, B.; et al. Mutations in the EGFR kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. J. Clin. Invest. 2007, 117, 3846–3856. [Google Scholar] [CrossRef]
- Mendelsohn, J.; Baselga, J. Status of epidermal growth factor receptor antagonists in the biology and treatment of cancer. J. Clin. Oncol. 2003, 21, 2787–2799. [Google Scholar] [CrossRef]
- Inoue, A.; Suzuki, T.; Fukuhara, T.; Maemondo, M.; Kimura, Y.; Morikawa, N.; Watanabe, H.; Saijo, Y.; Nukiwa, T. Prospective phase II study of gefitinib for chemotherapy-naive patients with advanced non-small-cell lung cancer with epidermal growth factor receptor gene mutations. J. Clin. Oncol. 2006, 24, 3340–3346. [Google Scholar] [CrossRef]
- Yu, H.A.; Arcila, M.E.; Rekhtman, N.; Sima, C.S.; Zakowski, M.F.; Pao, W.; Kris, M.G.; Miller, V.A.; Ladanyi, M.; Riely, G.J. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. 2013, 19, 2240–2247. [Google Scholar] [CrossRef]
- Kim, S.M.; Kwon, O.J.; Hong, Y.K.; Kim, J.H.; Solca, F.; Ha, S.J.; Soo, R.A.; Christensen, J.G.; Lee, J.H.; Cho, B.C. Activation of IL-6R/JAK1/STAT3 signaling induces de novo resistance to irreversible EGFR inhibitors in non-small cell lung cancer with T790M resistance mutation. Mol. Cancer Ther. 2012, 11, 2254–2264. [Google Scholar] [CrossRef]
- Slinger, E.; Maussang, D.; Schreiber, A.; Siderius, M.; Rahbar, A.; Fraile-Ramos, A.; Lira, S.A.; Soderberg-Naucler, C.; Smit, M.J. HCMV-encoded chemokine receptor US28 mediates proliferative signaling through the IL-6-STAT3 axis. Sci. Signal. 2010, 3, ra58. [Google Scholar]
- Blackwell, T.S.; Christman, J.W. The role of nuclear factor-kappa B in cytokine gene regulation. Am. J. Respir. Cell Mol. Biol. 1997, 17, 3–9. [Google Scholar] [CrossRef]
- Meads, M.B.; Gatenby, R.A.; Dalton, W.S. Environment-mediated drug resistance: A major contributor to minimal residual disease. Nat. Rev. Cancer 2009, 9, 665–674. [Google Scholar] [CrossRef]
- Pu, Y.S.; Hour, T.C.; Chuang, S.E.; Cheng, A.L.; Lai, M.K.; Kuo, M.L. Interleukin-6 is responsible for drug resistance and anti-apoptotic effects in prostatic cancer cells. Prostate 2004, 60, 120–129. [Google Scholar] [CrossRef]
- Conze, D.; Weiss, L.; Regen, P.S.; Bhushan, A.; Weaver, D.; Johnson, P.; Rincon, M. Autocrine production of interleukin 6 causes multidrug resistance in breast cancer cells. Cancer Res. 2001, 61, 8851–8858. [Google Scholar]
- Akira, S.; Taga, T.; Kishimoto, T. Interleukin-6 in biology and medicine. Adv. Immunol. 1993, 54, 1–78. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Yamamoto, Y.; Yokota, S.; Nakagawa, M.; Ito, M.; Ogura, T. Involvement of interleukin-6 in the elevation of plasma fibrinogen levels in lung cancer patients. Jpn. J. Clin. Oncol. 1998, 28, 740–744. [Google Scholar] [CrossRef]
- Yanagawa, H.; Sone, S.; Takahashi, Y.; Haku, T.; Yano, S.; Shinohara, T.; Ogura, T. Serum levels of interleukin 6 in patients with lung cancer. Br. J. Cancer 1995, 71, 1095–1098. [Google Scholar] [CrossRef]
- Harada, D.; Takigawa, N.; Ochi, N.; Ninomiya, T.; Yasugi, M.; Kubo, T.; Takeda, H.; Ichihara, E.; Ohashi, K.; Takata, S.; et al. JAK2-related pathway induces acquired erlotinib resistance in lung cancer cells harboring an epidermal growth factor receptor-activating mutation. Cancer Sci. 2012, 103, 1795–1802. [Google Scholar] [CrossRef]
- Li, R.; Hu, Z.; Sun, S.Y.; Chen, Z.G.; Owonikoko, T.K.; Sica, G.L.; Ramalingam, S.S.; Curran, W.J.; Khuri, F.R.; Deng, X. Niclosamide overcomes acquired resistance to erlotinib through suppression of STAT3 in non-small cell lung cancer. Mol. Cancer Ther. 2013, 12, 2200–2212. [Google Scholar] [CrossRef]
- Garcia, R.; Bowman, T.L.; Niu, G.; Yu, H.; Minton, S.; Muro-Cacho, C.A.; Cox, C.E.; Falcone, R.; Fairclough, R.; Parsons, S.; et al. Constitutive activation of Stat3 by the Src and JAK tyrosine kinases participates in growth regulation of human breast carcinoma cells. Oncogene 2001, 20, 2499–2513. [Google Scholar] [CrossRef]
- Huang, M.; Dorsey, J.F.; Epling-Burnette, P.K.; Nimmanapalli, R.; Landowski, T.H.; Mora, L.B.; Niu, G.; Sinibaldi, D.; Bai, F.; Kraker, A.; et al. Inhibition of Bcr-Abl kinase activity by PD180970 blocks constitutive activation of Stat5 and growth of CML cells. Oncogene 2002, 21, 8804–8816. [Google Scholar] [CrossRef]
- Levis, M.; Allebach, J.; Tse, K.F.; Zheng, R.; Baldwin, B.R.; Smith, B.D.; Jones-Bolin, S.; Ruggeri, B.; Dionne, C.; Small, D. A FLT3-targeted tyrosine kinase inhibitor is cytotoxic to leukemia cells in vitro and in vivo. Blood 2002, 99, 3885–3891. [Google Scholar] [CrossRef]
- Morishita, R.; Tomita, N.; Kaneda, Y.; Ogihara, T. Molecular therapy to inhibit NFkappaB activation by transcription factor decoy oligonucleotides. Curr. Opin. Pharmacol. 2004, 4, 139–146. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, J.; Wang, L.; Wei, H.; Tian, Z. Therapeutic effects of STAT3 decoy oligodeoxynucleotide on human lung cancer in xenograft mice. BMC Cancer 2007, 7, 149. [Google Scholar] [CrossRef]
- Leong, P.L.; Andrews, G.A.; Johnson, D.E.; Dyer, K.F.; Xi, S.; Mai, J.C.; Robbins, P.D.; Gadiparthi, S.; Burke, N.A.; Watkins, S.F.; et al. Targeted inhibition of Stat3 with a decoy oligonucleotide abrogates head and neck cancer cell growth. Proc. Natl. Acad. Sci. USA 2003, 100, 4138–4143. [Google Scholar] [CrossRef]
- Sen, M.; Thomas, S.M.; Kim, S.; Yeh, J.I.; Ferris, R.L.; Johnson, J.T.; Duvvuri, U.; Lee, J.; Sahu, N.; Joyce, S.; et al. First-in-human trial of a STAT3 decoy oligonucleotide in head and neck tumors: Implications for cancer therapy. Cancer Discov. 2012, 2, 694–705. [Google Scholar] [CrossRef]
- Hedvat, M.; Huszar, D.; Herrmann, A.; Gozgit, J.M.; Schroeder, A.; Sheehy, A.; Buettner, R.; Proia, D.; Kowolik, C.M.; Xin, H.; et al. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell 2009, 16, 487–497. [Google Scholar] [CrossRef]
- Puchalski, T.; Prabhakar, U.; Jiao, Q.; Berns, B.; Davis, H.M. Pharmacokinetic and pharmacodynamic modeling of an anti-interleukin-6 chimeric monoclonal antibody (siltuximab) in patients with metastatic renal cell carcinoma. Clin. Cancer Res. 2010, 16, 1652–1661. [Google Scholar]
- Kim, M.J.; Nam, H.J.; Kim, H.P.; Han, S.W.; Im, S.A.; Kim, T.Y.; Oh, D.Y.; Bang, Y.J. OPB-31121, a novel small molecular inhibitor, disrupts the JAK2/STAT3 pathway and exhibits an antitumor activity in gastric cancer cells. Cancer Lett. 2013, 335, 145–152. [Google Scholar] [CrossRef]
- Ioannidis, S.; Lamb, M.L.; Wang, T.; Almeida, L.; Block, M.H.; Davies, A.M.; Peng, B.; Su, M.; Zhang, H.J.; Hoffmann, E.; et al. Discovery of 5-chloro-N2-[(1s)-1-(5-fluoropyrimidin-2-yl)ethyl]-N4-(5-methyl-1h-pyrazol-3-yl)p yrimidine-2,4-diamine (AZD1480) as a novel inhibitor of the Jak/Stat pathway. J. Med. Chem. 2011, 54, 262–276. [Google Scholar] [CrossRef]
- Haghikia, A.; Hoch, M.; Stapel, B.; Hilfiker-Kleiner, D. STAT3 regulation of and by microRNAs in development and disease. JAKSTAT 2012, 1, 143–150. [Google Scholar]
- Lin, H.Y.; Chiang, C.H.; Hung, W.C. STAT3 upregulates miR-92a to inhibit RECK expression and to promote invasiveness of lung cancer cells. Br. J. Cancer 2013, 109, 731–738. [Google Scholar] [CrossRef]
- Dai, B.; Meng, J.; Peyton, M.; Girard, L.; Bornmann, W.G.; Ji, L.; Minna, J.D.; Fang, B.; Roth, J.A. STAT3 mediates resistance to MEK inhibitor through microRNA miR-17. Cancer Res. 2011, 71, 3658–3668. [Google Scholar] [CrossRef]
- Vishwamitra, D.; Li, Y.; Wilson, D.; Manshouri, R.; Curry, C.V.; Shi, B.; Tang, X.M.; Sheehan, A.M.; Wistuba, I.I.; Shi, P.; et al. MicroRNA 96 is a post-transcriptional suppressor of anaplastic lymphoma kinase expression. Am. J. Pathol. 2012, 180, 1772–1780. [Google Scholar] [CrossRef]
- Du, L.; Subauste, M.C.; DeSevo, C.; Zhao, Z.; Baker, M.; Borkowski, R.; Schageman, J.J.; Greer, R.; Yang, C.R.; Suraokar, M.; et al. miR-337–3p and its targets STAT3 and RAP1A modulate taxane sensitivity in non-small cell lung cancers. PLoS One 2012, 7, e39167. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Harada, D.; Takigawa, N.; Kiura, K. The Role of STAT3 in Non-Small Cell Lung Cancer. Cancers 2014, 6, 708-722. https://doi.org/10.3390/cancers6020708
AMA Style
Harada D, Takigawa N, Kiura K. The Role of STAT3 in Non-Small Cell Lung Cancer. Cancers. 2014; 6(2):708-722. https://doi.org/10.3390/cancers6020708
Chicago/Turabian StyleHarada, Daijiro, Nagio Takigawa, and Katsuyuki Kiura. 2014. "The Role of STAT3 in Non-Small Cell Lung Cancer" Cancers 6, no. 2: 708-722. https://doi.org/10.3390/cancers6020708