1. Introduction
Merkel Cell Polyomavirus (MCPyV) is the first human polyomavirus to be linked to a human cancer, Merkel Cell Carcinoma (MCC) [
1,
2,
3]. Since its discovery in 2008, this virus has been found clonally integrated in a majority of MCC tumors. In most MCPyV-related MCC tumors, the major viral protein, Large T antigen (LT), has been mutated such that it is expressed in a truncated form [
4]. These tumor-derived truncated LT proteins retain their binding sites for retinoblastoma protein (pRb) and DnaJ family heatshock proteins, thereby driving proliferation [
1,
4]. Indeed, at least one study has shown that tumors with knocked-down LT protein regress rapidly in a xenograft model [
5]. Both LT and the splice variant, small T antigen (sT) have been shown to have oncogenic properties [
4,
5,
6,
7], although the precise contribution of each protein to the process of tumorigenesis is still unclear.
MCPyV appears to be a natural resident of the skin microflora, and is acquired early in life [
8,
9,
10]. While considerable efforts have been made to better understand MCPyV’s oncogenic potential, especially in MCC tumors, comparatively little work has been done to better understand this virus’s basic life cycle. The lack of a relevant cell culture system for propagating virus has made investigations of its basic virology difficult; however, ectopic expression of MCPyV LT in cell lines such as HEK 293, C33A and U2OS has been valuable in enhancing our understanding of MCPyV’s interaction with host cells [
11,
12,
13,
14].
Our lab has previously characterized MCPyV LT’s interaction with the host cell to stimulate replication [
14]. In that study we showed by immunoflourescent staining (IF), fluorescent
in situ hybridization (FISH) and BrdU staining that MCPyV LT proteins form large nuclear foci which contain actively replicating plasmids carrying the viral origin of replication (Ori). We also showed that several cellular factors colocalize to these foci, including: the double bromodomain protein, Brd4, the PCNA loading protein replication factor 1 (RFC1), and the single-stranded DNA binding protein RPA70. In another study we demonstrated that full length MCPyV LT activates host DNA damage response (DDR) pathways and dramatically alters the host cell cycle [
12]. Additionally, members of the DDR pathway were seen to colocalize with nuclear foci containing actively replicating viral genomes, potentially contributing to viral replication [
13]. While it is still unclear whether DDR activation and recruitment upon LT expression is a side-effect of active viral replication and/or LT helicase activity, or if this activation is being actively subverted and manipulated by MCPyV, the link between MCPyV LT expression and DDR activation is well established. This DDR activity, coupled with LT’s ability to dramatically alter the host cell cycle, may provide enough low-level genomic instability to lead to integration of its genome into the host cell genome, which appears to occur in the majority of MCPyV-related MCC tumors studied to date.
Merkel cells may not represent the natural host cell of MCPyV and may pre-dispose MCPyV to randomly integrate its genome. Indeed, the prototypical polyomavirus, Simian Virus 40 (SV40) has a transforming phenotype in cell lines that are non-permissive for viral replication [
15]; Merkel cells may similarly represent a non-permissive host for MCPyV. A better understanding of how MCPyV replication is regulated would provide a clearer framework for understanding how infection may be altered in Merkel cells and lead to integration of the mutated viral genome.
SV40 LT has been a model for understanding eukaryotic replication for decades [
16]. SV40 LT is recruited to the viral Ori through its origin binding domain (OBD), which recognizes GAGGC pentanucleotide repeats arranged symmetrically within the Ori. LT then oligomerizes into two hexameric protein complexes arranged in a head-to-head fashion. The C-terminal helicase domains make non-specific contacts with an extended palindrome and an A/T rich tract flanking the central pentanucleotide repeats; these become the initial sites of unwinding. LT then acts as a helicase to unwind the viral genome and recruits cellular factors to begin replication [
16,
17].
Phosphorylation has been a well-established mechanism by which SV40 LT replication is regulated [
18]. T124 was identified as a critical residue for regulating SV40 LT-mediated viral replication; removal of this phosphorylation either biochemically or genetically abrogated replication [
19,
20,
21,
22]. Intensive biochemical studies demonstrated that this phosphorylation plays an important role in mediating interactions between both hexamers at the Ori. Alanine mutants are defective in forming double-hexamer complexes and unwinding the viral origin [
23,
24].
Somewhat paradoxically, early biochemical analyses of purified SV40 LT seemed to indicate that phosphatase treatment could actually stimulate viral replication [
25,
26]. It was later clarified that, in addition to phosphorylation at T124, there are serine phosphorylation modifications nearby which have an inhibitory effect on viral replication [
27]. These phosphorylation events seem to accumulate throughout the course of infection [
28]. These observations led to a model where T124 phosphorylation stimulates replication, while subsequent phosphorylation at neighboring serines dampen this effect, potentially altering LT’s activity on the viral genome to favor transcription of the capsid genes [
18].
No such analysis of MCPyV LT phosphorylation has yet been reported. We sought to provide an initial framework for understanding the regulation of MCPyV LT’s functions by performing a proteomic analysis to search for relevant phosphorylation sites. Our studies identify three phosphorylation marks on MCPyV LT; T271, T297 and T299. We found that T271 had no effect on replication, while T297 and T299 phosphorylation had antagonistic effects. Both T297 and T299 altered the binding affinity of MCPyV for the viral Ori while leaving unwinding and helicase functions largely intact. Taken together, our data reveal a dynamic interplay between multiple phosphorylation sites, which together regulate LT’s ability to initiate replication at the viral origin.
3. Experimental
3.1. Cell Lines, Cell Culture and Transfection
HEK 293 and C33A cell lines were maintained in Dulbecco’s Modified Eagle Medium (DMEM; Invitrogen, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (Hyclone, Logan, UT, USA). Cells were transfected using the calcium phosphate method as described previously [
14].
3.2. Recombinant Plasmids
Construction of the pcDNA4c-MCPyV LT and pcDNA4c-MCPyV Ori plasmids have been described previously [
14]. For the phospho-mutant constructs T271A, T297A and T299A, synthetic oligonucleotides containing the desired mutation were annealed with denatured template plasmid pcDNA4c-MCPyV LT and extended with
Pfu Turbo polymerase (Agilent Technologies, Santa Clara, CA, USA) by QuikChange PCR following the manufacturer’s instructions. Unmutated plasmid DNA templates were removed by DpnI digestion, and the remaining DNA was used to transform DH5α competent cells. To generate IgG-IgG-TEV (IIT) affinity-tagged constructs, a DNA sequence encoding two IgG binding domains of
Staphylococcus aureus protein A and a TEV protease cleavage site was fused in frame to the N terminus of MCPyV LT constructs using the KpnI site. All constructs were confirmed by DNA sequencing.
3.3. Antibodies
The following antibodies were used for immunoflourescent staining: mouse anti-Xpress (R910-25, Invitrogen), rabbit anti-RFC1 (H-300, Santa Cruz, Dallas, TX, USA), rabbit anti-RPA70 (2267, Cell Signaling, Mölndal, Sweden), Alexa Fluor 594 goat anti-rabbit IgG (A11012, Invitrogen), and Alexa Fluor 488 goat anti-mouse IgG (A11001, Invitrogen). The polyclonal rabbit anti-Brd4CA recognizes aa 1313–1362 [
14]. Antibodies used for western blotting include: mouse anti-MCPyV LT (CM2B4, Santa Cruz), mouse anti-glyceraldehyde 3-phosphate-dehydrogenase (GAPDH) (G8140-01, US Biological, Salem, MA, USA) and HRP-conjugated horse anti-mouse IgG (7076, Cell Signaling, Danvers, MA, USA).
3.5. Mass Spectrometry Analysis
The mass spectrometry analysis was provided by the Proteomics Core Facility, University of Pennsylvania. Protein samples were digested with trypsin as described by Strader
et al. [
34]. Digested peptides were then purified by liquid chromatography using standard purification techniques or with a titanium oxide column (GE Healthcare Biosciences, Pittsburgh, PA, USA). Peptides were analyzed by nanoLC/MS/MS with a LTQ Mass Spectrometer (Thermo Scientific, Waltham, MA, USA) equipped with a nano LC-2D HPLC system (Eksigent, Framingham, MA, USA). The data were analyzed with Sequest and Scaffold software.
3.6. Sequence Alignment of Polyomavirus LT Proteins
Clone Manager 9 software was used to align the amino acid sequences of various polyomavirus LT proteins. Amino acid sequences were aligned using the Mult-Way view and BLOSUM 62 scoring matrix. Polyomavirus LT sequences and their NCBI accession numbers are as follows: Merkel Cell Polyomavirus LT, R17a isolate (HM011555.1); Murine Polyomavirus LT (NC_001515.1); African Green Monkey Polyomavirus LT (NC_004763.2); WU Polyomavirus LT (NC_009539.1); KI Polyomavirus LT (NC_009238.1); BK Polyomavirus LT (NC_001538.1); JC Polyomavirus LT (NC_001699.1); SV40 LT (NC_001669.1).
3.7. Phyre2 Modeling of MCPyV LT Fragments
We modeled MCPyV LT amino acids 290–433 using Phyre2 software’s intensive mode [
35]. The PDB file was then visualized using PyMOL software [
36]. The model was oriented using the origin binding domain crystal structure reported by Harrison and colleagues as a reference [
30].
3.8. Immunoflourescent (IF) Staining
C33A cells were fixed with 3% paraformaldehyde in PBS for 20 min at 4 °C. Cells were incubated in blocking/permeabilization buffer (0.5% Triton X-100 and 3% BSA in PBS) for 10 min at room temperature and stained with specific primary antibodies (as described in the legends) at room temperature for 60 min. After incubation, the cells were washed three times with blocking/permeabilization buffer and incubated with Alexa Fluor 594 goat anti-rabbit IgG and 488 goat anti-mouse IgG (Invitrogen, Molecular Probes, Ashburn, VA, USA) for an additional 60 min. After incubation with the secondary antibodies, cells were counterstained with DAPI (4',6'-diamidino-2-phenylindol) and examined with an Olympus IX81 inverted fluorescence microscope.
3.9. Microscopy and Image Analysis
All immunofluorescent images were collected using an inverted fluorescence microscope (Olympus IX81) connected to a high-resolution charge-coupled device camera (QImaging, FAST1394). Images were analyzed and presented using SlideBook 5.0 software (Intelligent Imaging Innovations, Inc., Denver, CO, USA). The scale bars were added using ImageJ software.
3.10. Southern Blotting
Replication assays were performed as described previously [
13]. Briefly, MCPyV LT constructs were transfected into C33A cells using the calcium phosphate method. Forty-eight hours post-transfection, whole genomic DNA was extracted. 15 µg total DNA was digested with BamHI, treated with or without DpnI at 37 °C overnight, and separated on a 0.7% agarose gel. DNA was transferred to a Hybond-N+ nitrocellulose membrane (Amersham, Piscataway, NJ, USA) and hybridized with a pcDNA4c-MCPyV Ori probe labeled with [α-
32P] dCTP using Prime-It II random primer labeling kit (Agilent Technologies) per the manufacturer’s instructions. The results were analyzed using a Phosphorimager (Typhoon 9400; GE Healthcare).
3.11. Western Blotting
Cells were lysed in hypertonic lysis buffer (10 mM HEPES [pH 7.9], 500 mM NaCl, 3 mM MgCl2, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, and protease inhibitors) by passage through a 22-gauge needle 10 times. After a 20-min incubation on ice, the soluble and insoluble fractions were separated by centrifugation at 5,000 rpm for 10 min at 4 °C. The supernatants (20 μg) were resolved on an SDS-PAGE gel and transferred to PVDF membrane. Membranes were blocked in 5% PBST-milk for 1 h at room temperature and incubated in PBST-milk containing primary antibodies at room temperature for 1 h. After washing three times with PBST, membranes were then incubated with HRP-conjugated secondary antibodies in PBST-milk for 1 h at room temperature. Western blots were developed using ECL solution and images were captured using a Fuji imaging system.
3.12. Affinity Purification of MCPyV LT
HEK 293 cells were transfected with constructs expressing IIT-tagged MCPyV LT (wild-type or mutant) using the calcium phosphate method. Forty-eight hours post-transfection, nuclear extracts were prepared. Briefly, cells were resuspended in Buffer A (10 mM HEPES [pH 7.9], 10 mM KCl, 0.1 mM EDTA, 0.2 mM PMSF and protease inhibitors). Cells were swollen on ice for 10 min before NP-40 was added to a final concentration of 6% and then vortexed for 10 s. Nuclei were pelleted by centrifugation at 1200× g at 4 °C for 5 min. Nuclei were resuspended in Buffer B (20 mM HEPE [pH 7.9], 400 mM NaCl, 20% glycerol, 1 mM EDTA, 0.2mM PMSF and protease inhibitors) and lysed by passing through a 22-gauge needle 10 times. Lysates were clarified at 20,000× g at 4 °C for 15 min. The supernatant was then immunopurified with IgG-Sepharose 6 Fast Flow (GE Healthcare) beads pre-blocked with 1% BSA for 2 h at 4 °C. Bound immune complexes were washed three times with IP 150 buffer (10 mM Tris-HCl [pH 8.0], 150 mM NaCl, 0.1% NP-40) and once with TEV cleavage buffer (50 mM Tris-HCl [pH 8.0], 6% glycerol, 0.5 mM EDTA, 0.5 mM DTT, 1 mM PMSF). Bound proteins were then cleaved with TEV protease (Invitrogen) in TEV cleavage buffer, overnight at 4 °C. The beads were spun down and the supernatant collected for further biochemical analysis.
3.13. Electromobility Shift Assays (EMSA)
The EMSA probe was generated by PCR amplifying a portion of pcDNA4c-MCPyV Ori (Forward Primer: TTG GCA GAG GCT TGG GGC TCC, Reverse Primer: GCG GAA TTC TAA GCC TCT TAA GCC TC). The PCR product was purified using a Qiagen PCR Purification Kit (Cat# 28104) following the manufacturer’s instructions. The purified probe (100 ng) was then 5' labeled with [32P-γ] ATP with T4 Polynucleotide Kinase (New England Biolabs, Ipswich, MA, USA) following the manufacturer’s instructions. The labeled probe was then diluted 1:50 before being used in the EMSA.
Binding reactions (20 µL) were assembled on ice. Various amounts of affinity purified MCPyV LT was mixed with 40 fmol labeled probe in binding buffer (30 mM Tris-HCl [pH 8.0], 10% glycerol, 0.5 µg BSA, 10 ng poly d(I-C), 40 ng Sonicated Salmon Sperm DNA, 5 mM AMP-PNP, 1 mM DTT, and 1 mM PMSF). Reactions were incubated at room temperature for 25 min and then separated on a 4% non-denaturing polyacrylamide gel in 0.5 × TBE at 120 V for 3 h. The gel was dried and subjected to autoradiography. The non-hydrolyzable ATP analogue, adenylyl imidodiphosphate (AMP-PNP) was obtained from Sigma (St. Louis, MO, USA, Cat# A2647).
3.14. Unwinding Assay
The probe was generated by annealing two oligos (80 ng each) spanning the central portion of the MCPyV Ori sequence (
Figure 6A) (Top Strand Oligo: GTG ACT TTT TTT TTT CAA GTT GGC AGA GGC TTG GGG CTC CTA GCC TCC GAG GCC TCT GGA AAA AAA AGA GAG AGG CC; Bottom Strand Oligo: CAG AGG CCT CTC TCT TTT TTT TCC AGA GGC CTC GGA GGC TAG GAG CCC CAA GCC TCT GCC AAC TTG AAA AAA AAA AGT CAC). The four-nucleotide overhang was filled in using Klenow DNA Polymerase (New England Biolabs) by mixing the duplexed probe in a reaction containing 0.1 mM dTTP, dGTP, and [
32P-α] dCTP and incubating at room temperature for 20 min. After adding dATP to a final concentration of 0.1mM, reactions were incubated for another 20 min at room temperature.
Various amounts of purified MCPyV LT were combined with 60 pg of labeled probe in 20 µL unwinding reaction buffer (30 mM HEPES [pH 8.0], 0.1 mg/mL BSA, 7 mM MgCl2, 4 mM ATP, 40 mM creatine phosphate, 25 µg/mL creatine phosphate kinase). Reactions were carried out at room temperature for 1 h. Reactions were stopped by adding 5 µL 5 × Stop Buffer (2.5% SDS, and 100 mM EDTA). Loading dye (bromophenol blue, 4% sucrose and 1XTBE) was added and samples were separated on an 11% non-denaturing PAGE in 0.5XTBE. Gels were dried and exposed for autoradiography.
3.15. Helicase Assay
The helicase assay was performed as previously described with minor modification [
12]. Wild-type or mutant MCPyV LT fused to an IIT tag was expressed in HEK 293 cells and purified using IgG Sepharose 6 Fast Flow (GE Healthcare), which were preblocked with 1% BSA in PBS at 4 °C for >1 h. Beads with bound LT were split into two equal fractions for SDS-PAGE/Coomassie brilliant blue staining and helicase assays, respectively. To label the helicase assay substrate, 35 ng of a 31-mer oligo (5'-CCA GGG TTT TCC CAG TCA CGA CGT TGT AAA C-3') was annealed to 1 μg of M13mp18 DNA (New England BioLabs). The primer was then elongated using Klenow polymerase (New England BioLabs) in a 50 μL reaction containing 0.1 mM dCTP, dGTP, and [α-
32P] dATP. After 20 min of incubation at room temperature, 0.1 mM dATP was added for 20 min. Then, 0.5 μL of labeled substrate was used in each reaction. LT purified on IgG Sepharose was incubated with the substrate in helicase assay buffer (20 mM Tris-HCl [pH 7.5], 10 mM MgCl
2, 1 mM DTT, 0.1 mg/mL BSA, and 5 mM ATP) at 37 °C for 30 min. The reaction was stopped by adding SDS to a final concentration of 0.2% and EDTA to 50 mM. Total reaction mixtures were resolved by electrophoresis on 11% non-denaturing polyacrylamide gels. The gels were dried and subjected to autoradiography.
3.16. Statistical Analyses
Prism software was used to perform a one-way ANOVA test. A p < 0.05 was considered statistically significant.
4. Discussion
Merkel Cell Polyomavirus is the first human polyomavirus linked to a human cancer. As such, it has garnered a considerable amount of interest, especially with regards to its oncogenic potential and its causative role in MCC. Much of the basic virology of MCPyV, in contrast, has been lacking, in large part due to the difficulty in propagating the virus and the lack of a natural host cell line. Previous work from our lab has established that MCPyV LT interacts with the host DNA damage response machinery, potentially to regulate viral genome replication [
13]. Understanding how polyomavirus replication is regulated will be critical for understanding the very early steps of MCPyV-induced transformation and oncogenesis.
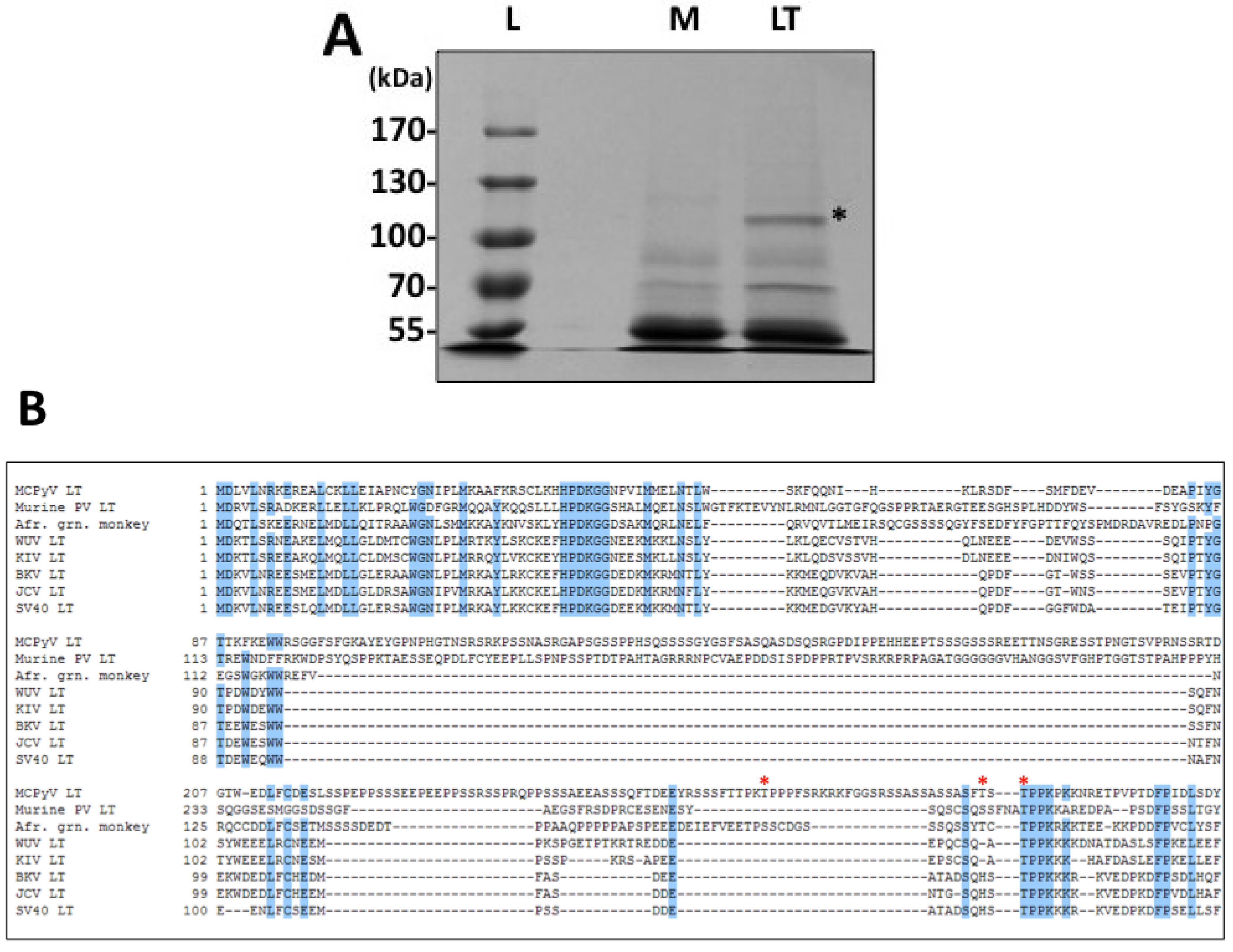
Phosphorylation has been a well-established mechanism of regulation for SV40 LT activities, especially for genome replication. In addition, MCPyV LT has a unique stretch of amino acids that is rich in serines and threonines, offering many new potential phosphorylation sites and therefore mechanisms of regulation. To search for relevant sites in a relatively unbiased fashion, we performed a proteomic analysis of ectopically expressed MCPyV LT (
Figure 1 and
Supplementary Table S1). This analysis identified three threonines that are likely phosphorylated when MCPyV LT is expressed: T271, T297 and T299. We generated alanine substitutions of these sites to probe their function.
T271 was immediately interesting to us for a variety of reasons. It was independently identified in multiple peptides in both standard and titanium oxide purifications (
Supplementary Table S1), providing us with a high degree of confidence that this site is phosphorylated in cells. More intriguing, this site is located in the unique region of MCPyV LT (aa 95–290). It does not have any homologies to other polyomavirus LT proteins analyzed (
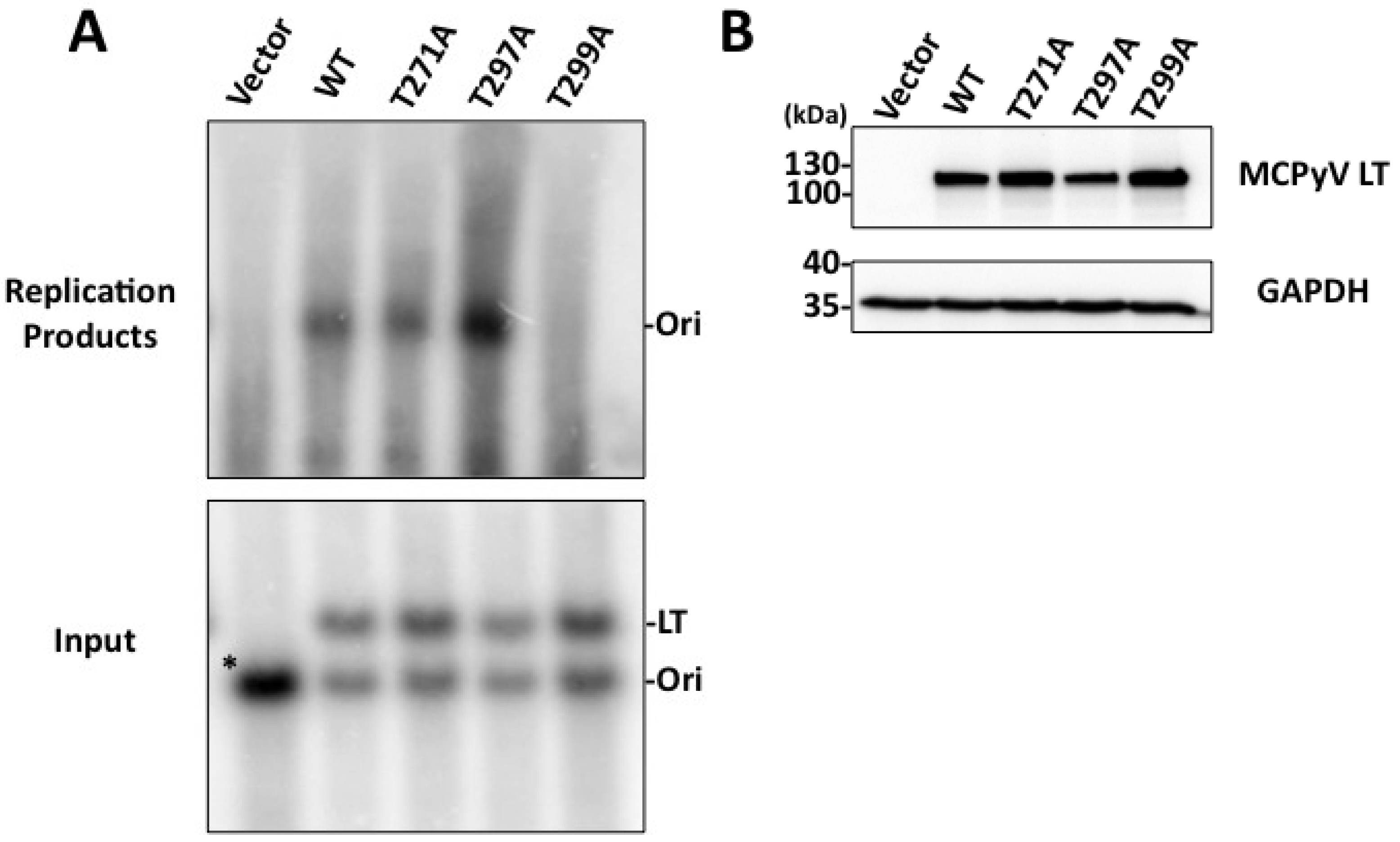
Figure 1B). We anticipate phosphorylation at this site may represent a novel function that MCPyV has acquired. Efforts thus far, however, have not revealed what those functions may be. The T271A mutant’s ability to bind Brd4 and activate the host DDR is similar to that seen for WT LT (data not shown). Additionally, this mutant was able to form replication foci and replicate plasmids containing the Ori almost as well as WT (
Figure 3 and
Figure 4). Additional experiments will be performed to identify its role in MCPyV infection.
T299, in contrast, is highly conserved among all polyomavirus LT proteins analyzed (
Figure 1B). This site is homologous to T124 in SV40 LT, which has been extensively studied for its role in regulating LT-mediated DNA replication. Alanine substitution of this site in SV40 LT completely abrogated replication. Biochemical analysis of T124A mutants showed that it had somewhat impaired double-hexamer interactions and unwinding activity [
23,
24]. More importantly, its ability to unwind duplex Ori DNA was abrogated while basic helicase activity remained unperturbed [
24]. In line with these findings, T299A in MCPyV LT also failed to replicate plasmids containing the viral Ori (
Figure 4) and did not form replication foci (
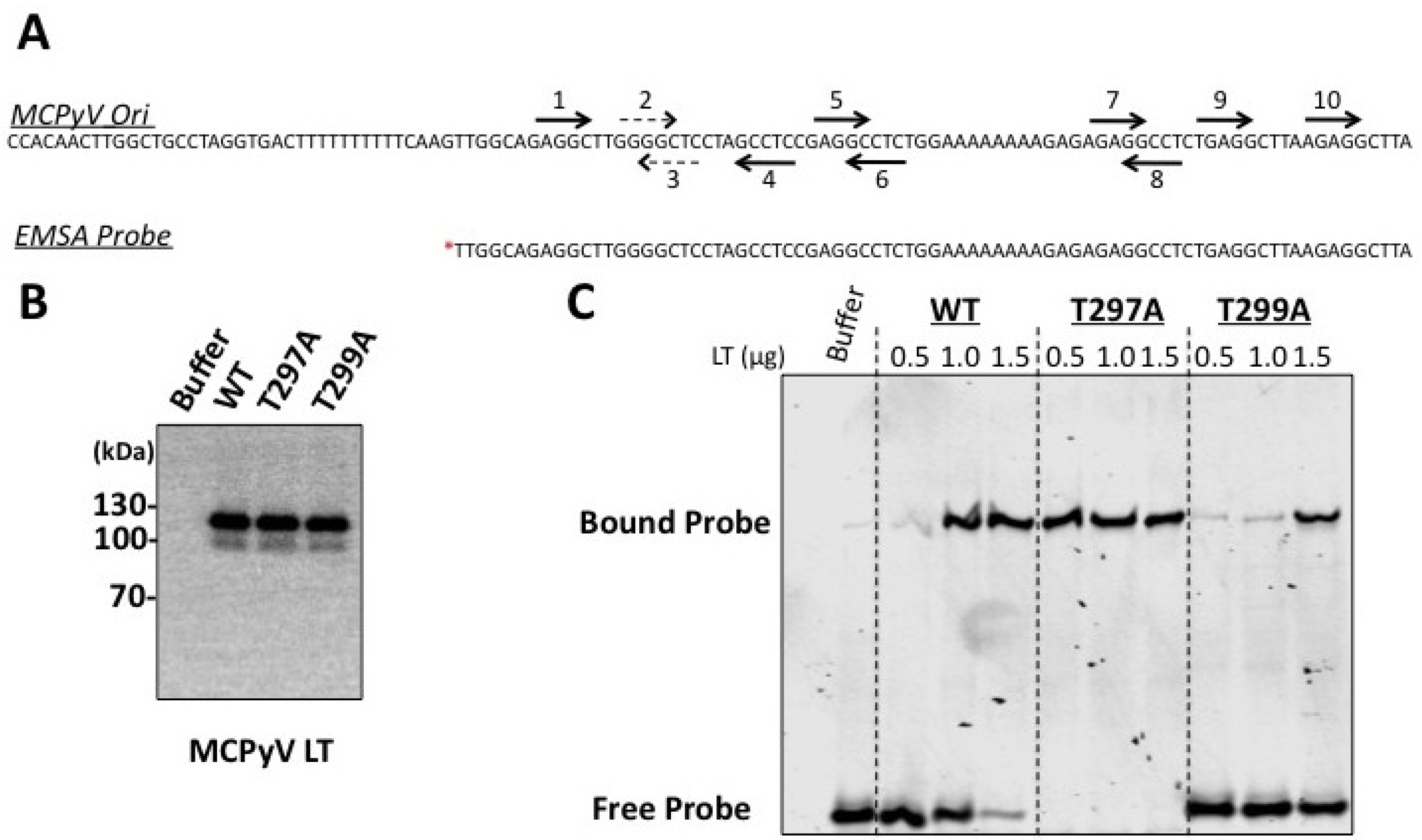
Figure 3). This mutant had a reduced capacity to bind the viral Ori in EMSA experiments (
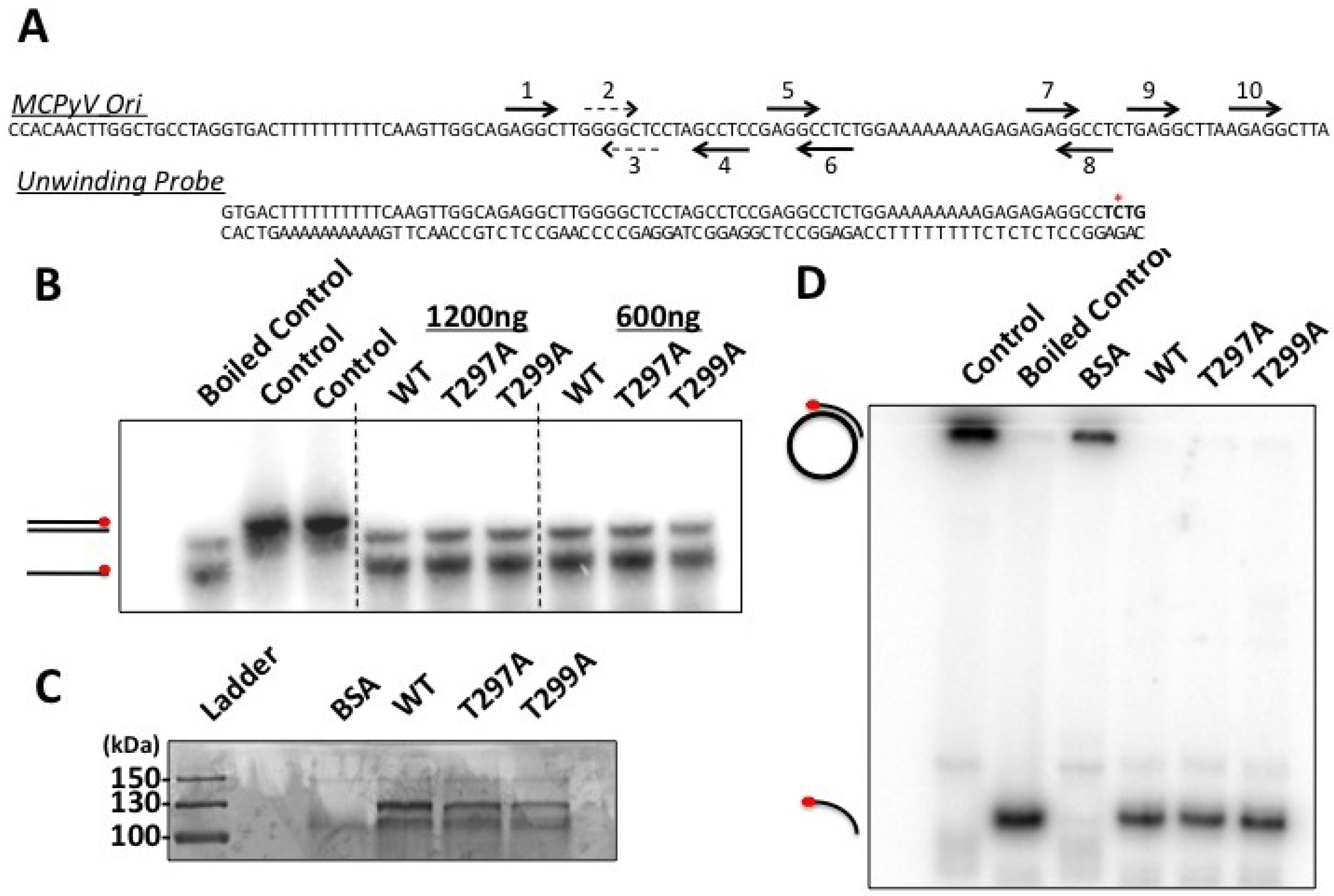
Figure 5). Although studies of the homologous LT mutant, T124A, in SV0 demonstrated that unwinding of the origin was attenuated, we were unable to reproduce this finding in our studies [
20]. While it is possible that MCPyV LT behaves differently from other polyomavirus LT’s, we believe technical limitations in our hands are more likely responsible for not seeing this phenotype in MCPyV T299A LT. Its helicase activity remained identical to wild-type (
Figure 6D), which has been reported for T124A LT in SV40 [
20]. Our EMSA studies did not indicate an attenuated double-hexamer phenotype as shown by Barbaro and colleagues for SV40 LT [
23], and the unwinding phenotype we observe is extremely subtle. It is possible our experimental conditions are not conducive for revealing these phenotypes, or (less likely) that T299 phosphorylation behaves in a slightly different biochemical manner than T124 in SV40 LT. We conclude that phosphorylation of T299 is required for MCPyV LT to initiate replication of its genome in ways similar to T124 phosphorylation in SV40.
T297 was not well conserved among the polyomavirus LT proteins analyzed. Modeling of this site seemed to indicate that this residue might face and even interact with DNA when the LT OBD engages the viral genome (
Figure 2A). We speculated this site might have an impact on DNA replication. Indeed, the T297A mutant had twice as many LT-positive nuclei exhibiting replication foci as WT LT (
Figure 3D). Supporting this observation, Southern blotting of
in cellulo replication products showed that this mutant replicated plasmids containing the viral origin to a very high degree (
Figure 4). Biochemical analyses revealed a strikingly robust affinity for the viral Ori (
Figure 5), while unwinding and helicase activities remained largely unaffected (
Figure 6). These data indicate that phosphorylation of this site would dramatically decrease LT’s capacity to bind the viral Ori, which would presumably limit its ability to initiate viral replication. These observations are in line with our structural model (
Figure 2) predicting that this site faces the OBD/DNA binding interface. The negative charge of a phosphate moiety at this site would presumably clash with the negatively charged phosphate-backbone of DNA, leading to reduced DNA binding. SV40 has also been reported to have phosphorylation sites that negatively impact replication. Phosphorylation at serines 120 and 123 was shown to have a negative effect on replication [
27]. Threonine 297 may provide a similar regulatory function for MCPyV LT. It is possible that phosphorylation at a site neighboring the stimulatory threonine (124 for SV40 LT, 299 for MCPyV LT) may be a general feature of polyomavirus LT proteins to limit Ori recognition and to provide a brake for viral replication.
We attempted to generate phosphomimetic mutants (threonine to aspartate or glutamate) of these sites to probe these dynamics more closely; however, these mutants behaved just like alanine substitutions (data not shown). Interestingly, a MCPyV LT expression construct containing both T297A and T299A mutations matched the T299A phenotype completely: it failed to form replication foci or replicate plasmids with viral Ori’s (data not shown). The T297A mutation would allow for enhanced binding of the origin (
Figure 5), but the T299A mutation, which likely acts at steps after Ori binding during initiation of replication, completely abrogated replication of this double mutant (data not shown).
Taken together, our data support a model where T299 and T297 phosphorylation act as antagonistic ON and OFF switches for replication, respectively. We would hypothesize that T299 is first phosphorylated to stimulate viral replication, while subsequent phosphorylation at threonine 297 would abrogate Ori recognition and presumably reduce viral genome replication, possibly in favor of late gene expression and/or packaging. Phosphatases may also play a role, either removing phosphates from T299 to halt replication or from T297 to allow replication to continue. Without antibodies specific for phosphorylation at these sites, it is difficult to track when these sites become phosphorylated during infection or to begin searching for the kinases that add these marks during infection. Analysis of the amino acid sequences of these sites offer some clues. For SV40, cdc2/CDK1 was shown
in vitro to be responsible for phosphorylation at T124, the homologue to MCPyV LT T299 [
19]. The residues surrounding both MCPyV LT T299 and SV40 LT T124 (TPPK for both viruses, see
Figure 1B) exhibit a classic cdc2/CDK1 consensus sequence (S-P-X-basic residue) [
37]. Although it was not directly tested here, it is likely that cdc2/CDK1 plays a role in phosphorylating T299 during MCPyV infection. Casein kinase II was also shown to phosphorylate nearby serine residues in SV40 LT
in vitro, which played a role in SV40 LT nuclear import [
38,
39,
40,
41]. Finally, ATM kinase has been shown to phosphorylate SV40 LT in this region as well, contributing to LT-mediated replication [
42]. The threnonines T271 and T297 identified in this study do not exhibit homologies to the known consensus sequence of either of these kinases, indicating that other kinases are likely involved. Finally, other MCPyV viral proteins, like sT antigen, 57 kT antigen and ALTO [
43], may affect when, where and how LT is phosphorylated during the viral life cycle. These questions should be explored further as more reagents and cell lines become available for MCPyV studies.
One of the hallmark features of MCPyV LT in MCC is that the protein frequently becomes mutated such that it is expressed in a truncated fashion [
4]. These truncations almost always delete the helicase domain and the OBD. Interestingly, the three phosphorylation sites identified in this study are almost always omitted from the truncated proteins as well. It has been hypothesized that LT becomes truncated to avoid replicating the integrated viral genome, which would presumably cause genomic instability [
1]. In line with this reasoning, at least one MCPyV related MCC tumor has been identified with a full-length LT protein but the integrated viral genome contains a mutated Ori that fails to support viral replication [
4,
31]. Given that mutation of T299 completely abolishes LT’s capacity to replicate the viral origin, it is interesting to note that this mutation has never been observed in any of the MCC cases studied thus far. It is possible that the OBD, helicase domain and/or extreme C-terminal domain contain additional activities beyond replication that are negatively selected out during MCC progression. Our previous studies have indicated that the C-terminal half of MCPyV LT interacts with the p53 pathway to maintain cells in a stalled S-phase, which may be conducive to viral genome replication but antithetical to tumorigenesis [
12]. Others have similarly postulated that the C-terminal domain contains activities beyond replication that are negatively selected during MCC oncogenesis [
44]. Further investigation of this region of the protein may provide a broader and more comprehensive understanding of how MCPyV LT manipulates the host cell, and how these activities become disrupted during MCC tumorigenesis.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





