Targeting MET Amplification as a New Oncogenic Driver

Abstract
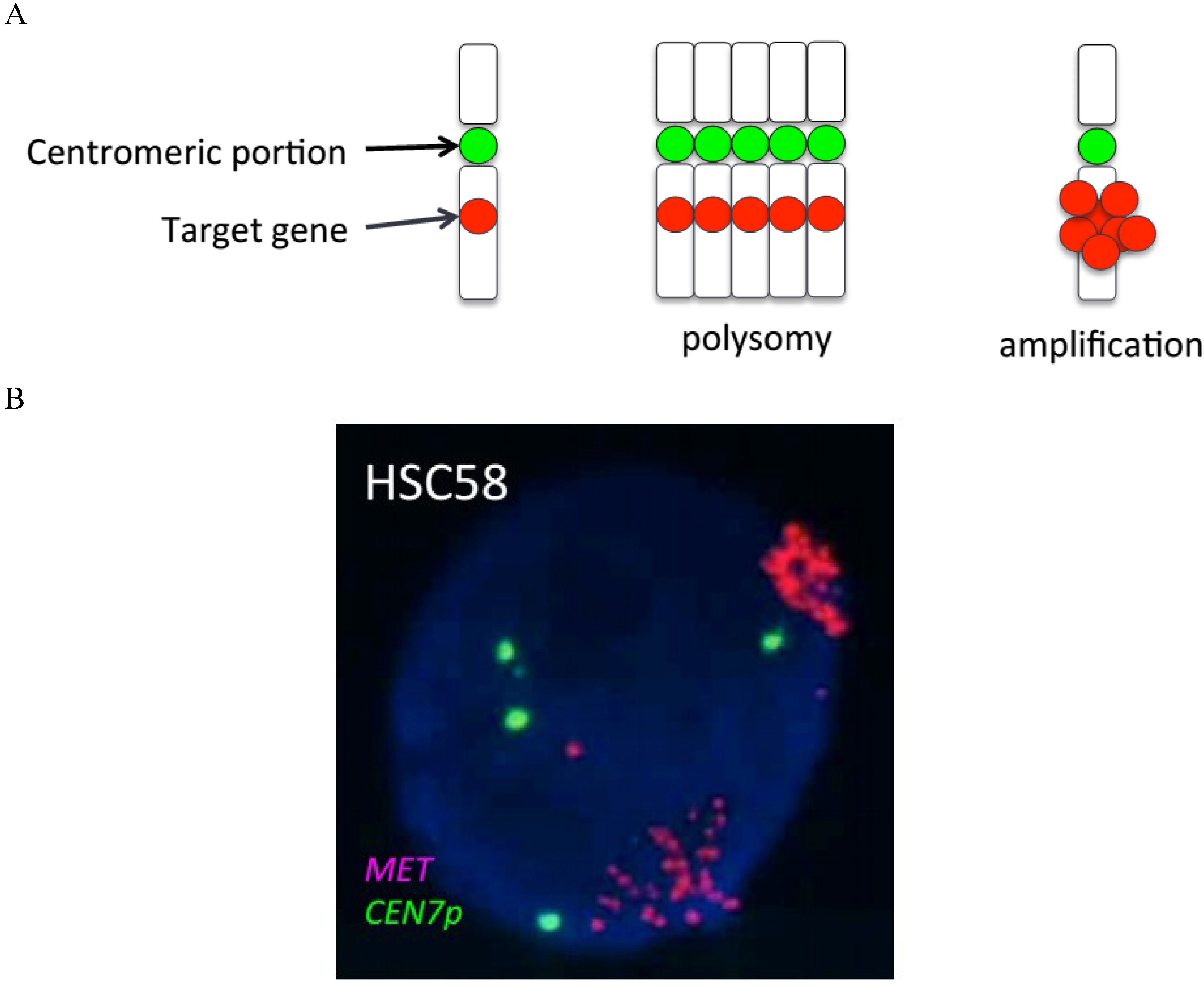
:1. Introduction
2. Preclinical Findings
3. Prevalence of MET Amplification in Cancer Patients

{kind=link}
| Study | Number of Patients | Technique | Classification | Positivity (%) |
|---|---|---|---|---|
| Camidge et al. (2010) [43] | 66 | FISH | MET/CEP7 ratio > 2.0 | 0 |
| Onozato et al. (2009) [33] | 148 | PCR based | GCN > 2 | 1.4 |
| Kubo et al. (2009) [34] | 100 | PCR based | GCN > 5 | 2.0 |
| Bean et al. (2007) [30] | 16 | PCR based | GCN > 5 | 3.0 |
| Go et al. (2010) [27] | 180 | FISH | MET/CEP7 ratio > 2.0 | 3.9 |
| Okamoto et al. (2014) [44] | 229 | FISH | MET/CEP7 ratio > 2.2 | 3.9 |
| Cappuzzo et al. (2009) [45] | 447 | FISH | MET/CEP7 ratio > 2.0 | 4.1 |
| Onitsuka et al. (2010) [32] | 183 | PCR based | GCN > 1.31 | 4.4 |
| Okuda et al. (2008) [31] | 213 | PCR based | GCN > 3 | 5.6 |
| Beau-Faller et al. (2008) [35] | 106 | PCR based | GCN > mean + 2SD of 30 normal lung DNA samples | 20.8 |
| Study | Number of Patients | Technique | Classification | Positivity (%) |
|---|---|---|---|---|
| Janjigian et al. (2011) [29] | 38 | FISH | MET/CEP7 ratio > 2.0 | 0 |
| Kawakami et al. (2013) [46] | 266 | FISH | MET/CEP7 ratio > 2.2 | 1.5 |
| Lennerz et al. (2011) [28] | 267 (junctional and gastric) | FISH | MET/CEP7 ratio > 2.2 | 2.2 |
| Hara et al. (1998) [20] | 154 | FISH | NA | 3.9 |
| Liu et al. (2014) [47] | 196 | FISH | MET/CEP7 ratio > 2.0 | 6.1 |
| Graziano et al. (2011) [40] | 216 | PCR based | GCN ≥ 5 | 9.7 |
| Tsugawa et al. (1998) [21] | 70 | Slot blot analysis | Ratio > 2 (relative to normal mucosa) | 10.0 |
| Nakajima et al. (1999) [19] | 128 | Southern blot analysis | Ratio > 2 (relative to normal mucosa) | 10.2 |
| Lee et al. (2011) [39] | 472 | PCR based | GCN ≥ 4 | 21.2 |
| Shi et al. (2012) [48] | 128 | PCR based | GCN ≥ 4 | 30.5 |

4. Clinical Response to Crizotinib in MET Amplification—Positive Cancer Patients
5. Conclusions
Acknowledgements
Conflicts of Interest
References
- Mok, T.S.; Wu, Y.L.; Thongprasert, S.; Yang, C.H.; Chu, D.T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef]
- Sequist, L.V.; Martins, R.G.; Spigel, D.; Grunberg, S.M.; Spira, A.; Janne, P.A.; Joshi, V.A.; McCollum, D.; Evans, T.L.; Muzikansky, A.; et al. First-line gefitinib in patients with advanced non-small-cell lung cancer harboring somatic EGFR mutations. J. Clin. Oncol. 2008, 26, 2442–2449. [Google Scholar]
- Porter, J. Small molecule c-Met kinase inhibitors: A review of recent patents. Expert Opin. Ther. Pat. 2010, 20, 159–177. [Google Scholar] [CrossRef]
- Christensen, J.G.; Schreck, R.; Burrows, J.; Kuruganti, P.; Chan, E.; Le, P.; Chen, J.; Wang, X.; Ruslim, L.; Blake, R.; et al. A selective small molecule inhibitor of c-Met kinase inhibits c-Met-dependent phenotypes in vitro and exhibits cytoreductive antitumor activity in vivo. Cancer Res. 2003, 63, 7345–7355. [Google Scholar]
- Christensen, J.G.; Burrows, J.; Salgia, R. c-Met as a target for human cancer and characterization of inhibitors for therapeutic intervention. Cancer Lett. 2005, 225, 1–26. [Google Scholar] [CrossRef]
- Davis, I.J.; McFadden, A.W.; Zhang, Y.; Coxon, A.; Burgess, T.L.; Wagner, A.J.; Fisher, D.E. Identification of the receptor tyrosine kinase c-Met and its ligand, hepatocyte growth factor, as therapeutic targets in clear cell sarcoma. Cancer Res. 2010, 70, 639–645. [Google Scholar]
- Di Renzo, M.F.; Olivero, M.; Martone, T.; Maffe, A.; Maggiora, P.; Stefani, A.D.; Valente, G.; Giordano, S.; Cortesina, G.; Comoglio, P.M. Somatic mutations of the Met oncogene are selected during metastatic spread of human HNSC carcinomas. Oncogene 2000, 19, 1547–1555. [Google Scholar] [CrossRef]
- Park, W.S.; Dong, S.M.; Kim, S.Y.; Na, E.Y.; Shin, M.S.; Pi, J.H.; Kim, B.J.; Bae, J.H.; Hong, Y.K.; Lee, K.S.; et al. Somatic mutations in the kinase domain of the Met/hepatocyte growth factor receptor gene in childhood hepatocellular carcinomas. Cancer Res. 1999, 59, 307–310. [Google Scholar]
- Schmidt, L.; Duh, F.M.; Chen, F.; Kishida, T.; Glenn, G.; Choyke, P.; Scherer, S.W.; Zhuang, Z.; Lubensky, I.; Dean, M.; et al. Germline and somatic mutations in the tyrosine kinase domain of the Met proto-oncogene in papillary renal carcinomas. Nat. Genet. 1997, 16, 68–73. [Google Scholar]
- Birchmeier, C.; Birchmeier, W.; Gherardi, E.; vande Woude, G.F. Met, metastasis, motility and more. Nat. Rev. Mol. Cell Biol 2003, 4, 915–925. [Google Scholar]
- Danilkovitch-Miagkova, A.; Zbar, B. Dysregulation of Met receptor tyrosine kinase activity in invasive tumors. J. Clin. Invest. 2002, 109, 863–867. [Google Scholar] [CrossRef]
- Tanizaki, J.; Okamoto, I.; Okamoto, K.; Takezawa, K.; Kuwata, K.; Yamaguchi, H.; Nakagawa, K. Met tyrosine kinase inhibitor crizotinib (PF-02341066) shows differential antitumor effects in non-small cell lung cancer according to Met alterations. J. Thorac. Oncol. 2011, 6, 1624–1631. [Google Scholar] [CrossRef]
- Zou, H.Y.; Li, Q.; Lee, J.H.; Arango, M.E.; Burgess, K.; Qiu, M.; Engstrom, L.D.; Yamazaki, S.; Parker, M.; Timofeevski, S.; et al. Sensitivity of selected human tumor models to PF-04217903, a novel selective c-Met kinase inhibitor. Mol. Cancer Ther. 2012, 11, 1036–1047. [Google Scholar]
- Zou, H.Y.; Li, Q.; Lee, J.H.; Arango, M.E.; McDonnell, S.R.; Yamazaki, S.; Koudriakova, T.B.; Alton, G.; Cui, J.J.; Kung, P.P.; et al. An orally available small-molecule inhibitor of c-Met, PF-2341066, exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic mechanisms. Cancer Res. 2007, 67, 4408–4417. [Google Scholar] [CrossRef]
- Timofeevski, S.L.; McTigue, M.A.; Ryan, K.; Cui, J.; Zou, H.Y.; Zhu, J.X.; Chau, F.; Alton, G.; Karlicek, S.; Christensen, J.G.; et al. Enzymatic characterization of c-Met receptor tyrosine kinase oncogenic mutants and kinetic studies with aminopyridine and triazolopyrazine inhibitors. Biochemistry 2009, 48, 5339–5349. [Google Scholar] [CrossRef]
- Park, W.S.; Oh, R.R.; Kim, Y.S.; Park, J.Y.; Shin, M.S.; Lee, H.K.; Lee, S.H.; Yoo, N.J.; Lee, J.Y. Absence of mutations in the kinase domain of the Met gene and frequent expression of Met and HGF/SF protein in primary gastric carcinomas. APMIS 2000, 108, 195–200. [Google Scholar]
- Lee, J.H.; Han, S.U.; Cho, H.; Jennings, B.; Gerrard, B.; Dean, M.; Schmidt, L.; Zbar, B.; vande Woude, G.F. A novel germ line juxtamembrane Met mutation in human gastric cancer. Oncogene 2000, 19, 4947–4953. [Google Scholar]
- Chen, J.D.; Kearns, S.; Porter, T.; Richards, F.M.; Maher, E.R.; Teh, B.T. Met mutation and familial gastric cancer. J. Med. Genet. 2001, 38, E26. [Google Scholar] [CrossRef]
- Nakajima, M.; Sawada, H.; Yamada, Y.; Watanabe, A.; Tatsumi, M.; Yamashita, J.; Matsuda, M.; Sakaguchi, T.; Hirao, T.; Nakano, H. The prognostic significance of amplification and overexpression of c-Met and c-Erb b-2 in human gastric carcinomas. Cancer 1999, 85, 1894–1902. [Google Scholar] [CrossRef]
- Hara, T.; Ooi, A.; Kobayashi, M.; Mai, M.; Yanagihara, K.; Nakanishi, I. Amplification of c-Myc, k-Sam, and c-Met in gastric cancers: Detection by fluorescence in situ hybridization. Lab. Invest. 1998, 78, 1143–1153. [Google Scholar]
- Tsugawa, K.; Yonemura, Y.; Hirono, Y.; Fushida, S.; Kaji, M.; Miwa, K.; Miyazaki, I.; Yamamoto, H. Amplification of the c-Met, c-Erbb-2 and epidermal growth factor receptor gene in human gastric cancers: Correlation to clinical features. Oncology 1998, 55, 475–481. [Google Scholar] [CrossRef]
- Smolen, G.A.; Sordella, R.; Muir, B.; Mohapatra, G.; Barmettler, A.; Archibald, H.; Kim, W.J.; Okimoto, R.A.; Bell, D.W.; Sgroi, D.C.; et al. Amplification of Met may identify a subset of cancers with extreme sensitivity to the selective tyrosine kinase inhibitor PHA-665752. Proc. Natl. Acad. Sci. USA 2006, 103, 2316–2321. [Google Scholar]
- Okamoto, W.; Okamoto, I.; Arao, T.; Kuwata, K.; Hatashita, E.; Yamaguchi, H.; Sakai, K.; Yanagihara, K.; Nishio, K.; Nakagawa, K. Antitumor action of the Met tyrosine kinase inhibitor crizotinib (PF-02341066) in gastric cancer positive for Met amplification. Mol. Cancer Ther. 2012, 11, 1557–1564. [Google Scholar] [CrossRef]
- Masuya, D.; Huang, C.; Liu, D.; Nakashima, T.; Kameyama, K.; Haba, R.; Ueno, M.; Yokomise, H. The tumour-stromal interaction between intratumoral c-Met and stromal hepatocyte growth factor associated with tumour growth and prognosis in non-small-cell lung cancer patients. Br. J. Cancer 2004, 90, 1555–1562. [Google Scholar] [CrossRef]
- Nakamura, Y.; Niki, T.; Goto, A.; Morikawa, T.; Miyazawa, K.; Nakajima, J.; Fukayama, M. c-Met activation in lung adenocarcinoma tissues: An immunohistochemical analysis. Cancer Sci. 2007, 98, 1006–1013. [Google Scholar] [CrossRef]
- Zhao, X.; Weir, B.A.; LaFramboise, T.; Lin, M.; Beroukhim, R.; Garraway, L.; Beheshti, J.; Lee, J.C.; Naoki, K.; Richards, W.G.; et al. Homozygous deletions and chromosome amplifications in human lung carcinomas revealed by single nucleotide polymorphism array analysis. Cancer Res. 2005, 65, 5561–5570. [Google Scholar] [CrossRef]
- Go, H.; Jeon, Y.K.; Park, H.J.; Sung, S.W.; Seo, J.W.; Chung, D.H. High Met gene copy number leads to shorter survival in patients with non-small cell lung cancer. J. Thorac. Oncol. 2010, 5, 305–313. [Google Scholar]
- Lennerz, J.K.; Kwak, E.L.; Ackerman, A.; Michael, M.; Fox, S.B.; Bergethon, K.; Lauwers, G.Y.; Christensen, J.G.; Wilner, K.D.; Haber, D.A.; et al. Met amplification identifies a small and aggressive subgroup of esophagogastric adenocarcinoma with evidence of responsiveness to crizotinib. J. Clin. Oncol. 2011, 29, 4803–4810. [Google Scholar] [CrossRef]
- Janjigian, Y.Y.; Tang, L.H.; Coit, D.G.; Kelsen, D.P.; Francone, T.D.; Weiser, M.R.; Jhanwar, S.C.; Shah, M.A. Met expression and amplification in patients with localized gastric cancer. Cancer Epidemiol. Biomark. Prev. 2011, 20, 1021–1027. [Google Scholar] [CrossRef]
- Bean, J.; Brennan, C.; Shih, J.Y.; Riely, G.; Viale, A.; Wang, L.; Chitale, D.; Motoi, N.; Szoke, J.; Broderick, S.; et al. Met amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc. Natl. Acad. Sci. USA 2007, 104, 20932–20937. [Google Scholar] [CrossRef]
- Okuda, K.; Sasaki, H.; Yukiue, H.; Yano, M.; Fujii, Y. Met gene copy number predicts the prognosis for completely resected non-small cell lung cancer. Cancer Sci. 2008, 99, 2280–2285. [Google Scholar] [CrossRef]
- Onitsuka, T.; Uramoto, H.; Nose, N.; Takenoyama, M.; Hanagiri, T.; Sugio, K.; Yasumoto, K. Acquired resistance to gefitinib: The contribution of mechanisms other than the T790M, Met, and HGF status. Lung Cancer 2010, 68, 198–203. [Google Scholar] [CrossRef]
- Onozato, R.; Kosaka, T.; Kuwano, H.; Sekido, Y.; Yatabe, Y.; Mitsudomi, T. Activation of Met by gene amplification or by splice mutations deleting the juxtamembrane domain in primary resected lung cancers. J. Thorac. Oncol. 2009, 4, 5–11. [Google Scholar]
- Kubo, T.; Yamamoto, H.; Lockwood, W.W.; Valencia, I.; Soh, J.; Peyton, M.; Jida, M.; Otani, H.; Fujii, T.; Ouchida, M.; et al. Met gene amplification or EGFR mutation activate Met in lung cancers untreated with EGFR tyrosine kinase inhibitors. Int. J. Cancer 2009, 124, 1778–1784. [Google Scholar] [CrossRef]
- Beau-Faller, M.; Ruppert, A.M.; Voegeli, A.C.; Neuville, A.; Meyer, N.; Guerin, E.; Legrain, M.; Mennecier, B.; Wihlm, J.M.; Massard, G.; et al. Met gene copy number in non-small cell lung cancer: Molecular analysis in a targeted tyrosine kinase inhibitor naive cohort. J. Thorac. Oncol. 2008, 3, 331–339. [Google Scholar] [CrossRef]
- Kuniyasu, H.; Yasui, W.; Kitadai, Y.; Yokozaki, H.; Ito, H.; Tahara, E. Frequent amplification of the c-Met gene in scirrhous type stomach cancer. Biochem. Biophys. Res. Commun. 1992, 189, 227–232. [Google Scholar] [CrossRef]
- Tsujimoto, H.; Sugihara, H.; Hagiwara, A.; Hattori, T. Amplification of growth factor receptor genes and DNA ploidy pattern in the progression of gastric cancer. Virchows Arch. 1997, 431, 383–389. [Google Scholar] [CrossRef]
- Seruca, R.; Suijkerbuijk, R.F.; Gartner, F.; Criado, B.; Veiga, I.; Olde-Weghuis, D.; David, L.; Castedo, S.; Sobrinho-Simoes, M. Increasing levels of Myc and Met co-amplification during tumor progression of a case of gastric cancer. Cancer Genet. Cytogenet. 1995, 82, 140–145. [Google Scholar]
- Lee, J.; Seo, J.W.; Jun, H.J.; Ki, C.S.; Park, S.H.; Park, Y.S.; Lim, H.Y.; Choi, M.G.; Bae, J.M.; Sohn, T.S.; et al. Impact of Met amplification on gastric cancer: Possible roles as a novel prognostic marker and a potential therapeutic target. Oncol. Rep. 2011, 25, 1517–1524. [Google Scholar]
- Graziano, F.; Galluccio, N.; Lorenzini, P.; Ruzzo, A.; Canestrari, E.; D'Emidio, S.; Catalano, V.; Sisti, V.; Ligorio, C.; Andreoni, F.; et al. Genetic activation of the Met pathway and prognosis of patients with high-risk, radically resected gastric cancer. J. Clin. Oncol. 2011, 29, 4789–4795. [Google Scholar] [CrossRef]
- Albertson, D.G. Gene amplification in cancer. Trends Genet. 2006, 22, 447–455. [Google Scholar] [CrossRef]
- Vanden Bempt, I.; van Loo, P.; Drijkoningen, M.; Neven, P.; Smeets, A.; Christiaens, M.R.; Paridaens, R.; de Wolf-Peeters, C. Polysomy 17 in breast cancer: Clinicopathologic significance and impact on Her-2 testing. J. Clin. Oncol. 2008, 26, 4869–4874. [Google Scholar]
- Camidge, D.R.; Kono, S.A.; Flacco, A.; Tan, A.C.; Doebele, R.C.; Zhou, Q.; Crino, L.; Franklin, W.A.; Varella-Garcia, M. Optimizing the detection of lung cancer patients harboring anaplastic lymphoma kinase (ALK) gene rearrangements potentially suitable for ALK inhibitor treatment. Clin. Cancer Res. 2010, 16, 5581–5590. [Google Scholar] [CrossRef]
- Okamoto, I.; Sakai, K.; Morita, S.; Yoshioka, H.; Kaneda, H.; Takeda, K.; Hirashima, T.; Kogure, Y.; Kimura, T.; Takahashi, T.; et al. Multiplex genomic profiling of non–small cell lung cancers from the LETS phase III trial of first-line S-1/carboplatin versus paclitaxel/carboplatin: Results of a west Japan oncology group study. Oncotarget 2014, 5, 2293–2304. [Google Scholar]
- Cappuzzo, F.; Marchetti, A.; Skokan, M.; Rossi, E.; Gajapathy, S.; Felicioni, L.; del Grammastro, M.; Sciarrotta, M.G.; Buttitta, F.; Incarbone, M.; et al. Increased Met gene copy number negatively affects survival of surgically resected non-small-cell lung cancer patients. J. Clin. Oncol. 2009, 27, 1667–1674. [Google Scholar] [CrossRef]
- Kawakami, H.; Okamoto, I.; Arao, T.; Okamoto, W.; Matsumoto, K.; Taniguchi, H.; Kuwata, K.; Yamaguchi, H.; Nishio, K.; Nakagawa, K.; et al. Met amplification as a potential therapeutic target in gastric cancer. Oncotarget 2013, 4, 9–17. [Google Scholar]
- Liu, Y.J.; Shen, D.; Yin, X.; Gavine, P.; Zhang, T.; Su, X.; Zhan, P.; Xu, Y.; Lv, J.; Qian, J.; et al. Her2, Met and FGFR2 oncogenic driver alterations define distinct molecular segments for targeted therapies in gastric carcinoma. Br. J. Cancer 2014, 110, 1169–1178. [Google Scholar]
- Shi, J.; Yao, D.; Liu, W.; Wang, N.; Lv, H.; He, N.; Shi, B.; Hou, P.; Ji, M. Frequent gene amplification predicts poor prognosis in gastric cancer. Int. J. Mol. Sci. 2012, 13, 4714–4726. [Google Scholar]
- Albertson, D.G.; Collins, C.; McCormick, F.; Gray, J.W. Chromosome aberrations in solid tumors. Nat. Genet. 2003, 34, 369–376. [Google Scholar]
- Wolff, A.C.; Hammond, M.E.; Hicks, D.G.; Dowsett, M.; McShane, L.M.; Allison, K.H.; Allred, D.C.; Bartlett, J.M.; Bilous, M.; Fitzgibbons, P.; et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J. Clin. Oncol. 2013, 31, 3997–4013. [Google Scholar] [CrossRef]
- Ma, Y.; Lespagnard, L.; Durbecq, V.; Paesmans, M.; Desmedt, C.; Gomez-Galdon, M.; Veys, I.; Cardoso, F.; Sotiriou, C.; di Leo, A.; et al. Polysomy 17 in Her-2/Neu status elaboration in breast cancer: Effect on daily practice. Clin. Cancer Res. 2005, 11, 4393–4399. [Google Scholar] [CrossRef]
- Zhu, K.; Kong, X.; Zhao, D.; Liang, Z.; Luo, C. c-Met kinase inhibitors: A patent review (2011–2013). Expert Opin. Ther. Pat. 2014, 24, 217–230. [Google Scholar] [CrossRef]
- Elisei, R.; Schlumberger, M.J.; Muller, S.P.; Schoffski, P.; Brose, M.S.; Shah, M.H.; Licitra, L.; Jarzab, B.; Medvedev, V.; Kreissl, M.C.; et al. Cabozantinib in progressive medullary thyroid cancer. J. Clin. Oncol. 2013, 31, 3639–3646. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Vaishampayan, U.; Rosenberg, J.E.; Logan, T.F.; Harzstark, A.L.; Bukowski, R.M.; Rini, B.I.; Srinivas, S.; Stein, M.N.; Adams, L.M.; et al. Phase II and biomarker study of the dual Met/VEGFR2 inhibitor foretinib in patients with papillary renal cell carcinoma. J. Clin. Oncol. 2013, 31, 181–186. [Google Scholar] [CrossRef]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. Met amplification leads to gefitinib resistance in lung cancer by activating Erbb3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef]
- Katayama, R.; Aoyama, A.; Yamori, T.; Qi, J.; Oh-hara, T.; Song, Y.; Engelman, J.A.; Fujita, N. Cytotoxic activity of tivantinib (ARQ 197) is not due solely to c-Met inhibition. Cancer Res. 2013, 73, 3087–3096. [Google Scholar] [CrossRef]
- Ou, S.H.; Kwak, E.L.; Siwak-Tapp, C.; Dy, J.; Bergethon, K.; Clark, J.W.; Camidge, D.R.; Solomon, B.J.; Maki, R.G.; Bang, Y.J.; et al. Activity of crizotinib (PF02341066), a dual mesenchymal-epithelial transition (MET) and anaplastic lymphoma kinase (ALK) inhibitor, in a non-small cell lung cancer patient with de novo MET amplification. J. Thorac. Oncol. 2011, 6, 942–946. [Google Scholar] [CrossRef]
- Chi, A.S.; Batchelor, T.T.; Kwak, E.L.; Clark, J.W.; Wang, D.L.; Wilner, K.D.; Louis, D.N.; Iafrate, A.J. Rapid radiographic and clinical improvement after treatment of a Met-amplified recurrent glioblastoma with a mesenchymal-epithelial transition inhibitor. J. Clin. Oncol. 2012, 30, e30–e33. [Google Scholar] [CrossRef]
- Schwab, R.; Petak, I.; Kollar, M.; Pinter, F.; Varkondi, E.; Kohanka, A.; Barti-Juhasz, H.; Schonleber, J.; Brauswetter, D.; Kopper, L.; et al. Major partial response to crizotinib, a dual Met/ALK inhibitor, in a squamous cell lung (SCC) carcinoma patient with de novo c-Met amplification in the absence of ALK rearrangement. Lung Cancer 2014, 83, 109–111. [Google Scholar]
- Camidge, D.R.; Ou, S.-H.I.; Shapiro, G.; Otterson, G.A.; Villaruz, L.C.; Villalona-Calero, M.A.; Iafrate, A.J.; Varella-Garcia, M.; Dacic, S.; Cardarella, S.; et al. Efficacy and safety of crizotinib in patients with advanced c-MET-amplified non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2014, 32, 5. [Google Scholar] [CrossRef]
- Qi, J.; McTigue, M.A.; Rogers, A.; Lifshits, E.; Christensen, J.G.; Janne, P.A.; Engelman, J.A. Multiple mutations and bypass mechanisms can contribute to development of acquired resistance to Met inhibitors. Cancer Res. 2011, 71, 1081–1091. [Google Scholar]
- McDermott, U.; Pusapati, R.V.; Christensen, J.G.; Gray, N.S.; Settleman, J. Acquired resistance of non-small cell lung cancer cells to Met kinase inhibition is mediated by a switch to epidermal growth factor receptor dependency. Cancer Res. 2010, 70, 1625–1634. [Google Scholar]
- Lee, N.V.; Lira, M.E.; Pavlicek, A.; Ye, J.; Buckman, D.; Bagrodia, S.; Srinivasa, S.P.; Zhao, Y.; Aparicio, S.; Rejto, P.A.; et al. A novel SND1-BRAF fusion confers resistance to c-Met inhibitor PF-04217903 in GTl16 cells through MAPK activation. PLoS One 2012, 7, e39653. [Google Scholar]
- Cepero, V.; Sierra, J.R.; Corso, S.; Ghiso, E.; Casorzo, L.; Perera, T.; Comoglio, P.M.; Giordano, S. Met and Kras gene amplification mediates acquired resistance to Met tyrosine kinase inhibitors. Cancer Res. 2010, 70, 7580–7590. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kawakami, H.; Okamoto, I.; Okamoto, W.; Tanizaki, J.; Nakagawa, K.; Nishio, K. Targeting MET Amplification as a New Oncogenic Driver. Cancers 2014, 6, 1540-1552. https://doi.org/10.3390/cancers6031540
Kawakami H, Okamoto I, Okamoto W, Tanizaki J, Nakagawa K, Nishio K. Targeting MET Amplification as a New Oncogenic Driver. Cancers. 2014; 6(3):1540-1552. https://doi.org/10.3390/cancers6031540
Chicago/Turabian StyleKawakami, Hisato, Isamu Okamoto, Wataru Okamoto, Junko Tanizaki, Kazuhiko Nakagawa, and Kazuto Nishio. 2014. "Targeting MET Amplification as a New Oncogenic Driver" Cancers 6, no. 3: 1540-1552. https://doi.org/10.3390/cancers6031540