The c-Met Inhibitor MSC2156119J Effectively Inhibits Tumor Growth in Liver Cancer Models

Abstract
:1. Introduction
2. Results and Discussion
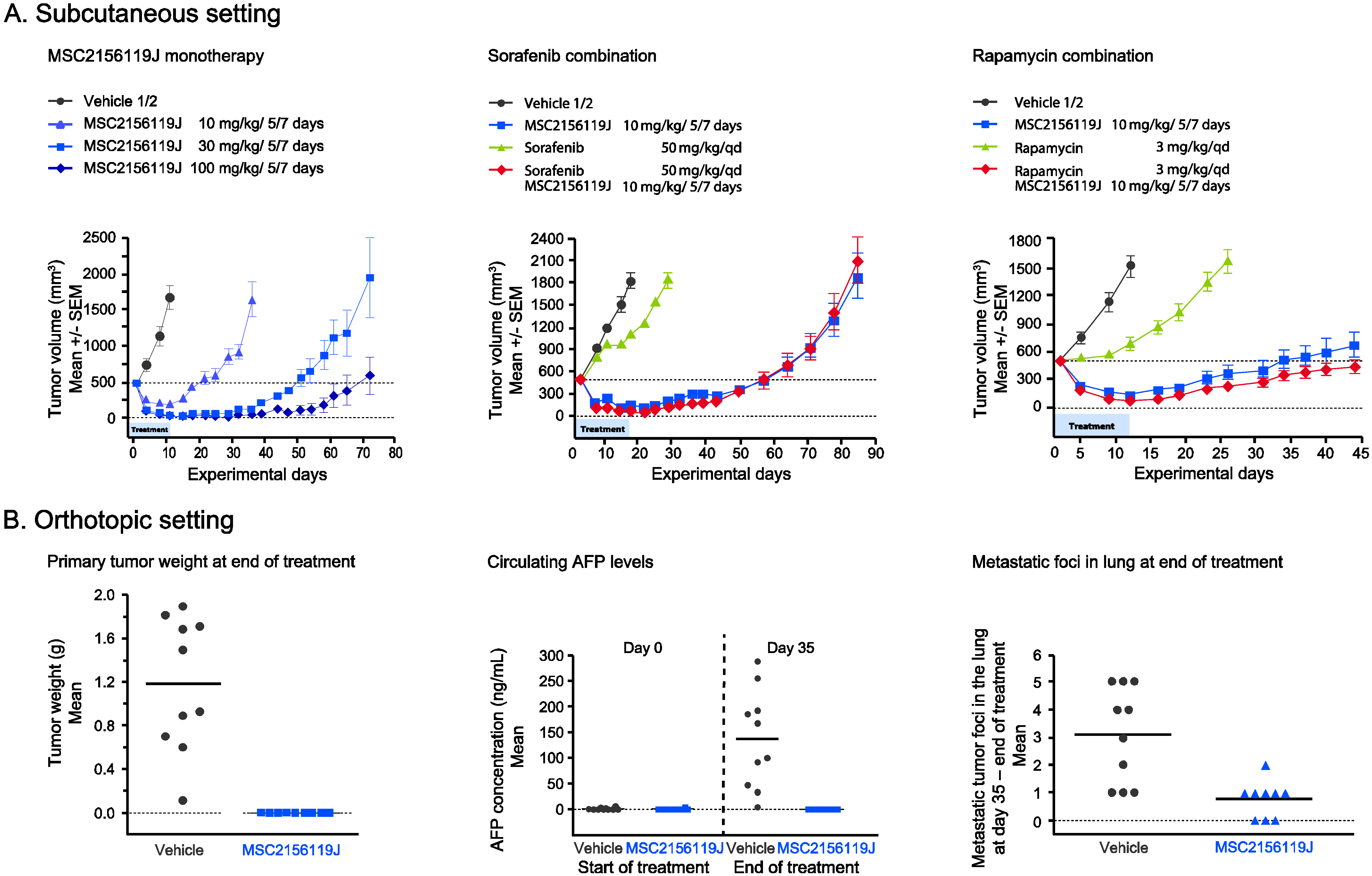
2.1. Efficacy of MSC2156119J as Monotherapy or in Combination with Sorafenib or Rapamycin in MHCC97H Xenografts
Effect of MSC2156119J on Primary Tumors, Circulating AFP Levels, and Metastases Formation
2.2. Efficacy of MSC2156119J and Sorafenib in HuPrime Primary Explant Xenograft Models
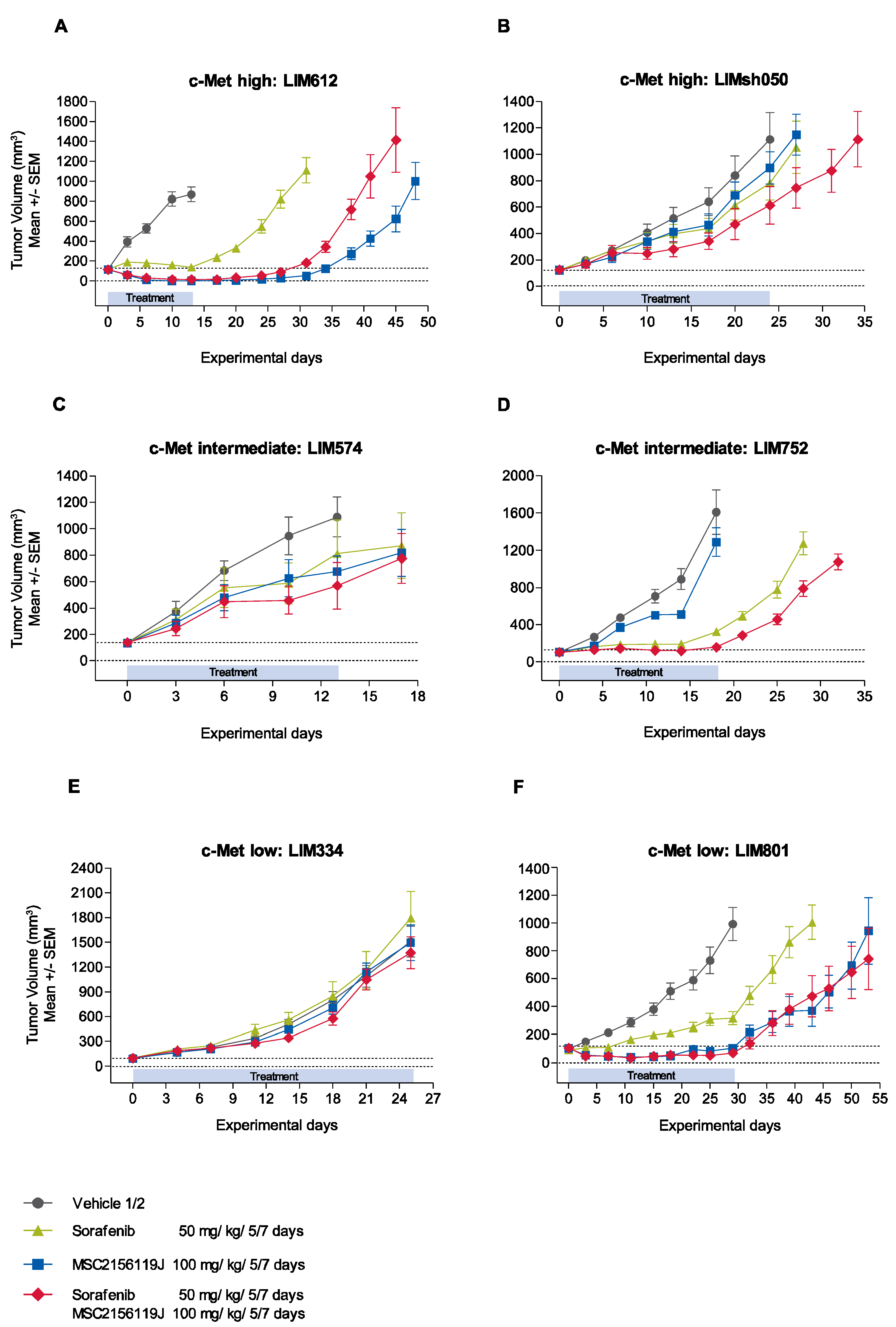
2.2.1. Kinetics of Tumor Growth after MSC2156119J and Sorafenib Treatment



2.2.2. Treatment Tolerability
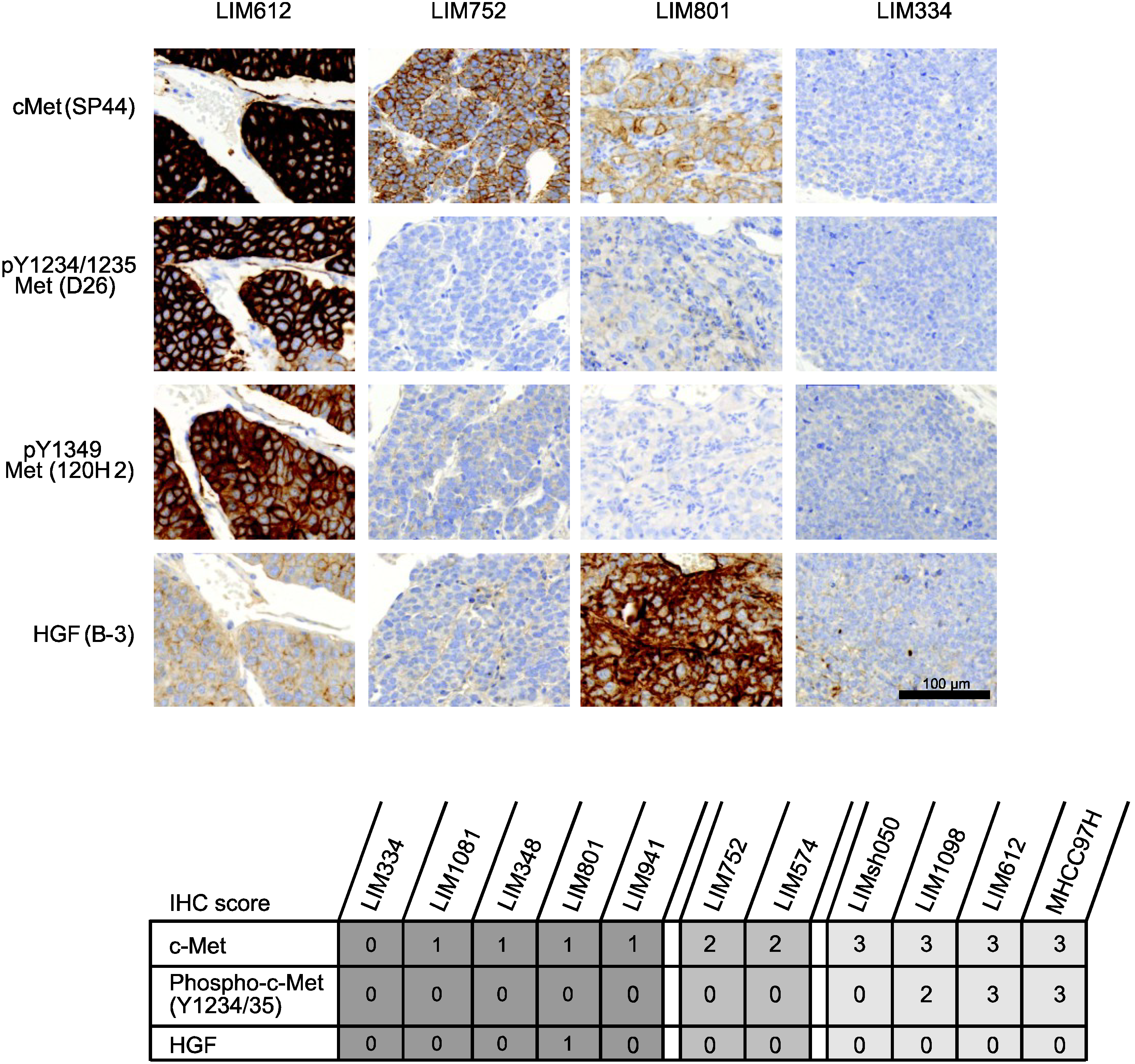
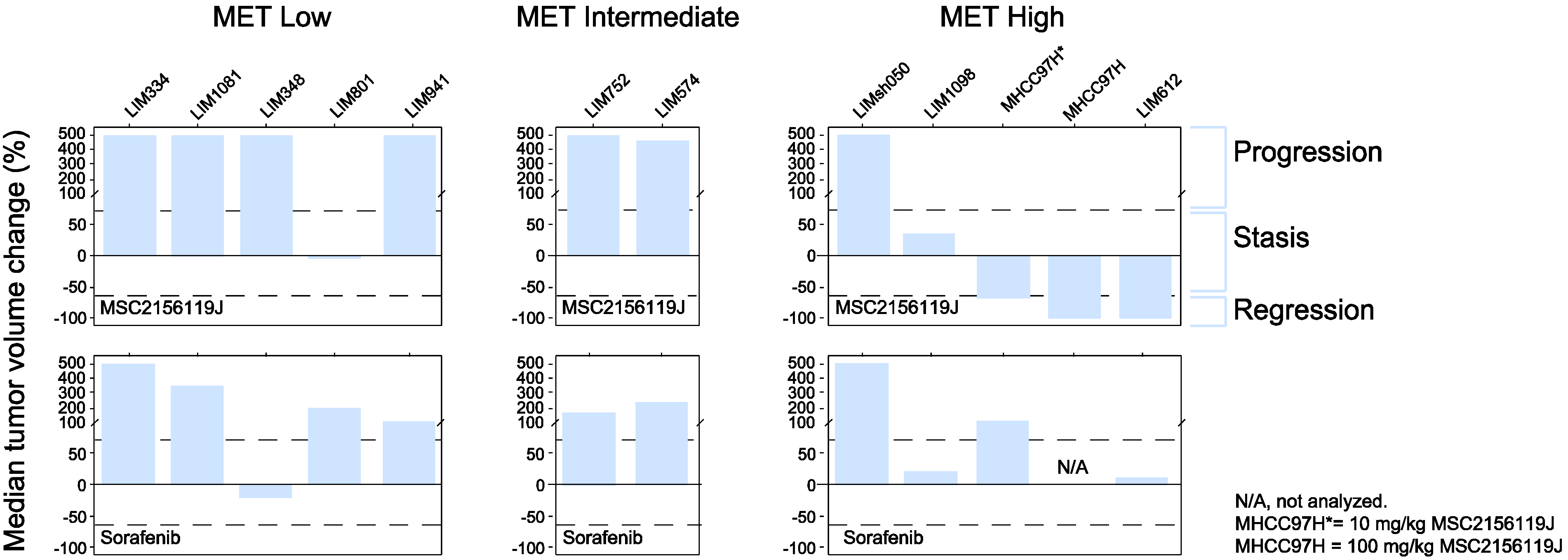
2.2.3. Tumor Response According to c-Met Expression Status
2.3. Discussion


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Efficacy | |||
|---|---|---|---|
| Regression | Stasis | Progression | |
| Activated c-Met receptor (phospho-c-Met positive) | |||
| c-Met low | 0/0 | 0/0 | 0/0 |
| c-Met intermediate | 0/0 | 0/0 | 0/0 |
| c-Met high | 2/3 | 1/3 | 0/3 |
| Nonactivated c-Met receptor (phospho-c-Met negative) | |||
| c-Met low | 0/5 | 1/5 | 4/5 |
| c-Met intermediate | 0/2 | 0/2 | 2/2 |
| c-Met high | 0/1 | 0/1 | 1/1 |
3. Experimental
3.1. Compounds
3.2. Animals and Housing
3.3. MHCC97H Human HCC Xenograft Model
3.4. HuPrime Model with Human HCC Explants
3.5. Immunohistochemistry
3.6. Statistical Analyses
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Borowiak, M.; Garratt, A.N.; Wüstefeld, T.; Strehle, M.; Trautwein, C.; Birchmeier, C. Met provides essential signals for liver regeneration. Proc. Natl. Acad. Sci. USA 2004, 101, 10608–10613. [Google Scholar]
- Sakata, H.; Takayama, H.; Sharp, R.; Rubin, J.S.; Merlino, G.; LaRochelle, W.J. Hepatocyte growth factor/scatter factor overexpression induces growth, abnormal development, and tumor formation in transgenic mouse livers. Cell Growth Differ. 1996, 7, 1513–1523. [Google Scholar]
- Jung, K.H.; Park, B.H.; Hong, S.S. Progress in cancer therapy targeting c-Met signaling pathway. Arch. Pharm. Res. 2012, 35, 595–604. [Google Scholar]
- You, H.; Ding, W.; Dang, H.; Jiang, Y.; Rountree, C.B. c-Met represents a potential therapeutic target for personalized treatment in hepatocellular carcinoma. Hepatology 2011, 54, 879–889. [Google Scholar]
- Eder, J.P.; vande Woude, G.F.; Boerner, S.A.; LoRusso, P.M. Novel therapeutic inhibitors of the c-Met signaling pathway in cancer. Clin. Cancer Res. 2009, 15, 2207–2214. [Google Scholar] [CrossRef]
- Wang, R.; Ferrell, L.D.; Faouzi, S.; Maher, J.J.; Bishop, J.M. Activation of the Met receptor by cell attachment induces and sustains hepatocellular carcinomas in transgenic mice. J. Cell Biol. 2001, 153, 1023–1034. [Google Scholar] [CrossRef]
- Tward, A.D.; Jones, K.D.; Yant, S.; Cheung, S.T.; Fan, S.T.; Chen, X.; Kay, M.A.; Wang, R.; Bishop, J.M. Distinct pathways of genomic progression to benign and malignant tumors of the liver. Proc. Natl. Acad. Sci. USA 2007, 104, 14771–14776. [Google Scholar] [CrossRef]
- Chu, J.S.; Ge, F.J.; Zhang, B.; Wang, Y.; Silvestris, N.; Liu, L.J.; Zhao, C.H.; Lin, L.; Brunetti, A.E.; Fu, Y.L.; et al. Expression and prognostic value of VEGFR-2, PDGFR-ß, and c-Met in advanced hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2013, 32, 16. [Google Scholar]
- Goyal, L.; Muzumdar, M.D.; Zhu, A.X. Targeting the HGF/c-Met pathway in hepatocellular carcinoma. Clin. Cancer Res. 2013, 19, 2310–2318. [Google Scholar] [CrossRef]
- Inagaki, Y.; Qi, F.; Gao, J.; Qu, X.; Hasegawa, K.; Sugawara, Y.; Tang, W.; Kokudo, N. Effect of c-Met inhibitor SU11274 on hepatocellular carcinoma cell growth. Biosci. Trends 2011, 5, 52–56. [Google Scholar] [CrossRef]
- Bladt, F.; Faden, B.; Friese-Hamim, M.; Knuehl, C.; Wilm, C.; Fittschen, C.; Grädler, U.; Meyring, M.; Dorsch, D.; Jaehrling, F.; et al. EMD 1214063 and EMD 1204831 constitute a new class of potent and highly selective c-Met inhibitors. Clin. Cancer Res. 2013, 19, 2941–2951. [Google Scholar] [CrossRef]
- Rubio-Vigueira, B.; Hidalgo, M. Direct in vivo xenograft tumor model for predicting chemotherapeutic drug response in cancer patients. Clin. Pharmacol. Ther. 2009, 85, 217–221. [Google Scholar] [CrossRef]
- Bertotti, A.; Migliardi, G.; Galimi, F.; Sassi, F.; Torti, D.; Isella, C.; Corà, D.; di Nicolantonio, F.; Buscarino, M.; Petti, C.; et al. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 2011, 1, 508–523. [Google Scholar] [CrossRef]
- Gentile, A.; Trusolino, L.; Comoglio, P.M. The Met tyrosine kinase receptor in development and cancer. Cancer Metastasis Rev. 2008, 27, 85–94. [Google Scholar] [CrossRef]
- Ogunwobi, O.O.; Puszyk, W.; Dong, H.J.; Liu, C. Epigenetic upregulation of HGF and c-Met drives metastasis in hepatocellular carcinoma. PLoS One 2013, 8, e63765. [Google Scholar] [CrossRef]
- Qiu, J.; Huang, P.; Liu, Q.; Hong, J.; Li, B.; Lu, C.; Wang, L.; Wang, J.; Yuan, Y. Identification of MACC1 as a novel prognostic marker in hepatocellular carcinoma. J. Transl. Med. 2011, 9, 166. [Google Scholar] [CrossRef]
- Corso, S.; Giordano, S. Cell-autonomous and non-cell-autonomous mechanisms of HGF/MET-driven resistance to targeted therapies: From basic research to a clinical perspective. Cancer Discov. 2013, 3, 978–992. [Google Scholar] [CrossRef]
- Schmidt, C.; Bladt, F.; Goedecke, S.; Brinkmann, V.; Zschiesche, W.; Sharpe, M.; Gherardi, E.; Birchmeier, C. Scatter factor/hepatocyte growth factor is essential for liver development. Nature 1995, 373, 699–702. [Google Scholar] [CrossRef]
- Bladt, F.; Riethmacher, D.; Isenmann, S.; Aguzzi, A.; Birchmeier, C. Essential role for the c-met receptor in the migration of myogenic precursor cells into the limb bud. Nature 1995, 376, 768–771. [Google Scholar] [CrossRef]
- Zhang, S.Z.; Pan, F.Y.; Xu, J.F.; Yuan, J.; Guo, S.Y.; Dai, G.; Xue, B.; Shen, W.G.; Wen, C.J.; Zhao, D.H.; et al. Knockdown of c-Met by adenovirus-delivered small interfering RNA inhibits hepatocellular carcinoma growth in vitro and in vivo. Mol. Cancer Ther. 2005, 4, 1577–1584. [Google Scholar] [CrossRef]
- Xie, B.; Xing, R.; Chen, P.; Gou, Y.; Li, S.; Xiao, J.; Dong, J. Down-regulation of c-Met expression inhibits human HCC cells growth and invasion by RNA interference. J. Surg. Res. 2010, 162, 231–238. [Google Scholar] [CrossRef]
- Navab, R.; Liu, J.; Seiden-Long, I.; Shih, W.; Li, M.; Bandarchi, B.; Chen, Y.; Lau, D.; Zu, Y.F.; Cescon, D.; et al. Co-expression of Met and hepatocyte growth factor promotes systemic metastasis in NCI-H460 non-small cell lung carcinoma cells. Neoplasia 2009, 11, 1292–1300. [Google Scholar]
- Koochekpour, S.; Jeffers, M.; Rulong, S.; Taylor, G.; Klineberg, E.; Hudson, E.A.; Resau, J.H.; vande Woude, G.F. Met and hepatocyte growth factor/scatter factor expression in human gliomas. Cancer Res. 1997, 57, 5391–5398. [Google Scholar]
- Xie, Q.; Bradley, R.; Kang, L.; Koeman, J.; Ascierto, M.L.; Worschech, A.; de Giorgi, V.; Wang, E.; Kefene, L.; Su, Y.; et al. Hepatocyte growth factor (HGF) autocrine activation predicts sensitivity to MET inhibition in glioblastoma. Proc. Natl. Acad. Sci. USA 2012, 2, 570–575. [Google Scholar]
- Munshi, N.; Jeay, S.; Li, Y.; Chen, C.R.; France, D.S.; Ashwell, M.A.; Hill, J.; Moussa, M.M.; Leggett, D.S.; Li, C.J. ARQ 197, a novel and selective inhibitor of the human c-Met receptor tyrosine kinase with antitumor activity. Mol. Cancer Ther. 2010, 9, 1544–1553. [Google Scholar] [CrossRef]
- Santoro, A.; Rimassa, L.; Borbath, I.; Daniele, B.; Salvagni, S.; van Laethem, J.L.; van Vlierberghe, H.; Trojan, J.; Kolligs, F.T.; Weiss, A.; et al. Tivantinib for second-line treatment of advanced hepatocellular carcinoma: A randomised, placebo-controlled phase 2 study. Lancet Oncol. 2013, 14, 55–63. [Google Scholar]
- Katayama, R.; Aoyama, A.; Yamori, T.; Qi, J.; Oh-hara, T.; Song, Y.; Engelman, J.A.; Fujita, N. Cytotoxic activity of tivantinib (ARQ197) is not due solely to c-Met inhibition. Cancer Res. 2013, 73, 3087–3096. [Google Scholar] [CrossRef]
- Basilico, C.; Pennacchietti, S.; Vigna, E.; Chiriaco, C.; Arena, S.; Bardelli, A.; Valdembri, D.; Serini, G.; Michieli, P. Tivantinib (ARQ197) displays cytotoxic activity that is independent of its ability to bind MET. Clin. Cancer Res. 2013, 19, 2381–2392. [Google Scholar] [CrossRef]
- Spigel, D.R.; Edelman, M.J.; Mok, T.; O’Byrne, K.; Paz-Ares, L.; Yu, W.; Rittweger, K.; Thurm, H.; MetLung Phase III Study Group. Treatment rationale study design for the MetLung trial: A randomized, double-blind Phase III study of onartuzumab (MetMAb) in combination with erlotinib versus erlotinib alone in patients who have received standard chemotherapy for stage IIIB or IV Met-positive non-small-cell lung cancer. Clin. Lung Cancer 2012, 13, 500–504. [Google Scholar]
- Xie, Q.; Su, Y.; Dykema, K.; Johnson, J.; Koeman, J.; de Giorgi, V.; Huang, A.; Schlegel, R.; Essenburg, C.; Kang, L.; et al. Overexpression of HGF promotes HBV-induced hepatocellular carcinoma progression and is an effective indicator for Met-targeting therapy. Genes Cancer 2013, 4, 247–260. [Google Scholar] [CrossRef]
- Chisari, F.V.; Klopchin, K.; Moriyama, T.; Pasquinelli, C.; Dunsford, H.A.; Sell, S.; Pinkert, C.A.; Brinster, R.L.; Palmiter, R.D. Molecular pathogenesis of hepatocellular carcinoma in hepatitis B virus transgenic mice. Cell 1989, 59, 1145–1156. [Google Scholar] [CrossRef]
- Takeda, S.; Liu, H.; Sasagawa, S.; Dong, Y.; Trainor, A.P.; Cheng, E.H.; Hsieh, J.J. HGF-MET signals via the MLL-ETS2 complex in hepatocellular carcinoma. J. Clin. Investig. 2013, 123, 3154–3165. [Google Scholar] [CrossRef]
- Marquardt, J.U.; Thorgeirsson, S.S. Linking MLL and the HGF-MET signaling pathway in liver cancer. J. Clin. Investig. 2013, 123, 2780–2783. [Google Scholar] [CrossRef]
- Li, Y.; Tang, Z.Y.; Ye, S.L.; Liu, Y.K.; Chen, J.; Xue, Q.; Chen, J.; Gao, D.M.; Bao, W.H. Establishment of cell clones with different metastatic potential from the metastatic hepatocellular carcinoma cell line MHCC97. World J. Gastroenterol. 2001, 7, 630–636. [Google Scholar]
- Wu, F.S.; Zheng, S.S.; Wu, L.J.; Teng, L.S.; Ma, Z.M.; Zhao, W.H.; Wu, W. Calcitriol inhibits the growth of MHCC97 hepatocellular cell lines by down-modulating c-met and ERK expressions. Liver Int. 2007, 27, 700–707. [Google Scholar] [CrossRef]
- Therasse, P.; Arbuck, S.G.; Eisenhauer, E.A.; Wanders, J.; Kaplan, R.S.; Rubinstein, L.; Verweij, J.; van Glabbeke, M.; van Oosterom, A.T.; Christian, M.C.; et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for research and treatment of cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J. Natl. Cancer Inst. 2000, 92, 205–216. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bladt, F.; Friese-Hamim, M.; Ihling, C.; Wilm, C.; Blaukat, A. The c-Met Inhibitor MSC2156119J Effectively Inhibits Tumor Growth in Liver Cancer Models. Cancers 2014, 6, 1736-1752. https://doi.org/10.3390/cancers6031736
Bladt F, Friese-Hamim M, Ihling C, Wilm C, Blaukat A. The c-Met Inhibitor MSC2156119J Effectively Inhibits Tumor Growth in Liver Cancer Models. Cancers. 2014; 6(3):1736-1752. https://doi.org/10.3390/cancers6031736
Chicago/Turabian StyleBladt, Friedhelm, Manja Friese-Hamim, Christian Ihling, Claudia Wilm, and Andree Blaukat. 2014. "The c-Met Inhibitor MSC2156119J Effectively Inhibits Tumor Growth in Liver Cancer Models" Cancers 6, no. 3: 1736-1752. https://doi.org/10.3390/cancers6031736