
Epigenomic Regulation of Androgen Receptor Signaling: Potential Role in Prostate Cancer Therapy

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
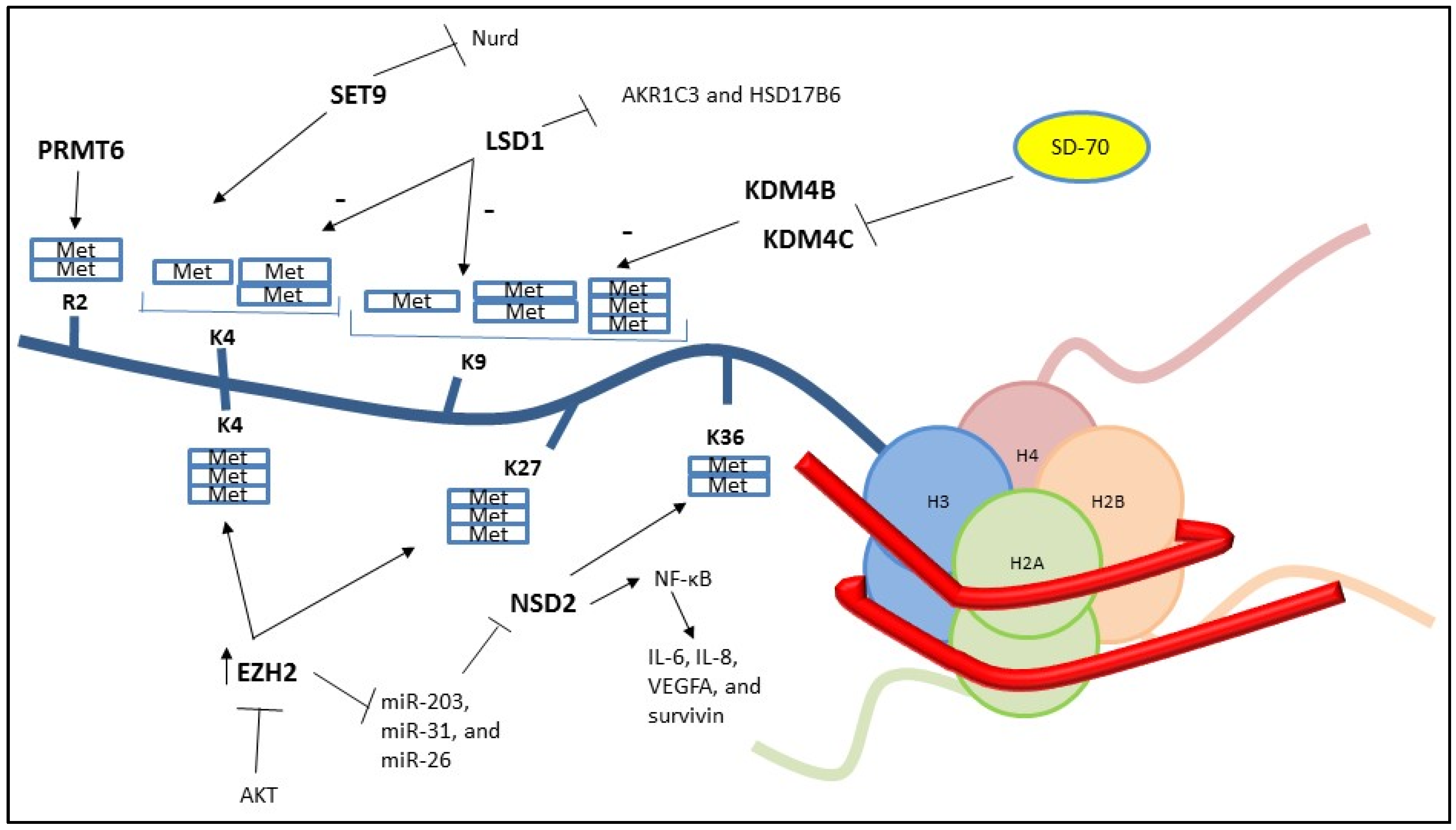
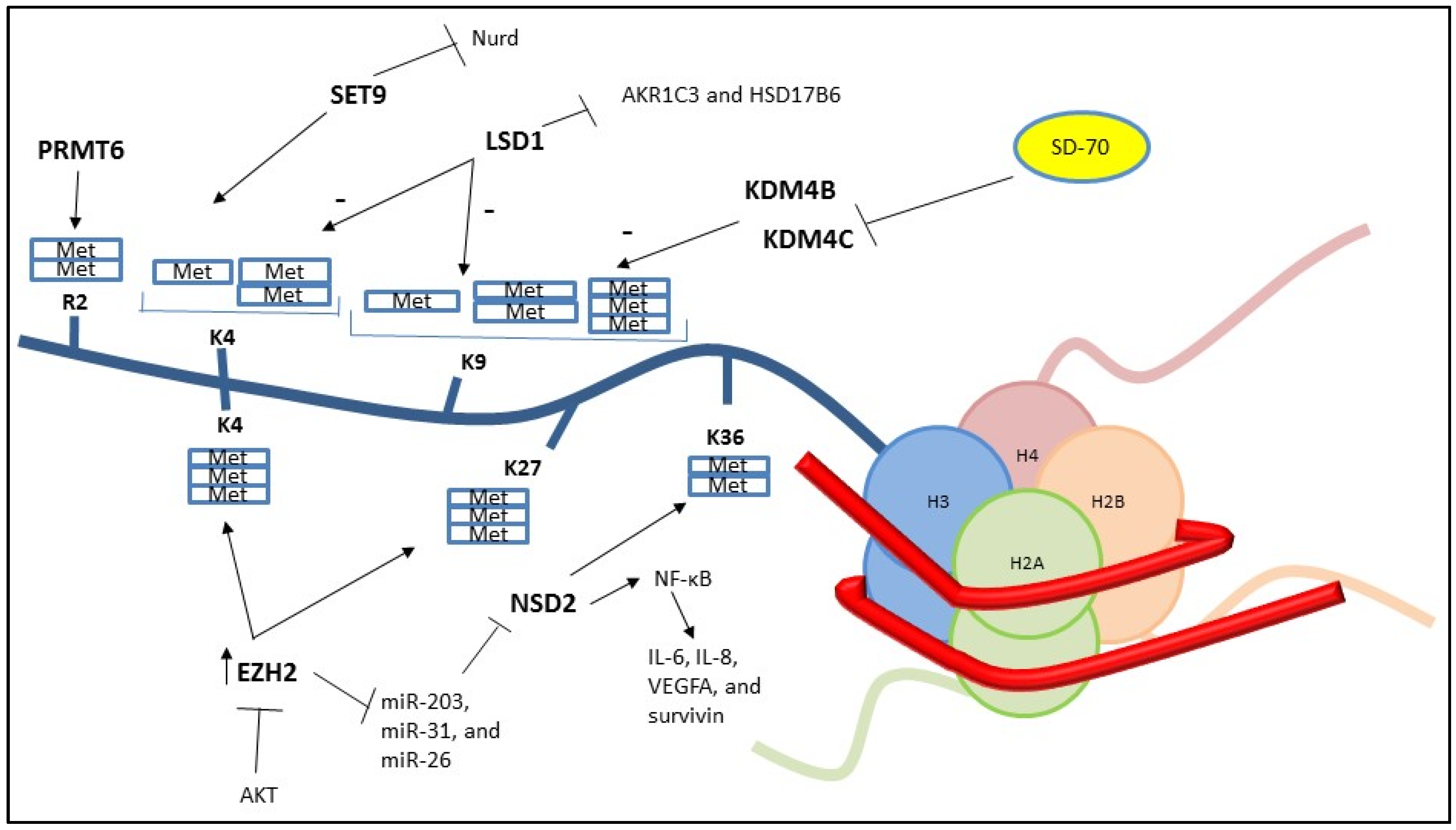
2. Histone Methylation
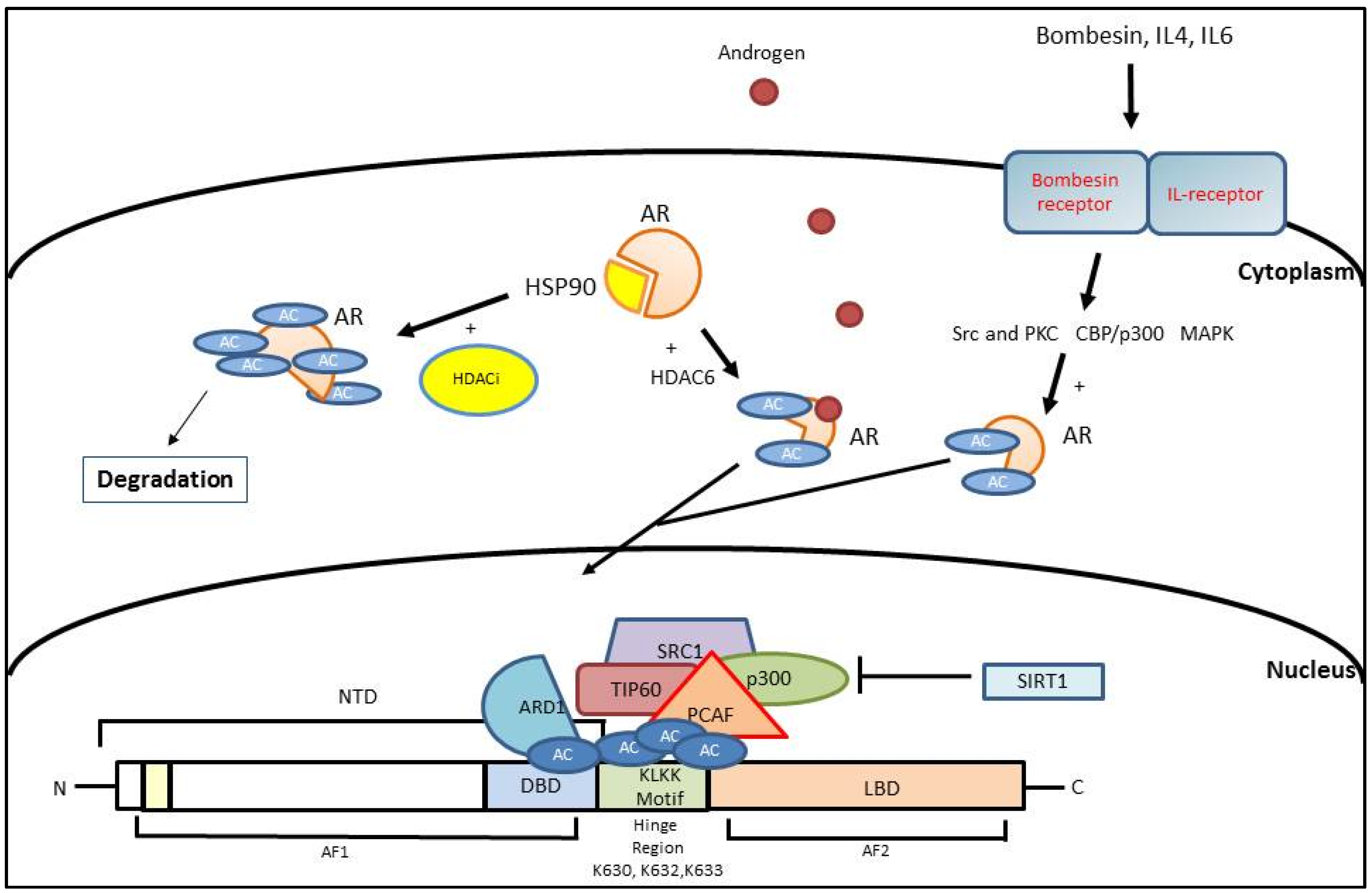
3. Histone Acetylation
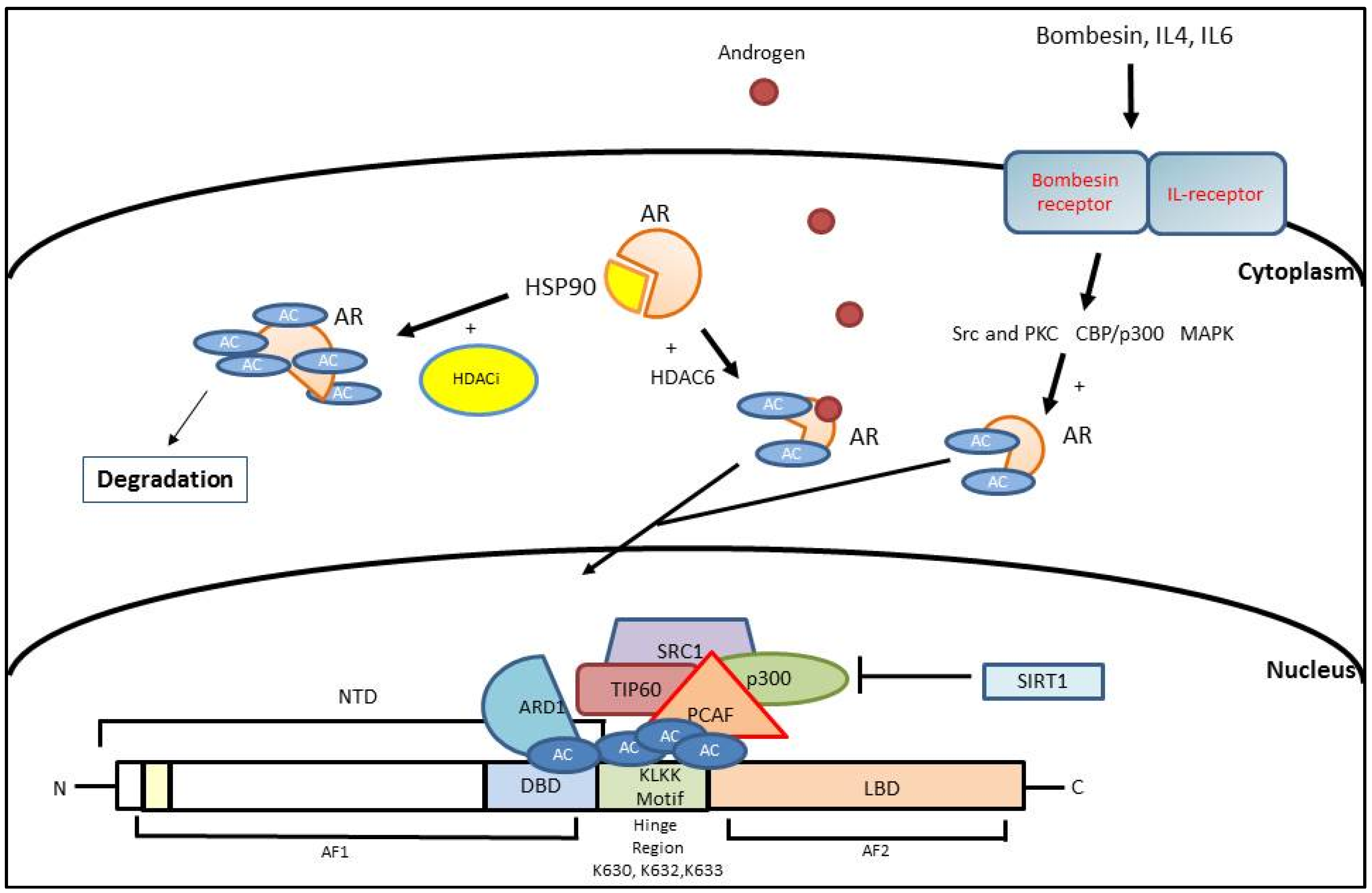
3.1. AR Activation Mediated by Histone Acetylation
3.2. AR Inhibition Mediated by Histone Acetylation
4. Non-Coding RNA
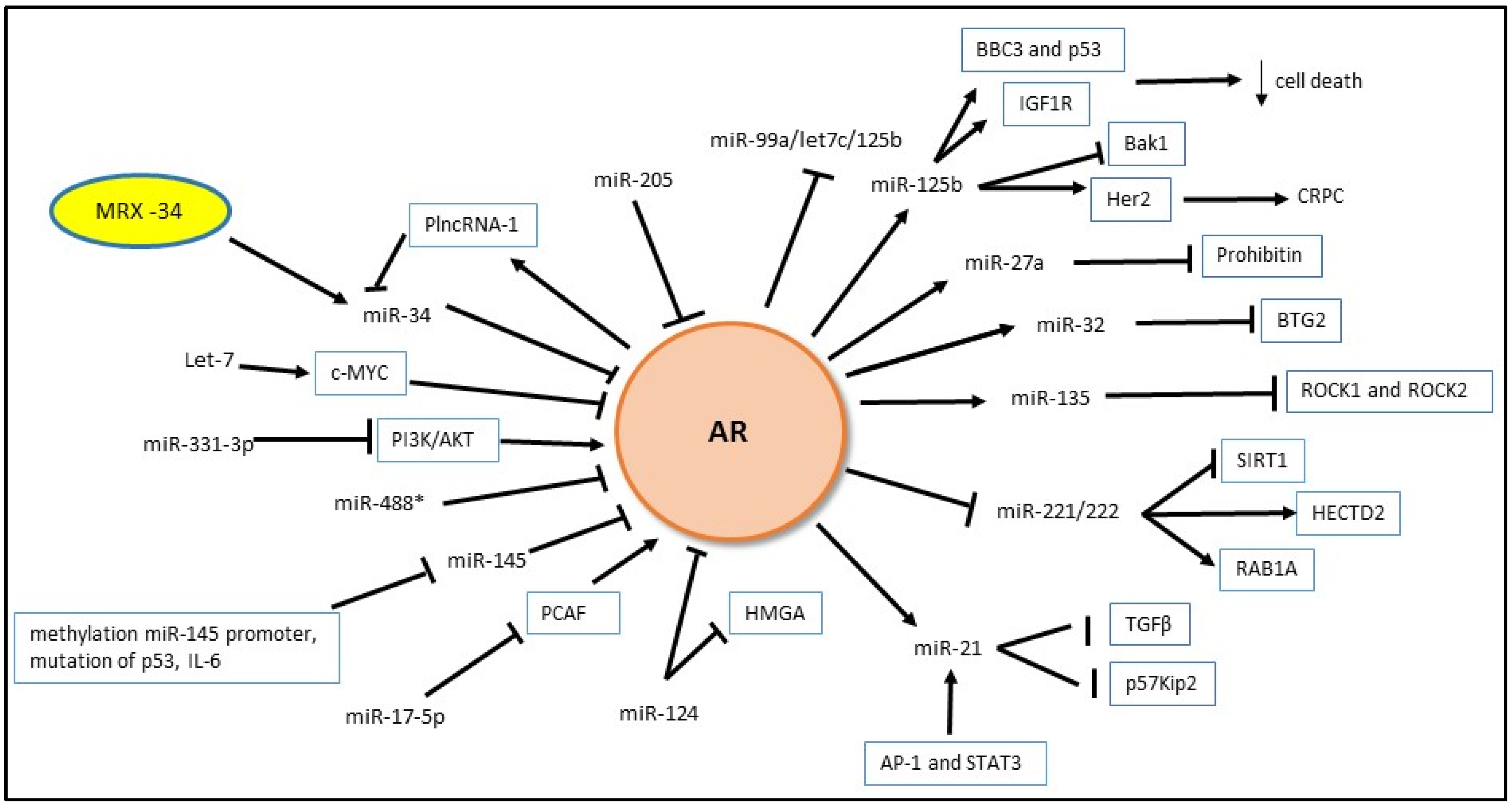
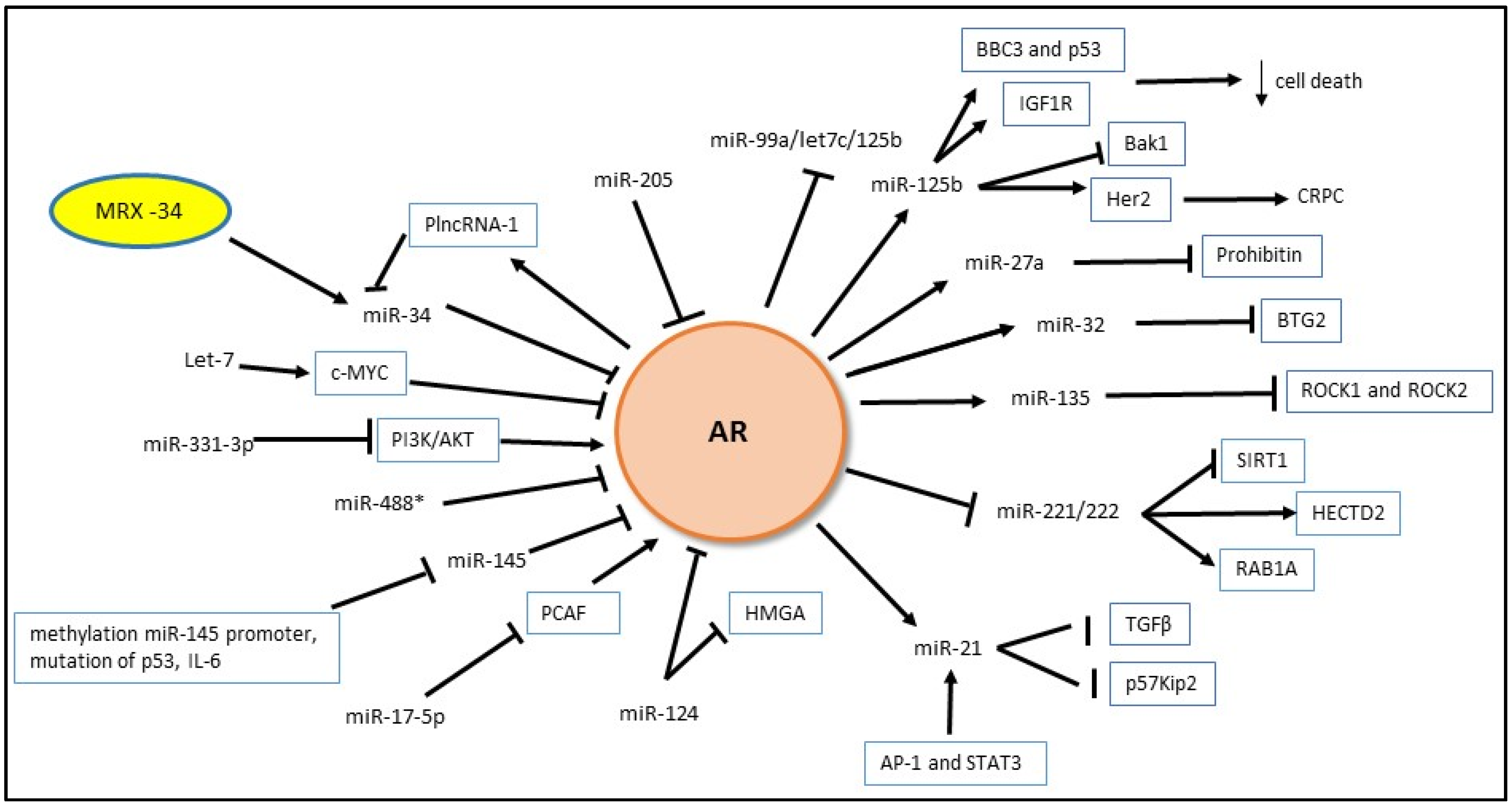
4.1. MicroRNA and AR
4.1.1. Androgen Regulation of miRNA Expression
4.1.2. MiRNA Regulation of Androgen Signaling
4.2. Long Non Coding RNA and AR
5. Novel PCa Biomarkers
5.1. Epigenetic Signature as Biomarkers
5.2. Long Non-Coding RNA as Biomarkers
5.3. MicroRNA as Biomarkers
6. Novel Treatments
6.1. Demethylase Inhibitor
6.2. Deacetylase Inhibitor
6.3. Non Coding RNA Therapy
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Van der Steen, T.; Tindall, D.J.; Huang, H. Posttranslational modification of the androgen receptor in prostate cancer. Int. J. Mol. Sci. 2013, 14, 14833–14859. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, N.; Gulley, J.L.; Dahut, W.L. Androgen deprivation therapy for prostate cancer. J. Am. Med. Assoc. 2005, 294, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Mills, I.G. Maintaining and reprogramming genomic androgen receptor activity in prostate cancer. Nat. Rev. Cancer 2014, 14, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Huggins, C.; Stevens, R.E., Jr.; Hodges, C.V. Studies on prostatic cancer: The effects of castration on advanced carcinoma of the prostate gland. Arch. Surg. 1941, 43, 209–223. [Google Scholar] [CrossRef]
- Masson, S.; Bahl, A. Metastatic castrate-resistant prostate cancer: Dawn of a new age of management. BJU Int. 2012, 110, 1110–1114. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Meissner, A.; Lander, E.S. The mammalian epigenome. Cell 2007, 128, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Probst, A.V.; Dunleavy, E.; Almouzni, G. Epigenetic inheritance during the cell cycle. Nat. Rev. Mol. Cell Biol. 2009, 10, 192–206. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, R.; Gupta, S. Epigenetics and cancer. J. Appl. Physiol. (1985) 2010, 109, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Peterson, C.L.; Laniel, M.A. Histones and histone modifications. Curr. Biol. 2004, 14, R546–R551. [Google Scholar] [CrossRef] [PubMed]
- Strahl, B.D.; Ohba, R.; Cook, R.G.; Allis, C.D. Methylation of histone H3 at lysine 4 is highly conserved and correlates with transcriptionally active nuclei in tetrahymena. Proc. Natl. Acad. Sci. USA 1999, 96, 14967–14972. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Schneider, R.; Myers, F.A.; Thorne, A.W.; Crane-Robinson, C.; Kouzarides, T. Spatial distribution of Di- and Tri-methyl Lysine 36 of histone H3 at active genes. J. Biol. Chem. 2005, 280, 17732–17736. [Google Scholar] [CrossRef] [PubMed]
- Heard, E.; Rougeulle, C.; Arnaud, D.; Avner, P.; Allis, C.D.; Spector, D.L. Methylation of histone H3 at Lys-9 is an early mark on the X chromosome during X inactivation. Cell 2001, 107, 727–738. [Google Scholar] [CrossRef]
- Rougeulle, C.; Chaumeil, J.; Sarma, K.; Allis, C.D.; Reinberg, D.; Avner, P.; Heard, E. Differential histone H3 Lys-9 and Lys-27 methylation profiles on the X chromosome. Mol. Cell. Biol. 2004, 24, 5475–5484. [Google Scholar] [CrossRef] [PubMed]
- Obianyo, O.; Thompson, P.R. Kinetic mechanism of protein arginine methyltransferase 6 (PRMT6). J. Biol. Chem. 2012, 287, 6062–6071. [Google Scholar] [CrossRef] [PubMed]
- Schurter, B.T.; Koh, S.S.; Chen, D.; Bunick, G.J.; Harp, J.M.; Hanson, B.L.; Henschen-Edman, A.; Mackay, D.R.; Stallcup, M.R.; Aswad, D.W. Methylation of histone H3 by coactivator-associated arginine methyltransferase 1. Biochemistry 2001, 40, 5747–5756. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Yuan, X.; Balk, S.P. Androgen receptor epigenetics. Transl. Androl. Urology 2013, 2, 148–157. [Google Scholar]
- Nishioka, K.; Chuikov, S.; Sarma, K.; Erdjument-Bromage, H.; Allis, C.D.; Tempst, P.; Reinberg, D. Set9, a novel histone H3 methyltransferase that facilitates transcription by precluding histone tail modifications required for heterochromatin formation. Genes Dev. 2002, 16, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Sonderstrup, I.M.; Nygard, S.B.; Poulsen, T.S.; Linnemann, D.; Stenvang, J.; Nielsen, H.J.; Bartek, J.; Brunner, N.; Norgaard, P.; Riis, L. Topoisomerase-1 and -2a gene copy numbers are elevated in mismatch repair-proficient colorectal cancers. Mol. Oncol. 2015, 9, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.R.; Jing, C.; Walker, P.A.; Martin, S.R.; Howell, S.A.; Blackburn, G.M.; Gamblin, S.J.; Xiao, B. Crystal structure and functional analysis of the histone methyltransferase set7/9. Cell 2002, 111, 105–115. [Google Scholar] [CrossRef]
- Gaughan, L.; Stockley, J.; Wang, N.; McCracken, S.R.; Treumann, A.; Armstrong, K.; Shaheen, F.; Watt, K.; McEwan, I.J.; Wang, C.; et al. Regulation of the androgen receptor by SET9-mediated methylation. Nucleic Acids Res. 2011, 39, 1266–1279. [Google Scholar] [CrossRef] [PubMed]
- Ko, S.; Ahn, J.; Song, C.S.; Kim, S.; Knapczyk-Stwora, K.; Chatterjee, B. Lysine methylation and functional modulation of androgen receptor by SET9 methyltransferase. Mol. Endocrinol. 2011, 25, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Langley, E.; Kemppainen, J.A.; Wilson, E.M. Intermolecular NH2-/carboxyl-terminal interactions in androgen receptor dimerization revealed by mutations that cause androgen insensitivity. J. Biol. Chem. 1998, 273, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Langley, E.; Zhou, Z.X.; Wilson, E.M. Evidence for an anti-parallel orientation of the ligand-activated human androgen receptor dimer. J. Biol. Chem. 1995, 270, 29983–29990. [Google Scholar] [PubMed]
- He, B.; Gampe, R.T., Jr.; Kole, A.J.; Hnat, A.T.; Stanley, T.B.; An, G.; Stewart, E.L.; Kalman, R.I.; Minges, J.T.; Wilson, E.M. Structural basis for androgen receptor interdomain and coactivator interactions suggests a transition in nuclear receptor activation function dominance. Mol. Cell 2004, 16, 425–438. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.B.; Choi, Y.; Lee, J.M.; Choi, K.C.; Kim, H.C.; Yoo, J.Y.; Lee, Y.H.; Yoon, H.G. The histone methyltransferase, NSD2, enhances androgen receptor-mediated transcription. FEBS Lett. 2009, 583, 1880–1886. [Google Scholar] [CrossRef] [PubMed]
- Asangani, I.A.; Ateeq, B.; Cao, Q.; Dodson, L.; Pandhi, M.; Kunju, L.P.; Mehra, R.; Lonigro, R.J.; Siddiqui, J.; Palanisamy, N.; et al. Characterization of the EZH2-MMSET histone methyltransferase regulatory axis in cancer. Mol. Cell 2013, 49, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Guo, L.; Duan, Z.J.; Tepper, C.G.; Xue, L.; Chen, X.; Kung, H.J.; Gao, A.C.; Zou, J.X.; Chen, H.W. Histone methyltransferase NSD2/Mmset mediates constitutive NF-κB signaling for cancer cell proliferation, survival, and tumor growth via a feed-forward loop. Mol. Cell. Biol. 2012, 32, 3121–3131. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.C.; Yu, J.; Runkle, C.; Wu, L.; Hu, M.; Wu, D.; Liu, J.S.; Wang, Q.; Qin, Z.S.; Yu, J. Cooperation between polycomb and androgen receptor during oncogenic transformation. Genome Res. 2012, 22, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.; Otte, A.P.; et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Dardenne, E.; Beltran, H.; Benelli, M.; Gayvert, K.; Berger, A.; Puca, L.; Cyrta, J.; Sboner, A.; Noorzad, Z.; MacDonald, T.; et al. N-Myc induces an EZH2-mediated transcriptional program driving neuroendocrine prostate cancer. Cancer Cell 2016, 30, 563–577. [Google Scholar] [CrossRef] [PubMed]
- Tolkach, Y.; Merseburger, A.; Herrmann, T.; Kuczyk, M.; Serth, J.; Imkamp, F. Signatures of adverse pathological features, androgen insensitivity and metastatic potential in prostate cancer. Anticancer Res. 2015, 35, 5443–5451. [Google Scholar] [PubMed]
- Cha, T.L.; Zhou, B.P.; Xia, W.; Wu, Y.; Yang, C.C.; Chen, C.T.; Ping, B.; Otte, A.P.; Hung, M.C. Akt-mediated phosphorylation of EZH2 suppresses methylation of Lysine 27 in histone H3. Science 2005, 310, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Wu, Z.J.; Groner, A.C.; He, H.H.; Cai, C.; Lis, R.T.; Wu, X.; Stack, E.C.; Loda, M.; Liu, T.; et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is polycomb-independent. Science 2012, 338, 1465–1469. [Google Scholar] [CrossRef] [PubMed]
- Hyllus, D.; Stein, C.; Schnabel, K.; Schiltz, E.; Imhof, A.; Dou, Y.; Hsieh, J.; Bauer, U.M. PRMT6-mediated methylation of R2 in histone H3 antagonizes H3 K4 trimethylation. Genes Dev. 2007, 21, 3369–3380. [Google Scholar] [CrossRef] [PubMed]
- Vieira, F.Q.; Costa-Pinheiro, P.; Ramalho-Carvalho, J.; Pereira, A.; Menezes, F.D.; Antunes, L.; Carneiro, I.; Oliveira, J.; Henrique, R.; Jeronimo, C. Deregulated expression of selected histone methylases and demethylases in prostate carcinoma. Endocr. Relat. Cancer 2014, 21, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Almeida-Rios, D.; Graca, I.; Vieira, F.Q.; Ramalho-Carvalho, J.; Pereira-Silva, E.; Martins, A.T.; Oliveira, J.; Goncalves, C.S.; Costa, B.M.; Henrique, R.; et al. Histone methyltransferase PRMT6 plays an oncogenic role of in prostate cancer. Oncotarget 2016, 7, 53018–53028. [Google Scholar] [CrossRef] [PubMed]
- Metzger, E.; Wissmann, M.; Yin, N.; Muller, J.M.; Schneider, R.; Peters, A.H.; Gunther, T.; Buettner, R.; Schule, R. Lsd1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 2005, 437, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Metzger, E.; Yin, N.; Wissmann, M.; Kunowska, N.; Fischer, K.; Friedrichs, N.; Patnaik, D.; Higgins, J.M.; Potier, N.; Scheidtmann, K.H.; et al. Phosphorylation of histone H3 at threonine 11 establishes a novel chromatin mark for transcriptional regulation. Nat. Cell Biol. 2008, 10, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Metzger, E.; Imhof, A.; Patel, D.; Kahl, P.; Hoffmeyer, K.; Friedrichs, N.; Muller, J.M.; Greschik, H.; Kirfel, J.; Ji, S.; et al. Phosphorylation of histone H3T6 by PKCbeta(i) controls demethylation at histone H3K4. Nature 2010, 464, 792–796. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhang, J.; Zhang, Y.; Wan, X.; Zhang, C.; Huang, X.; Huang, W.; Pu, H.; Pei, C.; Wu, H.; et al. Kdm1a triggers androgen-induced miRNA transcription via H3K4me2 demethylation and DNA oxidation. Prostate 2015, 75, 936–946. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; He, H.H.; Chen, S.; Coleman, I.; Wang, H.; Fang, Z.; Chen, S.; Nelson, P.S.; Liu, X.S.; Brown, M.; et al. Androgen receptor gene expression in prostate cancer is directly suppressed by the androgen receptor through recruitment of lysine-specific demethylase 1. Cancer Cell 2011, 20, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Stanbrough, M.; Bubley, G.J.; Ross, K.; Golub, T.R.; Rubin, M.A.; Penning, T.M.; Febbo, P.G.; Balk, S.P. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006, 66, 2815–2825. [Google Scholar] [CrossRef] [PubMed]
- Wako, K.; Kawasaki, T.; Yamana, K.; Suzuki, K.; Jiang, S.; Umezu, H.; Nishiyama, T.; Takahashi, K.; Hamakubo, T.; Kodama, T.; et al. Expression of androgen receptor through androgen-converting enzymes is associated with biological aggressiveness in prostate cancer. J. Clin. Pathol. 2008, 61, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Lou, W.; Zhu, Y.; Yang, J.C.; Nadiminty, N.; Gaikwad, N.W.; Evans, C.P.; Gao, A.C. Intracrine androgens and AKR1C3 activation confer resistance to enzalutamide in prostate cancer. Cancer Res. 2015, 75, 1413–1422. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Armstrong, C.M.; Lou, W.; Lombard, A.; Evans, C.P.; Gao, A.C. Inhibition of AKR1C3 activation overcomes resistance to abiraterone in advanced prostate cancer. Mol. Cancer Ther. 2016, 16, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Coffey, K.; Rogerson, L.; Ryan-Munden, C.; Alkharaif, D.; Stockley, J.; Heer, R.; Sahadevan, K.; O’Neill, D.; Jones, D.; Darby, S.; et al. The lysine demethylase, KDM4B, is a key molecule in androgen receptor signalling and turnover. Nucleic Acids Res. 2013, 41, 4433–4446. [Google Scholar] [CrossRef] [PubMed]
- Wissmann, M.; Yin, N.; Muller, J.M.; Greschik, H.; Fodor, B.D.; Jenuwein, T.; Vogler, C.; Schneider, R.; Gunther, T.; Buettner, R.; et al. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat. Cell Biol. 2007, 9, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Yamane, K.; Toumazou, C.; Tsukada, Y.; Erdjument-Bromage, H.; Tempst, P.; Wong, J.; Zhang, Y. JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell 2006, 125, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.W.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R.; et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005, 310, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Haffner, M.C.; de Marzo, A.M.; Meeker, A.K.; Nelson, W.G.; Yegnasubramanian, S. Transcription-induced DNA double strand breaks: Both oncogenic force and potential therapeutic target? Clin. Cancer Res. 2011, 17, 3858–3864. [Google Scholar] [CrossRef] [PubMed]
- Haffner, M.C.; Aryee, M.J.; Toubaji, A.; Esopi, D.M.; Albadine, R.; Gurel, B.; Isaacs, W.B.; Bova, G.S.; Liu, W.; Xu, J.; et al. Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements. Nat. Genet. 2010, 42, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Yu, J.; Mani, R.S.; Cao, Q.; Brenner, C.J.; Cao, X.; Wang, X.; Wu, L.; Li, J.; Hu, M.; et al. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell 2010, 17, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Metzger, E.; Willmann, D.; McMillan, J.; Forne, I.; Metzger, P.; Gerhardt, S.; Petroll, K.; von Maessenhausen, A.; Urban, S.; Schott, A.K.; et al. Assembly of methylated KDM1A and CHD1 drives androgen receptor-dependent transcription and translocation. Nat. Struct. Mol. Biol. 2016, 23, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Rao, M.; Wu, K.; Wang, C.; Zhang, X.; Hessien, M.; Yeung, Y.G.; Gioeli, D.; Weber, M.J.; Pestell, R.G. The androgen receptor acetylation site regulates cAMP and Akt but not ERK-induced activity. J. Biol. Chem. 2004, 279, 29436–29449. [Google Scholar] [CrossRef] [PubMed]
- Grant, P.A.; Berger, S.L. Histone acetyltransferase complexes. Semin. Cell Dev. Biol. 1999, 10, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Wang, C.; Reutens, A.T.; Wang, J.; Angeletti, R.H.; Siconolfi-Baez, L.; Ogryzko, V.; Avantaggiati, M.L.; Pestell, R.G. P300 and p300/cAMP-response element-binding protein-associated factor acetylate the androgen receptor at sites governing hormone-dependent transactivation. J. Biol. Chem. 2000, 275, 20853–20860. [Google Scholar] [CrossRef] [PubMed]
- Coffey, K.; Robson, C.N. Regulation of the androgen receptor by post-translational modifications. J. Endocrinol. 2012, 215, 221–237. [Google Scholar] [CrossRef] [PubMed]
- Lavery, D.N.; Bevan, C.L. Androgen receptor signalling in prostate cancer: The functional consequences of acetylation. J. Biomed. Biotechnol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Culig, Z. Androgen receptor coactivators in regulation of growth and differentiation in prostate cancer. J. Cell. Physiol. 2016, 231, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Rao, M.; Wang, C.; Sakamaki, T.; Wang, J.; di Vizio, D.; Zhang, X.; Albanese, C.; Balk, S.; Chang, C.; et al. Acetylation of androgen receptor enhances coactivator binding and promotes prostate cancer cell growth. Mol. Cell. Biol. 2003, 23, 8563–8575. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Shimelis, H.; Linn, D.E.; Jiang, R.; Yang, X.; Sun, F.; Guo, Z.; Chen, H.; Li, W.; Chen, H.; et al. Regulation of androgen receptor transcriptional activity and specificity by rnf6-induced ubiquitination. Cancer Cell 2009, 15, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wu, R.C.; O’Malley, B.W. Normal and cancer-related functions of the p160 steroid receptor co-activator (SRC) family. Nat. Rev. Cancer 2009, 9, 615–630. [Google Scholar] [CrossRef] [PubMed]
- Gaughan, L.; Logan, I.R.; Cook, S.; Neal, D.E.; Robson, C.N. Tip60 and histone deacetylase 1 regulate androgen receptor activity through changes to the acetylation status of the receptor. J. Biol. Chem. 2002, 277, 25904–25913. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, Z.; Guo, J.; Li, Y.; Bavarva, J.H.; Qian, C.; Brahimi-Horn, M.C.; Tan, D.; Liu, W. Inactivation of androgen-induced regulator ard1 inhibits androgen receptor acetylation and prostate tumorigenesis. Proc. Natl. Acad. Sci. USA 2012, 109, 3053–3058. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. The CBP co-activator is a histone acetyltransferase. Nature 1996, 384, 641–643. [Google Scholar] [CrossRef] [PubMed]
- Ogryzko, V.V.; Schiltz, R.L.; Russanova, V.; Howard, B.H.; Nakatani, Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell 1996, 87, 953–959. [Google Scholar] [CrossRef]
- Zhong, J.; Ding, L.; Bohrer, L.R.; Pan, Y.; Liu, P.; Zhang, J.; Sebo, T.J.; Karnes, R.J.; Tindall, D.J.; van Deursen, J.; et al. P300 acetyltransferase regulates androgen receptor degradation and pten-deficient prostate tumorigenesis. Cancer Res. 2014, 74, 1870–1880. [Google Scholar] [CrossRef] [PubMed]
- Levy, L.; Wei, Y.; Labalette, C.; Wu, Y.; Renard, C.A.; Buendia, M.A.; Neuveut, C. Acetylation of beta-catenin by p300 regulates beta-catenin-Tcf4 interaction. Mol. Cell. Biol. 2004, 24, 3404–3414. [Google Scholar] [CrossRef] [PubMed]
- Spencer, T.E.; Jenster, G.; Burcin, M.M.; Allis, C.D.; Zhou, J.; Mizzen, C.A.; McKenna, N.J.; Onate, S.A.; Tsai, S.Y.; Tsai, M.J.; et al. Steroid receptor coactivator-1 is a histone acetyltransferase. Nature 1997, 389, 194–198. [Google Scholar] [PubMed]
- McKenna, N.J.; Lanz, R.B.; O’Malley, B.W. Nuclear receptor coregulators: Cellular and molecular biology. Endocr. Rev. 1999, 20, 321–344. [Google Scholar] [CrossRef] [PubMed]
- Nakka, M.; Agoulnik, I.U.; Weigel, N.L. Targeted disruption of the p160 coactivator interface of androgen receptor (AR) selectively inhibits AR activity in both androgen-dependent and castration-resistant ar-expressing prostate cancer cells. Int. J. Biochem. Cell Biol. 2013, 45, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Zhu, J.; Goodman, O.B., Jr.; Pestell, R.G.; Schlegel, P.N.; Nanus, D.M.; Shen, R. Activation of p300 histone acetyltransferase activity and acetylation of the androgen receptor by bombesin in prostate cancer cells. Oncogene 2006, 25, 2011–2021. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.O.; Chun, J.Y.; Nadiminty, N.; Lou, W.; Feng, S.; Gao, A.C. Interleukin-4 activates androgen receptor through CBP/p300. Prostate 2009, 69, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Malinowska, K.; Neuwirt, H.; Cavarretta, I.T.; Bektic, J.; Steiner, H.; Dietrich, H.; Moser, P.L.; Fuchs, D.; Hobisch, A.; Culig, Z. Interleukin-6 stimulation of growth of prostate cancer in vitro and in vivo through activation of the androgen receptor. Endocr.-Relat. Cancer 2009, 16, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Ueda, T.; Mawji, N.R.; Bruchovsky, N.; Sadar, M.D. Ligand-independent activation of the androgen receptor by interleukin-6 and the role of steroid receptor coactivator-1 in prostate cancer cells. J. Biol. Chem. 2002, 277, 38087–38094. [Google Scholar] [CrossRef] [PubMed]
- Brady, M.E.; Ozanne, D.M.; Gaughan, L.; Waite, I.; Cook, S.; Neal, D.E.; Robson, C.N. Tip60 is a nuclear hormone receptor coactivator. J. Biol. Chem. 1999, 274, 17599–17604. [Google Scholar] [CrossRef] [PubMed]
- Gaughan, L.; Logan, I.R.; Neal, D.E.; Robson, C.N. Regulation of androgen receptor and histone deacetylase 1 by mdm2-mediated ubiquitylation. Nucleic Acids Res. 2005, 33, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Shiota, M.; Yokomizo, A.; Masubuchi, D.; Tada, Y.; Inokuchi, J.; Eto, M.; Uchiumi, T.; Fujimoto, N.; Naito, S. Tip60 promotes prostate cancer cell proliferation by translocation of androgen receptor into the nucleus. Prostate 2010, 70, 540–554. [Google Scholar] [CrossRef] [PubMed]
- Park, E.C.; Szostak, J.W. Ard1 and nat1 proteins form a complex that has N-terminal acetyltransferase activity. EMBO J. 1992, 11, 2087–2093. [Google Scholar] [PubMed]
- Hua, K.T.; Tan, C.T.; Johansson, G.; Lee, J.M.; Yang, P.W.; Lu, H.Y.; Chen, C.K.; Su, J.L.; Chen, P.B.; Wu, Y.L.; et al. N-alpha-acetyltransferase 10 protein suppresses cancer cell metastasis by binding PIX proteins and inhibiting Cdc42/Rac1 activity. Cancer Cell 2011, 19, 218–231. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.W.; Bae, M.K.; Ahn, M.Y.; Kim, S.H.; Sohn, T.K.; Bae, M.H.; Yoo, M.A.; Song, E.J.; Lee, K.J.; Kim, K.W. Regulation and destabilization of HIF-1alpha by ARD1-mediated acetylation. Cell 2002, 111, 709–720. [Google Scholar] [CrossRef]
- Lee, C.F.; Ou, D.S.; Lee, S.B.; Chang, L.H.; Lin, R.K.; Li, Y.S.; Upadhyay, A.K.; Cheng, X.; Wang, Y.C.; Hsu, H.S.; et al. HNaa10p contributes to tumorigenesis by facilitating DNMT1-mediated tumor suppressor gene silencing. J. Clin. Investig. 2010, 120, 2920–2930. [Google Scholar] [CrossRef] [PubMed]
- DePaolo, J.S.; Wang, Z.; Guo, J.; Zhang, G.; Qian, C.; Zhang, H.; Zabaleta, J.; Liu, W. Acetylation of androgen receptor by ARD1 promotes dissociation from HSP90 complex and prostate tumorigenesis. Oncotarget 2016, 7, 71417–71428. [Google Scholar] [CrossRef] [PubMed]
- Urbanucci, A.; Marttila, S.; Janne, O.A.; Visakorpi, T. Androgen receptor overexpression alters binding dynamics of the receptor to chromatin and chromatin structure. Prostate 2012, 72, 1223–1232. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Shen, H.C.; Wantroba, M.; Khalid, O.; Liang, G.; Wang, Q.; Gentzschein, E.; Pinski, J.K.; Stanczyk, F.Z.; Jones, P.A.; et al. Locus-wide chromatin remodeling and enhanced androgen receptor-mediated transcription in recurrent prostate tumor cells. Mol. Cell. Biol. 2006, 26, 7331–7341. [Google Scholar] [CrossRef] [PubMed]
- Karvonen, U.; Janne, O.A.; Palvimo, J.J. Androgen receptor regulates nuclear trafficking and nuclear domain residency of corepressor HDAC7 in a ligand-dependent fashion. Exp. Cell Res. 2006, 312, 3165–3183. [Google Scholar] [CrossRef] [PubMed]
- Ai, J.; Wang, Y.; Dar, J.A.; Liu, J.; Liu, L.; Nelson, J.B.; Wang, Z. Hdac6 regulates androgen receptor hypersensitivity and nuclear localization via modulating HSP90 acetylation in castration-resistant prostate cancer. Mol. Endocrinol. 2009, 23, 1963–1972. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Liu, M.; Sauve, A.A.; Jiao, X.; Zhang, X.; Wu, X.; Powell, M.J.; Yang, T.; Gu, W.; Avantaggiati, M.L.; et al. Hormonal control of androgen receptor function through SIRT1. Mol. Cell. Biol. 2006, 26, 8122–8135. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Zhang, J.; Kaipainen, A.; Lucas, J.M.; Yang, H. Long non-coding RNA: A newly deciphered “code” in prostate cancer. Cancer Lett. 2016, 375, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Brosnan, C.A.; Voinnet, O. The long and the short of noncoding RNAs. Curr. Opin. Cell Biol. 2009, 21, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Tay, Y.; Zhang, J.; Thomson, A.M.; Lim, B.; Rigoutsos, I. MicroRNAs to nanog, Oct4 and Sox2 coding regions modulate embryonic stem cell differentiation. Nature 2008, 455, 1124–1128. [Google Scholar] [CrossRef] [PubMed]
- Shih, J.W.; Wang, L.Y.; Hung, C.L.; Kung, H.J.; Hsieh, C.L. Non-coding RNAs in castration-resistant prostate cancer: Regulation of androgen receptor signaling and cancer metabolism. Int. J. Mol. Sci. 2015, 16, 28943–28978. [Google Scholar] [CrossRef] [PubMed]
- Waltering, K.K.; Porkka, K.P.; Jalava, S.E.; Urbanucci, A.; Kohonen, P.J.; Latonen, L.M.; Kallioniemi, O.P.; Jenster, G.; Visakorpi, T. Androgen regulation of micro-RNAs in prostate cancer. Prostate 2011, 71, 604–614. [Google Scholar] [CrossRef] [PubMed]
- Mo, W.; Zhang, J.; Li, X.; Meng, D.; Gao, Y.; Yang, S.; Wan, X.; Zhou, C.; Guo, F.; Huang, Y.; et al. Identification of novel AR-targeted microRNAs mediating androgen signalling through critical pathways to regulate cell viability in prostate cancer. PLoS ONE 2013, 8, e56592. [Google Scholar] [CrossRef] [PubMed]
- Murata, T.; Takayama, K.; Katayama, S.; Urano, T.; Horie-Inoue, K.; Ikeda, K.; Takahashi, S.; Kawazu, C.; Hasegawa, A.; Ouchi, Y.; et al. MiR-148a is an androgen-responsive microRNA that promotes LNCaP prostate cell growth by repressing its target cand1 expression. Prostate Cancer Prostatic Dis. 2010, 13, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, C.E.; Dart, D.A.; Sita-Lumsden, A.; Cheng, H.; Rennie, P.S.; Bevan, C.L. Androgen-regulated processing of the oncomir miR-27a, which targets prohibitin in prostate cancer. Hum. Mol. Genet. 2012, 21, 3112–3127. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.; Tsutsumi, S.; Katayama, S.; Okayama, T.; Horie-Inoue, K.; Ikeda, K.; Urano, T.; Kawazu, C.; Hasegawa, A.; Ikeo, K.; et al. Integration of cap analysis of gene expression and chromatin immunoprecipitation analysis on array reveals genome-wide androgen receptor signaling in prostate cancer cells. Oncogene 2011, 30, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Layer, R.; Mueller, A.C.; Cichewicz, M.A.; Negishi, M.; Paschal, B.M.; Dutta, A. Regulation of several androgen-induced genes through the repression of the miR-99a/let-7c/miR-125b-2 miRNA cluster in prostate cancer cells. Oncogene 2014, 33, 1448–1457. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.; Young, C.Y.; Yuan, H. MicroRNAs and prostate cancer. Acta Biochim. Biophys. Sin. 2010, 42, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Catto, J.W.; Alcaraz, A.; Bjartell, A.S.; de Vere White, R.; Evans, C.P.; Fussel, S.; Hamdy, F.C.; Kallioniemi, O.; Mengual, L.; Schlomm, T.; et al. MicroRNA in prostate, bladder, and kidney cancer: A systematic review. Eur. Urol. 2011, 59, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Bemis, L.; Su, L.J.; Gao, D.; Flaig, T.W. Mir-125b regulation of androgen receptor signaling via modulation of the receptor complex co-repressor NCOR2. Biores. Open Access 2012, 1, 55–62. [Google Scholar] [CrossRef] [PubMed]
- ChunJiao, S.; Huan, C.; ChaoYang, X.; GuoMei, R. Uncovering the roles of miRNAs and their relationship with androgen receptor in prostate cancer. IUBMB Life 2014, 66, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Lv, Y.G.; Yan, C.Y.; Yi, J.; Ling, R. Enforced expression of hsa-miR-125a-3p in breast cancer cells potentiates docetaxel sensitivity via modulation of BRCA1 signaling. Biochem. Biophys. Res. Commun. 2016, 479, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Kroiss, A.; Vincent, S.; Decaussin-Petrucci, M.; Meugnier, E.; Viallet, J.; Ruffion, A.; Chalmel, F.; Samarut, J.; Allioli, N. Androgen-regulated microRNA-135a decreases prostate cancer cell migration and invasion through downregulating rock1 and rock2. Oncogene 2015, 34, 2846–2855. [Google Scholar] [CrossRef] [PubMed]
- Coarfa, C.; Fiskus, W.; Eedunuri, V.K.; Rajapakshe, K.; Foley, C.; Chew, S.A.; Shah, S.S.; Geng, C.; Shou, J.; Mohamed, J.S.; et al. Comprehensive proteomic profiling identifies the androgen receptor axis and other signaling pathways as targets of microRNAs suppressed in metastatic prostate cancer. Oncogene 2016, 35, 2345–2356. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Pu, H.; Huang, W.; Yang, S.; Zhang, Y.; Kong, Z.; Yang, Z.; Zhao, P.; Li, A.; Li, T.; et al. Androgen-induced miR-135a acts as a tumor suppressor through downregulating RBAK and MMP11, and mediates resistance to androgen deprivation therapy. Oncotarget 2016, 7, 51284–51300. [Google Scholar] [CrossRef] [PubMed]
- Jalava, S.E.; Urbanucci, A.; Latonen, L.; Waltering, K.K.; Sahu, B.; Janne, O.A.; Seppala, J.; Lahdesmaki, H.; Tammela, T.L.; Visakorpi, T. Androgen-regulated miR-32 targets BTG2 and is overexpressed in castration-resistant prostate cancer. Oncogene 2012, 31, 4460–4471. [Google Scholar] [CrossRef] [PubMed]
- Mao, B.; Zhang, Z.; Wang, G. BTG2: A rising star of tumor suppressors (review). Int. J. Oncol. 2015, 46, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Ribas, J.; Ni, X.; Haffner, M.; Wentzel, E.A.; Salmasi, A.H.; Chowdhury, W.H.; Kudrolli, T.A.; Yegnasubramanian, S.; Luo, J.; Rodriguez, R.; et al. MiR-21: An androgen receptor-regulated microRNA that promotes hormone-dependent and hormone-independent prostate cancer growth. Cancer Res. 2009, 69, 7165–7169. [Google Scholar] [CrossRef] [PubMed]
- Fujita, S.; Ito, T.; Mizutani, T.; Minoguchi, S.; Yamamichi, N.; Sakurai, K.; Iba, H. MiR-21 gene expression triggered by AP-1 is sustained through a double-negative feedback mechanism. J. Mol. Biol. 2008, 378, 492–504. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Jaeger, S.A.; Hirsch, H.A.; Bulyk, M.L.; Struhl, K. STAT3 activation of miR-21 and miR-181b-1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Mol. Cell 2010, 39, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Deng, J.J.; Gowda, P.S.; Rao, M.K.; Lin, C.L.; Chen, C.L.; Huang, T.; Sun, L.Z. Androgen receptor and microRNA-21 axis downregulates transforming growth factor beta receptor II (TGFBR2) expression in prostate cancer. Oncogene 2014, 33, 4097–4106. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Lin, C.L.; Huang, T.H.; Bouamar, H.; Sun, L.Z. MicroRNA-21 inhibits p57kip2 expression in prostate cancer. Mol. Cancer 2014. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, M.; Quintavalle, C.; Romano, G.; Croce, C.M.; Condorelli, G. MiR221/222 in cancer: Their role in tumor progression and response to therapy. Curr. Mol. Med. 2012, 12, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Gordanpour, A.; Stanimirovic, A.; Nam, R.K.; Moreno, C.S.; Sherman, C.; Sugar, L.; Seth, A. MiR-221 is down-regulated in tmprss2:Erg fusion-positive prostate cancer. Anticancer Res. 2011, 31, 403–410. [Google Scholar] [PubMed]
- Li, T.; Li, R.S.; Li, Y.H.; Zhong, S.; Chen, Y.Y.; Zhang, C.M.; Hu, M.M.; Shen, Z.J. MiR-21 as an independent biochemical recurrence predictor and potential therapeutic target for prostate cancer. J. Urol. 2012, 187, 1466–1472. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yang, Y.; Gan, R.; Zhao, L.; Li, W.; Zhou, H.; Wang, X.; Lu, J.; Meng, Q.H. Down-regulation of miR-221 and miR-222 restrain prostate cancer cell proliferation and migration that is partly mediated by activation of sirt1. PLoS ONE 2014, 9, e98833. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Wang, X.; He, H.H.; Sweeney, C.J.; Liu, S.X.; Brown, M.; Balk, S.; Lee, G.S.; Kantoff, P.W. MiR-221 promotes the development of androgen independence in prostate cancer cells via downregulation of HECTD2 and RAB1A. Oncogene 2014, 33, 2790–2800. [Google Scholar] [CrossRef] [PubMed]
- Ostling, P.; Leivonen, S.K.; Aakula, A.; Kohonen, P.; Makela, R.; Hagman, Z.; Edsjo, A.; Kangaspeska, S.; Edgren, H.; Nicorici, D.; et al. Systematic analysis of microRNAs targeting the androgen receptor in prostate cancer cells. Cancer Res. 2011, 71, 1956–1967. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Alumkal, J. Epigenetic regulation of androgen receptor signaling in prostate cancer. Epigenetics 2010, 5, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Hagman, Z.; Haflidadottir, B.S.; Ceder, J.A.; Larne, O.; Bjartell, A.; Lilja, H.; Edsjo, A.; Ceder, Y. MiR-205 negatively regulates the androgen receptor and is associated with adverse outcome of prostate cancer patients. Br. J. Cancer 2013, 108, 1668–1676. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Lin, C.P.; Ho, J.J.; He, X.; Okada, N.; Bu, P.; Zhong, Y.; Kim, S.Y.; Bennett, M.J.; Chen, C.; et al. MiR-34 miRNAs provide a barrier for somatic cell reprogramming. Nat. Cell Biol. 2011, 13, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Hagman, Z.; Larne, O.; Edsjo, A.; Bjartell, A.; Ehrnstrom, R.A.; Ulmert, D.; Lilja, H.; Ceder, Y. MiR-34c is downregulated in prostate cancer and exerts tumor suppressive functions. Int. J. Cancer 2010, 127, 2768–2776. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Heath, E.; Chen, W.; Cher, M.; Powell, I.; Heilbrun, L.; Li, Y.; Ali, S.; Sethi, S.; Hassan, O.; et al. Epigenetic silencing of miR-34a in human prostate cancer cells and tumor tissue specimens can be reversed by br-dim treatment. Am. J. Transl. Res. 2012, 4, 14–23. [Google Scholar] [PubMed]
- Cannell, I.G.; Kong, Y.W.; Johnston, S.J.; Chen, M.L.; Collins, H.M.; Dobbyn, H.C.; Elia, A.; Kress, T.R.; Dickens, M.; Clemens, M.J.; et al. P38 mapk/mk2-mediated induction of miR-34c following DNA damage prevents myc-dependent DNA replication. Proc. Natl. Acad. Sci. USA 2010, 107, 5375–5380. [Google Scholar] [CrossRef] [PubMed]
- Corney, D.C.; Flesken-Nikitin, A.; Godwin, A.K.; Wang, W.; Nikitin, A.Y. MicroRNA-34b and microRNA-34c are targets of p53 and cooperate in control of cell proliferation and adhesion-independent growth. Cancer Res. 2007, 67, 8433–8438. [Google Scholar] [CrossRef] [PubMed]
- Toyota, M.; Suzuki, H.; Sasaki, Y.; Maruyama, R.; Imai, K.; Shinomura, Y.; Tokino, T. Epigenetic silencing of microRNA-34b/c and B-cell translocation gene 4 is associated with cpg island methylation in colorectal cancer. Cancer Res. 2008, 68, 4123–4132. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Xu, C.; Li, Y.; Cai, X.; Ren, S.; Liu, H.; Wang, Y.; Wang, F.; Chen, R.; Qu, M.; et al. A feed-forward regulatory loop between androgen receptor and PlncRNA-1 promotes prostate cancer progression. Cancer Lett. 2016, 374, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Liu, C.G.; Sevignani, C.; Ferracin, M.; Felli, N.; Dumitru, C.D.; Shimizu, M.; Cimmino, A.; Zupo, S.; Dono, M.; et al. MicroRNA profiling reveals distinct signatures in B cell chronic lymphocytic leukemias. Proc. Natl. Acad. Sci. USA 2004, 101, 11755–11760. [Google Scholar] [CrossRef] [PubMed]
- Ozen, M.; Creighton, C.J.; Ozdemir, M.; Ittmann, M. Widespread deregulation of microRNA expression in human prostate cancer. Oncogene 2008, 27, 1788–1793. [Google Scholar] [CrossRef]
- Johnson, S.M.; Grosshans, H.; Shingara, J.; Byrom, M.; Jarvis, R.; Cheng, A.; Labourier, E.; Reinert, K.L.; Brown, D.; Slack, F.J. RAS is regulated by the let-7 microRNA family. Cell 2005, 120, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.S.; Lu, J.; Mercer, K.L.; Golub, T.R.; Jacks, T. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat. Genet. 2007, 39, 673–677. [Google Scholar] [CrossRef] [PubMed]
- Nadiminty, N.; Tummala, R.; Lou, W.; Zhu, Y.; Zhang, J.; Chen, X.; eVere White, R.W.; Kung, H.J.; Evans, C.P.; Gao, A.C. MicroRNA let-7c suppresses androgen receptor expression and activity via regulation of myc expression in prostate cancer cells. J. Biol. Chem. 2012, 287, 1527–1537. [Google Scholar] [CrossRef]
- Gao, L.; Schwartzman, J.; Gibbs, A.; Lisac, R.; Kleinschmidt, R.; Wilmot, B.; Bottomly, D.; Coleman, I.; Nelson, P.; McWeeney, S.; et al. Androgen receptor promotes ligand-independent prostate cancer progression through c-Myc upregulation. PLoS ONE 2013, 8, e63563. [Google Scholar] [CrossRef] [PubMed]
- Epis, M.R.; Giles, K.M.; Barker, A.; Kendrick, T.S.; Leedman, P.J. Mir-331–3p regulates erbb-2 expression and androgen receptor signaling in prostate cancer. J. Biol. Chem. 2009, 284, 24696–24704. [Google Scholar] [CrossRef] [PubMed]
- Sikand, K.; Slaibi, J.E.; Singh, R.; Slane, S.D.; Shukla, G.C. Mir 488* inhibits androgen receptor expression in prostate carcinoma cells. Int. J. Cancer 2011, 129, 810–819. [Google Scholar] [CrossRef] [PubMed]
- Gong, A.Y.; Eischeid, A.N.; Xiao, J.; Zhao, J.; Chen, D.; Wang, Z.Y.; Young, C.Y.; Chen, X.M. Mir-17–5p targets the p300/cbp-associated factor and modulates androgen receptor transcriptional activity in cultured prostate cancer cells. BMC Cancer 2012. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.M.; Castillo, L.; Mahon, K.L.; Chiam, K.; Lee, B.Y.; Nguyen, Q.; Boyer, M.J.; Stockler, M.R.; Pavlakis, N.; Marx, G.; et al. Circulating microRNAs are associated with docetaxel chemotherapy outcome in castration-resistant prostate cancer. Br. J. Cancer 2014, 110, 2462–2471. [Google Scholar] [CrossRef] [PubMed]
- Lujambio, A.; Ropero, S.; Ballestar, E.; Fraga, M.F.; Cerrato, C.; Setien, F.; Casado, S.; Suarez-Gauthier, A.; Sanchez-Cespedes, M.; Git, A.; et al. Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Res. 2007, 67, 1424–1429. [Google Scholar] [CrossRef] [PubMed]
- Agirre, X.; Vilas-Zornoza, A.; Jimenez-Velasco, A.; Martin-Subero, J.I.; Cordeu, L.; Garate, L.; San Jose-Eneriz, E.; Abizanda, G.; Rodriguez-Otero, P.; Fortes, P.; et al. Epigenetic silencing of the tumor suppressor microRNA Hsa-miR-124a regulates CDK6 expression and confers a poor prognosis in acute lymphoblastic leukemia. Cancer Res. 2009, 69, 4443–4453. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.B.; Xue, L.; Ma, A.H.; Tepper, C.G.; Gandour-Edwards, R.; Kung, H.J.; deVere White, R.W. Tumor suppressive miR-124 targets androgen receptor and inhibits proliferation of prostate cancer cells. Oncogene 2013, 32, 4130–4138. [Google Scholar] [CrossRef] [PubMed]
- Chu, M.; Chang, Y.; Guo, Y.; Wang, N.; Cui, J.; Gao, W.Q. Regulation and methylation of tumor suppressor miR-124 by androgen receptor in prostate cancer cells. PLoS ONE 2015, 10, e0116197. [Google Scholar] [CrossRef] [PubMed]
- Larne, O.; Martens-Uzunova, E.; Hagman, Z.; Edsjo, A.; Lippolis, G.; den Berg, M.S.; Bjartell, A.; Jenster, G.; Ceder, Y. Miq—A novel microRNA based diagnostic and prognostic tool for prostate cancer. Int. J. Cancer 2013, 132, 2867–2875. [Google Scholar] [CrossRef] [PubMed]
- Wach, S.; Nolte, E.; Szczyrba, J.; Stohr, R.; Hartmann, A.; Orntoft, T.; Dyrskjot, L.; Eltze, E.; Wieland, W.; Keck, B.; et al. MicroRNA profiles of prostate carcinoma detected by multiplatform microRNA screening. Int. J. Cancer 2012, 130, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Larne, O.; Hagman, Z.; Lilja, H.; Bjartell, A.; Edsjo, A.; Ceder, Y. Mir-145 suppress the androgen receptor in prostate cancer cells and correlates to prostate cancer prognosis. Carcinogenesis 2015, 36, 858–866. [Google Scholar] [CrossRef] [PubMed]
- Gutschner, T.; Diederichs, S. The hallmarks of cancer: A long non-coding RNA point of view. RNA Biol. 2012, 9, 703–719. [Google Scholar] [CrossRef] [PubMed]
- Prensner, J.R.; Chinnaiyan, A.M. The emergence of lncRNAs in cancer biology. Cancer Discov. 2011, 1, 391–407. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Rinn, J.L. Modular regulatory principles of large non-coding RNAs. Nature 2012, 482, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Srikantan, V.; Zou, Z.; Petrovics, G.; Xu, L.; Augustus, M.; Davis, L.; Livezey, J.R.; Connell, T.; Sesterhenn, I.A.; Yoshino, K.; et al. PCGEM1, a prostate-specific gene, is overexpressed in prostate cancer. Proc. Natl. Acad. Sci. USA 2000, 97, 12216–12221. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Lin, C.; Jin, C.; Yang, J.C.; Tanasa, B.; Li, W.; Merkurjev, D.; Ohgi, K.A.; Meng, D.; Zhang, J.; et al. LncRNA-dependent mechanisms of androgen-receptor-regulated gene activation programs. Nature 2013, 500, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Petrovics, G.; Zhang, W.; Makarem, M.; Street, J.P.; Connelly, R.; Sun, L.; Sesterhenn, I.A.; Srikantan, V.; Moul, J.W.; Srivastava, S. Elevated expression of PCGEM1, a prostate-specific gene with cell growth-promoting function, is associated with high-risk prostate cancer patients. Oncogene 2004, 23, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Ravindranath, L.; Tran, N.; Petrovics, G.; Srivastava, S. Regulation of apoptosis by a prostate-specific and prostate cancer-associated noncoding gene, PCGEM1. DNA Cell Biol. 2006, 25, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Romanuik, T.L.; Wang, G.; Morozova, O.; Delaney, A.; Marra, M.A.; Sadar, M.D. LNCaP Atlas: Gene expression associated with in vivo progression to castration-recurrent prostate cancer. BMC Med. Genom. 2010. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.; Nakagawa, H.; Uemura, M.; Piao, L.; Ashikawa, K.; Hosono, N.; Takata, R.; Akamatsu, S.; Kawaguchi, T.; Morizono, T.; et al. Association of a novel long non-coding RNA in 8q24 with prostate cancer susceptibility. Cancer Sci. 2011, 102, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Parolia, A.; Crea, F.; Xue, H.; Wang, Y.; Mo, F.; Ramnarine, V.R.; Liu, H.H.; Lin, D.; Saidy, N.R.; Clermont, P.L.; et al. The long non-coding RNA PCGEM1 is regulated by androgen receptor activity in vivo. Mol. Cancer 2015. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.L.; Wang, L.Y.; Yu, Y.L.; Chen, H.W.; Srivastava, S.; Petrovics, G.; Kung, H.J. A long noncoding RNA connects c-Myc to tumor metabolism. Proc. Natl. Acad. Sci. USA 2014, 111, 18697–18702. [Google Scholar] [CrossRef] [PubMed]
- Prensner, J.R.; Sahu, A.; Iyer, M.K.; Malik, R.; Chandler, B.; Asangani, I.A.; Poliakov, A.; Vergara, I.A.; Alshalalfa, M.; Jenkins, R.B.; et al. The IncRNAs PCGEM1 and PRNCR1 are not implicated in castration resistant prostate cancer. Oncotarget 2014, 5, 1434–1438. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.T.; Huang, J.; Zhou, N.; Zhang, Z.; Koirala, P.; Zhou, X.; Wu, F.; Ding, X.; Mo, Y.Y. Regulation of PCGEM1 by p54/nrb in prostate cancer. Sci. Rep. 2016. [Google Scholar] [CrossRef] [PubMed]
- Hessels, D.; Klein Gunnewiek, J.M.; van Oort, I.; Karthaus, H.F.; van Leenders, G.J.; van Balken, B.; Kiemeney, L.A.; Witjes, J.A.; Schalken, J.A. DD3(PCA3)-based molecular urine analysis for the diagnosis of prostate cancer. Eur. Urol. 2003, 44, 8–15, discussion 15–16. [Google Scholar] [CrossRef]
- Popa, I.; Fradet, Y.; Beaudry, G.; Hovington, H.; Beaudry, G.; Tetu, B. Identification of PCA3 (DD3) in prostatic carcinoma by in situ hybridization. Mod. Pathol. 2007, 20, 1121–1127. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.B.; Palumbo, A.; de Mello, K.D.; Sternberg, C.; Caetano, M.S.; de Oliveira, F.L.; Neves, A.F.; Nasciutti, L.E.; Goulart, L.R.; Gimba, E.R. PCA3 noncoding RNA is involved in the control of prostate-cancer cell survival and modulates androgen receptor signaling. BMC Cancer 2012. [Google Scholar] [CrossRef] [PubMed]
- Lemos, A.E.; Ferreira, L.B.; Batoreu, N.M.; de Freitas, P.P.; Bonamino, M.H.; Gimba, E.R. PCA3 long noncoding RNA modulates the expression of key cancer-related genes in lncap prostate cancer cells. Tumour Biol. 2016, 37, 11339–11348. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.; Horie-Inoue, K.; Katayama, S.; Suzuki, T.; Tsutsumi, S.; Ikeda, K.; Urano, T.; Fujimura, T.; Takagi, K.; Takahashi, S.; et al. Androgen-responsive long noncoding RNA CTBP1-AS promotes prostate cancer. EMBO J. 2013, 32, 1665–1680. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Asangani, I.A.; Chakravarthi, B.V.; Ateeq, B.; Lonigro, R.J.; Cao, Q.; Mani, R.S.; Camacho, D.F.; McGregor, N.; Schumann, T.E.; et al. Role of transcriptional corepressor CtBP1 in prostate cancer progression. Neoplasia 2012, 14, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Ren, S.; Lu, J.; Wang, F.; Xu, W.; Sun, Y.; Wei, M.; Chen, J.; Gao, X.; Xu, C.; et al. The prostate cancer-up-regulated long noncoding RNA PlncRNA-1 modulates apoptosis and proliferation through reciprocal regulation of androgen receptor. Urol. Oncol. 2013, 31, 1117–1123. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Zhao, J.C.; Kim, J.; Fong, K.W.; Yang, Y.A.; Chakravarti, D.; Mo, Y.Y.; Yu, J. LncRNA hotair enhances the androgen-receptor-mediated transcriptional program and drives castration-resistant prostate cancer. Cell Rep. 2015, 13, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Crea, F.; Watahiki, A.; Quagliata, L.; Xue, H.; Pikor, L.; Parolia, A.; Wang, Y.; Lin, D.; Lam, W.L.; Farrar, W.L.; et al. Identification of a long non-coding RNA as a novel biomarker and potential therapeutic target for metastatic prostate cancer. Oncotarget 2014, 5, 764–774. [Google Scholar] [CrossRef] [PubMed]
- Malik, R.; Patel, L.; Prensner, J.R.; Shi, Y.; Iyer, M.K.; Subramaniyan, S.; Carley, A.; Niknafs, Y.S.; Sahu, A.; Han, S.; et al. The lncRNA PCAT29 inhibits oncogenic phenotypes in prostate cancer. Mol. Cancer Res. 2014, 12, 1081–1087. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, K.; Reon, B.J.; Anaya, J.; Dutta, A. The IncRNA DRAIC/PCAT29 locus constitutes a tumor-suppressive nexus. Mol. Cancer Res. 2015, 13, 828–838. [Google Scholar] [CrossRef] [PubMed]
- Saini, S. Psa and beyond: Alternative prostate cancer biomarkers. Cell. Oncol. (Dordr.) 2016, 39, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Cary, K.C.; Cooperberg, M.R. Biomarkers in prostate cancer surveillance and screening: Past, present, and future. Ther. Adv. Urol. 2013, 5, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Walter, L.C.; Bertenthal, D.; Lindquist, K.; Konety, B.R. PSA screening among elderly men with limited life expectancies. J. Am. Med. Assoc. 2006, 296, 2336–2342. [Google Scholar] [CrossRef] [PubMed]
- Strope, S.A.; Andriole, G.L. Prostate cancer screening: Current status and future perspectives. Nat. Rev. Urol. 2010, 7, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Jeronimo, C.; Henrique, R. Epigenetic biomarkers in urological tumors: A systematic review. Cancer Lett. 2014, 342, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Massie, C.E.; Mills, I.G.; Lynch, A.G. The importance of DNA methylation in prostate cancer development. J. Steroid Biochem. Mol. Biol. 2016, 37, 11339–11348. [Google Scholar] [CrossRef] [PubMed]
- Henrique, R.; Jeronimo, C. Molecular detection of prostate cancer: A role for GSTP1 hypermethylation. Eur. Urol. 2004, 46, 660–669, discussion 669. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Giovannucci, E.; Welge, J.; Mallick, P.; Tang, W.Y.; Ho, S.M. Measurement of GSTP1 promoter methylation in body fluids may complement PSA screening: A meta-analysis. Br. J. Cancer 2011, 105, 65–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blute, M.L., Jr.; Damaschke, N.A.; Jarrard, D.F. The epigenetics of prostate cancer diagnosis and prognosis: Update on clinical applications. Curr. Opin. Urol. 2015, 25, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Strand, S.H.; Orntoft, T.F.; Sorensen, K.D. Prognostic DNA methylation markers for prostate cancer. Int. J. Mol. Sci. 2014, 15, 16544–16576. [Google Scholar] [CrossRef] [PubMed]
- Baden, J.; Green, G.; Painter, J.; Curtin, K.; Markiewicz, J.; Jones, J.; Astacio, T.; Canning, S.; Quijano, J.; Guinto, W.; et al. Multicenter evaluation of an investigational prostate cancer methylation assay. J. Urol. 2009, 182, 1186–1193. [Google Scholar] [CrossRef] [PubMed]
- Baden, J.; Adams, S.; Astacio, T.; Jones, J.; Markiewicz, J.; Painter, J.; Trust, C.; Wang, Y.; Green, G. Predicting prostate biopsy result in men with prostate specific antigen 2.0 to 10.0 ng/mL using an investigational prostate cancer methylation assay. J. Urol. 2011, 186, 2101–2106. [Google Scholar] [CrossRef] [PubMed]
- Stewart, G.D.; Van Neste, L.; Delvenne, P.; Delree, P.; Delga, A.; McNeill, S.A.; O’Donnell, M.; Clark, J.; Van Criekinge, W.; Bigley, J.; et al. Clinical utility of an epigenetic assay to detect occult prostate cancer in histopathologically negative biopsies: Results of the matloc study. J. Urol. 2013, 189, 1110–1116. [Google Scholar] [CrossRef] [PubMed]
- Partin, A.W.; Van Neste, L.; Klein, E.A.; Marks, L.S.; Gee, J.R.; Troyer, D.A.; Rieger-Christ, K.; Jones, J.S.; Magi-Galluzzi, C.; Mangold, L.A.; et al. Clinical validation of an epigenetic assay to predict negative histopathological results in repeat prostate biopsies. J. Urol. 2014, 192, 1081–1087. [Google Scholar] [CrossRef] [PubMed]
- Carroll, P.R.; Parsons, J.K.; Andriole, G.; Bahnson, R.R.; Castle, E.P.; Catalona, W.J.; Dahl, D.M.; Davis, J.W.; Epstein, J.I.; Etzioni, R.B.; et al. NCCN guidelines insights: Prostate cancer early detection, version 2.2016. J. Natl. Compr. Cancer Netw. 2016, 14, 509–519. [Google Scholar]
- Aryee, M.J.; Liu, W.; Engelmann, J.C.; Nuhn, P.; Gurel, M.; Haffner, M.C.; Esopi, D.; Irizarry, R.A.; Getzenberg, R.H.; Nelson, W.G.; et al. DNA methylation alterations exhibit intraindividual stability and interindividual heterogeneity in prostate cancer metastases. Sci. Transl. Med. 2013. [Google Scholar] [CrossRef] [PubMed]
- Romero Otero, J.; Garcia Gomez, B.; Campos Juanatey, F.; Touijer, K.A. Prostate cancer biomarkers: An update. Urol. Oncol. 2014, 32, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Bussemakers, M.J.; van Bokhoven, A.; Verhaegh, G.W.; Smit, F.P.; Karthaus, H.F.; Schalken, J.A.; Debruyne, F.M.; Ru, N.; Isaacs, W.B. DD3: A new prostate-specific gene, highly overexpressed in prostate cancer. Cancer Res. 1999, 59, 5975–5979. [Google Scholar] [PubMed]
- Sartori, D.A.; Chan, D.W. Biomarkers in prostate cancer: What’s new? Curr. Opin. Oncol. 2014, 26, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Haese, A.; de la Taille, A.; van Poppel, H.; Marberger, M.; Stenzl, A.; Mulders, P.F.; Huland, H.; Abbou, C.C.; Remzi, M.; Tinzl, M.; et al. Clinical utility of the PCA3 urine assay in european men scheduled for repeat biopsy. Eur. Urol. 2008, 54, 1081–1088. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Yang, H.; Yang, H. Diagnostic value of urine prostate cancer antigen 3 test using a cutoff value of 35 ug/L in patients with prostate cancer. Tumour Biol. 2014, 35, 8573–8580. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Gou, X.; Huang, P.; Mou, C. The PCA3 test for guiding repeat biopsy of prostate cancer and its cut-off score: A systematic review and meta-analysis. Asian J. Androl. 2014, 16, 487–492. [Google Scholar] [PubMed]
- Crawford, E.D.; Rove, K.O.; Trabulsi, E.J.; Qian, J.; Drewnowska, K.P.; Kaminetsky, J.C.; Huisman, T.K.; Bilowus, M.L.; Freedman, S.J.; Glover, W.L., Jr.; et al. Diagnostic performance of PCA3 to detect prostate cancer in men with increased prostate specific antigen: A prospective study of 1962 cases. J. Urol. 2012, 188, 1726–1731. [Google Scholar] [CrossRef] [PubMed]
- Merola, R.; Tomao, L.; Antenucci, A.; Sperduti, I.; Sentinelli, S.; Masi, S.; Mandoj, C.; Orlandi, G.; Papalia, R.; Guaglianone, S.; et al. PCA3 in prostate cancer and tumor aggressiveness detection on 407 high-risk patients: A national cancer institute experience. J. Exp. Clin. Cancer Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Chevli, K.K.; Duff, M.; Walter, P.; Yu, C.; Capuder, B.; Elshafei, A.; Malczewski, S.; Kattan, M.W.; Jones, J.S. Urinary PCA3 as a predictor of prostate cancer in a cohort of 3073 men undergoing initial prostate biopsy. J. Urol. 2014, 191, 1743–1748. [Google Scholar] [CrossRef] [PubMed]
- Hessels, D.; van Gils, M.P.; van Hooij, O.; Jannink, S.A.; Witjes, J.A.; Verhaegh, G.W.; Schalken, J.A. Predictive value of PCA3 in urinary sediments in determining clinico-pathological characteristics of prostate cancer. Prostate 2010, 70, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Van Gils, M.P.; Hessels, D.; Hulsbergen-van de Kaa, C.A.; Witjes, J.A.; Jansen, C.F.; Mulders, P.F.; Rittenhouse, H.G.; Schalken, J.A. Detailed analysis of histopathological parameters in radical prostatectomy specimens and PCA3 urine test results. Prostate 2008, 68, 1215–1222. [Google Scholar] [CrossRef] [PubMed]
- Vlaeminck-Guillem, V.; Ruffion, A.; Andre, J.; Devonec, M.; Paparel, P. Urinary prostate cancer 3 test: Toward the age of reason? Urology 2010, 75, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Erho, N.; Crisan, A.; Vergara, I.A.; Mitra, A.P.; Ghadessi, M.; Buerki, C.; Bergstralh, E.J.; Kollmeyer, T.; Fink, S.; Haddad, Z.; et al. Discovery and validation of a prostate cancer genomic classifier that predicts early metastasis following radical prostatectomy. PLoS ONE 2013, 8, e66855. [Google Scholar] [CrossRef] [PubMed]
- Moschini, M.; Spahn, M.; Mattei, A.; Cheville, J.; Karnes, R.J. Incorporation of tissue-based genomic biomarkers into localized prostate cancer clinics. BMC Med. 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karnes, R.J.; Bergstralh, E.J.; Davicioni, E.; Ghadessi, M.; Buerki, C.; Mitra, A.P.; Crisan, A.; Erho, N.; Vergara, I.A.; Lam, L.L.; et al. Validation of a genomic classifier that predicts metastasis following radical prostatectomy in an at risk patient population. J. Urol. 2013, 190, 2047–2053. [Google Scholar] [CrossRef] [PubMed]
- Ross, A.E.; Feng, F.Y.; Ghadessi, M.; Erho, N.; Crisan, A.; Buerki, C.; Sundi, D.; Mitra, A.P.; Vergara, I.A.; Thompson, D.J.; et al. A genomic classifier predicting metastatic disease progression in men with biochemical recurrence after prostatectomy. Prostate Cancer Prostatic Dis. 2014, 17, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Den, R.B.; Feng, F.Y.; Showalter, T.N.; Mishra, M.V.; Trabulsi, E.J.; Lallas, C.D.; Gomella, L.G.; Kelly, W.K.; Birbe, R.C.; McCue, P.A.; et al. Genomic prostate cancer classifier predicts biochemical failure and metastases in patients after postoperative radiation therapy. Int. J. Radiat. Oncol. Biol. Phys. 2014, 89, 1038–1046. [Google Scholar] [CrossRef] [PubMed]
- Den, R.B.; Yousefi, K.; Trabulsi, E.J.; Abdollah, F.; Choeurng, V.; Feng, F.Y.; Dicker, A.P.; Lallas, C.D.; Gomella, L.G.; Davicioni, E.; et al. Genomic classifier identifies men with adverse pathology after radical prostatectomy who benefit from adjuvant radiation therapy. J. Clin. Oncol. 2015, 33, 944–951. [Google Scholar] [CrossRef] [PubMed]
- Mohler, J.L.; Armstrong, A.J.; Bahnson, R.R.; D’Amico, A.V.; Davis, B.J.; Eastham, J.A.; Enke, C.A.; Farrington, T.A.; Higano, C.S.; Horwitz, E.M.; et al. Prostate cancer, version 1.2016. J. Natl. Compr. Cancer Network 2016, 14, 19–30. [Google Scholar]
- Fabris, L.; Ceder, Y.; Chinnaiyan, A.M.; Jenster, G.W.; Sorensen, K.D.; Tomlins, S.; Visakorpi, T.; Calin, G.A. The potential of microRNAs as prostate cancer biomarkers. Eur. Urol. 2016, 70, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Schubert, M.; Junker, K.; Heinzelmann, J. Prognostic and predictive miRNA biomarkers in bladder, kidney and prostate cancer: Where do we stand in biomarker development? J. Cancer Res. Clin. Oncol. 2016, 142, 1673–1695. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Hruby, G.W.; McKiernan, J.M.; Gurvich, I.; Lipsky, M.J.; Benson, M.C.; Santella, R.M. Dysregulation of circulating microRNAs and prediction of aggressive prostate cancer. Prostate 2012, 72, 1469–1477. [Google Scholar] [CrossRef] [PubMed]
- Melbo-Jorgensen, C.; Ness, N.; Andersen, S.; Valkov, A.; Donnem, T.; Al-Saad, S.; Kiselev, Y.; Berg, T.; Nordby, Y.; Bremnes, R.M.; et al. Stromal expression of MiR-21 predicts biochemical failure in prostate cancer patients with gleason score 6. PLoS ONE 2014, 9, e113039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, T.; Yang, M.; Chen, S.; Balk, S.; Pomerantz, M.; Hsieh, C.L.; Brown, M.; Lee, G.S.; Kantoff, P.W. The altered expression of MiR-221/-222 and MiR-23b/-27b is associated with the development of human castration resistant prostate cancer. Prostate 2012, 72, 1093–1103. [Google Scholar] [CrossRef] [PubMed]
- Kneitz, B.; Krebs, M.; Kalogirou, C.; Schubert, M.; Joniau, S.; van Poppel, H.; Lerut, E.; Kneitz, S.; Scholz, C.J.; Strobel, P.; et al. Survival in patients with high-risk prostate cancer is predicted by MiR-221, which regulates proliferation, apoptosis, and invasion of prostate cancer cells by inhibiting IRF2 and SOCS3. Cancer Res. 2014, 74, 2591–2603. [Google Scholar] [CrossRef] [PubMed]
- Spahn, M.; Joniau, S.; Gontero, P.; Fieuws, S.; Marchioro, G.; Tombal, B.; Kneitz, B.; Hsu, C.Y.; van Der Eeckt, K.; Bader, P.; et al. Outcome predictors of radical prostatectomy in patients with prostate-specific antigen greater than 20 ng/mL: A european multi-institutional study of 712 patients. Eur. Urol. 2010, 58, 1–7, discussion 10–11. [Google Scholar] [CrossRef] [PubMed]
- Denis, L.; Murphy, G.P. Overview of phase III trials on combined androgen treatment in patients with metastatic prostate cancer. Cancer 1993, 72, 3888–3895. [Google Scholar] [CrossRef]
- Hellerstedt, B.A.; Pienta, K.J. The truth is out there: An overall perspective on androgen deprivation. Urol. Oncol. 2003, 21, 272–281. [Google Scholar] [CrossRef]
- Rojas, A.; Liu, G.; Coleman, I.; Nelson, P.S.; Zhang, M.; Dash, R.; Fisher, P.B.; Plymate, S.R.; Wu, J.D. Il-6 promotes prostate tumorigenesis and progression through autocrine cross-activation of IGF-IR. Oncogene 2011, 30, 2345–2355. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Desano, J.; Tang, W.; Meng, X.; Meng, Y.; Burstein, E.; Lawrence, T.S.; Xu, L. Natural proteasome inhibitor celastrol suppresses androgen-independent prostate cancer progression by modulating apoptotic proteins and nf-kappab. PLoS ONE 2010, 5, e14153. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekar, T.; Yang, J.C.; Gao, A.C.; Evans, C.P. Mechanisms of resistance in castration-resistant prostate cancer (crpc). Transl. Androl. Urol. 2015, 4, 365–380. [Google Scholar] [PubMed]
- Anders, L.; Guenther, M.G.; Qi, J.; Fan, Z.P.; Marineau, J.J.; Rahl, P.B.; Loven, J.; Sigova, A.A.; Smith, W.B.; Lee, T.I.; et al. Genome-wide localization of small molecules. Nat. Biotechnol. 2014, 32, 92–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, C.; Yang, L.; Xie, M.; Lin, C.; Merkurjev, D.; Yang, J.C.; Tanasa, B.; Oh, S.; Zhang, J.; Ohgi, K.A.; et al. Chem-seq permits identification of genomic targets of drugs against androgen receptor regulation selected by functional phenotypic screens. Proc. Natl. Acad. Sci. USA 2014, 111, 9235–9240. [Google Scholar] [CrossRef] [PubMed]
- King, O.N.; Li, X.S.; Sakurai, M.; Kawamura, A.; Rose, N.R.; Ng, S.S.; Quinn, A.M.; Rai, G.; Mott, B.T.; Beswick, P.; et al. Quantitative high-throughput screening identifies 8-hydroxyquinolines as cell-active histone demethylase inhibitors. PLoS ONE 2010, 5, e15535. [Google Scholar] [CrossRef] [PubMed]
- Cloos, P.A.; Christensen, J.; Agger, K.; Maiolica, A.; Rappsilber, J.; Antal, T.; Hansen, K.H.; Helin, K. The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature 2006, 442, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Whetstine, J.R.; Nottke, A.; Lan, F.; Huarte, M.; Smolikov, S.; Chen, Z.; Spooner, E.; Li, E.; Zhang, G.; Colaiacovo, M.; et al. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell 2006, 125, 467–481. [Google Scholar] [CrossRef] [PubMed]
- Piekarz, R.L.; Frye, R.; Turner, M.; Wright, J.J.; Allen, S.L.; Kirschbaum, M.H.; Zain, J.; Prince, H.M.; Leonard, J.P.; Geskin, L.J.; et al. Phase ii multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous t-cell lymphoma. J. Clin. Oncol. 2009, 27, 5410–5417. [Google Scholar] [CrossRef] [PubMed]
- Wheler, J.J.; Janku, F.; Falchook, G.S.; Jackson, T.L.; Fu, S.; Naing, A.; Tsimberidou, A.M.; Moulder, S.L.; Hong, D.S.; Yang, H.; et al. Phase I study of anti-VEGF monoclonal antibody bevacizumab and histone deacetylase inhibitor valproic acid in patients with advanced cancers. Cancer Chemother. Pharmacol. 2014, 73, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Welsbie, D.S.; Xu, J.; Chen, Y.; Borsu, L.; Scher, H.I.; Rosen, N.; Sawyers, C.L. Histone deacetylases are required for androgen receptor function in hormone-sensitive and castrate-resistant prostate cancer. Cancer Res. 2009, 69, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Rokhlin, O.W.; Glover, R.B.; Guseva, N.V.; Taghiyev, A.F.; Kohlgraf, K.G.; Cohen, M.B. Mechanisms of cell death induced by histone deacetylase inhibitors in androgen receptor-positive prostate cancer cells. Mol. Cancer Res. 2006, 4, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Fliss, A.E.; Robins, D.M.; Caplan, A.J. Hsp90 regulates androgen receptor hormone binding affinity in vivo. J. Biol. Chem. 1996, 271, 28697–28702. [Google Scholar] [CrossRef] [PubMed]
- Solit, D.B.; Scher, H.I.; Rosen, N. Hsp90 as a therapeutic target in prostate cancer. Semin. Oncol. 2003, 30, 709–716. [Google Scholar] [CrossRef]
- Kaushik, D.; Vashistha, V.; Isharwal, S.; Sediqe, S.A.; Lin, M.F. Histone deacetylase inhibitors in castration-resistant prostate cancer: Molecular mechanism of action and recent clinical trials. Ther. Adv. Urol. 2015, 7, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Guo, Z.S.; Marcu, M.G.; Neckers, L.; Nguyen, D.M.; Chen, G.A.; Schrump, D.S. Modulation of p53, ErbB1, ErbB2, and Raf-1 expression in lung cancer cells by depsipeptide FR901228. J. Natl. Cancer Inst. 2002, 94, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Molife, L.R.; Attard, G.; Fong, P.C.; Karavasilis, V.; Reid, A.H.; Patterson, S.; Riggs, C.E., Jr.; Higano, C.; Stadler, W.M.; McCulloch, W.; et al. Phase II, two-stage, single-arm trial of the histone deacetylase inhibitor (HDACi) romidepsin in metastatic castration-resistant prostate cancer (CRPC). Ann. Oncol. 2010, 21, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Rathkopf, D.; Wong, B.Y.; Ross, R.W.; Anand, A.; Tanaka, E.; Woo, M.M.; Hu, J.; Dzik-Jurasz, A.; Yang, W.; Scher, H.I. A phase I study of oral panobinostat alone and in combination with docetaxel in patients with castration-resistant prostate cancer. Cancer Chemother. Pharmacol. 2010, 66, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Rathkopf, D.E.; Picus, J.; Hussain, A.; Ellard, S.; Chi, K.N.; Nydam, T.; Allen-Freda, E.; Mishra, K.K.; Porro, M.G.; Scher, H.I.; et al. A phase 2 study of intravenous panobinostat in patients with castration-resistant prostate cancer. Cancer Chemother. Pharmacol. 2013, 72, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Garzon, R.; Marcucci, G.; Croce, C.M. Targeting microRNAs in cancer: Rationale, strategies and challenges. Nature reviews. Drug Discov. 2010, 9, 775–789. [Google Scholar] [CrossRef] [PubMed]
- Li, X.J.; Ren, Z.J.; Tang, J.H. MicroRNA-34a: A potential therapeutic target in human cancer. Cell Death Dis. 2014, 5, e1327. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.G.; Zheng, J.N.; Pei, D.S. P53/microRNA-34-induced metabolic regulation: New opportunities in anticancer therapy. Mol. Cancer 2014. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, S.; Saini, S.; Majid, S.; Hirata, H.; Ueno, K.; Deng, G.; Dahiya, R. MicroRNA-34a modulates c-myc transcriptional complexes to suppress malignancy in human prostate cancer cells. PLoS ONE 2012, 7, e29722. [Google Scholar] [CrossRef] [PubMed]
- Bouchie, A. First microRNA mimic enters clinic. Nat. Biotechnol. 2013. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cucchiara, V.; Yang, J.C.; Mirone, V.; Gao, A.C.; Rosenfeld, M.G.; Evans, C.P. Epigenomic Regulation of Androgen Receptor Signaling: Potential Role in Prostate Cancer Therapy. Cancers 2017, 9, 9. https://doi.org/10.3390/cancers9010009
Cucchiara V, Yang JC, Mirone V, Gao AC, Rosenfeld MG, Evans CP. Epigenomic Regulation of Androgen Receptor Signaling: Potential Role in Prostate Cancer Therapy. Cancers. 2017; 9(1):9. https://doi.org/10.3390/cancers9010009
Chicago/Turabian StyleCucchiara, Vito, Joy C. Yang, Vincenzo Mirone, Allen C. Gao, Michael G. Rosenfeld, and Christopher P. Evans. 2017. "Epigenomic Regulation of Androgen Receptor Signaling: Potential Role in Prostate Cancer Therapy" Cancers 9, no. 1: 9. https://doi.org/10.3390/cancers9010009