STAT3 but Not HIF-1α Is Important in Mediating Hypoxia-Induced Chemoresistance in MDA-MB-231, a Triple Negative Breast Cancer Cell Line

 , ,
, ,
Abstract
:1. Introduction
2. Results
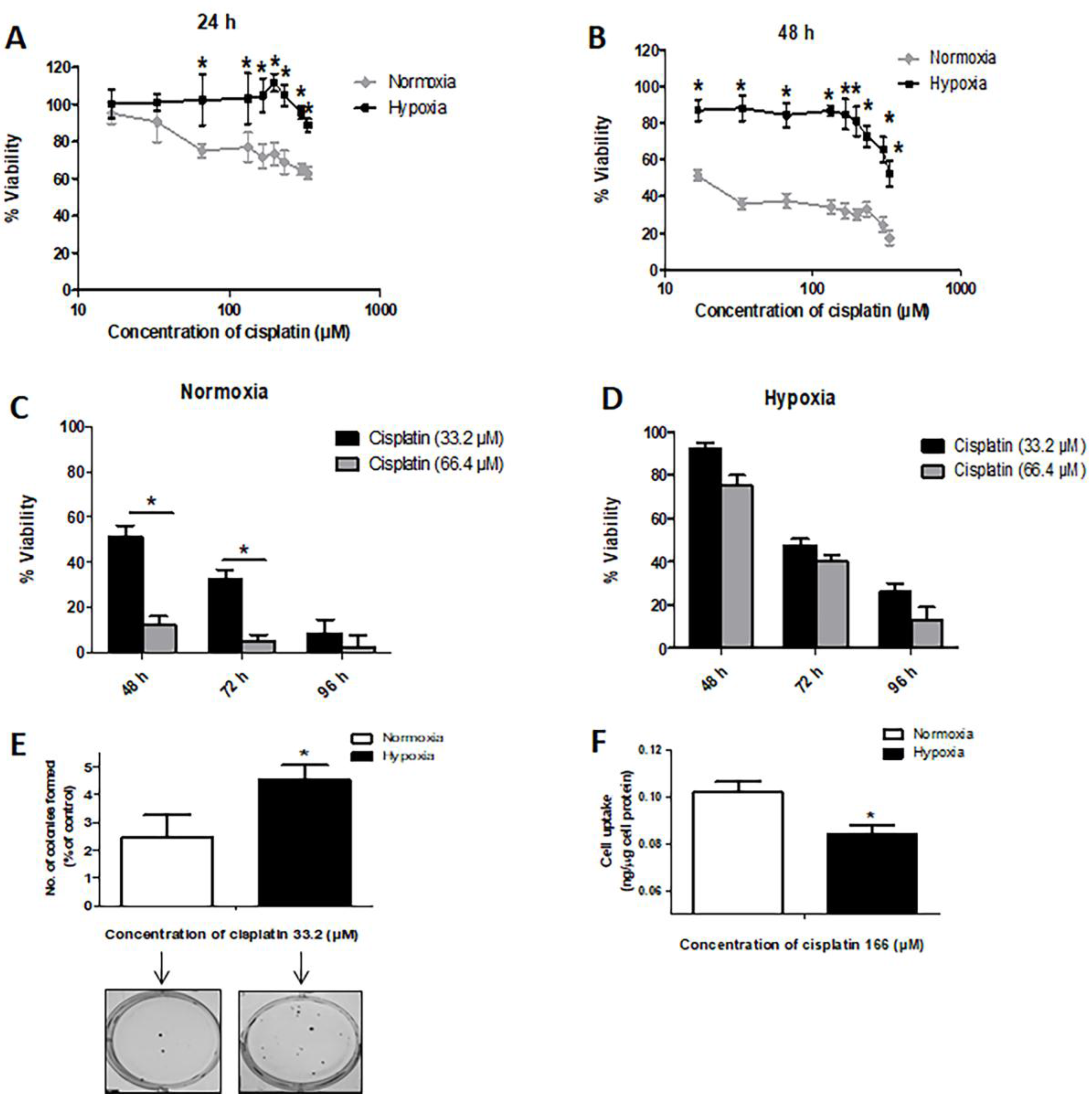
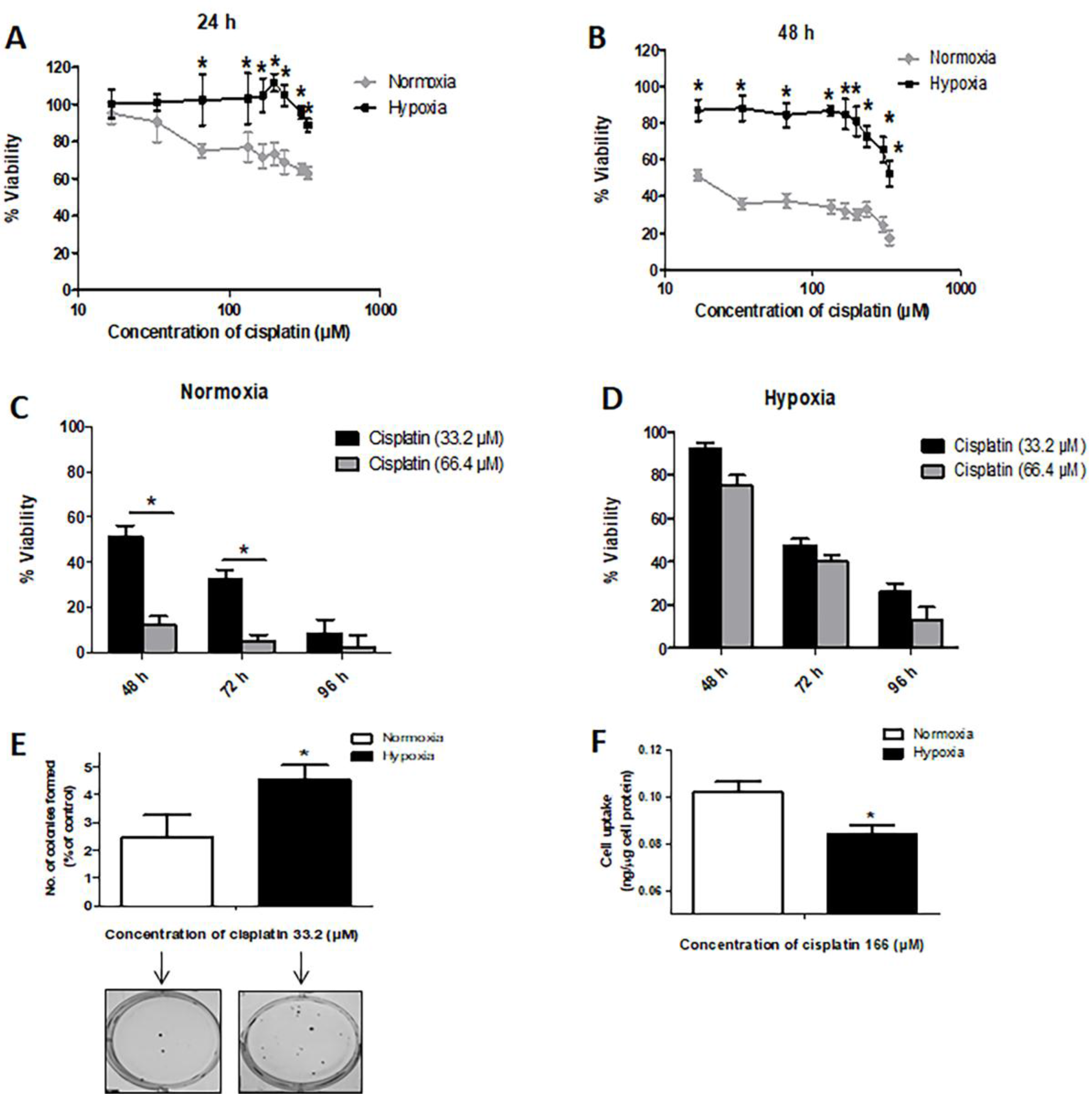
2.1. Hypoxia Induces Resistance to Cisplatin in MDA-MB-231 Cells
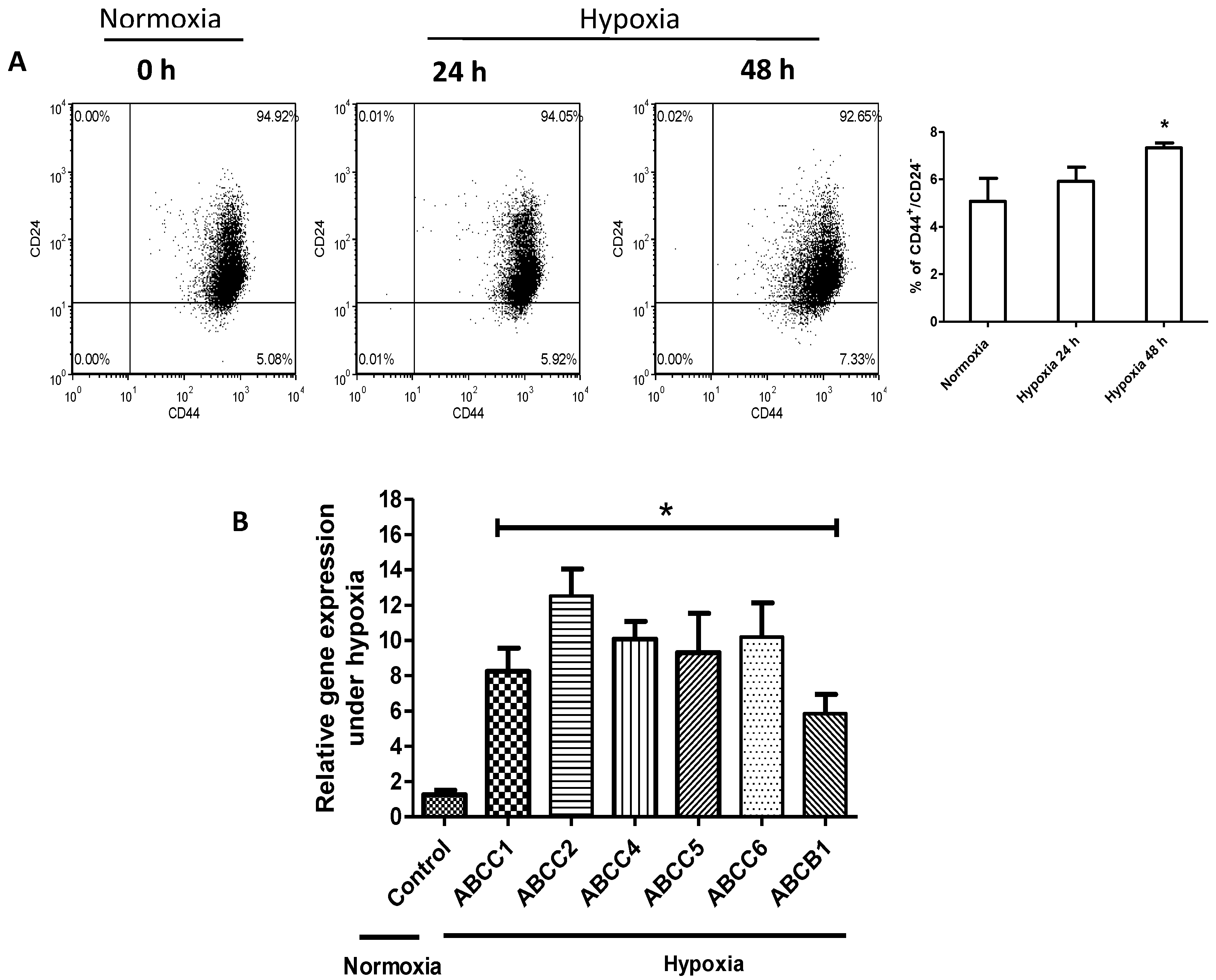
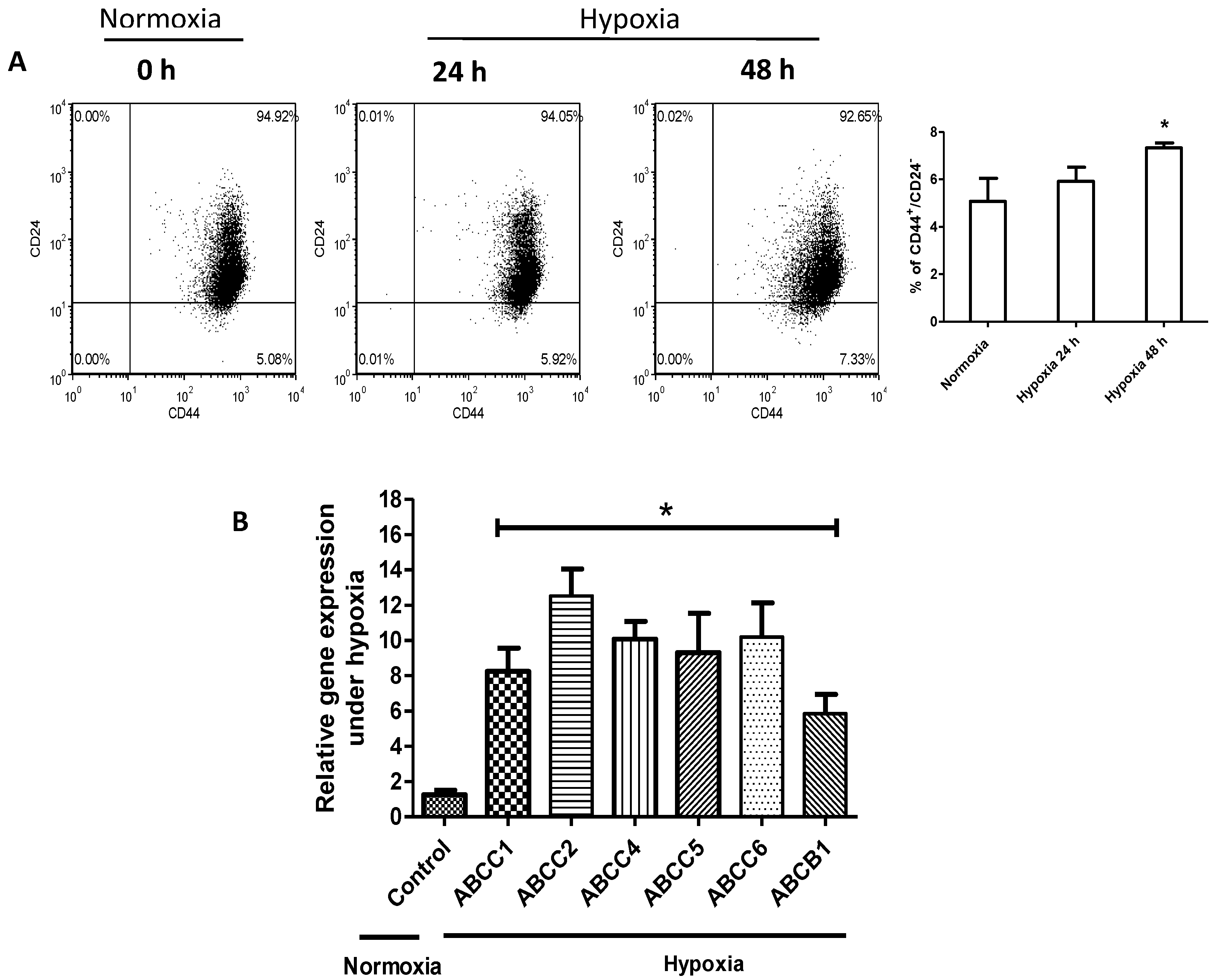
2.2. Hypoxia Confers Stem-Like Features to Cells
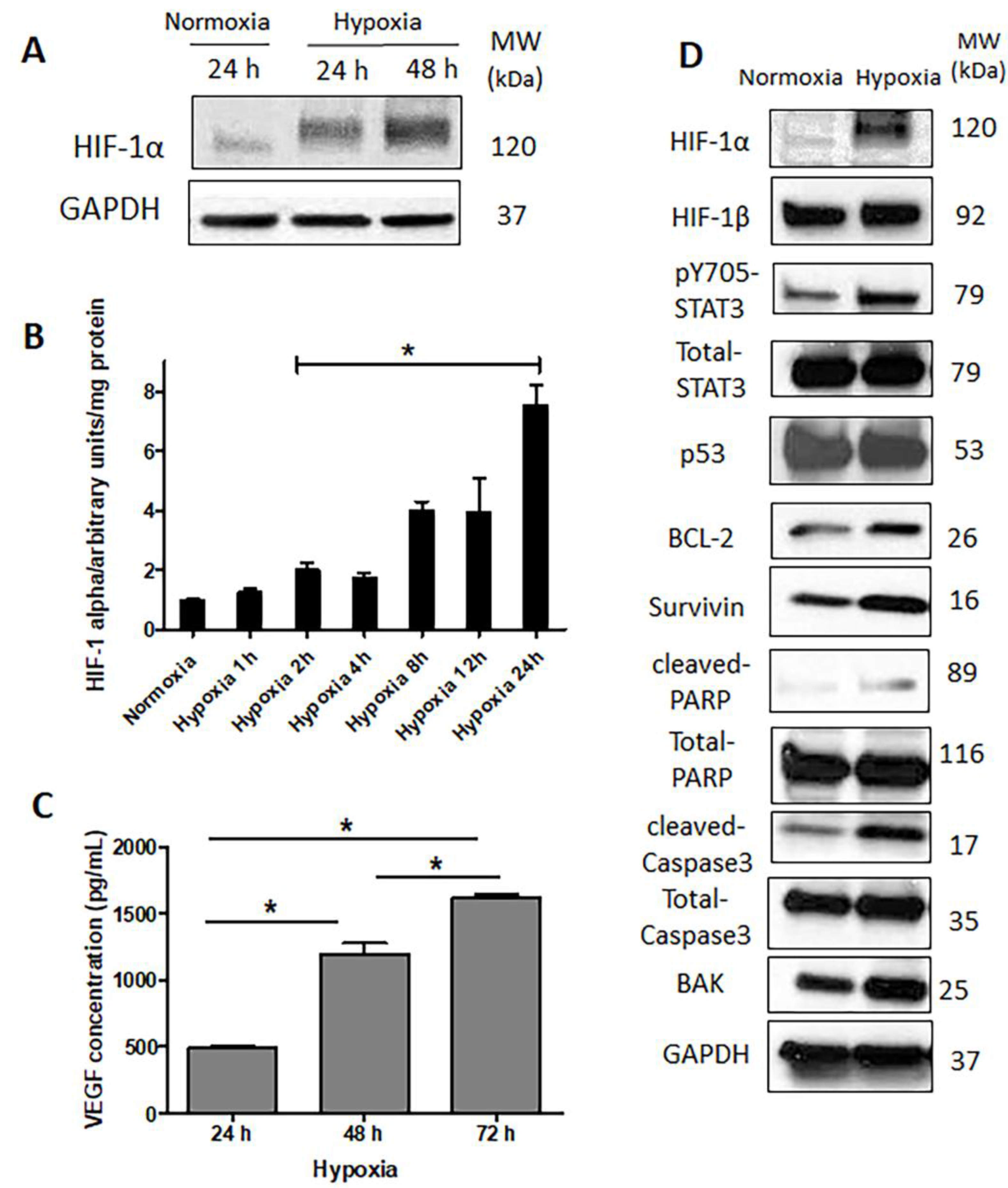
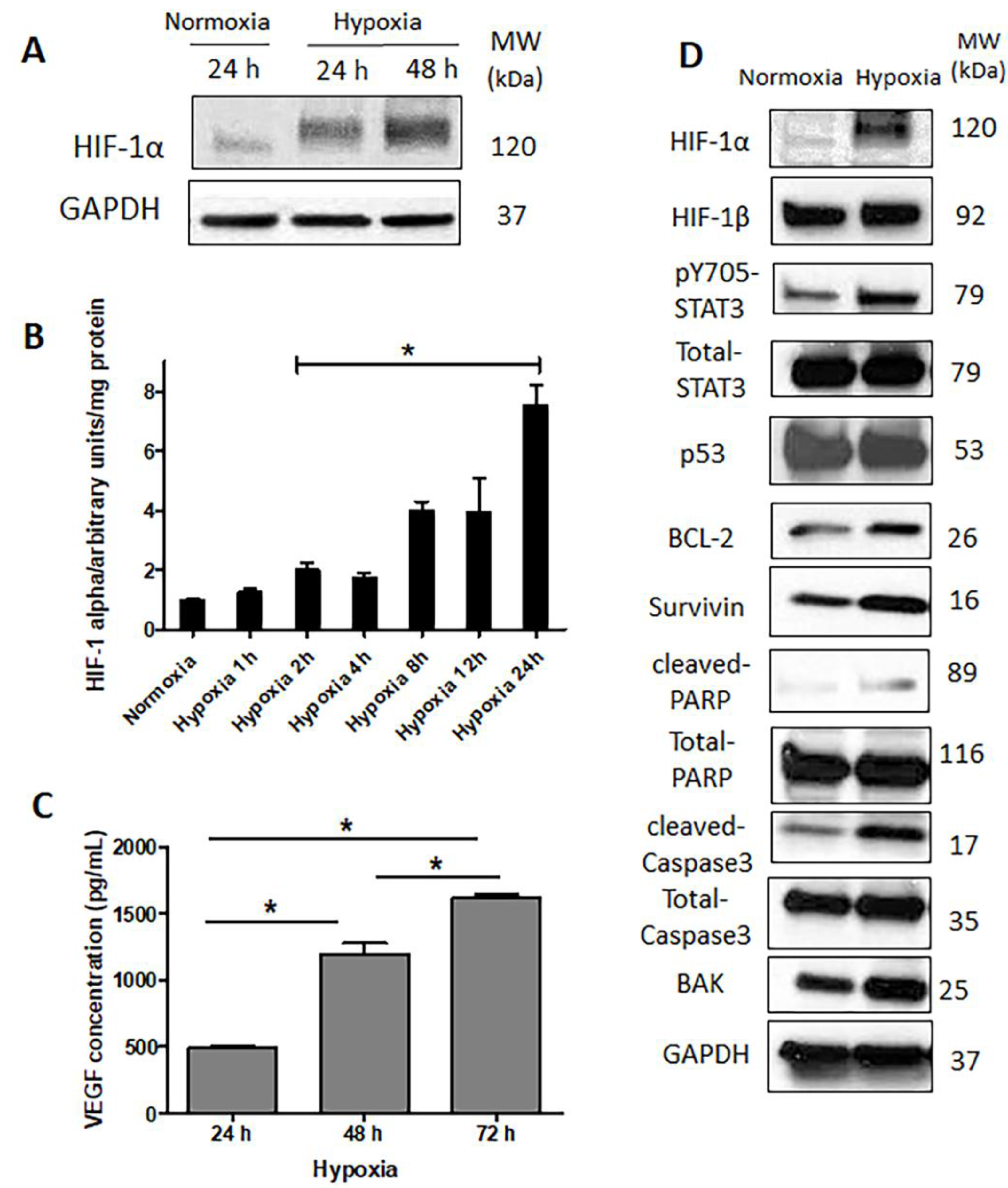
2.3. HIF-1α Is Upregulated and Functionally Active in Response to Hypoxia
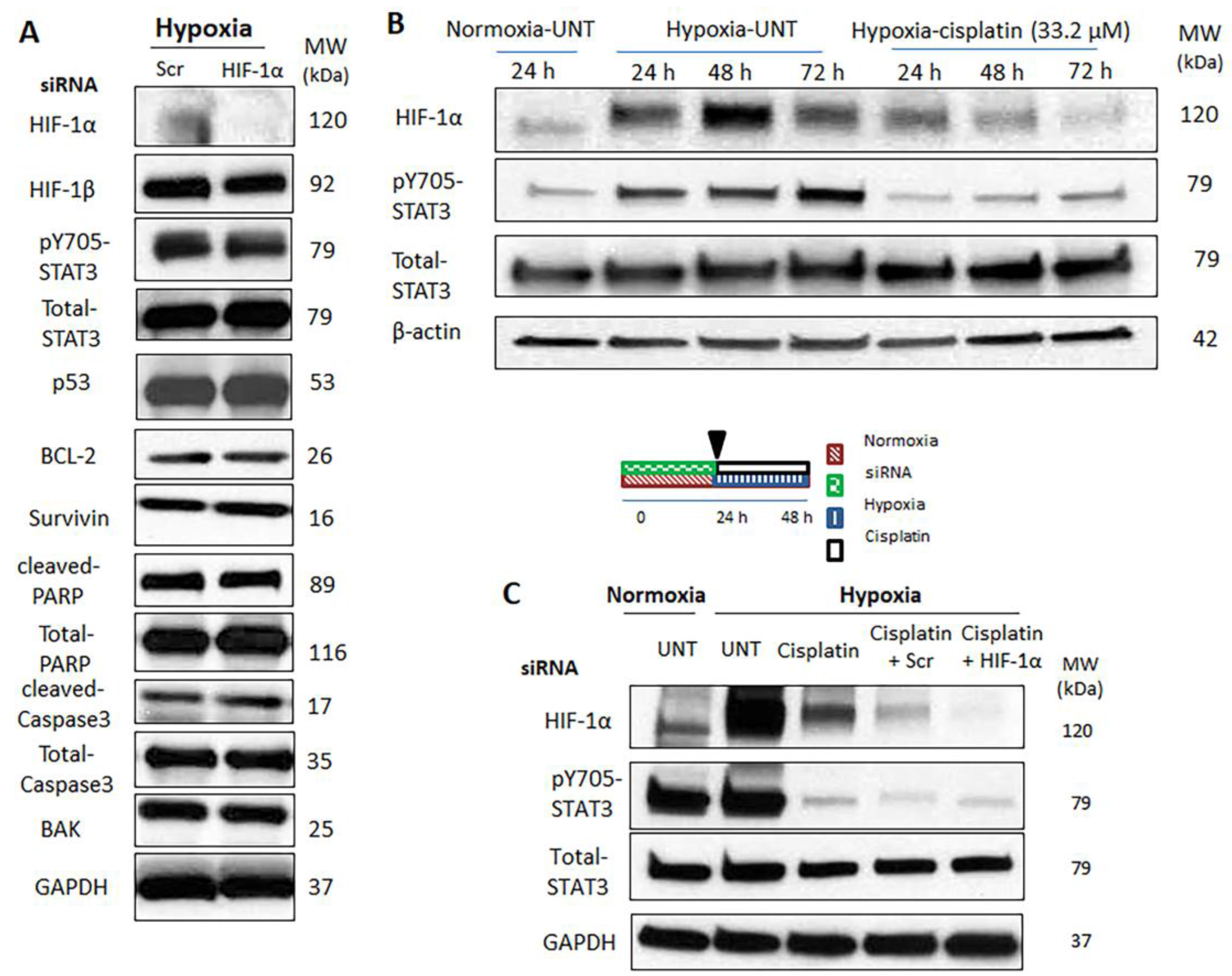
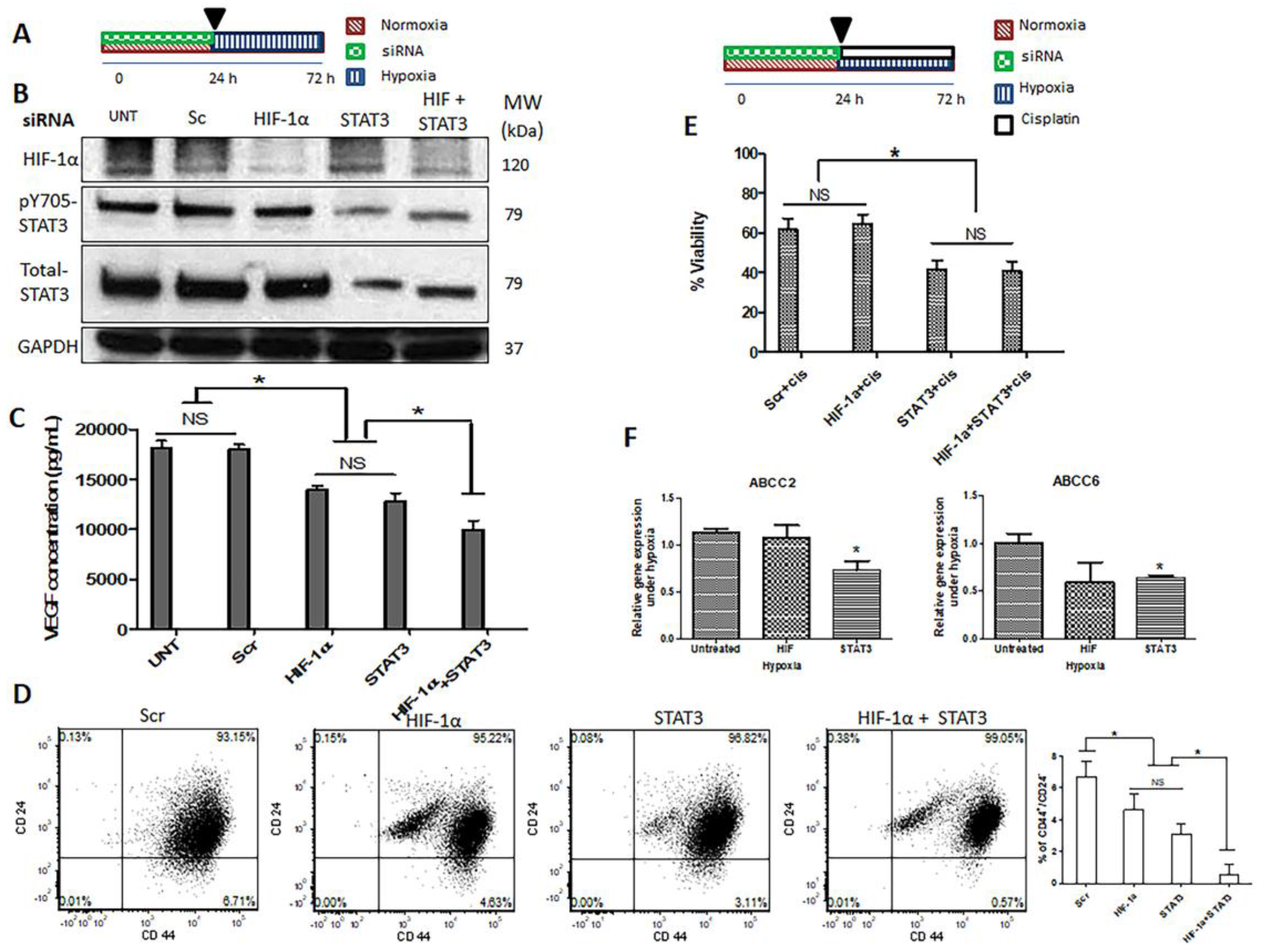
2.4. Hypoxia-Induced STAT3 Activation Is Independent of HIF-1α
2.5. Simultaneous Inhibition of HIF-1α and STAT3 Proteins Is More Efficient in Suppressing the Acquisition of Cancer Stemness Induced by Hypoxia
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Small Interfering RNAs (siRNAs) Complex Preparation
4.3. Trypan Blue Assay
4.4. MTT Assay
4.5. Colony Formation Assay
4.6. Flow Cytometry Analyses for CD44+/CD24− Expression
4.7. RNA Extraction, cDNA Synthesis, Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
4.8. Western Blot
4.9. Vascular Endothelial Growth Factor Enzyme-Linked Immunosorbent (VEGF Elisa) Assay
4.10. HIF-1α DNA Binding Activity
4.11. Flow Cytometric Detection of Apoptosis Using Annexin V-FITC and Propidium Iodide
4.12. Cell Uptake
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tredan, O.; Galmarini, C.M.; Patel, K.; Tannock, I.F. Drug resistance and the solid tumor microenvironment. J. Natl. Cancer Inst. 2007, 99, 1441–1454. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 2010, 29, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Brahimi-Horn, C.; Pouyssegur, J. The role of the hypoxia-inducible factor in tumor metabolism growth and invasion. Bull. Cancer 2006, 93, E73–E80. [Google Scholar] [PubMed]
- Rohwer, N.; Cramer, T. Hypoxia-mediated drug resistance: Novel insights on the functional interaction of hifs and cell death pathways. Drug Resist. Updates 2011, 14, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, R.; Pare, G.C.; Frederiksen, L.J.; Semenza, G.L.; Graham, C.H. Hypoxia-induced resistance to anticancer drugs is associated with decreased senescence and requires hypoxia-inducible factor-1 activity. Mol. Cancer Ther. 2008, 7, 1961–1973. [Google Scholar] [CrossRef] [PubMed]
- Erler, J.T.; Cawthorne, C.J.; Williams, K.J.; Koritzinsky, M.; Wouters, B.G.; Wilson, C.; Miller, C.; Demonacos, C.; Stratford, I.J.; Dive, C. Hypoxia-mediated down-regulation of bid and bax in tumors occurs via hypoxia-inducible factor 1-dependent and -independent mechanisms and contributes to drug resistance. Mol. Cell. Biol. 2004, 24, 2875–2889. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.W.; Su, Y.; Zhu, H.; Cao, J.; Ding, W.J.; Zhao, Y.C.; He, Q.J.; Yang, B. HIF-1alpha-dependent autophagy protects HeLa cells from fenretinide (4-HPR)-induced apoptosis in hypoxia. Pharmacol. Res. 2010, 62, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.M.; Jiang, Z.F.; Ding, P.S.; Shao, L.J.; Liu, R.Y. Hypoxia-induced autophagy mediates cisplatin resistance in lung cancer cells. Sci. Rep. 2015, 5, 12291. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.M.; Cowen, R.L.; Debray, C.; Eustace, A.; Erler, J.T.; Sheppard, F.C.; Parker, C.A.; Stratford, I.J.; Williams, K.J. Reversing hypoxic cell chemoresistance in vitro using genetic and small molecule approaches targeting hypoxia inducible factor-1. Mol. Pharmacol. 2006, 69, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Hussein, D.; Estlin, E.J.; Dive, C.; Makin, G.W. Chronic hypoxia promotes hypoxia-inducible factor-1alpha-dependent resistance to etoposide and vincristine in neuroblastoma cells. Mol. Cancer Ther. 2006, 5, 2241–2250. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Ao, Q.; Zhang, Q.; Yang, X.; Xing, H.; Li, F.; Chen, G.; Zhou, J.; Wang, S.; Xu, G.; et al. Hypoxia induced paclitaxel resistance in human ovarian cancers via hypoxia-inducible factor 1alpha. J. Cancer Res. Clin. Oncol. 2010, 136, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Fu, P.; Du, F.; Chen, W.; Yao, M.; Lv, K.; Liu, Y. Tanshinone iia blocks epithelial-mesenchymal transition through HIF-1alpha downregulation, reversing hypoxia-induced chemotherapy resistance in breast cancer cell lines. Oncol. Rep. 2014, 31, 2561–2568. [Google Scholar] [CrossRef] [PubMed]
- Cosse, J.P.; Ronvaux, M.; Ninane, N.; Raes, M.J.; Michiels, C. Hypoxia-induced decrease in p53 protein level and increase in c-jun DNA binding activity results in cancer cell resistance to etoposide. Neoplasia 2009, 11, 976–986. [Google Scholar] [CrossRef] [PubMed]
- Achison, M.; Hupp, T.R. Hypoxia attenuates the p53 response to cellular damage. Oncogene 2003, 22, 3431–3440. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Biju, M.P.; Wang, M.H.; Haase, V.H.; Dong, Z. Cytoprotective effects of hypoxia against cisplatin-induced tubular cell apoptosis: Involvement of mitochondrial inhibition and p53 suppression. J. Am. Soc. Nephrol. 2006, 17, 1875–1885. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, K.; Fidler, I.J. Hypoxia increases resistance of human pancreatic cancer cells to apoptosis induced by gemcitabine. Clin. Cancer Res. 2004, 10, 2299–2306. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Kobayashi, M.; Darmanin, S.; Qiao, Y.; Gully, C.; Zhao, R.; Yeung, S.C.; Lee, M.H. Pim-1 plays a pivotal role in hypoxia-induced chemoresistance. Oncogene 2009, 28, 2581–2592. [Google Scholar] [CrossRef] [PubMed]
- Piret, J.P.; Cosse, J.P.; Ninane, N.; Raes, M.; Michiels, C. Hypoxia protects HepG2 cells against etoposide-induced apoptosis via a HIF-1-independent pathway. Exp. Cell Res. 2006, 312, 2908–2920. [Google Scholar] [CrossRef] [PubMed]
- Rohwer, N.; Dame, C.; Haugstetter, A.; Wiedenmann, B.; Detjen, K.; Schmitt, C.A.; Cramer, T. Hypoxia-inducible factor 1alpha determines gastric cancer chemosensitivity via modulation of p53 and NF-kappab. PLoS ONE 2010, 5, e12038. [Google Scholar] [CrossRef] [PubMed]
- Schnitzer, S.E.; Schmid, T.; Zhou, J.; Brune, B. Hypoxia and HIF-1alpha protect a549 cells from drug-induced apoptosis. Cell Death Differ. 2006, 13, 1611–1613. [Google Scholar] [CrossRef] [PubMed]
- Selvendiran, K.; Bratasz, A.; Kuppusamy, M.L.; Tazi, M.F.; Rivera, B.K.; Kuppusamy, P. Hypoxia induces chemoresistance in ovarian cancer cells by activation of signal transducer and activator of transcription 3. Int. J. Cancer 2009, 125, 2198–2204. [Google Scholar] [CrossRef] [PubMed]
- Adamski, J.; Price, A.; Dive, C.; Makin, G. Hypoxia-induced cytotoxic drug resistance in osteosarcoma is independent of HIF-1alpha. PLoS ONE 2013, 8, e65304. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Venkatachalam, M.A.; Wang, J.; Patel, Y.; Saikumar, P.; Semenza, G.L.; Force, T.; Nishiyama, J. Up-regulation of apoptosis inhibitory protein IAP-2 by hypoxia. HIF-1-independent mechanisms. J. Biol. Chem. 2001, 276, 18702–18709. [Google Scholar] [CrossRef] [PubMed]
- Boller, Y.C.; Brandes, L.M.; Russell, R.L.; Lin, Z.P.; Patierno, S.R.; Kennedy, K.A. Prostaglandin A1 inhibits stress-induced NF-kappab activation and reverses resistance to topoisomerase II inhibitors. Oncol. Res. 2000, 12, 383–395. [Google Scholar] [PubMed]
- O'Reilly, E.A.; Gubbins, L.; Sharma, S.; Tully, R.; Guang, M.H.; Weiner-Gorzel, K.; McCaffrey, J.; Harrison, M.; Furlong, F.; Kell, M.; et al. The fate of chemoresistance in triple negative breast cancer (TNBC). BBA Clin. 2015, 3, 257–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.R.; Chen, A. Poly(adenosine diphosphate-ribose) polymerase inhibitors in cancer treatment. Hematol. Oncol. Clin. N. Am. 2012, 26, 649–670. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Xiao, Y.; Zhu, X.Y.; Ning, Z.Y.; Xu, H.F.; Wu, H.M. Hypoxia regulates stemness of breast cancer MDA-MB-231 cells. Med. Oncol. 2016, 33, 42. [Google Scholar] [CrossRef] [PubMed]
- Jaggupilli, A.; Elkord, E. Significance of CD44 and CD24 as cancer stem cell markers: An enduring ambiguity. Clin. Dev. Immunol. 2012, 2012, 708036. [Google Scholar] [CrossRef] [PubMed]
- Dean, M. ABC transporters, drug resistance, and cancer stem cells. J. Mammary Gland Biol. Neoplasia 2009, 14, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.E.; Lee, H.G.; Cho, I.H.; Chung, D.H.; Yoon, S.H.; Yang, Y.M.; Lee, J.W.; Choi, S.; Park, J.W.; Ye, S.K.; et al. STAT3 is a potential modulator of HIF-1-mediated VEGF expression in human renal carcinoma cells. FASEB J. 2005, 19, 1296–1298. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Briggs, J.; Park, S.; Niu, G.; Kortylewski, M.; Zhang, S.; Gritsko, T.; Turkson, J.; Kay, H.; Semenza, G.L.; et al. Targeting STAT3 blocks both HIF-1 and VEGF expression induced by multiple oncogenic growth signaling pathways. Oncogene 2005, 24, 5552–5560. [Google Scholar] [CrossRef] [PubMed]
- Duyndam, M.C.; van Berkel, M.P.; Dorsman, J.C.; Rockx, D.A.; Pinedo, H.M.; Boven, E. Cisplatin and doxorubicin repress vascular endothelial growth factor expression and differentially down-regulate hypoxia-inducible factor I activity in human ovarian cancer cells. Biochem. Pharmacol. 2007, 74, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.I.; Haber, M.; Henderson, M.J.; Norris, M.D. ABC transporters in cancer: More than just drug efflux pumps. Nat. Rev. Cancer 2010, 10, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.H.; Chen, M.T.; Wang, M.L.; Lee, Y.Y.; Chiou, G.Y.; Chien, C.S.; Huang, P.I.; Chen, Y.W.; Huang, M.C.; Chiou, S.H.; et al. Cisplatin-selected resistance is associated with increased motility and stem-like properties via activation of STAT3/Snail axis in atypical teratoid/rhabdoid tumor cells. Oncotarget 2015, 6, 1750–1768. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.P.; Dong, X.; Lin, L.; Jiang, X.; Wei, Z.; Zhai, B.; Sun, B.; Zhang, Q.; Wang, X.; Jiang, H.; et al. Up-regulation of survivin by Akt and hypoxia-inducible factor 1alpha contributes to cisplatin resistance in gastric cancer. FEBS J. 2014, 281, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Mamede, A.C.; Abrantes, A.M.; Pedrosa, L.; Casalta-Lopes, J.E.; Pires, A.S.; Teixo, R.J.; Goncalves, A.C.; Sarmento-Ribeiro, A.B.; Maia, C.J.; Botelho, M.F. Beyond the limits of oxygen: Effects of hypoxia in a hormone-independent prostate cancer cell line. ISRN Oncol. 2013, 2013, 918207. [Google Scholar] [CrossRef] [PubMed]
- Flamant, L.; Notte, A.; Ninane, N.; Raes, M.; Michiels, C. Anti-apoptotic role of HIF-1 and AP-1 in paclitaxel exposed breast cancer cells under hypoxia. Mol. Cancer 2010, 9, 191. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, R.; Graham, C.H. Hypoxia prevents etoposide-induced DNA damage in cancer cells through a mechanism involving hypoxia-inducible factor 1. Mol. Cancer Ther. 2009, 8, 1702–1713. [Google Scholar] [CrossRef] [PubMed]
- Notte, A.; Ninane, N.; Arnould, T.; Michiels, C. Hypoxia counteracts taxol-induced apoptosis in MDA-MB-231 breast cancer cells: Role of autophagy and jnk activation. Cell. Death Dis. 2013, 4, e638. [Google Scholar] [CrossRef] [PubMed]
- Ravizza, R.; Molteni, R.; Gariboldi, M.B.; Marras, E.; Perletti, G.; Monti, E. Effect of HIF-1 modulation on the response of two- and three-dimensional cultures of human colon cancer cells to 5-fluorouracil. Eur. J. Cancer 2009, 45, 890–898. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Liu, X.; Chi, W.; Liu, Y.; Wei, L.; Wang, X.; Yu, J. Hypoxia-induced resistance to cisplatin and doxorubicin in non-small cell lung cancer is inhibited by silencing of HIF-1alpha gene. Cancer Chemother. Pharmacol. 2006, 58, 776–784. [Google Scholar] [CrossRef] [PubMed]
- Kilic, M.; Kasperczyk, H.; Fulda, S.; Debatin, K.M. Role of hypoxia inducible factor-1 alpha in modulation of apoptosis resistance. Oncogene 2007, 26, 2027–2038. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Ning, X.; Sun, L.; Zhang, H.; Shi, Y.; Guo, C.; Han, S.; Liu, J.; Sun, S.; Han, Z.; et al. Hypoxia-inducible factor-1 alpha contributes to hypoxia-induced chemoresistance in gastric cancer. Cancer Sci. 2008, 99, 121–128. [Google Scholar] [PubMed]
- Roberts, D.L.; Williams, K.J.; Cowen, R.L.; Barathova, M.; Eustace, A.J.; Brittain-Dissont, S.; Tilby, M.J.; Pearson, D.G.; Ottley, C.J.; Stratford, I.J.; et al. Contribution of HIF-1 and drug penetrance to oxaliplatin resistance in hypoxic colorectal cancer cells. Br. J. Cancer 2009, 101, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Sermeus, A.; Cosse, J.P.; Crespin, M.; Mainfroid, V.; de Longueville, F.; Ninane, N.; Raes, M.; Remacle, J.; Michiels, C. Hypoxia induces protection against etoposide-induced apoptosis: Molecular profiling of changes in gene expression and transcription factor activity. Mol. Cancer 2008, 7, 27. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Bernauer, A.M.; Yingling, C.M.; Belinsky, S.A. Hif1alpha regulated expression of XPA contributes to cisplatin resistance in lung cancer. Carcinogenesis 2012, 33, 1187–1192. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Qu, Z.; Guo, X.; Zhao, Q.; Zhao, X.; Gao, L.; Sun, K.; Shen, F.; Wu, M.; Wei, L. Hypoxia-induced autophagy contributes to the chemoresistance of hepatocellular carcinoma cells. Autophagy 2009, 5, 1131–1144. [Google Scholar] [CrossRef] [PubMed]
- Koch, S.; Mayer, F.; Honecker, F.; Schittenhelm, M.; Bokemeyer, C. Efficacy of cytotoxic agents used in the treatment of testicular germ cell tumours under normoxic and hypoxic conditions in vitro. Br. J. Cancer 2003, 89, 2133–2139. [Google Scholar] [CrossRef] [PubMed]
- Louie, E.; Nik, S.; Chen, J.S.; Schmidt, M.; Song, B.; Pacson, C.; Chen, X.F.; Park, S.; Ju, J.; Chen, E.I. Identification of a stem-like cell population by exposing metastatic breast cancer cell lines to repetitive cycles of hypoxia and reoxygenation. Breast Cancer Res. 2010, 12, R94. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, A.L.; Ribeiro, V. Drug metabolism and transport under hypoxia. Curr. Drug Metab. 2013, 14, 969–975. [Google Scholar] [CrossRef] [PubMed]
- Crowder, S.W.; Balikov, D.A.; Hwang, Y.S.; Sung, H.J. Cancer stem cells under hypoxia as a chemoresistance factor in breast and brain. Curr. Pathobiol. Rep. 2014, 2, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Song, X.; Song, B.; Liu, Y.; Wei, L.; Wang, X.; Yu, J. Effects of lentivirus-mediated HIF-1alpha knockdown on hypoxia-related cisplatin resistance and their dependence on p53 status in fibrosarcoma cells. Cancer Gene Ther. 2008, 15, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Gu, F.; Ma, Y.; Zhang, Z.; Zhao, J.; Kobayashi, H.; Zhang, L.; Fu, L. Expression of Stat3 and Notch1 is associated with cisplatin resistance in head and neck squamous cell carcinoma. Oncol. Rep. 2010, 23, 671–676. [Google Scholar] [PubMed]
- Sheng, W.J.; Jiang, H.; Wu, D.L.; Zheng, J.H. Early responses of the STAT3 pathway to platinum drugs are associated with cisplatin resistance in epithelial ovarian cancer. Braz. J. Med. Biol. Res. 2013, 46, 650–658. [Google Scholar] [CrossRef] [PubMed]
- Eckstein, N. Platinum resistance in breast and ovarian cancer cell lines. J. Exp. Clin. Cancer Res. 2011, 30, 91. [Google Scholar] [CrossRef] [PubMed]
- Falamarzian, A.; Aliabadi, H.M.; Molavi, O.; Seubert, J.M.; Lai, R.; Uludag, H.; Lavasanifar, A. Effective down-regulation of signal transducer and activator of transcription 3 (Stat3) by polyplexes of sirna and lipid-substituted polyethyleneimine for sensitization of breast tumor cells to conventional chemotherapy. J. Biomed. Mater. Res. A 2014, 102, 3216–3228. [Google Scholar] [CrossRef] [PubMed]
- Pawlus, M.R.; Wang, L.; Hu, C.J. Stat3 and HIF1alpha cooperatively activate hif1 target genes in MDA-MB-231 and RCC4 cells. Oncogene 2014, 33, 1670–1679. [Google Scholar] [CrossRef] [PubMed]
- Oh, M.K.; Park, H.J.; Kim, N.H.; Park, S.J.; Park, I.Y.; Kim, I.S. Hypoxia-inducible factor-1alpha enhances haptoglobin gene expression by improving binding of Stat3 to the promoter. J. Biol. Chem. 2011, 286, 8857–8865. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, A.; Kugimiya, N.; Hosoyama, T.; Enoki, T.; Li, T.S.; Hamano, K. HIF-1alpha activation under glucose deprivation plays a central role in the acquisition of anti-apoptosis in human colon cancer cells. Int. J. Oncol. 2014, 44, 2077–2084. [Google Scholar] [CrossRef] [PubMed]
- Castaldo, S.A.; Freitas, J.R.; Conchinha, N.V.; Madureira, P.A. The tumorigenic roles of the cellular redox regulatory systems. Oxid. Med. Cell. Longev. 2016, 2016, 8413032. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, G.; Zhao, Y.; Liu, X.; Ding, Q.; Shi, J.; Ding, Y.; Wang, S. STAT3 mediates resistance of CD44(+)CD24(−/low) breast cancer stem cells to tamoxifen in vitro. J. Biomed. Res. 2012, 26, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.S.; Aroh, C.; Vadgama, J.V. Constitutive activation of STAT3 signaling regulates hTERT and promotes stem cell-like traits in human breast cancer cells. PLoS ONE 2013, 8, e83971. [Google Scholar] [CrossRef] [PubMed]
- Marotta, L.L.; Almendro, V.; Marusyk, A.; Shipitsin, M.; Schemme, J.; Walker, S.R.; Bloushtain-Qimron, N.; Kim, J.J.; Choudhury, S.A.; Maruyama, R.; et al. The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(−) stem cell-like breast cancer cells in human tumors. J. Clin. Investig. 2011, 121, 2723–2735. [Google Scholar] [CrossRef] [PubMed]
- Romain, B.; Hachet-Haas, M.; Rohr, S.; Brigand, C.; Galzi, J.L.; Gaub, M.P.; Pencreach, E.; Guenot, D. Hypoxia differentially regulated CXCR4 and CXCR7 signaling in colon cancer. Mol. Cancer 2014, 13, 58. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primers | Reverse Primers |
|---|---|---|
| ABCC1 | 5′-CTCTATCTCTCCCGACATGACC-3′ | 5′-AGCAGACGATCCACAGCAAAA-3′ |
| ABCC2 | 5′-CCCTGCTGTTCGATATACCAATC-3′ | 5′-TCGAGAGAATCCAGAATAGGGAC-3′ |
| ABCC4 | 5′-AGCTGAGAATGACGCACAGAA-3′ | 5′-ATATGGGCTGGATTACTTTGGC-3′ |
| ABCC5 | 5′-AGTCCTGGGTATAGAAGTGTGAG-3′ | 5′-ATTCCAACGGTCGAGTTCTCC-3′ |
| ABCC6 | 5′-AAGGAGGTACTAGGTGGGCTT-3 | 5′-CCAGTAGGACCCTTCGAGC-3′ |
| ABCB1 | 5′-TTGCTGCTTACATTCAGGTTTCA-3 | 5′-AGCCTATCTCCTGTCGCATTA-3 |
| GAPDH | 5′-GGAGCGAGATCCCTCCAAAAT-3′ | 5′-GGCTGTTGTCATACTTCTCATGG-3′ |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soleymani Abyaneh, H.; Gupta, N.; Radziwon-Balicka, A.; Jurasz, P.; Seubert, J.; Lai, R.; Lavasanifar, A. STAT3 but Not HIF-1α Is Important in Mediating Hypoxia-Induced Chemoresistance in MDA-MB-231, a Triple Negative Breast Cancer Cell Line. Cancers 2017, 9, 137. https://doi.org/10.3390/cancers9100137
Soleymani Abyaneh H, Gupta N, Radziwon-Balicka A, Jurasz P, Seubert J, Lai R, Lavasanifar A. STAT3 but Not HIF-1α Is Important in Mediating Hypoxia-Induced Chemoresistance in MDA-MB-231, a Triple Negative Breast Cancer Cell Line. Cancers. 2017; 9(10):137. https://doi.org/10.3390/cancers9100137
Chicago/Turabian StyleSoleymani Abyaneh, Hoda, Nidhi Gupta, Aneta Radziwon-Balicka, Paul Jurasz, John Seubert, Raymond Lai, and Afsaneh Lavasanifar. 2017. "STAT3 but Not HIF-1α Is Important in Mediating Hypoxia-Induced Chemoresistance in MDA-MB-231, a Triple Negative Breast Cancer Cell Line" Cancers 9, no. 10: 137. https://doi.org/10.3390/cancers9100137