
Evolving Significance and Future Relevance of Anti-Angiogenic Activity of mTOR Inhibitors in Cancer Therapy


Department of Visceral Surgery, Lausanne University Hospital, Pavillon 4, avenue de Beaumont, 1011 Lausanne, Switzerland
*
Author to whom correspondence should be addressed.
Cancers 2017, 9(11), 152; https://doi.org/10.3390/cancers9110152
Submission received: 4 September 2017
/
Revised: 23 October 2017
/
Accepted: 27 October 2017
/
Published: 1 November 2017
(This article belongs to the Special Issue mTOR Pathway in Cancer)
{kind=link}
{kind=link}
{kind=link}
Abstract
:mTOR inhibitors have demonstrated remarkable anti-tumor activity in experimental models, mainly by reducing cancer cell growth and tumor angiogenesis. Their use in cancer patients as monotherapy has, however, generated only limited benefits, increasing median overall survival by only a few months. Likewise, in other targeted therapies, cancer cells develop resistance mechanisms to overcome mTOR inhibition. Hence, novel therapeutic strategies have to be designed to increase the efficacy of mTOR inhibitors in cancer. In this review, we discuss the present and future relevance of mTOR inhibitors in cancer therapy by focusing on their effects on tumor angiogenesis.
1. Introduction
The mechanistic target of rapamycin (mTOR) is a serine/threonine kinase that exerts its effect by forming an integral part of two structurally and functionally distinct protein complexes, named mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) [1,2]. mTORC1 coordinates cell growth in favorable extracellular conditions by stimulating protein, lipid, and nucleotide synthesis, and by inhibiting autophagy [3]. mTORC2 is primarily activated by growth factors, and stimulates cell proliferation and survival by activating members of the group of AGC protein kinases, such as AKT [4,5]. The mTOR signaling pathway is frequently overactivated in cancer cells, either by mutations of upstream components of the pathway or by mutations of mTOR itself [6,7]. In addition, the mTOR pathway participates in tumor angiogenesis [8]. Hence, targeting mTOR has the potential to slow down tumor progression.
Three different types of chemical inhibitors of mTOR have been tested in various cancer models (Figure 1) [9,10]. Firstly, rapamycin and its analogs—generally termed rapalogs—which bind, together with FKBP12 (FK506-binding 12 kDa protein), to the FRB domain of mTOR and exert a specific inhibition of mTORC1. This inhibition is, however, incomplete, as some epitopes phosphorylated by mTORC1 are resistant to rapalogs [11]. In addition, rapalogs do not directly block mTORC2, but might inhibit it in certain cell types following prolonged treatment [12]. Secondly, ATP-competitive inhibitors of mTOR that target the kinase domain of mTOR [13]. In contrast to rapalogs, these inhibitors completely block the activity of mTORC1 and mTORC2. Some of these inhibitors block PI3K as well, and are then named dual PI3K/mTOR inhibitors. Thirdly, and more recently, a compound composed of rapamycin cross-linked with a kinase inhibitor of mTOR has been generated, aiming to overcome resistance mutations to rapalogs or kinase inhibitors of mTOR [14].
In 1981, the anti-cancer activity of rapamycin was reported in various cancer cell lines [15]. Since then, numerous pre-clinical studies have confirmed that blocking mTOR impairs tumor progression [16,17]. Decreased cancer cell proliferation and reduced tumor angiogenesis are frequently associated with this effect. mTOR inhibitors have also demonstrated anti-cancer activity in patients, albeit limited, increasing median overall survival by a few months [18,19,20]. Hence, mTOR inhibitors used as monotherapy do not provide the expected anti-cancer efficacy. Several resistance mechanisms that dampen the effects of mTOR inhibitors have been identified [21]. The place of mTOR inhibitors in cancer needs, therefore, to be reconsidered, and novel therapeutic strategies based on mTOR inhibition have to be established. In this review, we discuss and speculate about the future use of therapies that target mTOR in cancer by focusing mainly on their effects on tumor endothelial cells.
2. mTOR Inhibitors and Tumor Angiogenesis
Blood supply in tumors is primarily established by formation of new blood vessels from pre-existing vascular networks in a process called angiogenesis [22]. A variety of cells, including tumor and tumor-associated stromal cells, participate in this process, in part by secreting growth factors and cytokines that stimulate tumor endothelial cells [23]. Tumor hypoxia is a key driver of angiogenesis, as hypoxic tumor cells secrete vascular endothelial growth factor (VEGF), which represents a major angiogenic factor [24,25]. Starving cancers by blocking tumor angiogenesis has been developed extensively over the past two decades [26]. Based hereon, therapeutic strategies targeting VEGF have shown clinical benefits that are, however, frequently not long-lasting [27].
Similarly to anti-VEGF treatments, mTOR inhibitors display anti-angiogenic properties [8]. This observation is supported by several in vitro and in vivo studies (Figure 2). In vitro, rapamycin was shown to markedly reduce spontaneous and growth factor-mediated endothelial cell proliferation [28,29,30,31]. This is associated with decreased cyclin D1 expression and a consequent reduction of S-phase entry by endothelial cells [31,32]. MicroRNAs (miRs) also influence the anti-proliferative effects of mTOR inhibitors in endothelial cells. Rapamycin increases expression of miR-21 in endothelial cells, and a downregulation of miR-21 abolishes the anti-proliferative effects of rapamycin [33]. Importantly, inhibition of mTOR by rapamycin also inhibits hypoxia-mediated endothelial cell proliferation [34].
Besides endothelial cell proliferation, rapamycin further influences other cell functions relevant to tumor angiogenesis. For instance, growth factor stimulated endothelial cell sprout formation in rat or mouse aorta is reduced by rapamycin [34]. In addition, rapamycin decreases endothelial cell survival [35,36,37]. In serum and growth-factor deprived conditions, VEGF-induced endothelial cell survival is inhibited by rapamycin as evidenced by flow cytometry analysis of annexin stainings or cell cycle profile of endothelial cells [35,37]. Pro-apoptotic effects of rapamycin in endothelial cells are mediated by its ability to inhibit mTORC2 activity and consequently AKT phosphorylation and activation [37]. Rapamycin also reduces endothelial cell migration [36,37,38]. Molecular mechanisms involved in this matter include increased expression of the cyclin-dependent kinase inhibitor p27, as well as miR-21 by rapamycin [33,39]. Finally, rapamycin further reduces the ability of endothelial cells to form tubular structures in vitro [40,41].
In addition to rapalogs, the effects of kinase inhibitors of mTOR on endothelial cell proliferation, survival, migration, and tube formation have been tested. These inhibitors possess similar but stronger activities than rapalogs on endothelial cells in vitro [42].
The anti-angiogenic properties of mTOR inhibitors have also been illustrated in various cancer models in vivo. For instance, rapamycin decreases angiogenesis in dorsal skin fold chambers transplanted with tumor cells, and in tumor xeno- and allografts [40]. The anti-angiogenic potential of the rapalog CCI-779 was demonstrated in the matrigel plug assay, where CCI-779 inhibited VEGF-stimulated vessel formation [43]. The latter further reduced microvessel density in two different rhabdomyosarcoma xenografts [44]. Reduced vessel density in these models was associated with decreased level of HIF-1 (hypoxia-inducible factor 1) and VEGF, confirming the role of mTOR in hypoxic tumor response. Besides tumor xeno- and allografts, rapalogs also demonstrated anti-angiogenic efficacy in a transgenic mouse model characterized by the development of ovarian serous adenocarcinomas [45]. In addition, rapalogs further decreased vascular density in patient-derived hepatocellular carcinoma xenografts [46]. Interestingly, the rapalog RAD001 reduced the growth of tumor xenografts generated from cancer cell lines that are either sensitive or insensitive to RAD001 in vitro. In either case, RAD001 reduced the number of tumor blood vessels in tumor xenografts, highlighting the anti-angiogenic effect of rapalogs as a major mechanism to decrease tumor xenograft growth [47]. The amount of intra-tumoral VEGF was also reduced by RAD001. Besides histological analysis, the anti-angiogenic activity of rapamycin has been evidenced by magnetic resonance imaging [48].
Finally, and more importantly, the anti-angiogenic effect of rapalogs has been reported in tumor patients [49]. Lymph node biopsies retrieved before and after treatment of a patient suffering from mantle cell lymphoma with CCI-779 revealed a decrease of tumor blood-vessel density.
Similarly to rapalogs, several studies have demonstrated anti-angiogenic effects of ATP-competitive inhibitors of mTOR. The dual PI3K/mTOR inhibitor NVP-BEZ235 showed anti-angiogenic activity in tumor mouse models of breast and renal cell cancers and glioma [50,51,52]. NVP-BEZ235 also reduced intra-tumoral levels of VEGF [52]. The anti-angiogenic effects of NVP-BEZ235 were more pronounced than with rapalogs [42,51,53]. Similarly, selective kinase inhibitors of mTOR reduced vessel density in various models [42,54,55]. For instance, the mTORC1/mTORC2 kinase inhibitor OXA-01 decreased tumor blood vessels and intra-tumoral levels of VEGF more potently than rapamycin [55]. Analogous findings were reported for PP242 [56].
Despite a clear anti-angiogenic activity of mTOR inhibitors in tumor mouse models, few studies have investigated their effects on tumor endothelial cells in vivo. Nevertheless, it was reported that rapamycin increased tumor endothelial cell apoptosis in orthotopic pancreatic tumors using terminal deoxynucleotidyl transferase nick end labeling (TUNEL) and CD31 double staining [35]. This was associated with damaged vessels containing thromboses. Formation of vessel thrombosis following rapamycin treatment was furthermore reported in lung cancer tumor xenografts [57].
Conditional cell depletion studies have partially confirmed the role of mTOR in endothelial cells and tumor angiogenesis. Specific ablation of tuberous sclerosis complex-1 (TSC1), a negative regulator of mTORC1, in endothelial cells resulted in the formation of lymphangiosarcoma characterized by sustained proliferation of endothelial cells [58]. Deletion of rictor, a component of mTORC2, in endothelial cells reduced VEGF-mediated endothelial cell proliferation. It also decreased the growth of tumor allografts, which was associated with diminished tumor angiogenesis [59]. The role of mTORC2 in VEGF signaling in endothelial cells was further confirmed using a phosphoproteomic approach [60]. Similarly, down-regulation of mTORC2 reduced prostaglandin E2-mediated endothelial cell responses and sprouting angiogenesis in vitro [61,62].
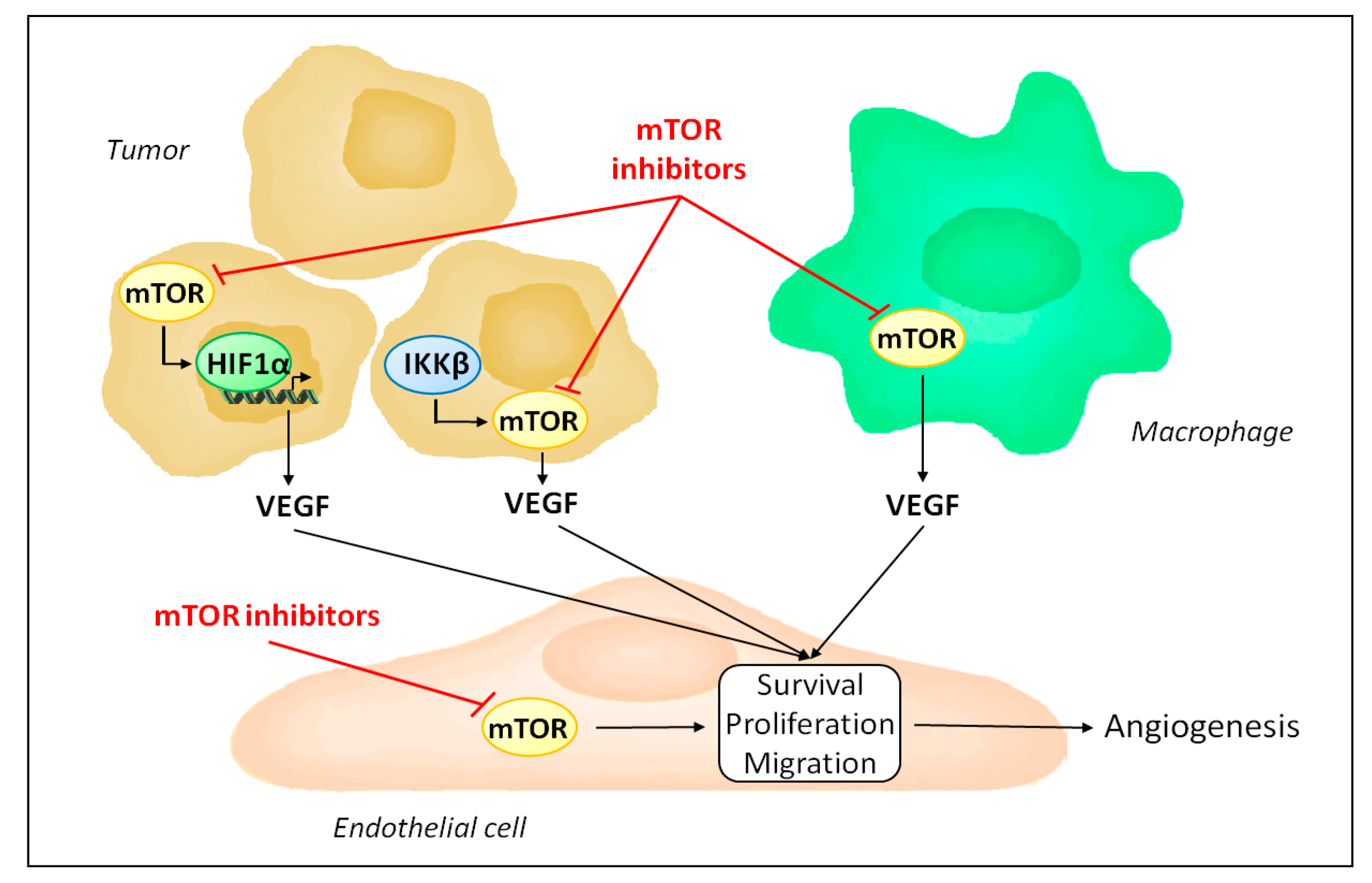
mTOR is ubiquitously expressed and its inhibition is not limited to endothelial cells. Accordingly, besides acting directly on endothelial cells, mTOR inhibitors influence angiogenesis by regulating the production of pro-angiogenic factors (Figure 2). Indeed, mTOR participates in the hypoxic tumor response by stabilizing hypoxia-inducible factor 1α and serves as a signaling intermediary in inflammation-mediated angiogenesis [63,64,65,66,67]. Consequently, mTOR inhibitors reduce the expression of VEGF [40,55].
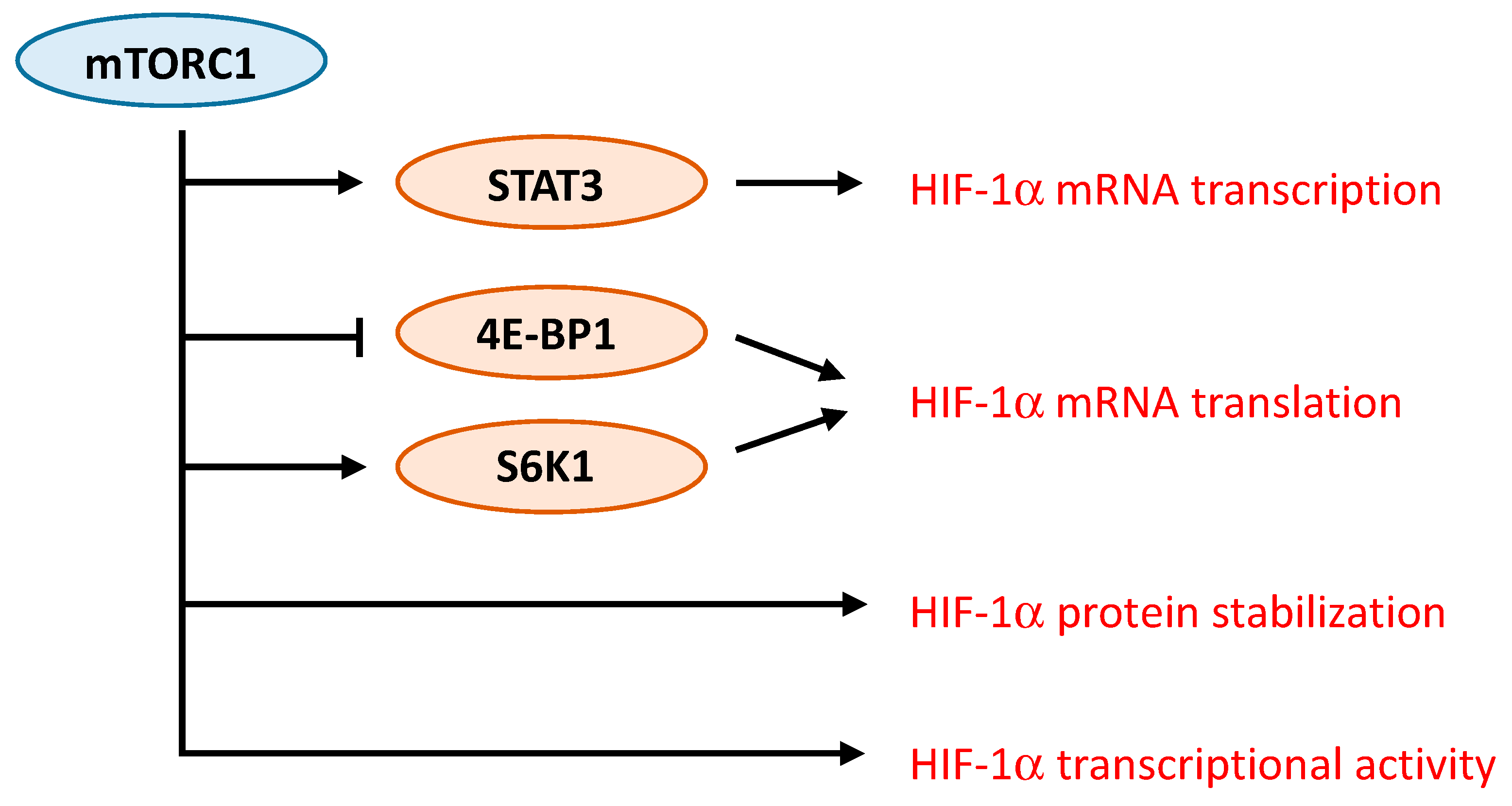
Several studies have demonstrated the complex interaction between mTORC1 and HIF-1α. Different mechanisms responsible for the regulation of HIF-1α by mTORC1 have been proposed (Figure 3). For instance, in prostate cancer cells, rapamycin decreases protein levels of HIF-1α by interfering with processes that promote HIF-1α protein stabilization [63]. In contrast, activation of HER2 receptor in breast cancer cells increases HIF-1α synthesis via stimulation of HIF-1α mRNA translation in a rapamycin-sensitive manner [68]. Up-regulation of HIF-1α mRNA translation was further observed either as a result of increased cap-dependent translation following 4E-BP1 phosphorylation or via ribosomal protein S6 kinase-1 [69,70]. An additional mechanism involves the promotion of the transcriptional activity of HIF-1α by mTORC1 [71]. Finally, more recently, one study specifically addressed the mechanisms driven by mTORC1 that induce HIF-1α signaling [72]. The role of mTORC1 and its downstream substrates 4E-BP1 and S6K1 in regulating HIF-1α mRNA translation was confirmed. In addition, the regulation of HIF-1α mRNA transcription by mTORC1 was demonstrated, and appears to involve STAT3 [72].
Tumor angiogenesis is also influenced by tumor associated stromal cells [23]. In particular, tumor associated macrophages play an important role in shaping the angiogenic response in tumors, and can either sustain or, in contrast, repress angiogenesis [73,74]. Interestingly, mTOR activity in macrophages has been shown to be an important factor in promoting the ability of macrophages to stimulate angiogenesis [75]. Production of VEGF and interleukin-10 by human monocytes following lipopolysaccharide stimulation was significantly reduced when monocytes were treated with rapamycin, compared to untreated monocytes. Furthermore, in tumor xenograft models, depletion of macrophages is sufficient to inhibit the anti-angiogenic activity of rapamycin [75]. In contrast, infusion of monocytes with increased mTORC1 activity following genetic ablation of TSC2 results in increased tumor growth and angiogenesis in host mice bearing tumor xenografts [75]. Further evidence exists for a role of mTOR as a regulator of macrophages polarization [76].
Finally, besides sprouting angiogenesis, five other modes of vessel formation in tumors have been identified [77]. One is vascular mimicry, a process by which tumor cells acquire endothelial-like characteristics and line tumor vessels [25,78,79]. The occurrence of vascular mimicry is not frequent, but is nevertheless correlated with poor clinical outcome [80]. The observations that vascular mimicry correlates with mTOR expression and that rapamycin inhibits the expression of endothelial cell markers by tumor cells in vitro suggest that mTOR might further contribute to tumor blood supply by regulating vascular mimicry [81,82]. Additional studies are, however, needed to fully characterize the consequences of mTOR inhibition in this process.
3. Resistances to the Anti-Angiogenic Effects of mTOR Inhibitors
Several resistance mechanisms to anti-VEGF therapies have been characterized. For instance, the stimulation of tumor endothelial cells by other growth factors than VEGF has been identified [83,84]. As mentioned above, alternate modes of vascularization to sprouting angiogenesis are employed by tumors [85]. In addition to vascular mimicry, cancer cells can grow along pre-existing vessels by co-opting blood vessels [77]. Additionally, new blood vessels can be formed by intussusceptions, the splitting of pre-existing vessels to give rise to two daughter vessels.
In contrast to anti-VEGF treatments, resistances to the anti-angiogenic effects of mTOR inhibitors have barely been investigated. Nevertheless, emerging studies show that tumors are still able to maintain blood supply despite mTOR inhibition. In this context, lack of anti-angiogenic effects by mTOR inhibitors has been reported. Treatment of mice bearing human cervical carcinoma xenografts with rapamycin does not decrease intra-tumoral mean vessel density despite decreasing tumor growth [86]. It is of note that rapamycin treatment has no significant effect on plasma levels of VEGF in this study. Likewise, rapamycin fails to alter microvessel density in a transgenic mouse model of human epidermal growth factor receptor 2 (HER2)-positive breast cancer, even though mTORC1 inhibition in tumor endothelial cells was documented by immunohistochemistry [87]. Absence of anti-angiogenic effect has also been reported for the dual PI3K/mTOR inhibitor NVP-BEZ235, as well as for the mTORC1/mTORC2 inhibitor KU-0063794 [88,89]. In all these studies, tumor analysis was performed at the end of treatment. It is therefore not possible to differentiate whether tumor blood vessels were intrinsically resistant to rapamycin or, following an initial reduction of mean vessel density, alternate signaling pathways were engaged to compensate for the inhibition of mTOR and hence restore the formation of tumor blood vessels. Consistent with this latter hypothesis, it has been shown that treatment of endothelial cells with rapamycin increases the activity of mitogen-activated protein kinase (MAPK), which counteracts the anti-angiogenic efficacy of mTOR inhibitors [42]. Likewise, treatment of cultured endothelial cells with rapamycin increases the expression of the serine/threonine-protein kinase Pim-1, which reduces the anti-proliferative efficacy of rapamycin [90]. Interestingly, compensatory mechanisms of tumor blood supply have been detected upon inhibition of sprouting angiogenesis by rapalogs. In a rat model of hepatocellular carcinoma, electron microscopy analysis of tumors revealed that RAD001 reduces sprouting angiogenesis, and that under these circumstances, the main vascular growth mode is intussusception [91]. Hence, as for anti-VEGF therapies, resistance mechanisms to the anti-angiogenic activity of mTOR inhibitors exist and need to be thoroughly characterized.
4. Combined Therapies to Increase the Anti-Angiogenic Efficacy of mTOR Inhibitors
Since the anti-cancer efficacy of mTOR inhibitors used as monotherapy was limited in cancer patients, pre-clinical studies have tested therapeutic approaches that combine mTOR inhibitors with other anti-cancer agents. Several reports have investigated the effects of such combined treatments on tumor angiogenesis. In this regard, the use of radiotherapy combined with mTOR inhibitors seems particularly interesting. Radiation increases mTORC1 activity in endothelial cells, suggesting that mTORC1 might counteract the effects of radiation [41]. Consistent with this hypothesis, rapalogs sensitize endothelial cells to radiation in culture by decreasing cell survival [41]. Combining rapalogs with radiation increases endothelial cell apoptosis as demonstrated by increased cleaved caspase-3 expression. Tubule formation by endothelial cells is inhibited to a greater extent by rapalogs in combination with radiation than by rapamycin or radiation alone [41]. Also, tumor growth of glioma allografts is significantly reduced by rapamycin in combination with radiation compared to either treatment alone. This effect is associated with reduced mean vessel density. Similar findings were reported in models of colon and pancreatic cancers, where disruption of VEGF production in cancer cells and VEGF-mediated signaling activation in endothelial cells induced by rapalogs were proposed as the underlying mechanisms [92]. Likewise, in sarcoma and non-small cell lung tumor xenografts, rapamycin treatment results in radio-sensitization, and reduction of tumor vessels is maximal under combined rapamycin-radiation treatment [93,94]. Interestingly, rapalogs further sensitize radio-resistant human oral squamous cell carcinoma tumor xenografts to fractionated radiation [95]. Compared to single treatments, combining rapalogs with fractionated radiation induces tumor endothelial cell apoptosis, which is associated with thrombus formation and tumor necrosis [95]. Dual PI3K/mTOR inhibitors demonstrated similar effects to rapalogs and radio-sensitized endothelial cells in vitro [96]. Based on these encouraging pre-clinical reports, phase I clinical trials combining rapalogs with radiation are performed.
Besides radiotherapy, combining mTOR inhibitors with chemotherapies shows additional anti-angiogenic activity. In the chick embryo chorioallantoic membrane, angiogenic response induced by neuroblastoma cells derived from cell lines or patients is maximally inhibited when rapamycin is combined with vinblastine [32]. Rapamycin also displays increased anti-angiogenic effects when administered with doxorubicin in a rat model of hepatoma [97].
Additionally, mTOR inhibitors have been tested in combination with anti-VEGF treatments. Co-administration of rapamycin and bevacizumab, a humanized recombinant monoclonal antibody that targets VEGF, was tested in mice bearing hepatocellular carcinoma tumor xenografts [98]. Combined treatments significantly reduced mean vessel density in tumor xenografts generated from six different cell lines. Importantly, the combination also decreased mean vessel density in a tumor xenograft that did not respond to single treatment, suggesting that the combination could overcome resistances to either treatment. The combination was also more efficient in decreasing VEGF levels [98]. Such combinations exhibited substantial activity and reasonable toxicity in advanced renal cell carcinoma and pancreatic neuroendocrine tumors in phase II trials [99,100]. In contrast, it was also reported that bevacizumab combined with CCI-779 did not provide any survival benefits compared to bevacizumab alone in advanced renal cell carcinoma [101].
Rapalogs were further tested with sorafenib and sunitinib, two small tyrosine kinase inhibitors that non-specifically target VEGF receptors. Formation of capillary tubes by endothelial cells was significantly more decreased by RAD001 in combination with sorafenib compared to single therapy [102]. Co-administration of sorafenib and RAD001 also decreased angiogenesis in osteosarcoma xenografts grown onto the chick embryo chorioallantoic membrane or in NOD/SCID mice. This effect was, however, not significantly different from treatment with sorafenib alone [102]. Other investigators reported that combined sorafenib/RAD001 neither decreased capillary tube formation nor significantly reduced endothelial cell proliferation compared to RAD001 alone [103]. However, combined treatment, when administered sequentially, significantly decreased endothelial cell sprouting from aortic rings compared to single treatment [103]. Absence of sprouting angiogenesis induced by combined sorafenib/RAD001 was further noted in a rat model of hepatocellular carcinoma [103]. Additional studies have revealed the potentiated anti-angiogenic effects of rapalogs combined with sunitinib. Association of rapamycin and sunitinib showed greater anti-angiogenic effects than rapamycin or sunitinib alone, both in vitro and in vivo [104]. The anti-angiogenic effect of RAD001 was also significantly increased in combination with TKI-258, another small tyrosine kinase inhibitor, in hepatocellular carcinoma tumor xenografts [105]. It is of note that a phase I clinical study revealed that sorafenib in combination with CCI-779 was associated with significant toxicity in metastatic melanoma patients [106]. Sunitinib combined with RAD001 in advanced renal cell carcinoma was also associated with toxicity and was only tolerated at attenuated doses.
As mentioned previously, treatment of endothelial cells with mTOR inhibitors results in increased MEK/MAPK pathway activity, which counteracts the anti-angiogenic efficacy of mTOR inhibitors [42]. Hence, combining MEK inhibitors with mTOR inhibitors provides greater anti-angiogenic effects, as evidenced in colon cancer and hepatocellular tumor xenografts [42,107]. Patients treated with such a therapeutic approach showed however non-negligible side effects that greatly limit its application in clinic [108].
Combining rapalogs with inhibitors of the insulin-like growth factor 1 receptor (IGF-1R) has also been tested. The rationale for such a combination is the observation that blocking IGF-1R abrogates rapamycin-mediated AKT activation in cancer cells [109]. Further evidence indicates that inhibitors of IGF-1R potentiate the anti-angiogenic efficacy of rapalogs by decreasing VEGF levels [110]. Clinical trials show contrasting results. While combining IGF1-R inhibitors with rapalogs in advanced sarcoma appears safe and provides anti-tumor activity [111,112,113], similar drug associations are no more effective than exemestane, an oral steroidal aromatase inhibitor, in estrogen receptor positive advanced breast cancer but exhibit more adverse effects [114].
Vascular disrupting agents target established tumor vasculature, which is distinct from anti-angiogenic agents that block neovascularization [115]. Hence, combining vascular disrupting agents with anti-angiogenic drugs is meaningful, and should result in increased anti-tumor activity. In this context, the effects of mTOR inhibitors combined with vascular disrupting agents have been tested [116,117]. In a three-dimensional spheroid sprouting assay, co-treatment of RAD001 with the vascular disrupting agent ASA404 significantly increased disruption of endothelial sprouts compared to single treatments. In vivo, ASA404 combined with RAD001 markedly increased tumor necrosis in a renal cell carcinoma model [116]. Similarly, NVP-BEZ235 combined with vascular-targeted photodynamic therapy showed a strong synergism characterized by increased endothelial cell apoptosis in vitro [117].
Finally, further pre-clinical studies showed that combined therapies can increase the anti-angiogenic effects of mTOR inhibitors. For example, co-administration of the histone deacetylase inhibitor LBH589 with rapamycin provides stronger anti-angiogenic effects in tumor xenografts compared to LBH589 or rapamycin alone. At a molecular level, this therapeutic strategy significantly reduces HIF-1α expression [118]. Likewise, methylnaltrexone, a peripheral-acting mu-opioid receptor antagonist, exerts a synergistic effect with CCI-779 or rapamycin on VEGF-induced endothelial cell proliferation and migration in cell culture and on angiogenesis in the matrigel plug assay [119]. Also, Toll-like receptor 9 agonist combined with RAD001 reduces VEGF production by renal cell carcinoma cells and impairs endothelial cell functions [120].
5. mTOR Inhibitors and Normalization of Tumor Vasculature
Due to excessive growth stimulation, tumor blood vessels display an aberrant morphology and poor functionality [121,122]. As a consequence, intra-tumoral fluid pressure is increased with areas of hypoxia that contribute to resistance to chemo- and radiotherapy. Hence, normalization of vascular abnormalities represents a therapeutic approach aiming to restore tumor blood perfusion and, thus, increased drug accessibility and reduced resistances mediated by hypoxia [123,124]. Tumor vessel normalization by anti-angiogenic drugs was initially observed with bevacizumab [125] and further investigated for mTOR inhibitors. Rapamycin reduces vessel permeability in a tumor xenograft model as evidenced by fluorescence tomography [126]. Similarly, rapamycin increases tumor perfusion and oxygenation in a model of rhabdomyosarcoma and potentiates the efficacy of radiotherapy [127]. Hence, rapamycin administration before irradiation to normalize the tumor vasculature represents a potential therapeutic strategy that needs to be precisely characterized. The observation that mTOR inhibitors sensitize various tumor xenografts to radiotherapy and chemotherapy further support such a therapeutic approach [93,128,129]. In addition, kinase inhibitors of mTOR provide similar effects on tumor vasculature normalization as rapalogs. NVP-BEZ235 decreases vascular permeability and accordingly intra-tumoral fluid pressure in a rat breast cancer model [50]. It further improves tumor oxygenation and response to radiotherapy [96]. Also, the mTORC1/mTORC2 inhibitor Palomid 529 inhibits VEGF-mediated increase of vascular permeability [54]. It is of note that the absence of effects of rapalogs on vascular permeability has also been reported, suggesting that the exact settings in which mTOR inhibitors induce vessel normalization have to be clearly identified [47,116].
6. mTOR Inhibitors and Tumor Endothelial Barrier
Tumor endothelium, by its unique position, regulates the trafficking of leukocytes into tumors by controlling the expression of adhesion molecules such as intercellular adhesion molecule-1 (ICAM-1) or vascular cell adhesion molecule-1 (VCAM-1) [130]. In addition, tumor endothelial cells are able to modify the activity of T lymphocytes as they express MHC major histocompatibility complex (MHC) class I and II as well as co-stimulatory and co-inhibitory molecules [131]. Hence, the tumor vasculature actively participates in the host tumor immune response. In the context of cancer, the endothelium is, however, most frequently anergic, failing to upregulate adhesion molecules and to properly recruit cytotoxic T cells. Moreover, tumor endothelium preferentially recruits T regulatory cells, further contributing to immune escape [132]. Furthermore, through the expression of Fas ligand, tumor endothelial cells are able to directly kill activated T lymphocytes [133]. Therefore, a therapeutic opportunity exists to shape tumor endothelium to promote an appropriate recruitment and activation of anti-tumor immune cells [134,135,136]. Emerging evidence suggests that mTOR inhibitors influence functions of endothelial cells that are relevant to host immune response. Expression of inhibitory molecules programmed death-ligand 1 (PD-L1) and programmed death-ligand 2 (PD-L2) is upregulated on endothelial cells both in vitro and in vivo upon rapamycin treatment [137]. Similarly, rapamycin reduces the expression of VCAM-1 on endothelial cells [138]. Hence, future experiments are needed to identify the effects of mTOR inhibitors on tumor endothelial barrier.
7. Biomarkers of Efficacy of mTOR Inhibitors
The identification of biomarkers that predict sensitivity or resistance to mTOR inhibitors would be key to appropriately selecting patients likely to respond to these therapies. In this regard, molecular alterations of PTEN, PI3K, KRAS or Bcl-2 overexpression have been associated with either sensitivity or resistance to mTOR inhibitors [139,140,141]. The use of such biomarkers needs, however, to be validated in clinical trials. While these studies have mostly focused on one or few predefined molecules, the application of next generation sequencing represents a promising tool in the quest of biomarkers [142]. In fact, it has already been successfully applied in a patient with anaplastic thyroid cancer, who exhibited a near-complete response to the rapalog RAD001 for 18 months followed by disease progression [143]. Pre-treatment whole exome sequencing revealed the presence of a non sense mutation of TSC2, a negative regulator of mTORC1, resulting in overactivation of mTORC1. Similar analysis following tumor progression demonstrated mTOR mutations that render mTORC1 resistant to rapalogs. Likewise, activating mutations of mTOR were detected in a patient with metastatic urothelial carcinoma who had a fourteen months complete response to RAD001 [144]. It would be interesting to further test whether such mutations can be detected in liquid biopsies, which would provide an easy follow-up [145]. In addition, to date, no endothelial specific biomarker exists that specifically predicts anti-angiogenic response to mTOR inhibitors. Nevertheless, inhibition of endothelial Akt signaling has been identified as an important process responsible for the anti-angiogenic effects of rapalogs [146]. Indeed, high levels of endothelial Akt activity is associated with reduced anti-cancer effects of rapamycin.
8. Conclusions
mTOR inhibitors delay tumor progression in part by reducing tumor angiogenesis. This effect is, however, limited, as resistance mechanisms developed by cancer cells assure tumor blood supply despite mTOR inhibition. Thus, identification of these mechanisms is warranted to develop therapeutic strategies that may increase the efficacy of mTOR inhibitors. In this context, combinatory strategies have demonstrated interesting efficacy in pre-clinical studies. Translating these observations into clinical trials might, however, be associated with significant toxicity. In addition, mTOR inhibitors are able to normalize tumor blood vessels, suggesting a potential use as neo-adjuvant therapy prior to chemo- or radiotherapy. The precise settings in which mTOR inhibitors provide vasculature normalization effects need to be fully characterized. Finally, emerging evidence suggests that mTOR inhibitors might influence endothelial functions that participate in the tumor immune response. Future investigations are necessary to clarify this interrelation.
Acknowledgments
This work was supported by the Swiss National Science Foundation (310030_160125).
Author Contributions
Seraina Faes designed the review and drafted the manuscript. Tania Santoro drafted the manuscript. Nicolas Demartines and Olivier Dormond designed the review and revised the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Saxton, R.A.; Sabatini, D.M. Mtor signaling in growth, metabolism, and disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Shimobayashi, M.; Hall, M.N. Making new contacts: The mtor network in metabolism and signalling crosstalk. Nat. Rev. Mol. Cell Biol. 2014, 15, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. Regulation of mtorc1 and its impact on gene expression at a glance. J. Cell Sci. 2013, 126, 1713–1719. [Google Scholar] [CrossRef] [PubMed]
- Gaubitz, C.; Prouteau, M.; Kusmider, B.; Loewith, R. Torc2 structure and function. Trends Biochem. Sci. 2016, 41, 532–545. [Google Scholar] [CrossRef] [PubMed]
- Oh, W.J.; Jacinto, E. Mtor complex 2 signaling and functions. Cell Cycle 2011, 10, 2305–2316. [Google Scholar] [CrossRef] [PubMed]
- Grabiner, B.C.; Nardi, V.; Birsoy, K.; Possemato, R.; Shen, K.; Sinha, S.; Jordan, A.; Beck, A.H.; Sabatini, D.M. A diverse array of cancer-associated mtor mutations are hyperactivating and can predict rapamycin sensitivity. Cancer Discov. 2014, 4, 554–563. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Montalto, G.; Cervello, M.; Nicoletti, F.; Fagone, P.; Malaponte, G.; Mazzarino, M.C.; et al. Mutations and deregulation of RAS/RAF/MEK/ERK and PI3K/PTEN/AKT/mTOR cascades which alter therapy response. Oncotarget 2012, 3, 954–987. [Google Scholar] [CrossRef] [PubMed]
- Dormond-Meuwly, A.; Dufour, M.; Demartines, N.; Dormond, O. Mtor inhibition and the tumor vasculature. Current Angiogenesis 2012, 1, 11–19. [Google Scholar] [CrossRef]
- Xie, J.; Wang, X.; Proud, C.G. Mtor inhibitors in cancer therapy. F1000Res 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Zaytseva, Y.Y.; Valentino, J.D.; Gulhati, P.; Evers, B.M. Mtor inhibitors in cancer therapy. Cancer Lett. 2012, 319, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Thoreen, C.C.; Kang, S.A.; Chang, J.W.; Liu, Q.; Zhang, J.; Gao, Y.; Reichling, L.J.; Sim, T.; Sabatini, D.M.; Gray, N.S. An atp-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mtorc1. J. Biol. Chem. 2009, 284, 8023–8032. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mtorc2 assembly and akt/pkb. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, D.; Colombi, M.; Moroni, C.; Hall, M.N. Rapamycin passes the torch: A new generation of mtor inhibitors. Nat. Rev. Drug Discov. 2011, 10, 868–880. [Google Scholar] [CrossRef] [PubMed]
- Rodrik-Outmezguine, V.S.; Okaniwa, M.; Yao, Z.; Novotny, C.J.; McWhirter, C.; Banaji, A.; Won, H.; Wong, W.; Berger, M.; de Stanchina, E.; et al. Overcoming mtor resistance mutations with a new-generation mtor inhibitor. Nature 2016, 534, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Douros, J.; Suffness, M. New antitumor substances of natural origin. Cancer Treat. Rev. 1981, 8, 63–87. [Google Scholar] [CrossRef]
- Populo, H.; Lopes, J.M.; Soares, P. The mtor signalling pathway in human cancer. Int. J. Mol. Sci. 2012, 13, 1886–1918. [Google Scholar] [CrossRef] [PubMed]
- Dufour, M.; Dormond-Meuwly, A.; Demartines, N.; Dormond, O. Targeting the mammalian target of rapamycin (mtor) in cancer therapy: Lessons from past and future perspectives. Cancers 2011, 3, 2478–2500. [Google Scholar] [CrossRef] [PubMed]
- Hudes, G.; Carducci, M.; Tomczak, P.; Dutcher, J.; Figlin, R.; Kapoor, A.; Staroslawska, E.; Sosman, J.; McDermott, D.; Bodrogi, I.; et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 2271–2281. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.C.; Shah, M.H.; Ito, T.; Bohas, C.L.; Wolin, E.M.; Van Cutsem, E.; Hobday, T.J.; Okusaka, T.; Capdevila, J.; de Vries, E.G.; et al. Everolimus for advanced pancreatic neuroendocrine tumors. N. Engl. J. Med. 2011, 364, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Campone, M.; Piccart, M.; Burris, H.A., 3rd; Rugo, H.S.; Sahmoud, T.; Noguchi, S.; Gnant, M.; Pritchard, K.I.; Lebrun, F.; et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N. Engl. J. Med. 2012, 366, 520–529. [Google Scholar] [CrossRef] [PubMed]
- Faes, S.; Demartines, N.; Dormond, O. Resistance to mtorc1 inhibitors in cancer therapy: From kinase mutations to intratumoral heterogeneity of kinase activity. Oxid. Med. Cell. Longev. 2017, 2017, 1726078. [Google Scholar] [CrossRef] [PubMed]
- Potente, M.; Gerhardt, H.; Carmeliet, P. Basic and therapeutic aspects of angiogenesis. Cell 2011, 146, 873–887. [Google Scholar] [CrossRef] [PubMed]
- De Palma, M.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.R.; Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef] [PubMed]
- Dey, N.; De, P.; Brian, L.J. Evading anti-angiogenic therapy: Resistance to anti-angiogenic therapy in solid tumors. Am. J. Transl. Res. 2015, 7, 1675–1698. [Google Scholar] [CrossRef] [PubMed]
- Weis, S.M.; Cheresh, D.A. Tumor angiogenesis: Molecular pathways and therapeutic targets. Nat. Med. 2011, 17, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Adamis, A.P. Ten years of anti-vascular endothelial growth factor therapy. Nat. Rev. Drug Discov. 2016, 15, 385–403. [Google Scholar] [CrossRef] [PubMed]
- Akselband, Y.; Harding, M.W.; Nelson, P.A. Rapamycin inhibits spontaneous and fibroblast growth factor beta-stimulated proliferation of endothelial cells and fibroblasts. Transplant. Proc. 1991, 23, 2833–2836. [Google Scholar] [PubMed]
- Mohacsi, P.J.; Tuller, D.; Hulliger, B.; Wijngaard, P.L. Different inhibitory effects of immunosuppressive drugs on human and rat aortic smooth muscle and endothelial cell proliferation stimulated by platelet-derived growth factor or endothelial cell growth factor. J. Heart Lung Transplant. 1997, 16, 484–492. [Google Scholar] [PubMed]
- Yu, Y.; Sato, J.D. Map kinases, phosphatidylinositol 3-kinase, and p70 s6 kinase mediate the mitogenic response of human endothelial cells to vascular endothelial growth factor. J. Cell. Physiol. 1999, 178, 235–246. [Google Scholar] [CrossRef]
- Vinals, F.; Chambard, J.C.; Pouyssegur, J. P70 s6 kinase-mediated protein synthesis is a critical step for vascular endothelial cell proliferation. J. Biol. Chem. 1999, 274, 26776–26782. [Google Scholar] [CrossRef] [PubMed]
- Marimpietri, D.; Nico, B.; Vacca, A.; Mangieri, D.; Catarsi, P.; Ponzoni, M.; Ribatti, D. Synergistic inhibition of human neuroblastoma-related angiogenesis by vinblastine and rapamycin. Oncogene 2005, 24, 6785–6795. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Zhao, Y.; Yu, L.; Xu, S.; Fu, G. Microrna-21 mediates the rapamycin-induced suppression of endothelial proliferation and migration. FEBS Lett. 2013, 587, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Humar, R.; Kiefer, F.N.; Berns, H.; Resink, T.J.; Battegay, E.J. Hypoxia enhances vascular cell proliferation and angiogenesis in vitro via rapamycin (mtor)-dependent signaling. FASEB J. 2002, 16, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Bruns, C.J.; Koehl, G.E.; Guba, M.; Yezhelyev, M.; Steinbauer, M.; Seeliger, H.; Schwend, A.; Hoehn, A.; Jauch, K.W.; Geissler, E.K. Rapamycin-induced endothelial cell death and tumor vessel thrombosis potentiate cytotoxic therapy against pancreatic cancer. Clin. Cancer Res. 2004, 10, 2109–2119. [Google Scholar] [CrossRef] [PubMed]
- Barilli, A.; Visigalli, R.; Sala, R.; Gazzola, G.C.; Parolari, A.; Tremoli, E.; Bonomini, S.; Simon, A.; Closs, E.I.; Dall’Asta, V.; et al. In human endothelial cells rapamycin causes mtorc2 inhibition and impairs cell viability and function. Cardiovasc. Res. 2008, 78, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Dormond, O.; Madsen, J.C.; Briscoe, D.M. The effects of mtor-akt interactions on anti-apoptotic signaling in vascular endothelial cells. J. Biol. Chem. 2007, 282, 23679–23686. [Google Scholar] [CrossRef] [PubMed]
- Matter, C.M.; Rozenberg, I.; Jaschko, A.; Greutert, H.; Kurz, D.J.; Wnendt, S.; Kuttler, B.; Joch, H.; Grunenfelder, J.; Zund, G.; et al. Effects of tacrolimus or sirolimus on proliferation of vascular smooth muscle and endothelial cells. J. Cardiovasc. Pharmacol. 2006, 48, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Moss, S.C.; Lightell, D.J., Jr.; Marx, S.O.; Marks, A.R.; Woods, T.C. Rapamycin regulates endothelial cell migration through regulation of the cyclin-dependent kinase inhibitor p27kip1. J. Biol. Chem. 2010, 285, 11991–11997. [Google Scholar] [CrossRef] [PubMed]
- Guba, M.; von Breitenbuch, P.; Steinbauer, M.; Koehl, G.; Flegel, S.; Hornung, M.; Bruns, C.J.; Zuelke, C.; Farkas, S.; Anthuber, M.; et al. Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: Involvement of vascular endothelial growth factor. Nat. Med. 2002, 8, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, E.T.; Cao, C.; Niermann, K.; Mu, Y.; Zeng, F.; Hallahan, D.E.; Lu, B. Enhanced radiation damage of tumor vasculature by mtor inhibitors. Oncogene 2005, 24, 5414–5422. [Google Scholar] [CrossRef] [PubMed]
- Dormond-Meuwly, A.; Roulin, D.; Dufour, M.; Benoit, M.; Demartines, N.; Dormond, O. The inhibition of mapk potentiates the anti-angiogenic efficacy of mtor inhibitors. Biochem. Biophys. Res. Commun. 2011, 407, 714–719. [Google Scholar] [CrossRef] [PubMed]
- Del Bufalo, D.; Ciuffreda, L.; Trisciuoglio, D.; Desideri, M.; Cognetti, F.; Zupi, G.; Milella, M. Antiangiogenic potential of the mammalian target of rapamycin inhibitor temsirolimus. Cancer Res. 2006, 66, 5549–5554. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Shen, N.; Mendoza, A.; Khanna, C.; Helman, L.J. Cci-779 inhibits rhabdomyosarcoma xenograft growth by an antiangiogenic mechanism linked to the targeting of mTOR/HIF-1ALPHA/VEGF signaling. Neoplasia 2006, 8, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Mabuchi, S.; Altomare, D.A.; Connolly, D.C.; Klein-Szanto, A.; Litwin, S.; Hoelzle, M.K.; Hensley, H.H.; Hamilton, T.C.; Testa, J.R. Rad001 (everolimus) delays tumor onset and progression in a transgenic mouse model of ovarian cancer. Cancer Res. 2007, 67, 2408–2413. [Google Scholar] [CrossRef] [PubMed]
- Huynh, H.; Chow, K.H.; Soo, K.C.; Toh, H.C.; Choo, S.P.; Foo, K.F.; Poon, D.; Ngo, V.C.; Tran, E. Rad001 (everolimus) inhibits tumour growth in xenograft models of human hepatocellular carcinoma. J. Cell. Mol. Med. 2009, 13, 1371–1380. [Google Scholar] [CrossRef] [PubMed]
- Lane, H.A.; Wood, J.M.; McSheehy, P.M.; Allegrini, P.R.; Boulay, A.; Brueggen, J.; Littlewood-Evans, A.; Maira, S.M.; Martiny-Baron, G.; Schnell, C.R.; et al. Mtor inhibitor rad001 (everolimus) has antiangiogenic/vascular properties distinct from a VEGFR tyrosine kinase inhibitor. Clin. Cancer Res. 2009, 15, 1612–1622. [Google Scholar] [CrossRef] [PubMed]
- Guimaraes, A.R.; Ross, R.; Figuereido, J.L.; Waterman, P.; Weissleder, R. Mri with magnetic nanoparticles monitors downstream anti-angiogenic effects of mtor inhibition. Mol. Imaging Biol. 2011, 13, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Shi, W.Y.; Wu, Z.Y.; Varna, M.; Wang, A.H.; Zhou, L.; Chen, L.; Shen, Z.X.; Lu, H.; Zhao, W.L.; et al. Cytostatic and anti-angiogenic effects of temsirolimus in refractory mantle cell lymphoma. J. Hematol. Oncol. 2010, 3, 30. [Google Scholar] [CrossRef] [PubMed]
- Schnell, C.R.; Stauffer, F.; Allegrini, P.R.; O’Reilly, T.; McSheehy, P.M.; Dartois, C.; Stumm, M.; Cozens, R.; Littlewood-Evans, A.; Garcia-Echeverria, C.; et al. Effects of the dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor nvp-bez235 on the tumor vasculature: Implications for clinical imaging. Cancer Res. 2008, 68, 6598–6607. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.C.; Cohen, M.B.; Panka, D.J.; Collins, M.; Ghebremichael, M.; Atkins, M.B.; Signoretti, S.; Mier, J.W. The efficacy of the novel dual pi3-kinase/mtor inhibitor nvp-bez235 compared with rapamycin in renal cell carcinoma. Clin. Cancer Res. 2010, 16, 3628–3638. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.J.; Koul, D.; LaFortune, T.; Tiao, N.; Shen, R.J.; Maira, S.M.; Garcia-Echevrria, C.; Yung, W.K. Nvp-bez235, a novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor, elicits multifaceted antitumor activities in human gliomas. Mol. Cancer Ther. 2009, 8, 2204–2210. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Mao, J.H.; Qian, L.; Zhu, H.; Gu, D.H.; Pan, X.D.; Yi, F.; Ji, D.M. Pre-clinical evaluation of azd-2014, a novel mtorc1/2 dual inhibitor, against renal cell carcinoma. Cancer Lett. 2015, 357, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Xue, Q.; Hopkins, B.; Perruzzi, C.; Udayakumar, D.; Sherris, D.; Benjamin, L.E. Palomid 529, a novel small-molecule drug, is a torc1/torc2 inhibitor that reduces tumor growth, tumor angiogenesis, and vascular permeability. Cancer Res. 2008, 68, 9551–9557. [Google Scholar] [CrossRef] [PubMed]
- Falcon, B.L.; Barr, S.; Gokhale, P.C.; Chou, J.; Fogarty, J.; Depeille, P.; Miglarese, M.; Epstein, D.M.; McDonald, D.M. Reduced vegf production, angiogenesis, and vascular regrowth contribute to the antitumor properties of dual mtorc1/mtorc2 inhibitors. Cancer Res. 2011, 71, 1573–1583. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, X.; Qin, L.; Xu, T.; Zhu, Z.; Zhong, S.; Zhang, M.; Shen, Z. The dual mtorc1 and mtorc2 inhibitor pp242 shows strong antitumor activity in a pheochromocytoma pc12 cell tumor model. Urology 2015, 85, 273.e1–273.e7. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Qin, Y.; Huang, J.; Qin, J.; Gu, J.; Zhu, H.; Liu, H.; Cai, Y.; Wu, X.; Feng, J. Effect of rapamycin-induced tumor vessel thrombosis combined with docetaxel in non-small-cell lung cancer. Anticancer Drugs 2013, 24, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Chen, S.; Liu, F.; Wu, H.; McHugh, J.; Bergin, I.L.; Gupta, A.; Adams, D.; Guan, J.L. Constitutive activation of mtorc1 in endothelial cells leads to the development and progression of lymphangiosarcoma through vegf autocrine signaling. Cancer Cell 2015, 28, 758–772. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Amato, K.R.; Song, W.; Youngblood, V.; Lee, K.; Boothby, M.; Brantley-Sieders, D.M.; Chen, J. Regulation of endothelial cell proliferation and vascular assembly through distinct mtorc2 signaling pathways. Mol. Cell. Biol. 2015, 35, 1299–1313. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, G.; Yu, K.; Jiang, Z.; Chung, A.; Yao, J.; Ha, C.; Toy, K.; Soriano, R.; Haley, B.; Blackwood, E.; et al. Phosphoproteomic analysis implicates the mtorc2-foxo1 axis in vegf signaling and feedback activation of receptor tyrosine kinases. Sci. Signal. 2013, 6, ra25. [Google Scholar] [CrossRef] [PubMed]
- Dada, S.; Demartines, N.; Dormond, O. Mtorc2 regulates pge2-mediated endothelial cell survival and migration. Biochem. Biophys. Res. Commun. 2008, 372, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Farhan, M.A.; Carmine-Simmen, K.; Lewis, J.D.; Moore, R.B.; Murray, A.G. Endothelial cell mtor complex-2 regulates sprouting angiogenesis. PLoS ONE 2015, 10, e0135245. [Google Scholar] [CrossRef] [PubMed]
- Hudson, C.C.; Liu, M.; Chiang, G.G.; Otterness, D.M.; Loomis, D.C.; Kaper, F.; Giaccia, A.J.; Abraham, R.T. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol. Cell. Biol. 2002, 22, 7004–7014. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Chiles, K.; Feldser, D.; Laughner, E.; Hanrahan, C.; Georgescu, M.M.; Simons, J.W.; Semenza, G.L. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/pten/akt/frap pathway in human prostate cancer cells: Implications for tumor angiogenesis and therapeutics. Cancer Res. 2000, 60, 1541–1545. [Google Scholar] [PubMed]
- Lee, D.F.; Kuo, H.P.; Chen, C.T.; Hsu, J.M.; Chou, C.K.; Wei, Y.; Sun, H.L.; Li, L.Y.; Ping, B.; Huang, W.C.; et al. Ikk beta suppression of tsc1 links inflammation and tumor angiogenesis via the mtor pathway. Cell 2007, 130, 440–455. [Google Scholar] [CrossRef] [PubMed]
- Dormond, O.; Contreras, A.G.; Meijer, E.; Datta, D.; Flynn, E.; Pal, S.; Briscoe, D.M. Cd40-induced signaling in human endothelial cells results in mtorc2- and akt-dependent expression of vascular endothelial growth factor in vitro and in vivo. J. Immunol. 2008, 181, 8088–8095. [Google Scholar] [CrossRef] [PubMed]
- Dey, N.; Sun, Y.; Carlson, J.H.; Wu, H.; Lin, X.; Leyland-Jones, B.; De, P. Anti-tumor efficacy of bez235 is complemented by its anti-angiogenic effects via downregulation of Pi3k-mTOR-HIF1alpha signaling in HER2-defined breast cancers. Am. J. Cancer Res. 2016, 6, 714–746. [Google Scholar] [PubMed]
- Laughner, E.; Taghavi, P.; Chiles, K.; Mahon, P.C.; Semenza, G.L. Her2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (hif-1alpha) synthesis: Novel mechanism for hif-1-mediated vascular endothelial growth factor expression. Mol. Cell. Biol. 2001, 21, 3995–4004. [Google Scholar] [CrossRef] [PubMed]
- Duvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a metabolic gene regulatory network downstream of mtor complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Tandon, P.; Gallo, C.A.; Khatri, S.; Barger, J.F.; Yepiskoposyan, H.; Plas, D.R. Requirement for ribosomal protein s6 kinase 1 to mediate glycolysis and apoptosis resistance induced by pten deficiency. Proc. Natl. Acad. Sci. USA 2011, 108, 2361–2365. [Google Scholar] [CrossRef] [PubMed]
- Land, S.C.; Tee, A.R. Hypoxia-inducible factor 1alpha is regulated by the mammalian target of rapamycin (mtor) via an mtor signaling motif. J. Biol. Chem. 2007, 282, 20534–20543. [Google Scholar] [CrossRef] [PubMed]
- Dodd, K.M.; Yang, J.; Shen, M.H.; Sampson, J.R.; Tee, A.R. Mtorc1 drives hif-1alpha and vegf-a signalling via multiple mechanisms involving 4e-bp1, s6k1 and stat3. Oncogene 2015, 34, 2239–2250. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Squadrito, M.L.; De Palma, M. Macrophage regulation of tumor angiogenesis: Implications for cancer therapy. Mol. Aspects Med. 2011, 32, 123–145. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Ma, T.; Shen, X.N.; Xia, X.F.; Xu, G.D.; Bai, X.L.; Liang, T.B. Macrophage-induced tumor angiogenesis is regulated by the tsc2-mtor pathway. Cancer Res. 2012, 72, 1363–1372. [Google Scholar] [CrossRef] [PubMed]
- Mercalli, A.; Calavita, I.; Dugnani, E.; Citro, A.; Cantarelli, E.; Nano, R.; Melzi, R.; Maffi, P.; Secchi, A.; Sordi, V.; et al. Rapamycin unbalances the polarization of human macrophages to m1. Immunology 2013, 140, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Paulis, Y.W.; Soetekouw, P.M.; Verheul, H.M.; Tjan-Heijnen, V.C.; Griffioen, A.W. Signalling pathways in vasculogenic mimicry. Biochim. Biophys. Acta 2010, 1806, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Dunleavey, J.M.; Dudley, A.C. Vascular mimicry: Concepts and implications for anti-angiogenic therapy. Curr. Angiogenes 2012, 1, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Zhang, S.; Zhao, X.; Zhang, W.; Hao, X. Vasculogenic mimicry is associated with poor survival in patients with mesothelial sarcomas and alveolar rhabdomyosarcomas. Int. J. Oncol. 2004, 25, 1609–1614. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Ke, Y.; Sun, X.; Yu, L.; Yang, Z.; Zhang, Y.; Du, M.; Wang, J.; Liu, X.; Huang, S. Mammalian target of rapamycin signaling is involved in the vasculogenic mimicry of glioma via hypoxia-inducible factor-1alpha. Oncol. Rep. 2014, 32, 1973–1980. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Feng, Y.J.; Yao, L.Q.; Cheng, M.J.; Xu, C.J.; Huang, Y.; Zhao, Y.Q.; Jiang, H. Plasticity of ovarian cancer cell skov3ip and vasculogenic mimicry in vivo. Int. J. Gynecol. Cancer 2008, 18, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.M.; Hurwitz, H.I. Understanding and targeting resistance to anti-angiogenic therapies. J. Gastrointest. Oncol. 2013, 4, 253–263. [Google Scholar] [PubMed]
- Bergers, G.; Hanahan, D. Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 2008, 8, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K.; Carmeliet, P. Snapshot: Tumor angiogenesis. Cell 2012, 149, 1408. [Google Scholar] [CrossRef] [PubMed]
- Birle, D.C.; Hedley, D.W. Signaling interactions of rapamycin combined with erlotinib in cervical carcinoma xenografts. Mol. Cancer Ther. 2006, 5, 2494–2502. [Google Scholar] [CrossRef] [PubMed]
- Mosley, J.D.; Poirier, J.T.; Seachrist, D.D.; Landis, M.D.; Keri, R.A. Rapamycin inhibits multiple stages of c-Neu/Erbb2 induced tumor progression in a transgenic mouse model of HER2-positive breast cancer. Mol. Cancer Ther. 2007, 6, 2188–2197. [Google Scholar] [CrossRef] [PubMed]
- Cao, P.; Maira, S.M.; Garcia-Echeverria, C.; Hedley, D.W. Activity of a novel, dual pi3-kinase/mtor inhibitor nvp-bez235 against primary human pancreatic cancers grown as orthotopic xenografts. Br. J. Cancer 2009, 100, 1267–1276. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Berel, D.; Wang, Y.; Li, P.; Bhowmick, N.A.; Figlin, R.A.; Kim, H.L. A comparison of ku0063794, a dual mtorc1 and mtorc2 inhibitor, and temsirolimus in preclinical renal cell carcinoma models. PLoS ONE 2013, 8, e54918. [Google Scholar] [CrossRef] [PubMed]
- Walpen, T.; Kalus, I.; Schwaller, J.; Peier, M.A.; Battegay, E.J.; Humar, R. Nuclear PIM1 confers resistance to rapamycin-impaired endothelial proliferation. Biochem. Biophys. Res. Commun. 2012, 429, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Semela, D.; Piguet, A.C.; Kolev, M.; Schmitter, K.; Hlushchuk, R.; Djonov, V.; Stoupis, C.; Dufour, J.F. Vascular remodeling and antitumoral effects of mTOR inhibition in a rat model of hepatocellular carcinoma. J. Hepatol. 2007, 46, 840–848. [Google Scholar] [CrossRef] [PubMed]
- Manegold, P.C.; Paringer, C.; Kulka, U.; Krimmel, K.; Eichhorn, M.E.; Wilkowski, R.; Jauch, K.W.; Guba, M.; Bruns, C.J. Antiangiogenic therapy with mammalian target of rapamycin inhibitor rad001 (everolimus) increases radiosensitivity in solid cancer. Clin. Cancer Res. 2008, 14, 892–900. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.D.; Spalding, A.C.; Somnay, Y.R.; Markwart, S.; Ray, M.E.; Hamstra, D.A. Inhibition of mtor radiosensitizes soft tissue sarcoma and tumor vasculature. Clin. Cancer Res. 2009, 15, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.W.; Moretti, L.; Mitchell, L.R.; Jung, D.K.; Lu, B. Combined bcl-2/mammalian target of rapamycin inhibition leads to enhanced radiosensitization via induction of apoptosis and autophagy in non-small cell lung tumor xenograft model. Clin. Cancer Res. 2009, 15, 6096–6105. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, Y.; Mori, M.; Kitahara, S.; Fukumoto, M.; Ezaki, T.; Mori, S.; Echigo, S.; Ohkubo, Y.; Fukumoto, M. Targeting of tumor endothelial cells combining 2 gy/day of x-ray with everolimus is the effective modality for overcoming clinically relevant radioresistant tumors. Cancer Med. 2014, 3, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Fokas, E.; Yoshimura, M.; Prevo, R.; Higgins, G.; Hackl, W.; Maira, S.M.; Bernhard, E.J.; McKenna, W.G.; Muschel, R.J. Nvp-bez235 and nvp-bgt226, dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitors, enhance tumor and endothelial cell radiosensitivity. Radiat. Oncol. 2012, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Piguet, A.C.; Semela, D.; Keogh, A.; Wilkens, L.; Stroka, D.; Stoupis, C.; St-Pierre, M.V.; Dufour, J.F. Inhibition of mtor in combination with doxorubicin in an experimental model of hepatocellular carcinoma. J. Hepatol. 2008, 49, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Huynh, H.; Chow, P.K.; Palanisamy, N.; Salto-Tellez, M.; Goh, B.C.; Lee, C.K.; Somani, A.; Lee, H.S.; Kalpana, R.; Yu, K.; et al. Bevacizumab and rapamycin induce growth suppression in mouse models of hepatocellular carcinoma. J. Hepatol. 2008, 49, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Hainsworth, J.D.; Spigel, D.R.; Burris, H.A., 3rd; Waterhouse, D.; Clark, B.L.; Whorf, R. Phase ii trial of bevacizumab and everolimus in patients with advanced renal cell carcinoma. J. Clin. Oncol. 2010, 28, 2131–2136. [Google Scholar] [CrossRef] [PubMed]
- Hobday, T.J.; Qin, R.; Reidy-Lagunes, D.; Moore, M.J.; Strosberg, J.; Kaubisch, A.; Shah, M.; Kindler, H.L.; Lenz, H.J.; Chen, H.; et al. Multicenter phase ii trial of temsirolimus and bevacizumab in pancreatic neuroendocrine tumors. J. Clin. Oncol. 2015, 33, 1551–1556. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Manola, J.B.; Pins, M.; McDermott, D.F.; Atkins, M.B.; Dutcher, J.J.; George, D.J.; Margolin, K.A.; DiPaola, R.S. Best: A randomized phase ii study of vascular endothelial growth factor, raf kinase, and mammalian target of rapamycin combination targeted therapy with bevacizumab, sorafenib, and temsirolimus in advanced renal cell carcinoma—A trial of the ecog-acrin cancer research group (e2804). J. Clin. Oncol. 2015, 33, 2384–2391. [Google Scholar] [PubMed]
- Pignochino, Y.; Dell’Aglio, C.; Basirico, M.; Capozzi, F.; Soster, M.; Marchio, S.; Bruno, S.; Gammaitoni, L.; Sangiolo, D.; Torchiaro, E.; et al. The combination of sorafenib and everolimus abrogates mtorc1 and mtorc2 upregulation in osteosarcoma preclinical models. Clin. Cancer Res. 2013, 19, 2117–2131. [Google Scholar] [CrossRef] [PubMed]
- Piguet, A.C.; Saar, B.; Hlushchuk, R.; St-Pierre, M.V.; McSheehy, P.M.; Radojevic, V.; Afthinos, M.; Terracciano, L.; Djonov, V.; Dufour, J.F. Everolimus augments the effects of sorafenib in a syngeneic orthotopic model of hepatocellular carcinoma. Mol. Cancer Ther. 2011, 10, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Tong, L.J.; Ding, J.; Meng, L.H. Systematic combination screening reveals synergism between rapamycin and sunitinib against human lung cancer. Cancer Lett. 2014, 342, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.L.; Wong, C.H.; Lau, C.P.; Zhou, Q.; Hui, C.W.; Lui, V.W.; Ma, B.B.; Chan, A.T.; Yeo, W. Preclinical evaluation of combined tki-258 and rad001 in hepatocellular carcinoma. Cancer Chemother. Pharmacol. 2013, 71, 1417–1425. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.A.; Fox, P.S.; Papadopoulos, N.E.; Bedikian, A.Y.; Hwu, W.J.; Lazar, A.J.; Prieto, V.G.; Culotta, K.S.; Madden, T.L.; Xu, Q.; et al. Phase i study of the combination of sorafenib and temsirolimus in patients with metastatic melanoma. Clin. Cancer Res. 2012, 18, 1120–1128. [Google Scholar] [CrossRef] [PubMed]
- Huynh, H. Azd6244 (arry-142886) enhances the antitumor activity of rapamycin in mouse models of human hepatocellular carcinoma. Cancer 2010, 116, 1315–1325. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.W.; Bendell, J.C.; Papadopoulos, K.P.; Burris, H.A., 3rd; Patnaik, A.; Jones, S.F.; Rasco, D.; Cox, D.S.; Durante, M.; Bellew, K.M.; et al. A phase ib trial of the oral mek inhibitor trametinib (gsk1120212) in combination with everolimus in patients with advanced solid tumors. Ann. Oncol. 2015, 26, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Harkavy, B.; Shen, N.; Grohar, P.; Helman, L.J. Rapamycin induces feedback activation of akt signaling through an Igf-1r-dependent mechanism. Oncogene 2007, 26, 1932–1940. [Google Scholar] [CrossRef] [PubMed]
- Kurmasheva, R.T.; Dudkin, L.; Billups, C.; Debelenko, L.V.; Morton, C.L.; Houghton, P.J. The insulin-like growth factor-1 receptor-targeting antibody, cp-751,871, suppresses tumor-derived vegf and synergizes with rapamycin in models of childhood sarcoma. Cancer Res. 2009, 69, 7662–7671. [Google Scholar] [CrossRef] [PubMed]
- Quek, R.; Wang, Q.; Morgan, J.A.; Shapiro, G.I.; Butrynski, J.E.; Ramaiya, N.; Huftalen, T.; Jederlinic, N.; Manola, J.; Wagner, A.J.; et al. Combination mtor and igf-1r inhibition: Phase i trial of everolimus and figitumumab in patients with advanced sarcomas and other solid tumors. Clin. Cancer Res. 2011, 17, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.K.; Tap, W.D.; Qin, L.X.; Livingston, M.B.; Undevia, S.D.; Chmielowski, B.; Agulnik, M.; Schuetze, S.M.; Reed, D.R.; Okuno, S.H.; et al. Cixutumumab and temsirolimus for patients with bone and soft-tissue sarcoma: A multicentre, open-label, phase 2 trial. Lancet Oncol. 2013, 14, 371–382. [Google Scholar] [CrossRef]
- Naing, A.; LoRusso, P.; Fu, S.; Hong, D.S.; Anderson, P.; Benjamin, R.S.; Ludwig, J.; Chen, H.X.; Doyle, L.A.; Kurzrock, R. Insulin growth factor-receptor (IGF-1r) antibody cixutumumab combined with the mtor inhibitor temsirolimus in patients with refractory ewing’s sarcoma family tumors. Clin. Cancer Res. 2012, 18, 2625–2631. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Morales, S.M.; Awada, A.; Blum, J.L.; Tan, A.R.; Ewertz, M.; Cortes, J.; Moy, B.; Ruddy, K.J.; Haddad, T.; et al. A phase ii study of combined ridaforolimus and dalotuzumab compared with exemestane in patients with estrogen receptor-positive breast cancer. Breast Cancer Res. Treat. 2017, 163, 535–544. [Google Scholar] [CrossRef] [PubMed]
- McKeage, M.J.; Baguley, B.C. Disrupting established tumor blood vessels: An emerging therapeutic strategy for cancer. Cancer 2010, 116, 1859–1871. [Google Scholar] [CrossRef] [PubMed]
- Ellis, L.; Shah, P.; Hammers, H.; Lehet, K.; Sotomayor, P.; Azabdaftari, G.; Seshadri, M.; Pili, R. Vascular disruption in combination with mtor inhibition in renal cell carcinoma. Mol. Cancer Ther. 2012, 11, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Kraus, D.; Palasuberniam, P.; Chen, B. Targeting phosphatidylinositol 3-kinase signaling pathway for therapeutic enhancement of vascular-targeted photodynamic therapy. Mol. Cancer Ther. 2017. [Google Scholar] [CrossRef] [PubMed]
- Verheul, H.M.; Salumbides, B.; Van Erp, K.; Hammers, H.; Qian, D.Z.; Sanni, T.; Atadja, P.; Pili, R. Combination strategy targeting the hypoxia inducible factor-1 alpha with mammalian target of rapamycin and histone deacetylase inhibitors. Clin. Cancer Res. 2008, 14, 3589–3597. [Google Scholar] [CrossRef] [PubMed]
- Singleton, P.A.; Mambetsariev, N.; Lennon, F.E.; Mathew, B.; Siegler, J.H.; Moreno-Vinasco, L.; Salgia, R.; Moss, J.; Garcia, J.G. Methylnaltrexone potentiates the anti-angiogenic effects of mtor inhibitors. J. Angiogenes Res. 2010, 2, 5. [Google Scholar] [CrossRef] [PubMed]
- Damiano, V.; Rosa, R.; Formisano, L.; Nappi, L.; Gelardi, T.; Marciano, R.; Cozzolino, I.; Troncone, G.; Agrawal, S.; Veneziani, B.M.; et al. Toll-like receptor 9 agonist imo cooperates with everolimus in renal cell carcinoma by interfering with tumour growth and angiogenesis. Br. J. Cancer 2013, 108, 1616–1623. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, S.; Baluk, P.; Kaidoh, T.; Haskell, A.; Jain, R.K.; McDonald, D.M. Abnormalities in pericytes on blood vessels and endothelial sprouts in tumors. Am. J. Pathol. 2002, 160, 985–1000. [Google Scholar] [CrossRef]
- Baluk, P.; Morikawa, S.; Haskell, A.; Mancuso, M.; McDonald, D.M. Abnormalities of basement membrane on blood vessels and endothelial sprouts in tumors. Am. J. Pathol 2003, 163, 1801–1815. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat. Rev. Drug Discov. 2011, 10, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Normalizing tumor vasculature with anti-angiogenic therapy: A new paradigm for combination therapy. Nat. Med. 2001, 7, 987–989. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Antiangiogenesis strategies revisited: From starving tumors to alleviating hypoxia. Cancer Cell 2014, 26, 605–622. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bindokas, V.; Shen, J.; Fan, H.; Hoffman, R.M.; Xing, H.R. Time-course imaging of therapeutic functional tumor vascular normalization by antiangiogenic agents. Mol. Cancer Ther. 2011, 10, 1173–1184. [Google Scholar] [CrossRef] [PubMed]
- Myers, A.L.; Orr, W.S.; Denbo, J.W.; Ng, C.Y.; Zhou, J.; Spence, Y.; Wu, J.; Davidoff, A.M. Rapamycin-induced tumor vasculature remodeling in rhabdomyosarcoma xenografts increases the effectiveness of adjuvant ionizing radiation. J. Pediatr. Surg. 2012, 47, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Eshleman, J.S.; Carlson, B.L.; Mladek, A.C.; Kastner, B.D.; Shide, K.L.; Sarkaria, J.N. Inhibition of the mammalian target of rapamycin sensitizes u87 xenografts to fractionated radiation therapy. Cancer Res. 2002, 62, 7291–7297. [Google Scholar] [PubMed]
- Wu, L.; Birle, D.C.; Tannock, I.F. Effects of the mammalian target of rapamycin inhibitor cci-779 used alone or with chemotherapy on human prostate cancer cells and xenografts. Cancer Res. 2005, 65, 2825–2831. [Google Scholar] [CrossRef] [PubMed]
- Carman, C.V.; Martinelli, R. T lymphocyte-endothelial interactions: Emerging understanding of trafficking and antigen-specific immunity. Front. Immunol. 2015, 6, 603. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Enis, D.R.; Koh, K.P.; Shiao, S.L.; Pober, J.S. T lymphocyte-endothelial cell interactions. Annu Rev. Immunol. 2004, 22, 683–709. [Google Scholar] [CrossRef] [PubMed]
- Nummer, D.; Suri-Payer, E.; Schmitz-Winnenthal, H.; Bonertz, A.; Galindo, L.; Antolovich, D.; Koch, M.; Buchler, M.; Weitz, J.; Schirrmacher, V.; et al. Role of tumor endothelium in CD4+ CD25+ regulatory T cell infiltration of human pancreatic carcinoma. J. Natl. Cancer Inst. 2007, 99, 1188–1199. [Google Scholar] [CrossRef] [PubMed]
- Motz, G.T.; Santoro, S.P.; Wang, L.P.; Garrabrant, T.; Lastra, R.R.; Hagemann, I.S.; Lal, P.; Feldman, M.D.; Benencia, F.; Coukos, G. Tumor endothelium fasl establishes a selective immune barrier promoting tolerance in tumors. Nat. Med. 2014, 20, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Lanitis, E.; Irving, M.; Coukos, G. Targeting the tumor vasculature to enhance t cell activity. Curr. Opin. Immunol. 2015, 33, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Uldry, E.; Faes, S.; Demartines, N.; Dormond, O. Fine-tuning tumor endothelial cells to selectively kill cancer. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Hendry, S.A.; Farnsworth, R.H.; Solomon, B.; Achen, M.G.; Stacker, S.A.; Fox, S.B. The role of the tumor vasculature in the host immune response: Implications for therapeutic strategies targeting the tumor microenvironment. Front. Immunol. 2016, 7, 621. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yi, T.; Qin, L.; Maldonado, R.A.; von Andrian, U.H.; Kulkarni, S.; Tellides, G.; Pober, J.S. Rapamycin-treated human endothelial cells preferentially activate allogeneic regulatory T cells. J. Clin. Investig. 2013, 123, 1677–1693. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Qin, L.; Manes, T.D.; Kirkiles-Smith, N.C.; Tellides, G.; Pober, J.S. Rapamycin antagonizes tnf induction of vcam-1 on endothelial cells by inhibiting mTORc2. J. Exp. Med. 2014, 211, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Neshat, M.S.; Mellinghoff, I.K.; Tran, C.; Stiles, B.; Thomas, G.; Petersen, R.; Frost, P.; Gibbons, J.J.; Wu, H.; Sawyers, C.L. Enhanced sensitivity of pten-deficient tumors to inhibition of frap/mtor. Proc. Natl. Acad. Sci. USA 2001, 98, 10314–10319. [Google Scholar] [CrossRef] [PubMed]
- Di Nicolantonio, F.; Arena, S.; Tabernero, J.; Grosso, S.; Molinari, F.; Macarulla, T.; Russo, M.; Cancelliere, C.; Zecchin, D.; Mazzucchelli, L.; et al. Deregulation of the pi3k and kras signaling pathways in human cancer cells determines their response to everolimus. J. Clin. Investig. 2010, 120, 2858–2866. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, D.; Boya, P.; Bellet, D.; Faivre, S.; Troalen, F.; Benard, J.; Saulnier, P.; Hopkins-Donaldson, S.; Zangemeister-Wittke, U.; Kroemer, G.; et al. Bcl-2 and CCND1/CDK4 expression levels predict the cellular effects of mtor inhibitors in human ovarian carcinoma. Apoptosis 2004, 9, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Cummings, C.A.; Peters, E.; Lacroix, L.; Andre, F.; Lackner, M.R. The role of next-generation sequencing in enabling personalized oncology therapy. Clin. Transl. Sci. 2016, 9, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Wagle, N.; Grabiner, B.C.; Van Allen, E.M.; Amin-Mansour, A.; Taylor-Weiner, A.; Rosenberg, M.; Gray, N.; Barletta, J.A.; Guo, Y.; Swanson, S.J.; et al. Response and acquired resistance to everolimus in anaplastic thyroid cancer. N. Engl. J. Med. 2014, 371, 1426–1433. [Google Scholar] [CrossRef] [PubMed]
- Wagle, N.; Grabiner, B.C.; Van Allen, E.M.; Hodis, E.; Jacobus, S.; Supko, J.G.; Stewart, M.; Choueiri, T.K.; Gandhi, L.; Cleary, J.M.; et al. Activating mtor mutations in a patient with an extraordinary response on a phase i trial of everolimus and pazopanib. Cancer Discov. 2014, 4, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Phung, T.L.; Eyiah-Mensah, G.; O’Donnell, R.K.; Bieniek, R.; Shechter, S.; Walsh, K.; Kuperwasser, C.; Benjamin, L.E. Endothelial Akt signaling is rate-limiting for rapamycin inhibition of mouse mammary tumor progression. Cancer Res. 2007, 67, 5070–5075. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
mTOR inhibitors. Three different types of mTOR inhibitors have been developed. Rapalogs, such as rapamycin, bind together with FKBP12 to the FRB domain of mTOR and block some functions of mTORC1. Kinase inhibitors of mTOR bind to the kinase domain of mTOR and block both mTORC1 and mTORC2. Rapalinks are composed of rapamycin cross-linked to a kinase inhibitor of mTOR.
Figure 1.
mTOR inhibitors. Three different types of mTOR inhibitors have been developed. Rapalogs, such as rapamycin, bind together with FKBP12 to the FRB domain of mTOR and block some functions of mTORC1. Kinase inhibitors of mTOR bind to the kinase domain of mTOR and block both mTORC1 and mTORC2. Rapalinks are composed of rapamycin cross-linked to a kinase inhibitor of mTOR.
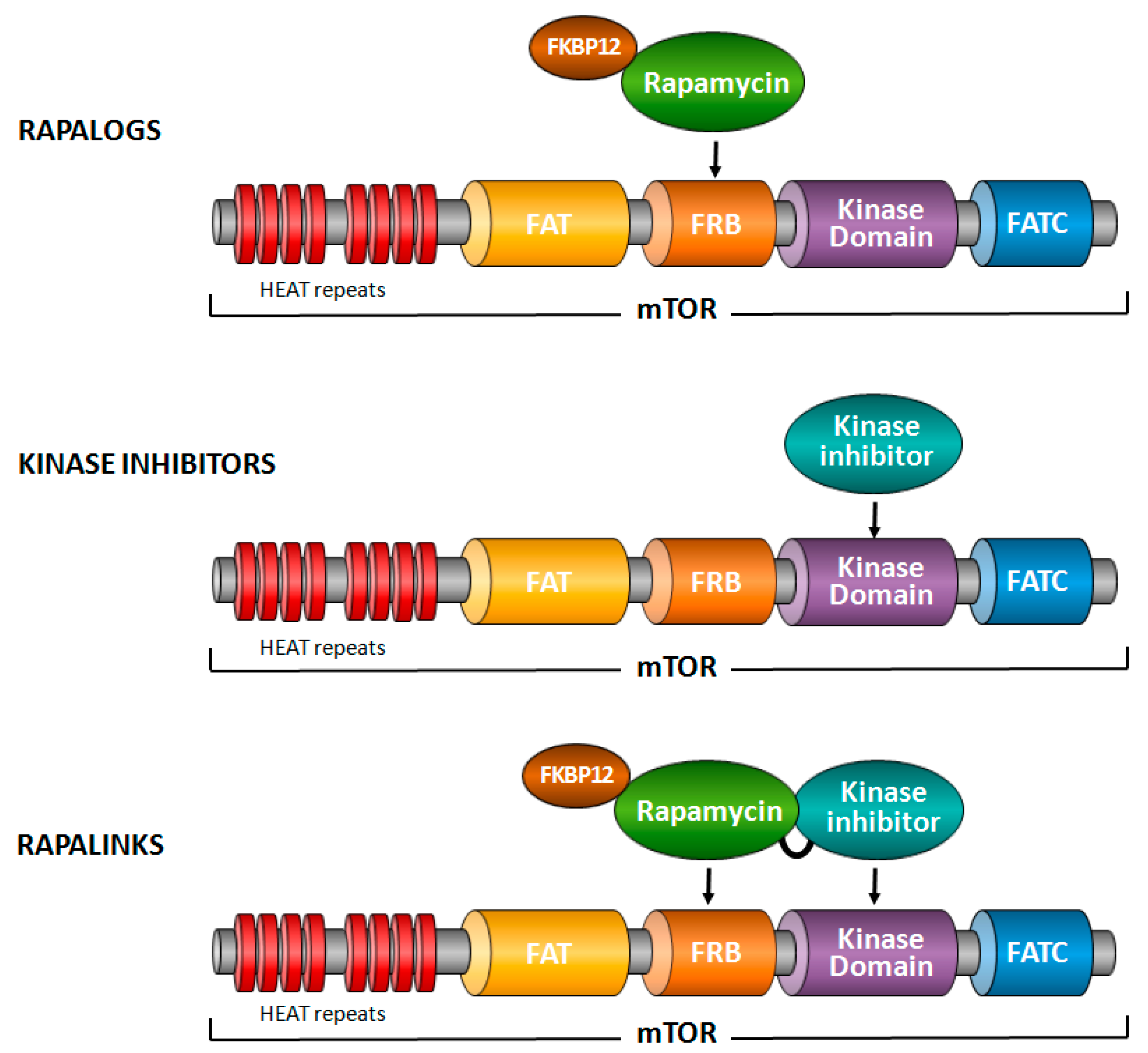
Figure 2.
Mechanisms by which mTOR inhibitors affect tumor angiogenesis. mTOR inhibitors reduce endothelial cell proliferation, survival and migration by blocking endothelial mTOR. In addition, they decrease VEGF production, as mTORC1 is required to stabilize HIF1α during the hypoxic tumor response and is activated by IKKβ in inflammation-mediated angiogenesis. Finally, mTOR inhibitors induce tumor associated macrophage polarization to an anti-angiogenic phenotype.
Figure 2.
Mechanisms by which mTOR inhibitors affect tumor angiogenesis. mTOR inhibitors reduce endothelial cell proliferation, survival and migration by blocking endothelial mTOR. In addition, they decrease VEGF production, as mTORC1 is required to stabilize HIF1α during the hypoxic tumor response and is activated by IKKβ in inflammation-mediated angiogenesis. Finally, mTOR inhibitors induce tumor associated macrophage polarization to an anti-angiogenic phenotype.
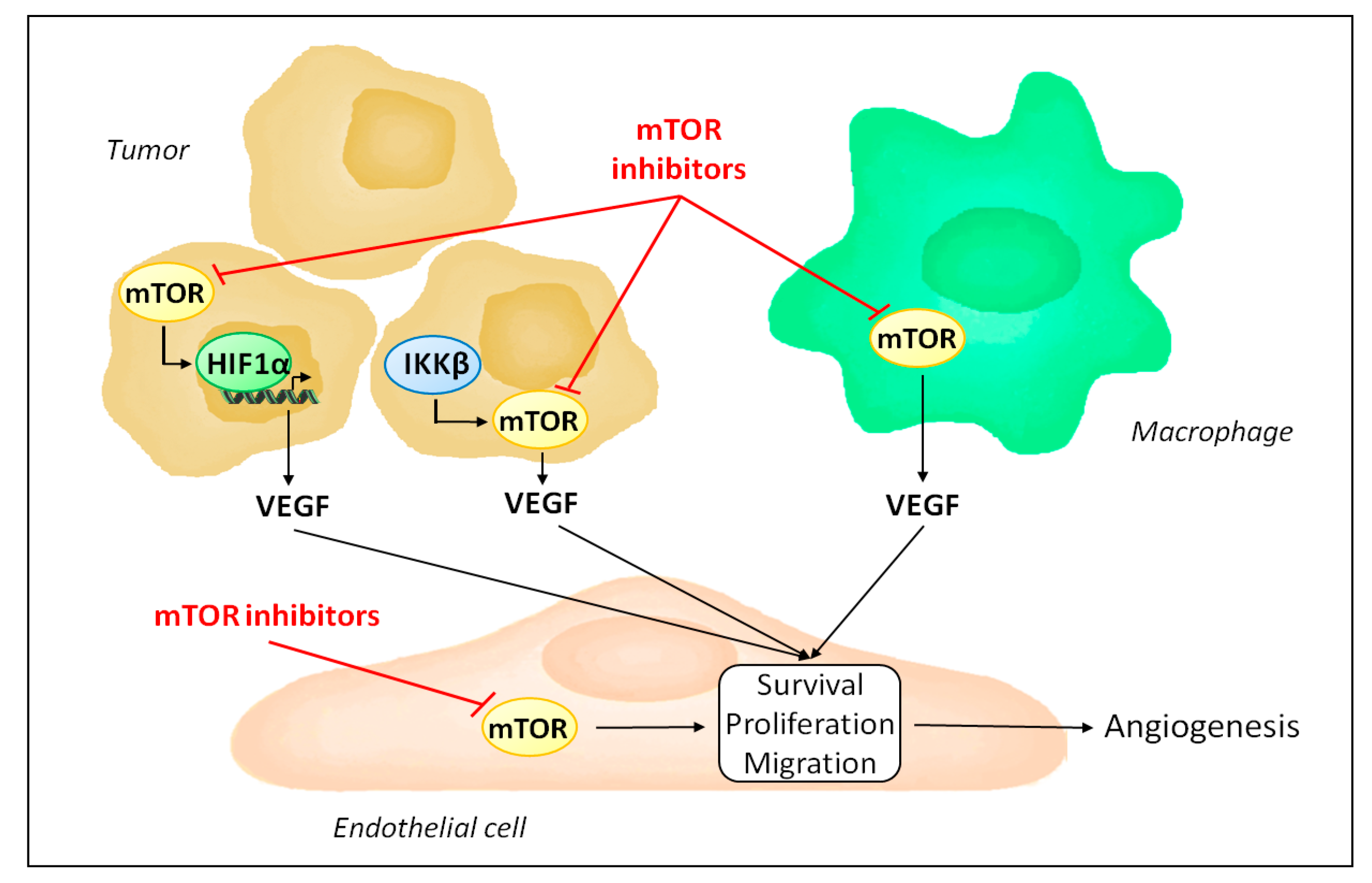
Figure 3.
mTORC1 regulates HIF-1α signaling via different mechanisms. mTORC1 controls HIF-1α mRNA transcription and translation, HIF-1α protein stability and HIF-1α transcriptional activity.
Figure 3.
mTORC1 regulates HIF-1α signaling via different mechanisms. mTORC1 controls HIF-1α mRNA transcription and translation, HIF-1α protein stability and HIF-1α transcriptional activity.

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Faes, S.; Santoro, T.; Demartines, N.; Dormond, O. Evolving Significance and Future Relevance of Anti-Angiogenic Activity of mTOR Inhibitors in Cancer Therapy. Cancers 2017, 9, 152. https://doi.org/10.3390/cancers9110152
AMA Style
Faes S, Santoro T, Demartines N, Dormond O. Evolving Significance and Future Relevance of Anti-Angiogenic Activity of mTOR Inhibitors in Cancer Therapy. Cancers. 2017; 9(11):152. https://doi.org/10.3390/cancers9110152
Chicago/Turabian StyleFaes, Seraina, Tania Santoro, Nicolas Demartines, and Olivier Dormond. 2017. "Evolving Significance and Future Relevance of Anti-Angiogenic Activity of mTOR Inhibitors in Cancer Therapy" Cancers 9, no. 11: 152. https://doi.org/10.3390/cancers9110152
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.