Genomic Destabilization Triggered by Replication Stress during Senescence

1
Division of Carcinogenesis and Cancer Prevention, National Cancer Center Research Institute, 5-1-1 Tsukiji, Chuo-ku, Tokyo 104-0045, Japan
2
Department of Biological Science and Technology, Tokyo University of Science, 6-3-1 Niijuku, Katsushika-ku, Tokyo 125-8585, Japan
3
Department of Biosciences, School of Science, Kitasato University, 1-15-1 Kitasato, Minami-ku, Sagamihara 252-0373, Japan
4
Department of Applied Chemistry, Tokyo University of Science, 1-3 Kagurazaka, Shinjuku-ku, Tokyo 162-8601, Japan
*
Author to whom correspondence should be addressed.
Cancers 2017, 9(11), 159; https://doi.org/10.3390/cancers9110159
Submission received: 25 September 2017
/
Revised: 13 November 2017
/
Accepted: 20 November 2017
/
Published: 21 November 2017
(This article belongs to the Special Issue Histone Modification in Cancer)
Abstract
:Most cancers develop after middle age, and are often associated with multiple mutations and genomic instability, implying that genomic destabilization is critical for age-related tumor development. In this manuscript, we review current knowledge regarding (1) the senescent cellular background, which is associated with a higher risk of genomic destabilization; and (2) the contributions of genomic destabilization to cancer development.
1. Introduction
Most cancers develop after middle age and are associated with various types of genomic instability, either chromosomal instability (CIN) or microsatellite instability (MSI) [1,2,3,4]. Cancer risk increases as a function of age [5,6]. Consistent with this, genomic destabilization is often observed in the senescent cellular state [7,8,9]. Such genomic instability is closely linked to cancer development. For example, mutations in the genes encoding breast cancer susceptibility 1 (BRCA1) and 2 (BRCA2) induce massive CIN due to a deficiency in homologous recombination, resulting in predisposition to cancer [10,11].
After serial proliferation, normal cells generally enter a growth-arrested state such as senescence or quiescence [12,13,14]. In quiescence, which is associated with low levels of H2AX, cells are protected from genomic destabilization [9,15,16]. Cells in this state are abundant in normal organs, and contribute to homeostasis [9]. By contrast, senescent cells accumulate persistent DNA double strand breaks (DSBs), and consequently contain numerous γH2AX foci [17,18]. This cellular state is associated with both aging-related diseases and precancerous states [18]. Senescence was long thought to be a mode of cancer prevention, partly because the tumor-suppressor protein p53 induces senescence when cells are damaged [19,20,21,22]. However, recent findings have revealed that senescence is often closely associated with genome destabilization and elevated cancer risk [23,24,25].
In this manuscript, we review the accumulated knowledge regarding the senescent cellular background associated with genomic destabilization, as well as its contributions to cancer development.
2. Genomic Instabilities in Cancer Cells and Cells Immortalized In Vitro
CIN and MSI are induced in cancer cells in a mutually exclusive manner, determined by mismatch repair (MMR) status (Table 1): MSI is induced in MMR-deficient backgrounds [26,27], whereas CIN is induced in most other cases [2,28]. MSI involves alterations in the lengths of microsatellite fragments, which contain short repetitive sequences (1–6 bases) [29]. CIN encompasses a wide variety of chromosomal abnormalities, including gene amplification [30], chromosomal deletions [31,32], chromosomal rearrangements [33], tetraploidy/aneuploidy [2], and loss of heterozygosity (LOH) [34] (Table 1). Recent work showed that CIN can also involve chromothripsis, i.e., multiple genomic rearrangements induced during a single catastrophic event, or chromoplexy, i.e., a complex DNA rearrangement caused by multiple DNA-strand breaks and their ligation into a new configuration [35,36]. Intriguingly, similar forms of genomic instability are also observed in cells immortalized or transformed in vitro [37,38]. Mouse embryonic fibroblast cells (MEFs) generally immortalize with destabilized genomes and mutations in the ARF/p53 module, whereas MEFs cultured under conditions that enable maintenance of genome stability never immortalize [9,39], suggesting that genomic instability abrogates the ARF/p53-dependent barrier.
3. Genomic Destabilization and Replication Stress
Genomic destabilization is generally caused by DSBs followed by erroneous repair [40,41]. It has been firmly established that ATM/ATR-mediated damage responses are massively activated in response to DSBs, leading to induction of repair or apoptosis, and thus serve as a barrier against genomic destabilization to prevent cancer [7,42,43]. In this context, a critical question is how such barrier systems are over-ridden during genomic destabilization. Several lines of evidence suggest that this phenomenon is associated with down-regulation of the variant histone H2AX. H2AX mediates efficient checkpoint responses and DSB repair [44,45], and is significantly down-regulated when the growth rate of normal cells decreases [9]. In fact, as shown in vivo and in vitro in both human and mouse cells, H2AX is down-regulated when normal cells enter a growth-arrested state. Although the mechanisms involved in down-regulation of H2AX in quiescent cells are not fully understood, it is clear at least that (1) H2AX down-regulation in undamaged cells is partly mediated by proteolytic degradation, mediated by the E3-ligase HUWE1 [46]; and (2) the formation of the H2AX-down-regulated state is dependent on both ARF and p53 and, hence, usually cannot occur after transformation and/or immortalization [9,15,39]. Unlike cells arrested in response to cell-cycle checkpoint signaling, cells in this state still progress through cell-cycle phases, especially under accelerated growth stimuli. Therefore, cells in this state are defective in repairing DSBs caused by replication stress, and are consequently vulnerable to genomic destabilization [47,48,49]. These issues are clearly illustrated in MEFs: MEFs grown under the 3T3 protocol maintain genome stability during primary growth, but become vulnerable to genomic destabilization when their growth rate decreases, even though the cultivation conditions are unchanged [39,50]. This risk of genomic destabilization is associated with replication stress; accordingly, genome stability is effectively maintained when exogenous growth stimuli are diminished [39,50]. Thus, normal cells generally become susceptible to genomic destabilization in the presence of high levels of growth stimuli.
Genomic destabilization commonly occurs when cells are subjected to replication stress (Figure 1), as is often observed in cells at precancerous stages [8,51,52,53]. By contrast, cells respond differently to DSBs directly caused by γ-rays [17,18,54], which are efficiently repaired through transient up-regulation of H2AX mediated by ATM and SIRT6 [46]. DSBs caused by replication stress do not induce H2AX expression and, therefore, persist [55]. The resultant cells usually accumulate a few γH2AX foci and express senescent characteristics [17,55]. Such cells still progress through the cell cycle in response to exogenous growth stimuli; consequently, DSBs are often carried over into mitosis, causing failures in cytokinesis and promoting the development of CIN with tetraploidy [37]. Since H2AX mediates checkpoint-response activation, such DSB carryover through the cell cycle is likely to be associated with down-regulation of H2AX. In support of this idea, the cellular H2AX level is associated with sensitivity to certain anti-cancer drugs: cancer cells treated with camptothecin accumulate H2AX and massively activate checkpoint responses, thereby effectively inducing apoptosis, whereas normal cells tend to survive in a H2AX-diminished state [50].
4. Genomic Destabilization and Induction of Cancer-Driver Mutations
How does genomic destabilization contribute to cancer development? Based on the knowledge accumulated to date, induction of mutations is the primary factor. In fact, as shown in colon, benign tumors mutated in Adenoma polyposis coli (APC) usually develop genomic instabilities [1,56]. The involvement of genomic destabilization in mutation induction is clearly illustrated in MEFs. Although immortalized MEFs, usually mutated in either ARF or p53, only appear after genomic destabilization, normal MEFs are prevented from immortalizing as long as genome stability is maintained in the quiescent state, which is established and maintained by both ARF and p53, concomitant with down-regulation of H2AX [9,39]. Thus, genomic destabilization might be a major cause of cancer-driver mutations.
Cancer risk is associated with the total number of stem cell divisions [57,58,59], which could be interpreted to mean that cancer risk rises in association with replication errors. However, this interpretation is unlikely to apply in multiple situations. In fact, cancers in colon invariably develop genomic instabilities (either CIN or MSI), except for cancer cells with hypermutation due to a proofreading-deficient mutation in Polε [60,61]. In addition, adenomas in colon often develop with tetraploidy, in which each gene is present in four copies, including the genes encoding APC, ARF, and p53 [62,63,64]. It is statistically impossible to stochastically mutate all four loci by simple replication errors; however, loss of all four copies could occur via genomic destabilization, e.g., through LOH. In this case, the elevated cancer risk in highly dividing stem cells could be due to the greater chance that replication stress will cause genomic destabilization, resulting in cancer-driving mutations.
As recently illustrated, hypermutation is often massively induced in association with kataegis, i.e., localized hypermutation in close proximity to genomic rearrangements [65]. This supports the aforementioned association between genomic destabilization and mutation induction, although the mechanism by which those mutations are induced remains elusive.
5. Oncogene Acceleration and Its Contribution to Cancer Development
Genomic destabilization is also caused by oncogene activation [7,8,37]. Oncogene-activated cells are widely subjected to accelerated S-phase entry because, as with exogenous growth stimuli, a major effect of oncogenes is cell-cycle progression [37,66]. Consequently, the resultant cells accumulate DSBs in association with replication stress, eventually express senescent characteristics [25,67], and are at higher risk of genomic destabilization [37].
Each of the four Yamanaka factors (c-Myc, Oct3/4, Sox2, and Klf4) was originally characterized as an oncogene [68,69,70]. However, in contrast to the effects of expressing any of them individually, simultaneous expression of all four factors leads to development of iPS cells [71]. This phenomenon implies that oncogene activation causes dedifferentiation effects, potentially contributing to the development of cancer stem cells (CSCs). Consistent with this idea, c-Myc, as well as oncogenic K-Ras, is widely used to achieve cellular transformation with tumorigenicity [72,73,74], a key characteristic of CSCs [75]. In fact, because immortalized MEFs do not exhibit CSC characteristics, they usually exhibit no or very limited tumor-formation ability; however, they can acquire tumor-formation ability after transformation with c-Myc, implying that this factor induces reprogramming and acquisition of CSC characteristics [67,76,77]. These lines of evidence suggest that c-Myc promotes cancer development via cellular reprograming, but the underlying mechanism remains unclear. In addition, it remains to be determined how c-Myc and K-Ras cause cellular transformation, unlike many other oncogenes, and how simultaneous expression of the four Yamanaka factors (as opposed to their individual expression) leads to cellular reprograming without causing replication stress and resultant genomic destabilization.
6. The Senescent Cellular Background and Its Contribution to Cancer Development
Genomic destabilization is generally triggered by erroneously repaired DSBs, and a large proportion of DSBs are associated with replication stress [40,41]. Consequently, cells in this state generally exhibit senescent phenotypes in response to accumulated DSBs [55] (Figure 2a). Cells containing accumulated DSBs that exhibit senescent phenotypes might be distinct from canonical senescent cells, notwithstanding the fact that DSBs accumulate in both types of cells. In fact, the canonical view is that senescence is induced as a form of cancer prevention, and represents a terminal fate [19,78]. However, multiple recent studies showed that cellular senescence can drive cancer development in some contexts [79]. In particular, DSB accumulation in cells with senescent phenotypes could trigger genomic destabilization, followed by immortalization and acquisition of CSC characteristics [38,80]. Since acquisition of such characteristics is dependent on cancer-driver mutations, such as loss of function in the ARF/p53 module, cells acquiring immortality or CSC characteristics must represent a minor fraction (Figure 2b); nonetheless, they inevitably appear, as demonstrated by multiple models including MEF immortalization [9,38,39].
Senescence with a higher risk of genomic destabilization is mainly caused by replication stress. In fact, cells at precancerous stages accumulate DSBs in association with replication stress and often exhibit senescent phenotype [8,45]. In addition, MEFs continuously cultivated under growth stimuli exhibit senescent phenotypes with DSB accumulation and develop genomic instability, and the resultant cells are subject to replication stress. By contrast, MEFs cultivated with reduced levels of growth stimuli are continuously quiescent and preserve their genome stability [9,39]. In support of these ideas, replication stress, genomic instability, and cellular senescence are simultaneously observed in cells exposed to aberrantly high levels of accelerated growth stimuli or oncogene activation [81]. Even in developed cancers harboring mutations in p53, a fraction of cells with senescent phenotypes is responsible for tumor progression [82]. Thus, senescent cellular states often drive cancer progression through genomic destabilization.
Senescent cells can promote cancer development in neighboring cells via multiple mechanisms. First, senescence in the stem cell niche can cause inappropriate stem cell differentiation, leading to CSC induction, as reviewed previously [16,83]. Second, Yamanaka factor expression in vivo induces senescence in some cells, which further contributes to reprogramming of neighboring cells via expression of IL6 and TNFα, leading to teratoma formation [84] (Figure 2b). Furthermore, IL6 and TNFα themselves mediate inflammation; therefore, cancer development can also be promoted in association with inflammation [85,86,87]. Specifically, cells subjected to inflammation express deaminase APOBEC3, thereby inducing deamination-mediated hypermutation [88,89]. Thus, the environmental effects caused by senescent cells contribute to an environment that promotes cancer. Among cells subjected to replication stress, those that develop immortality or CSC characteristics are dependent on mutations in cancer-driver genes and must, therefore, be a minor fraction; however, most of the surrounding cells still contribute to cancer progression via their environmental effects.
7. Age as a Risk Factor of Genomic Instability
Age is a risk factor for cancers that develop with genomic instability. Consistently, DSBs that risk genomic destabilization generally accumulate as a function of age [18]. Since cellular senescence is generally induced in response to DSBs [90,91,92] and is associated with organic aging [93,94], cells that accumulate DSBs are at a higher risk of cancer development through genomic destabilization. Although such DSB accumulation must be associated with a cellular state deficient in DSB repair, it remains unclear how cells in this state become repair-defective. This is partly because H2AX, which is essential for genome stability [45], is commonly down-regulated when normal cells enter a growth-arrested state [9]. However, this is probably not the only reason for the repair deficiency. In fact, senescent cells often form large γH2AX foci at DSB sites, even when the cellular H2AX level is low, but these lesions still do not undergo repair [18]. The mechanisms responsible for the repair defect remain to be elucidated, but this phenomenon might be due to dysfunction in sirtuin proteins SIRT1 and SIRT6. The sirtuin family regulates longevity and suppresses aging-related phenotypes [95,96]. In particular, SIRT1 and SIRT6 are localized in the nucleus, where they are involved in in multiple functions that include regulation of replication by SIRT1 [97] and mediation of DSB repair by SIRT6 [46,98,99].
Mammalian cells possess multiple types of barrier systems to defend themselves from cancer development. Based on recent knowledge, the risks of genome destabilization and the associated mutation are elevated when cells senesce. The induced mutations abrogate cancer-suppression systems, such as the ARF/p53 module.
8. Conclusions
Cancer is a disease associated with aging, and most cancers inevitably develop genomic instability. Based on recent knowledge, cellular senescence is tightly associated with cancer risk, primarily mediated by genomic destabilization, which is, in turn, associated with the induction of cancer-driver mutations. As cancer risk increases as a function of age, replication stress-associated DSBs that increase the risk of genomic destabilization also accumulate with age in vivo, causing cells to express senescent characteristics. Such DSB accumulation is induced in repair-deficient backgrounds partly because H2AX, which is required for recruitment of repair factors, is largely down-regulated when the growth rate of normal cells slows down. Consequently, the risk of genomic destabilization is generally elevated in such cellular backgrounds upon exposure to replication stress due to oxidation, aberrant growth stimuli, oncogene activation, or environmental factors that cause nucleotide adducts.
Acknowledgments
This work was supported by MEXT KAKENHI, no. 20770136.
Author Contributions
Yusuke Minakawa, Atsuhiro Shimizu, Yusuke Matsuno, and Ken-ichi Yoshioka wrote the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instability in colorectal cancers. Nature 1997, 386, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instabilities in human cancers. Nature 1998, 396, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Shih, I.M.; Zhou, W.; Goodman, S.N.; Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Evidence that genetic instability occurs at an early stage of colorectal tumorigenesis. Cancer Res. 2001, 61, 818–822. [Google Scholar] [PubMed]
- Hveem, T.S.; Merok, M.A.; Pretorius, M.E.; Novelli, M.; Bævre, M.S.; Sjo, O.H.; Clinch, N.; Liestøl, K.; Svindland, A.; Lothe, R.A.; et al. Prognostic impact of genomic instability in colorectal cancer. Br. J. Cancer 2014, 110, 2159–2164. [Google Scholar] [CrossRef] [PubMed]
- Edwards, B.K.; Howe, H.L.; Ries, L.A.; Thun, M.J.; Rosenberg, H.M.; Yancik, R.; Wingo, P.A.; Jemal, A.; Feigal, E.G. Annual report to the nation on the status of cancer, 1973–1999, featuring implications of age and aging on U.S. cancer burden. Cancer 2002, 94, 2766–2792. [Google Scholar] [CrossRef] [PubMed]
- Peto, J. Cancer epidemiology in the last century and the next decade. Nature 2001, 411, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Horejsí, Z.; Koed, K.; Krämer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.G.; Vassiliou, L.V.; Karakaidos, P.; Zacharatos, P.; Kotsinas, A.; Liloglou, T.; Venere, M.; Ditullio, R.A., Jr.; Kastrinakis, N.G.; Levy, B.; et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005, 434, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Atsumi, Y.; Fujimori, H.; Fukuda, H.; Inase, A.; Shinohe, K.; Yoshioka, Y.; Shikanai, M.; Ichijima, Y.; Unno, J.; Mizutani, S.; et al. Onset of quiescence following p53 mediated down-regulation of H2AX in normal cells. PLoS ONE 2011, 6, e23432. [Google Scholar] [CrossRef] [PubMed]
- Connor, F.; Bertwistle, D.; Mee, P.J.; Ross, G.M.; Swift, S.; Grigorieva, E.; Tybulewicz, V.L.; Ashworth, A. Tumorigenesis and a DNA repair defect in mice with a truncating Brca2 mutation. Nat. Genet. 1997, 17, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Moynahan, M.E.; Chiu, J.W.; Koller, B.H.; Jasin, M. BRCA1 controls homology-directed DNA repair. Mol. Cell 1999, 4, 511–518. [Google Scholar] [CrossRef]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Terzi, M.Y.; Izmirli, M.; Gogebakan, B. The cell fate: Senescence or quiescence. Mol. Biol. Rep. 2016, 43, 1213–1220. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, K.; Atsumi, Y.; Fukuda, H.; Masutani, M.; Teraoka, H. The quiescent cellular state is Arf/p53-dependent and associated with H2AX downregulation and genome stability. Int. J. Mol. Sci. 2012, 13, 6492–6506. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, K.; Atsumi, Y.; Nakagama, H.; Teraoka, H. Development of cancer-initiating cells and immortalized cells with genomic instability. World J. Stem Cells 2015, 7, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Grudzenski, S.; Raths, A.; Conrad, S.; Rübe, C.E.; Löbrich, M. Inducible response required for repair of low-dose radiation damage in human fibroblasts. Proc. Natl. Acad. Sci. USA 2010, 107, 14205–14210. [Google Scholar] [CrossRef] [PubMed]
- Sedelnikova, O.A.; Horikawa, I.; Zimonjic, D.B.; Popescu, N.C.; Bonner, W.M.; Barrett, J.C. Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat. Cell Biol. 2004, 6, 168–170. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [PubMed]
- Kulju, K.S.; Lehman, J.M. Increased p53 protein associated with aging in human-diploid fibroblasts. Exp. Cell Res. 1995, 217, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Sabin, R.J.; Anderson, R.M. Cellular Senescence—Its role in cancer and the response to ionizing radiation. Genome Integr. 2011, 2, 7. [Google Scholar] [CrossRef] [PubMed]
- Sager, R. Senescence as a mode of tumor suppression. Environ. Health Perspect. 1991, 93, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Lecot, P.; Alimirah, F.; Desprez, P.Y.; Campisi, J.; Wiley, C. Context-dependent effects of cellular senescence in cancer development. Br. J. Cancer 2016, 114, 1180–1184. [Google Scholar] [CrossRef] [PubMed]
- Mosieniak, G.; Sikora, E. Polyploidy: The link between senescence and cancer. Curr. Pharm. Des. 2010, 16, 734–740. [Google Scholar] [CrossRef] [PubMed]
- Van Riggelen, J.; Felsher, D.W. Myc and a Cdk2 senescence switch. Nat. Cell Biol. 2010, 12, 7–9. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Poulogiannis, G.; Frayling, I.M.; Arends, M.J. DNA mismatch repair deficiency in sporadic colorectal cancer and Lynch syndrome. Histopathology 2010, 56, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Gologan, A.; Sepulveda, A.R. Microsatellite instability and DNA mismatch repair deficiency testing in hereditary and sporadic gastrointestinal cancers. Clin. Lab. Med. 2005, 25, 179–196. [Google Scholar] [CrossRef] [PubMed]
- Woerner, S.M.; Kloor, M.; von Knebel Doeberitz, M.; Gebert, J.F. Microsatellite instability in the development of DNA mismatch repair deficient tumors. Cancer Biomark. 2006, 2, 69–86. [Google Scholar] [CrossRef] [PubMed]
- Geigl, J.B.; Obenauf, A.C.; Schwarzbraun, T.; Speicher, M.R. Defining ‘chromosomal instability’. Trends Genet. 2008, 24, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Almoguera, C.; Shibata, D.; Forrester, K.; Martin, J.; Arnheim, N.; Perucho, M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell 1988, 53, 549–554. [Google Scholar] [CrossRef] [Green Version]
- Williams, C.; Pontén, F.; Ahmadian, A.; Ren, Z.P.; Ling, G.; Rollman, O.; Ljung, A.; Jaspers, N.G.; Uhlén, M.; Lundeberg, J.; et al. Clones of normal keratinocytes and a variety of simultaneously present epidermal neoplastic lesions contain a multitude of p53 gene mutations in a xeroderma pigmentosum patient. Cancer Res. 1998, 58, 2449–2455. [Google Scholar] [PubMed]
- Nowell, P.C. Genetic alterations in leukemias and lymphomas: Impressive progress and continuing complexity. Cancer Genet. Cytogenet. 1997, 94, 13–19. [Google Scholar] [CrossRef]
- Thiagalingam, S.; Laken, S.; Willson, J.K.; Markowitz, S.D.; Kinzler, K.W.; Vogelstein, B.; Lengauer, C. Mechanisms underlying losses of heterozygosity in human colorectal cancers. Proc. Natl. Acad. Sci. USA 2001, 98, 2698–2702. [Google Scholar] [CrossRef] [PubMed]
- Baca, S.C.; Prandi, D.; Lawrence, M.S.; Mosquera, J.M.; Romanel, A.; Drier, Y.; Park, K.; Kitabayashi, N.; MacDonald, T.Y.; Ghandi, M.; et al. Punctuated evolution of prostate cancer genomes. Cell 2013, 153, 666–677. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Ichijima, Y.; Yoshioka, K.; Yoshioka, Y.; Shinohe, K.; Fujimori, H.; Unno, J.; Takagi, M.; Goto, H.; Inagaki, M.; Mizutani, S.; et al. DNA lesions induced by replication stress trigger mitotic aberration and tetraploidy development. PLoS ONE 2010, 5, e8821. [Google Scholar] [CrossRef] [PubMed]
- Fujimori, H.; Shikanai, M.; Teraoka, H.; Masutani, M.; Yoshioka, K. Induction of cancerous stem cells during embryonic stem cell differentiation. J. Biol. Chem. 2012, 287, 36777–36791. [Google Scholar] [CrossRef] [PubMed]
- Osawa, T.; Atsumi, Y.; Sugihara, E.; Saya, H.; Kanno, M.; Tashiro, F.; Masutani, M.; Yoshioka, K. Arf and p53 act as guardians of a quiescent cellular state by protecting against immortalization of cells with stable genomes. Biochem. Biophys. Res. Commun. 2013, 432, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H. DNA Damage, Aging, and Cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, R.; Raghavan, S.C. Induction of DNA damage and erroneous repair can explain genomic instability caused by endosulfan. Carcinogenesis 2016, 37, 929–940. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death by apoptosis. Trends Mol. Med. 2006, 12, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Stucki, M.; Jackson, S.P. gamma H2AX and MDC1: Anchoring the DNA-damage-response machinery to broken chromosomes. DNA Repair 2006, 5, 534–543. [Google Scholar] [CrossRef] [PubMed]
- Bonner, W.M.; Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Sedelnikova, O.A.; Solier, S.; Pommier, Y. gamma H2AX and cancer. Nat. Rev. Cancer 2008, 8, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Atsumi, Y.; Minakawa, Y.; Ono, M.; Dobashi, S.; Shinohe, K.; Shinohara, A.; Takeda, S.; Takagi, M.; Takamatsu, N.; Nakagama, H.; et al. ATM and SIRT6/SNF2H mediate transient H2AX stabilization when DSBs form by blocking HUWE1 to allow efficient gamma H2AX Foci formation. Cell Rep. 2015, 13, 2728–2740. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, A.; Garcia-Muse, T. Causes of genome instability. Annu. Rev. Genet. 2013, 47, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, H.; Garcia-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef] [PubMed]
- Mazouzi, A.; Velimezi, G.; Loizou, J.I. DNA replication stress: Causes, resolution and disease. Exp. Cell Res. 2014, 329, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Atsumi, Y.; Inase, A.; Osawa, T.; Sugihara, E.; Sakasai, R.; Fujimori, H.; Teraoka, H.; Saya, H.; Kanno, M.; Tashiro, F.; et al. The Arf/p53 protein module, which induces apoptosis, down-regulates histone H2AX to allow normal cells to survive in the presence of anti-cancer drugs. J. Biol. Chem. 2013, 288, 13269–13277. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.R.; Brenner, B.M.; Swede, H.; Chen, N.; Henry, W.M.; Conroy, J.M.; Karpenko, M.J.; Issa, J.P.; Bartos, J.D.; Brunelle, J.K.; et al. Intrachromosomal genomic instability in human sporadic colorectal cancer measured by genome-wide allelotyping and inter-(simple sequence repeat) PCR. Cancer Res. 2001, 61, 8274–8283. [Google Scholar] [PubMed]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Abbas, T.; Keaton, M.A.; Dutta, A. Genomic instability in cancer. Cold Spring Harb. Perspect. Biol. 2013, 3, a012914. [Google Scholar] [CrossRef] [PubMed]
- Sedelnikova, O.A.; Horikawa, I.; Redon, C.; Nakamura, A.; Zimonjic, D.B.; Popescu, N.C.; Bonner, W.M. Delayed kinetics of DNA double-strand break processing in normal and pathological aging. Aging Cell 2008, 7, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Minakawa, Y.; Atsumi, Y.; Shinohara, A.; Murakami, Y.; Yoshioka, K. Gamma-irradiated quiescent cells repair directly induced double-strand breaks but accumulate persistent double-strand breaks during subsequent DNA replication. Genes Cells 2016, 21, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Rao, C.V.; Yamada, H.Y. Genomic instability and colon carcinogenesis: From the perspective of genes. Front. Oncol. 2013, 3, 130. [Google Scholar] [CrossRef] [PubMed]
- Blokzijl, F.; de Ligt, J.; Jager, M.; Sasselli, V.; Roerink, S.; Sasaki, N.; Huch, M.; Boymans, S.; Kuijk, E.; Prins, P.; et al. Tissue-specific mutation accumulation in human adult stem cells during life. Nature 2016, 538, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Martincorena, I.; Campbell, P.J. Somatic mutation in cancer and normal cells. Science 2015, 349, 1483–1489. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Vogelstein, B. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 2015, 347, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Kane, D.P.; Shcherbakova, P.V. A common cancer-associated DNA polymerase epsilon mutation causes an exceptionally strong mutator phenotype, indicating fidelity defects distinct from loss of proofreading. Cancer Res. 2014, 74, 1895–1901. [Google Scholar] [CrossRef] [PubMed]
- Rayner, E.; van Gool, I.C.; Palles, C.; Kearsey, S.E.; Bosse, T.; Tomlinson, I.; Church, D.N. A panoply of errors: Polymerase proofreading domain mutations in cancer. Nat. Rev. Cancer 2016, 16, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, C.M.; Green, R.A.; Kaplan, K.B. APC mutations lead to cytokinetic failures in vitro and tetraploid genotypes in Min mice. J. Cell Biol. 2007, 178, 1109–1120. [Google Scholar] [CrossRef] [PubMed]
- Ceol, C.J.; Pellman, D.; Zon, L.I. APC and colon cancer: Two hits for one. Nat. Med. 2007, 13, 1286–1287. [Google Scholar] [CrossRef] [PubMed]
- Dikovskaya, D.; Schiffmann, D.; Newton, I.P.; Oakley, A.; Kroboth, K.; Sansom, O.; Jamieson, T.J.; Meniel, V.; Clarke, A.; Näthke, I.S. Loss of APC induces polyploidy as a result of a combination of defects in mitosis and apoptosis. J. Cell Biol. 2007, 176, 183–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nik-Zainal, S.; Alexandrov, L.B.; Wedge, D.C.; Van Loo, P.; Greenman, C.D.; Raine, K.; Jones, D.; Hinton, J.; Marshall, J.; Stebbings, L.A.; et al. Mutational processes molding the genomes of 21 breast cancers. Cell 2012, 149, 979–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartek, J.; Lukas, J. Pathways governing G1/S transition and their response to DNA damage. FEBS Lett. 2001, 490, 117–122. [Google Scholar] [CrossRef]
- Sasaki, R.; Narisawa-Saito, M.; Yugawa, T.; Fujita, M.; Tashiro, H.; Katabuchi, H.; Kiyono, T. Oncogenic transformation of human ovarian surface epithelial cells with defined cellular oncogenes. Carcinogenesis 2009, 30, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Bass, A.J.; Watanabe, H.; Mermel, C.H.; Yu, S.; Perner, S.; Verhaak, R.G.; Kim, S.Y.; Wardwell, L.; Tamayo, P.; Gat-Viks, I.; et al. SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat. Genet. 2009, 41, 1238–1242. [Google Scholar] [CrossRef] [PubMed]
- Felsher, D.W.; Bishop, J.M. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol. Cell 1999, 4, 199–207. [Google Scholar] [CrossRef]
- Gidekel, S.; Pizov, G.; Bergman, Y.; Pikarsky, E. Oct-3/4 is a dose-dependent oncogenic fate determinant. Cancer Cell 2003, 4, 361–370. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keath, E.J.; Caimi, P.G.; Cole, M.D. Fibroblast lines expressing activated c-myc oncogenes are tumorigenic in nude mice and syngeneic animals. Cell 1984, 39, 339–348. [Google Scholar] [CrossRef]
- D’Cruz, C.M.; Gunther, E.J.; Boxer, R.B.; Hartman, J.L.; Sintasath, L.; Moody, S.E.; Cox, J.D.; Ha, S.I.; Belka, G.K.; Golant, A.; et al. c-MYC induces mammary tumorigenesis by means of a preferred pathway involving spontaneous Kras2 mutations. Nat. Med. 2001, 7, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Lavialle, C.; Modjtahedi, N.; Cassingena, R.; Brison, O. High c-myc amplification level contributes to the tumorigenic phenotype of the human breast carcinoma cell line SW 613-S. Oncogene 1988, 3, 335–339. [Google Scholar] [PubMed]
- Pu, H.; Zheng, Q.; Li, H.; Wu, M.; An, J.; Gui, X.; Li, T.; Lu, D. CUDR promotes liver cancer stem cell growth through upregulating TERT and C-Myc. Oncotarget 2015, 6, 40775–40798. [Google Scholar] [CrossRef] [PubMed]
- Narisawa-Saito, M.; Handa, K.; Yugawa, T.; Ohno, S.; Fujita, M.; Kiyono, T. HPV16 E6-mediated stabilization of ErbB2 in neoplastic transformation of human cervical keratinocytes. Oncogene 2007, 26, 2988–2996. [Google Scholar] [CrossRef] [PubMed]
- Zindy, F.; Eischen, C.M.; Randle, D.H.; Kamijo, T.; Cleveland, J.L.; Sherr, C.J.; Roussel, M.F. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev. 1998, 12, 2424–2433. [Google Scholar] [CrossRef] [PubMed]
- Lasry, A.; Ben-Neriah, Y. Senescence-associated inflammatory responses: Aging and cancer perspectives. Trends Immunol. 2015, 36, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.; Serrano, M. Senescence in tumours: Evidence from mice and humans. Nat. Rev. Cancer 2010, 10, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Lamm, N.; Ben-David, U.; Golan-Lev, T.; Storchová, Z.; Benvenisty, N.; Kerem, B. Genomic instability in human pluripotent stem cells arises from replicative stress and chromosome condensation defects. Cell Stem Cell 2016, 18, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Courtois-Cox, S.; Jones, S.L.; Cichowski, K. Many roads lead to oncogene-induced senescence. Oncogene 2008, 27, 2801–2809. [Google Scholar] [CrossRef] [PubMed]
- Galanos, P.; Vougas, K.; Walter, D.; Polyzos, A.; Maya-Mendoza, A.; Haagensen, E.J.; Kokkalis, A.; Roumelioti, F.M.; Gagos, S.; Tzetis, M.; et al. Chronic p53-independent p21 expression causes genomic instability by deregulating replication licensing. Nat. Cell Biol. 2016, 18, 777–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knoepfler, P.S. Deconstructing stem cell tumorigenicity: A roadmap to safe regenerative medicine. Stem Cells 2009, 27, 1050–1056. [Google Scholar] [CrossRef] [PubMed]
- Mosteiro, L.; Pantoja, C.; Alcazar, N.; Marión, R.M.; Chondronasiou, D.; Rovira, M.; Fernandez-Marcos, P.J.; Muñoz-Martin, M.; Blanco-Aparicio, C.; Pastor, J.; et al. Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science 2016, 354. [Google Scholar] [CrossRef] [PubMed]
- Cooks, T.; Pateras, I.S.; Tarcic, O.; Solomon, H.; Schetter, A.J.; Wilder, S.; Lozano, G.; Pikarsky, E.; Forshew, T.; Rosenfeld, N.; et al. Mutant p53 prolongs NF-kappaB activation and promotes chronic inflammation and inflammation-associated colorectal cancer. Cancer Cell 2013, 23, 634–646. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Karin, M. Inflammatory cytokines in cancer: Tumour necrosis factor and interleukin 6 take the stage. Ann. Rheum. Dis. 2011, 70, I104–I108. [Google Scholar] [CrossRef] [PubMed]
- Sethi, G.; Sung, B.; Aggarwal, B.B. TNF: A master switch for inflammation to cancer. Front. Biosci. 2008, 13, 5094–5107. [Google Scholar] [CrossRef] [PubMed]
- Vartanian, J.P.; Henry, M.; Marchio, A.; Suspène, R.; Aynaud, M.M.; Guétard, D.; Cervantes-Gonzalez, M.; Battiston, C.; Mazzaferro, V.; Pineau, P.; et al. Massive APOBEC3 editing of hepatitis B viral DNA in cirrhosis. PLoS Pathog. 2010, 6, e1000928. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.J.; Nik-Zainal, S.; Wu, Y.L.; Stebbings, L.A.; Raine, K.; Campbell, P.J.; Rada, C.; Stratton, M.R.; Neuberger, M.S. DNA deaminases induce break-associated mutation showers with implication of APOBEC3B and 3A in breast cancer kataegis. Elife 2013, 16, e00534. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; La Rosa, S.; Hagos, E.G. Oxidative DNA damage causes premature senescence in mouse embryonic fibroblasts deficient for Krüppel-like factor 4. Mol. Carcinog. 2015, 54, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; Demaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 2015, 349. [Google Scholar] [CrossRef] [PubMed]
- White, R.R.; Vijg, J. Do DNA double-strand breaks drive aging? Mol. Cell 2016, 63, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zhang, X.; Zeng, M.; Yuan, J.; Liu, M.; Yin, Y.; Wu, X.; Keefe, D.L.; Liu, L. Increased DNA damage and repair deficiency in granulosa cells are associated with ovarian aging in rhesus monkey. J. Assist. Reprod. Genet. 2015, 32, 1069–1078. [Google Scholar] [CrossRef] [PubMed]
- Gorbunova, V.; Seluanov, A. DNA double strand break repair, aging and the chromatin connection. Mutat. Res. 2016, 788, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Giblin, W.; Skinner, M.E.; Lombard, D.B. Sirtuins: Guardians of mammalian healthspan. Trends Genet. 2014, 30, 271–286. [Google Scholar] [CrossRef] [PubMed]
- Haigis, M.C.; Sinclair, D.A. Mammalian sirtuins: Biological insights and disease relevance. Annu. Rev. Pathol. 2010, 5, 253–295. [Google Scholar] [CrossRef] [PubMed]
- Utani, K.; Fu, H.; Jang, S.M.; Marks, A.B.; Smith, O.K.; Zhang, Y.; Redon, C.E.; Shimizu, N.; Aladjem, M.I. Phosphorylated SIRT1 associates with replication origins to prevent excess replication initiation and preserve genomic stability. Nucleic Acids Res. 2017, 45, 7807–7824. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.; Hine, C.; Tina, X.; Van Meter, M.; Au, M.; Vaidya, A.; Seluanov, A.; Gorbunova, V. SIRT6 promotes DNA repair under stress by activating PARP1. Science 2011, 332, 1443–1446. [Google Scholar] [CrossRef] [PubMed]
- Toiber, D.; Erdel, F.; Bouazoune, K.; Silberman, D.M.; Zhong, L.; Mulligan, P.; Sebastian, C.; Cosentino, C.; Martinez-Pastor, B.; Giacosa, S.; et al. SIRT6 recruits SNF2H to DNA break sites, preventing genomic instability through chromatin remodeling. Mol. Cell 2013, 51, 454–468. [Google Scholar] [CrossRef] [PubMed]
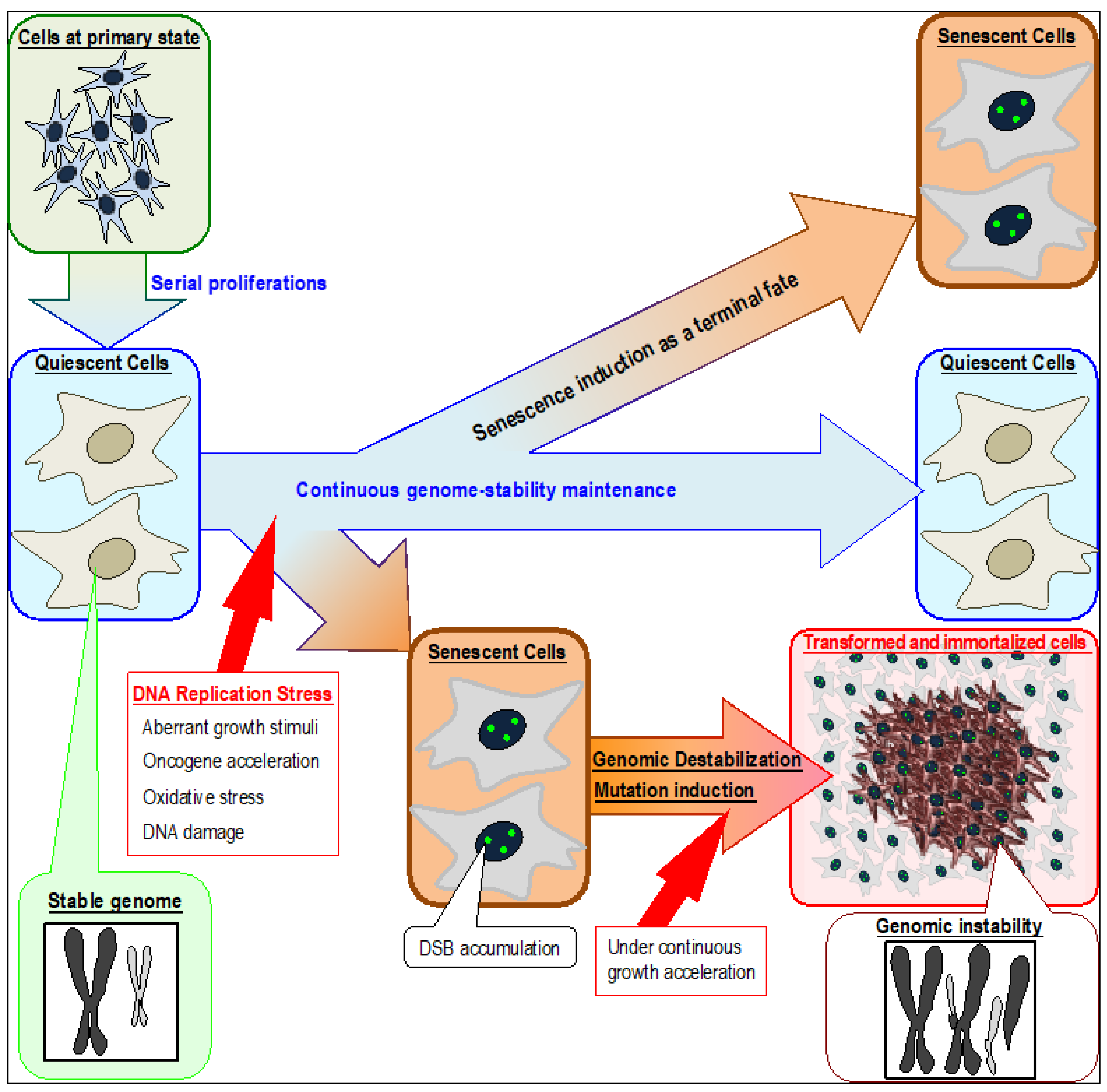
Figure 1.
Induction of senescent and transformed cells triggered by DNA replication stress in association with genomic destabilization. Although quiescent cells can continuously preserve their cellular state and maintain genome stability, senescence can be induced by stresses that cause replication-fork stalling in association with persistent DSB (DNA double strand break) accumulation. Normal quiescent cells are defective in repairing DSBs caused by replication stress and are, therefore, vulnerable to genomic destabilization. Replication stress occurs in association with fork stalling, which can arise by multiple reasons that include aberrant growth stimuli, oncogene activation, oxidative stress, and DNA damage. The resultant cells accumulate DSBs and exhibit senescent characteristics. Immortalized and transformed cells usually appear after genomic destabilization.
Figure 1.
Induction of senescent and transformed cells triggered by DNA replication stress in association with genomic destabilization. Although quiescent cells can continuously preserve their cellular state and maintain genome stability, senescence can be induced by stresses that cause replication-fork stalling in association with persistent DSB (DNA double strand break) accumulation. Normal quiescent cells are defective in repairing DSBs caused by replication stress and are, therefore, vulnerable to genomic destabilization. Replication stress occurs in association with fork stalling, which can arise by multiple reasons that include aberrant growth stimuli, oncogene activation, oxidative stress, and DNA damage. The resultant cells accumulate DSBs and exhibit senescent characteristics. Immortalized and transformed cells usually appear after genomic destabilization.
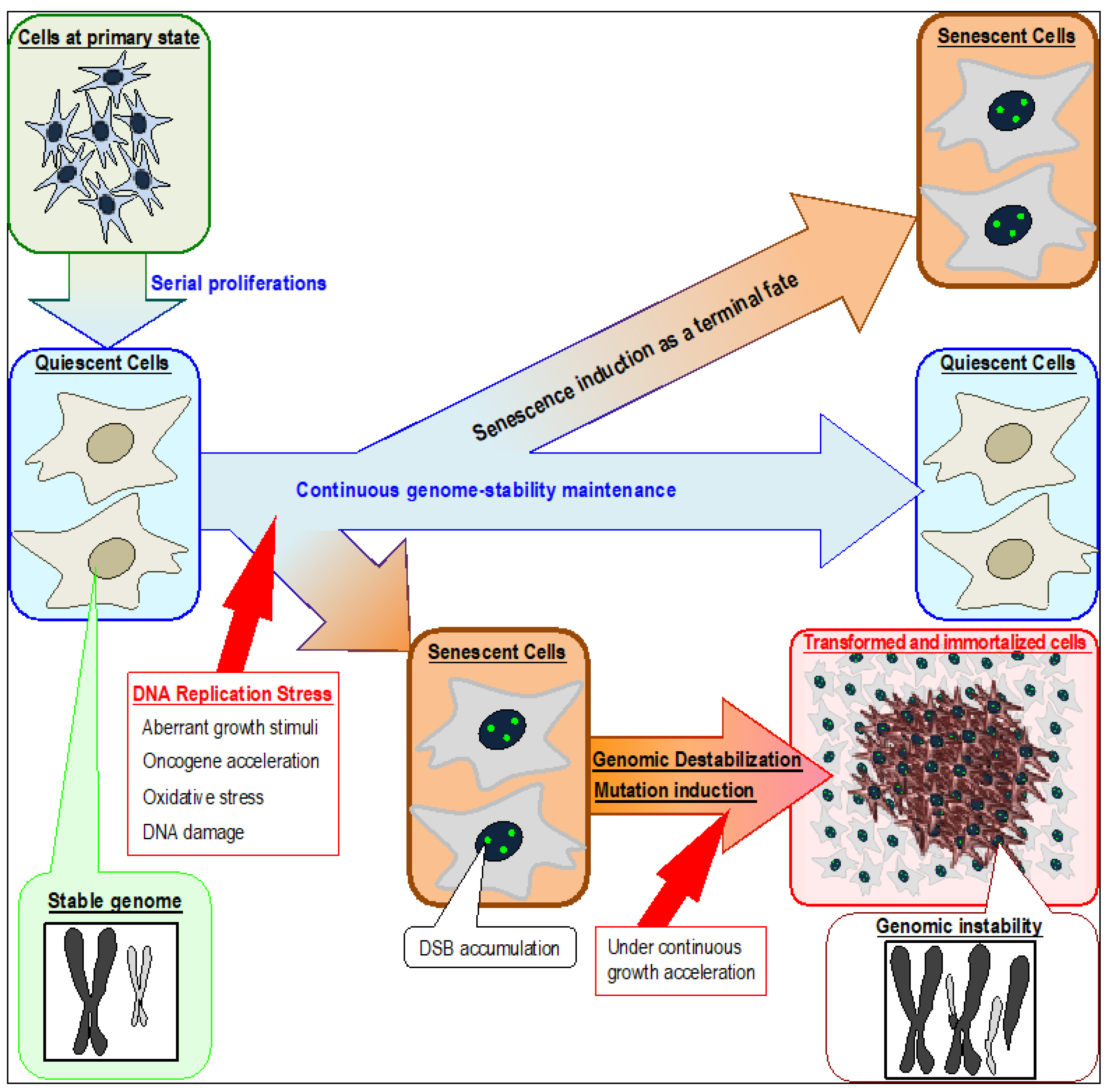
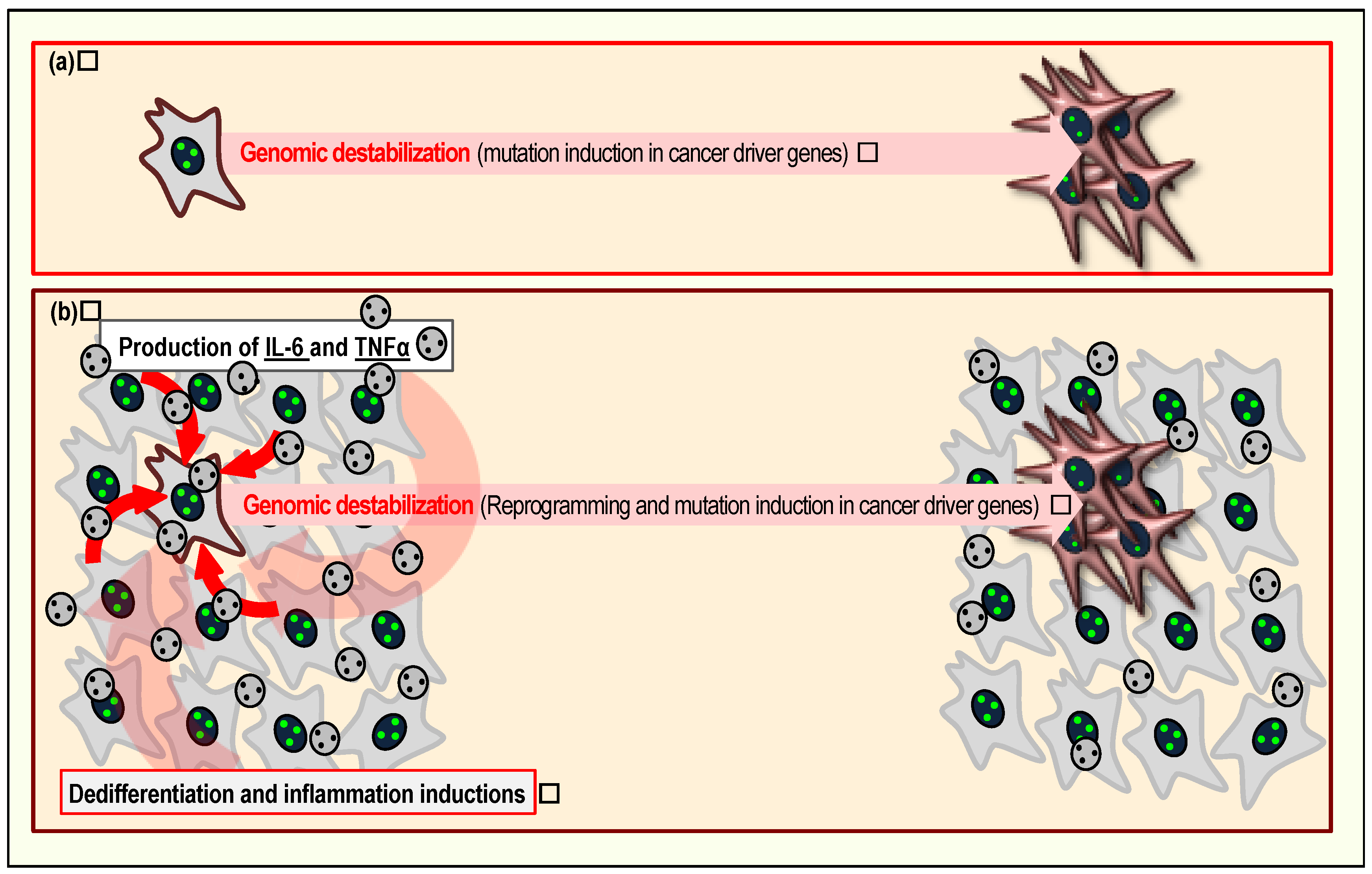
Figure 2.
Induction of senescent cells promotes cancer development. Cells in the senescent state can contribute to cancer development in at least two ways. First, under continuous growth stimuli, accumulated DSBs can trigger genomic destabilization, directly promoting immortalization and acquisition of CSC (cancer-stem cell) characteristics, thereby promoting tumor development (a). Second, senescent cells often produce factors associated with the senescence-associated secretory phenotype, including TNFα and IL6 (b). Secreted TNFα and IL6 induce dedifferentiation and inflammation, further contributing to cancer development.
Figure 2.
Induction of senescent cells promotes cancer development. Cells in the senescent state can contribute to cancer development in at least two ways. First, under continuous growth stimuli, accumulated DSBs can trigger genomic destabilization, directly promoting immortalization and acquisition of CSC (cancer-stem cell) characteristics, thereby promoting tumor development (a). Second, senescent cells often produce factors associated with the senescence-associated secretory phenotype, including TNFα and IL6 (b). Secreted TNFα and IL6 induce dedifferentiation and inflammation, further contributing to cancer development.

{kind=link}
{kind=link}
Table 1.
Types of genomic instability induced in cancer cells.
| MMR Status | Type | Mutation Rate | Genomic Alteration | Description |
|---|---|---|---|---|
| Proficient | CIN | Low | Gene amplification | |
| Chromosomal deletion | ||||
| Chromosomal rearrangement | Frequently occurs at common fragile sites | |||
| Tetraploidy/Aneuploidy | ||||
| Loss of heterozygousity | ||||
| Chromothripsis | Frequently occurs at common fragile sites | |||
| Chromoplexy | ||||
| Deficient | MSI | High | Alteration in the lengths of microsatellite fragments | Frequently occurs at common fragile sites |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Minakawa, Y.; Shimizu, A.; Matsuno, Y.; Yoshioka, K.-i. Genomic Destabilization Triggered by Replication Stress during Senescence. Cancers 2017, 9, 159. https://doi.org/10.3390/cancers9110159
AMA Style
Minakawa Y, Shimizu A, Matsuno Y, Yoshioka K-i. Genomic Destabilization Triggered by Replication Stress during Senescence. Cancers. 2017; 9(11):159. https://doi.org/10.3390/cancers9110159
Chicago/Turabian StyleMinakawa, Yusuke, Atsuhiro Shimizu, Yusuke Matsuno, and Ken-ichi Yoshioka. 2017. "Genomic Destabilization Triggered by Replication Stress during Senescence" Cancers 9, no. 11: 159. https://doi.org/10.3390/cancers9110159
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.