
Class (I) Phosphoinositide 3-Kinases in the Tumor Microenvironment


{kind=link}
Abstract
:1. Introduction
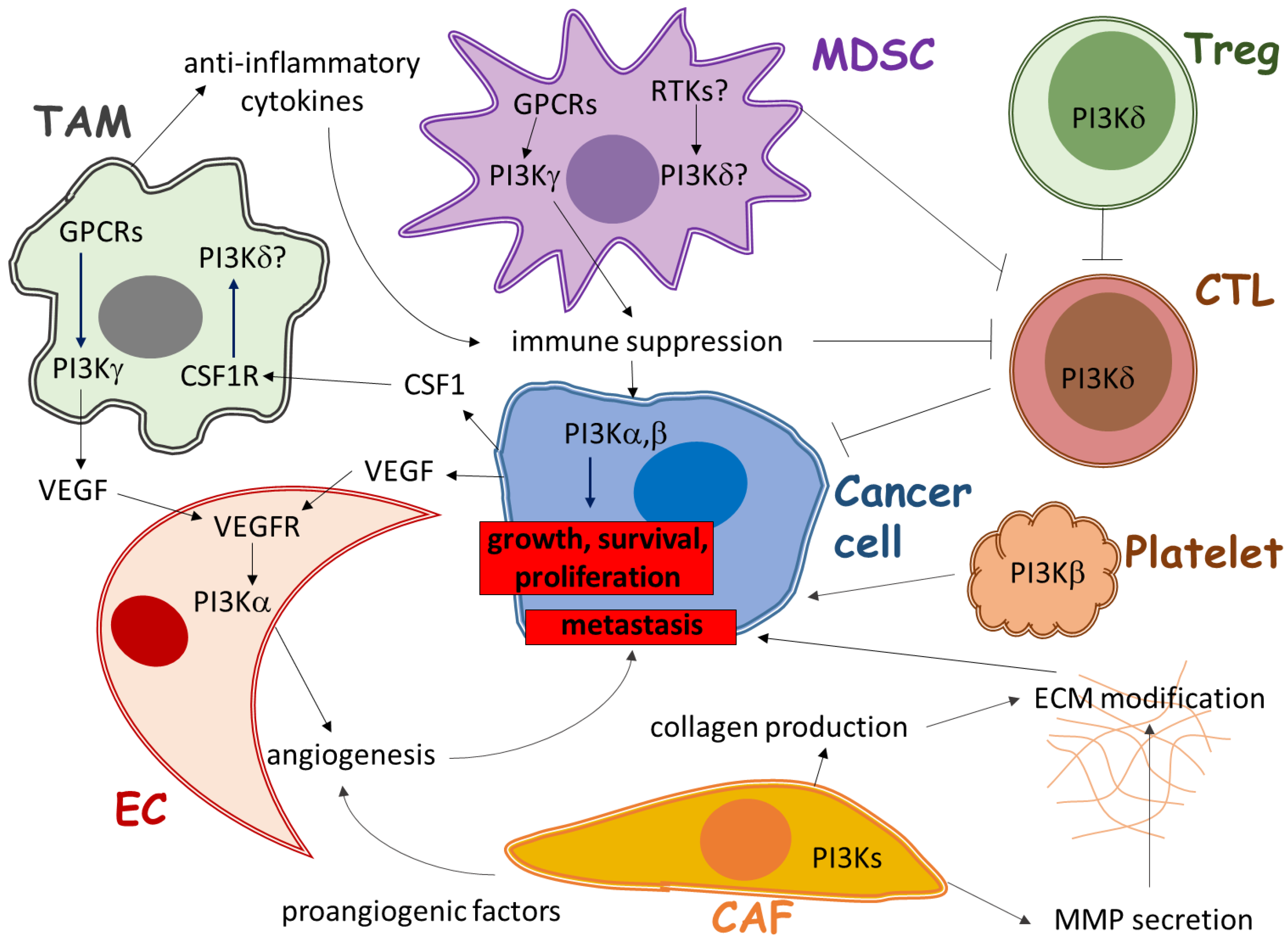
2. Role of PI3K in Immune Cells of the Tumor Microenvironment
2.1. The Role of PI3Kγ in Tumor-Associated Myeloid Cells
2.2. The Role of PI3Kδ in Regulatory T Cells
2.3. The Role of PI3Ks in Other Immune Cells
3. Role of PI3K in Angiogenesis in the Tumor Microenvironment
3.1. The Direct Role of PI3Kα in Angiogenesis
3.2. Indirect Role of PI3Ks in Angiogenesis
4. Role of PI3K in Stromal Fibroblasts of the Tumor Microenvironment
5. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hawkins, P.; Stephens, L.R. PI3K signalling in inflammation. Biochim. Biophys. Acta 2015, 1851, 882–897. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Stephens, L.; Hawkins, P. PI3K signalling: the path to discovery and understanding. Nat. Rev. Mol. Cell Biol. 2012, 13, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.; Anderson, K.E.; Davidson, K.; Stephens, L.R. Signalling through class I PI3Ks in mammalian cells. Biochem. Soc. Trans. 2006, 34, 647–662. [Google Scholar] [CrossRef] [PubMed]
- Okkenhaug, K. Signaling by the phosphoinositide 3-kinase family in immune cells. Annu. Rev. Immunol. 2013, 31, 675–704. [Google Scholar] [CrossRef] [PubMed]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.; Rommel, C. PI3K and cancer: lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, L.; Yuzugullu, H.; Zhao, J.J. PI3K in cancer: Divergent roles of isoforms, modes of activation and therapeutic targeting. Nat. Rev. Cancer 2015, 15, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Samuels, Y.; Diaz, L.A.; Schmidt-Kittler, O.; Cummins, J.M.; Delong, L.; Cheong, I.; Rago, C.; Huso, D.L.; Lengauer, C.; Kinzler, K.W.; et al. Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell 2005, 7, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Hill, K.; Kalifa, S.; Das, J.R.; Bhatti, T.; Gay, M.; Williams, D.; Taliferro-Smith, L.; De Marzo, A.M. The role of PI 3-kinase p110beta in AKT signally, cell survival, and proliferation in human prostate cancer cells. Prostate 2010, 70, 755–764. [Google Scholar] [PubMed]
- Dbouk, H.; Khalil, B.D.; Wu, H.; Shymanets, A.; Nürnberg, B.; Backer, J.M. Characterization of a tumor-associated activating mutation of the p110β PI 3-kinase. PLoS ONE 2013, 8, e63833. [Google Scholar]
- Wee, S.; Wiederschain, D.; Maira, S.M.; Loo, A.; Miller, C.; deBeaumont, R.; Stegmeier, F.; Yao, Y.M.; Lengauer, C. PTEN-deficient cancers depend on PIK3CB. Proc. Natl. Acad. Sci. USA 2008, 105, 13057–13062. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Chen, S.; Asara, J.M.; Balk, S.P. Phosphoinositide 3-kinase pathway activation in phosphate and tensin homolog (PTEN)-deficient prostate cancer cells is independent of receptor tyrosine kinases and mediated by the p110beta and p110delta catalytic subunits. J. Biol. Chem. 2010, 285, 14980–14989. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J. Targeting PI3K signalling in cancer: Opportunities, challenges and limitations. Nat. Rev. Cancer 2009, 9, 550–562. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.; Toker, A. The phosphoinositide 3-kinase pathway and therapy resistance in cancer. F1000Prime Rep. 2015, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Busaidy, N.; Farooki, A.; Dowlati, A.; Perentesis, J.P.; Dancey, J.E.; Doyle, L.A.; Brell, J.M.; Siu, L.L. Management of metabolic effects associated with anticancer agents targeting the PI3K-Akt-mTOR pathway. J. Clin. Oncol. 2012, 30, 2919–2928. [Google Scholar] [CrossRef] [PubMed]
- Safdari, Y.; Khalili, M.; Ebrahimzadeh, M.A.; Yazdani, Y.; Farajnia, S. Natural inhibitors of PI3K/AKT signaling in breast cancer: emphasis on newly-discovered molecular mechanisms of action. Pharmacol. Res. 2015, 93, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Dworak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar]
- McAllister, S.; Weinberg, R.A. The tumour-induced systemic environment as a critical regulator of cancer progression and metastasis. Nat. Cell Biol. 2014, 16, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Lucas, C.; Chandra, A.; Nejentsev, S.; Condliffe, A.M.; Okkenhaug, K. PI3Kδ and primary immunodeficiencies. Nat. Rev. Immunol. 2016, 16, 702–714. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, M.; Messer, K.S.; Ralainirina N Li, H.; Leem, C.J.; Gorjestani, S.; Woo, G.; Nguyen, A.V.; Figueiredo, C.C.; Foubert, P.; et al. PI3Kγ is a molecular switch that controls immune suppression. Nature 2016, 539, 437–442. [Google Scholar] [CrossRef] [PubMed]
- De Henau, O.; Rausch, M.; Winkler, D.; Campesato LF Liu, C.; Cymerman, D.H.; Budhu, S.; Ghosh, A.; Pink, M.; Tchaicha, J.; et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kγ in myeloid cells. Nature 2016, 539, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Ali, K.; Soond, D.R.; Pineiro, R.; Hagemann, T.; Pearce, W.; Lim, E.L.; Bouabe, H.; Scudamore, C.L.; Hancox, T.; Maecker, H.; et al. Inactivation of PI(3)K p110delta breaks regulatory T-cell-mediated immune tolerance to cancer. Nature 2014, 510, 407–411. [Google Scholar] [PubMed]
- Mosely, S.I.; Prime, J.E.; Sainson, R.C.; Koopmann, J.O.; Wang, D.Y.; Greenawalt, D.M.; Ahdesmaki, M.J.; Leyland, R.; Mullins, S.; Pacelli, L.; et al. Rational Selection of Syngeneic Preclinical Tumor Models for Immunotherapeutic Drug Discovery. Cancer Immunol. Res. 2017, 5, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Liu, L.; Gong, C.Y.; Shi, H.S.; Zeng, Y.H.; Wang, X.Z.; Zhao, Y.W.; Wei, Y.Q. Prognostic significance of tumor-associated macrophages in solid tumor: a meta-analysis of the literature. PLoS ONE 2012, 7, e50946. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Garcia, A.; Sanchez-Ruiz, J.; Flores, J.M.; Carrera, A.C. Phosphatidylinositol 3-kinase gamma inhibition ameliorates inflammation and tumor growth in a model of colitis-associated cancer. Gastroenterology 2010, 138, 1374–1383. [Google Scholar] [CrossRef] [PubMed]
- Schmid, M.C.; Avraamides, C.J.; Dippold, H.C.; Franco, I.; Foubert, P.; Ellies, L.G.; Acevedo, L.M.; Manglicmot, J.R.; Song, X.; Wrasidlo, W.; et al. Receptor tyrosine kinases and TLR/IL1Rs unexpectedly activate myeloid cell PI3kgamma, a single convergent point promoting tumor inflammation and progression. Cancer Cell 2011, 19, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Schmid, M.; Franco, I.; Kang, S.W.; Hirsch, E.; Quilliam, L.A.; Varner, J.A. PI3-kinase gamma promotes Rap1a-mediated activation of myeloid cell integrin alpha4beta1, leading to tumor inflammation and growth. PLoS ONE 2013, 8, e60226. [Google Scholar] [CrossRef] [PubMed]
- Khalil, D.; Smith, E.L.; Brentjens, R.J.; Wolchok, J.D. The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat. Rev. Clin. Oncol. 2016, 13, 273–290. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.; Stephens, L.R. PI3Kgamma is a key regulator of inflammatory responses and cardiovascular homeostasis. Science 2007, 318, 64–66. [Google Scholar] [CrossRef] [PubMed]
- Coffelt, S.B.; Kersten, K.; Doornebal, C.W.; Weiden, J.; Vrijland, K.; Hau, C.S.; Verstegen, N.J.; Ciampricotti, M.; Hawinkels, L.J.; Jonkers, J.; et al. IL-17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature 2015, 522, 345–348. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, A.; Brooks, M.W.; Houshyar, S.; Reinhardt, F.; Ardolino, M.; Fessler, E.; Chen, M.B.; Krall, J.A.; DeCock, J.; Zervantonakis, I.K.; et al. Neutrophils Suppress Intraluminal NK Cell-Mediated Tumor Cell Clearance and Enhance Extravasation of Disseminated Carcinoma Cells. Cancer Discov. 2016, 6, 630–649. [Google Scholar] [CrossRef] [PubMed]
- Okkenhaug, K.; Burger, J.A. PI3K signaling in normal B cells and chronic lymphocytic leukemia (CLL). Curr. Top Microbiol. Immunol. 2016, 393, 123–142. [Google Scholar] [PubMed]
- Chitu, V.; Stanley, E.R. Colony-stimulating factor-1 in immunity and inflammation. Curr. Opin. Immunol. 2006, 18, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Hayashi, S.; Kunisada, T.; Ogawa, M.; Nishikawa, S.; Okamura, H.; Sudo, T.; Shultz, L.D.; Nishikawa, S. The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature 1990, 345, 442–444. [Google Scholar] [CrossRef] [PubMed]
- Wiktor-Jedrzejczak, W.; Bartocci, A.; Ferrante, A.W., Jr.; Ahmed-Ansari, A.; Sell, K.W.; Pollard, J.W.; Stanley, E.R. Total absence of colony-stimulating factor 1 in the macrophage-deficient osteopetrotic (op/op) mouse. Proc. Natl. Acad. Sci. USA 1990, 87, 4828–4832. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.; Joyce, J.A. Molecular Pathways: Deciphering Mechanisms of Resistance to Macrophage-Targeted Therapies. Clin. Cancer Res. 2017, 23, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.; Bowman, R.L.; Akkari, L.; Quick, M.L.; Schuhmacher, A.J.; Huse, J.T.; Holland, E.C.; Sutton, J.C.; Joyce, J.A. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science 2016, 352, aad3018. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.; Zitvogel, L.; Palucka, A.K. Neutralizing tumor-promoting chronic inflammation: A magic bullet? Science 2013, 339, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Zebedin, E.; Simma, O.; Schuster, C.; Putz, E.M.; Fajmann, S.; Warsch, W.; Eckelhart, E.; Stoiber, D.; Weisz, E.; Schmid, J.A.; et al. Leukemic challenge unmasks a requirement for PI3Kdelta in NK cell-mediated tumor surveillance. Blood 2008, 112, 4655–4664. [Google Scholar] [CrossRef] [PubMed]
- Putz, E.; Prchal-Murphy, M.; Simma, O.A.; Forster, F.; Koenig, X.; Stockinger, H.; Piekorz, R.P.; Freissmuth, M.; Müller, M.; Sexl, V.; et al. PI3Kdelta is essential for tumor clearance mediated by cytotoxic T lymphocytes. PLoS ONE 2012, 7, e40852. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.; Katanaev, V.L.; Garlanda, C.; Azzolino, O.; Pirola, L.; Silengo, L.; Sozzani, S.; Mantovani, A.; Altruda, F.; Wymann, M.P. Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science 2000, 287, 1049–1053. [Google Scholar] [CrossRef] [PubMed]
- Spitzer, M.; Carmi, Y.; Reticker-Flynn, N.E.; Kwek, S.S.; Madhireddy, D.; Martins, M.M.; Gherardini, P.F.; Prestwood, T.R.; Chabon, J.; Bendall, S.C.; et al. Systemic Immunity Is Required for Effective Cancer Immunotherapy. Cell 2017, 168, 487–502. [Google Scholar] [CrossRef] [PubMed]
- Soler, A.; Angulo-Urarte, A.; Graupera, M. PI3K at the crossroads of tumor angiogenesis signaling pathways. Mol. Cell Oncol. 2015, 2, e975624. [Google Scholar] [CrossRef] [PubMed]
- Graupera, M.; Guillermet-Guibert, J.; Foukas, L.C.; Phng, L.K.; Cain, R.J.; Salpekar, A.; Pearce, W.; Meek, S.; Millan, J.; Cutillas, P.R.; et al. Angiogenesis selectively requires the p110alpha isoform of PI3K to control endothelial cell migration. Nature 2008, 453, 662–666. [Google Scholar] [CrossRef] [PubMed]
- Castel, P.; Carmona, F.J.; Grego-Bessa, J.; Berger, M.F.; Viale, A.; Anderson, K.V.; Bague, S.; Scaltriti, M.; Antonescu, C.R.; Baselga, E.; et al. Somatic PIK3CA mutations as a driver of sporadic venous malformations. Sci. Transl. Med. 2016, 8, 332ra342. [Google Scholar] [CrossRef] [PubMed]
- Stanczuk, L.; Martinez-Corral, I.; Ulvmar, M.H.; Zhang, Y.; Lavina, B.; Fruttiger, M.; Adams, R.H.; Saur, D.; Betsholtz, C.; Ortega, S.; et al. cKit lineage hemogenic endothelium-derived cells contribute to mesenteric lymphatic vessels. Cell Rep. 2015, 10, 1708–1721. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, T.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Stockmann, C.; Doedens, A.; Weidemann, A.; Zhang, N.; Takeda, N.; Greenberg, J.I.; Cheresh, D.A.; Johnson, R.S. Deletion of vascular endothelial growth factor in myeloid cells accelerates tumorigenesis. Nature 2008, 456, 814–818. [Google Scholar] [CrossRef] [PubMed]
- Rivera, L.; Meyronet, D.; Hervieu, V.; Frederick, M.J.; Bergsland, E.; Bergers, G. Intratumoral myeloid cells regulate responsiveness and resistance to antiangiogenic therapy. Cell Rep. 2015, 11, 577–591. [Google Scholar] [CrossRef] [PubMed]
- Schmid, M.; Varner, J.A. Myeloid cells in the tumor microenvironment: Modulation of tumor angiogenesis and tumor inflammation. J. Oncol. 2010, 2010, 201026. [Google Scholar] [CrossRef] [PubMed]
- Martin, V.; Guillermet-Guibert, J.; Chicanne, G.; Cabou, C.; Jandrot-Perrus, M.; Plantavid, M.; Vanhaesebroeck, B.; Payrastre, B.; Gratacap, M.P. Deletion of the p110beta isoform of phosphoinositide 3-kinase in platelets reveals its central role in Akt activation and thrombus formation in vitro and in vivo. Blood 2010, 115, 2008–2013. [Google Scholar] [CrossRef] [PubMed]
- Gay, L.; Felding-Habermann, B. Contribution of platelets to tumour metastasis. Nat. Rev. Cancer 2011, 11, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.; Ciraolo, E.; Franco, I.; Ghigo, A.; Martini, M. PI3K in cancer-stroma interactions: bad in seed and ugly in soil. Oncogene 2014, 33, 3083–3090. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, M.; Cappello, P.; Nguyen, A.V.; Ralainirina, N.; Hardamon, C.R.; Foubert, P.; Schmid, M.C.; Sun, P.; Mose, E.; Bouvet, M.; et al. Macrophage PI3Kgamma drives pancreatic ductal adenocarcinoma progression. Cancer Discov. 2016, 6, 870–885. [Google Scholar] [CrossRef] [PubMed]
- Awad, A.; Kandalam, V.; Chakrabarti, S.; Wang, X.; Penninger, J.M.; Davidge, S.T.; Oudit, G.Y.; Kassiri, Z. Tumor necrosis factor induces matrix metalloproteinases in cardiomyocytes and cardiofibroblasts differentially via superoxide production in a PI3Kgamma dependent manner. Am. J. Physiol. Cell Physiol. 2009, 298, C679–C692. [Google Scholar] [CrossRef] [PubMed]
- Trimboli, A.; Cantemir-Stone, C.Z.; Li, F.; Wallace, J.A.; Merchant, A.; Creasap, N.; Thompson, J.C.; Caserta, E.; Wang, H.; Chong, J.L.; et al. Pten in stromal fibroblasts suppresses mammary epithelial tumours. Nature 2009, 461, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Okkenhaug, K.; Graupera, M.; Vanhaesebroeck, B. Targeting PI3K in Cancer: Impact on Tumor Cells, Their Protective Stroma, Angiogenesis, and Immunotherapy. Cancer Discov. 2016, 6, 1090–1105. [Google Scholar] [CrossRef] [PubMed]
- Louie, C.; DiMaio, M.A.; Matsukuma, K.E.; Coutre, S.E.; Berry, G.J.; Longacre, T.A. Idelalisib-associated enterocolitis: Clinicopathologic features and distinction from other enterocolitides. Am. J. Surg. Pathol. 2015, 39, 1653–1660. [Google Scholar] [CrossRef] [PubMed]
- Weidner, A.; Panarelli, N.C.; Geyer, J.T.; Bhavsar, E.B.; Furman, R.R.; Leonard, J.P.; Jessurun, J.; Yantiss, R.K. Idelalisib-associated colitis: Histologic findings in 14 patients. Am. J. Surg. Pathol. 2015, 39, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Coutré, S.E.; Barrientos, J.C.; Brown, J.R.; de Vos, S.; Furman, R.R.; Keating, M.J.; Li, D.; O’Brien, S.M.; Pagel, J.M.; Poleski, M.H.; et al. Management of adverse events associated with idelalisib treatment: Expert panel opinion. Leuk. Lymphoma 2015, 56, 2779–2786. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. FDA Alerts Healthcare Professionals about Clinical Trials with Zydelig (idelalisib) in Combination with other Cancer Medicines. Center for Drug Evaluation and Research. Availiable online: http://www.fda.gov/Drugs/DrugSafety/ucm490618.htm (accessed on 14 March 2016).
- Hackl, H.; Charoentong, P.; Finotello, F.; Trajanoski, Z. Computational genomics tools for dissecting tumour-immune cell interactions. Nat. Rev. Genet. 2016, 17, 441–458. [Google Scholar] [CrossRef] [PubMed]
- Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017, 18, 248–262. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gyori, D.; Chessa, T.; Hawkins, P.T.; Stephens, L.R. Class (I) Phosphoinositide 3-Kinases in the Tumor Microenvironment. Cancers 2017, 9, 24. https://doi.org/10.3390/cancers9030024
Gyori D, Chessa T, Hawkins PT, Stephens LR. Class (I) Phosphoinositide 3-Kinases in the Tumor Microenvironment. Cancers. 2017; 9(3):24. https://doi.org/10.3390/cancers9030024
Chicago/Turabian StyleGyori, David, Tamara Chessa, Phillip T. Hawkins, and Len R. Stephens. 2017. "Class (I) Phosphoinositide 3-Kinases in the Tumor Microenvironment" Cancers 9, no. 3: 24. https://doi.org/10.3390/cancers9030024