
Epigenetic Regulation of the Epithelial to Mesenchymal Transition in Lung Cancer

1
Laboratoire Ecologie et Biologie des Interactions, Equipe SEVE, Université de Poitiers, UMR CNRS 7267, F-86073 Poitiers, France
2
Division of Hematology-Oncology, Medical University of South Carolina, 39 Sabin St., MSC 635, Charleston, SC 29425, USA
*
Author to whom correspondence should be addressed.
Cancers 2017, 9(7), 72; https://doi.org/10.3390/cancers9070072
Submission received: 29 May 2017
/
Revised: 17 June 2017
/
Accepted: 17 June 2017
/
Published: 24 June 2017
(This article belongs to the Special Issue The Epithelial-to-Mesenchymal Transition (EMT) in Cancer)
Abstract
:Lung cancer is the leading cause of cancer deaths worldwide. It is an aggressive and devastating cancer because of metastasis triggered by enhanced migration and invasion, and resistance to cytotoxic chemotherapy. The epithelial to mesenchymal transition (EMT) is a fundamental developmental process that is reactivated in wound healing and a variety of diseases including cancer where it promotes migration/invasion and metastasis, resistance to treatment, and generation and maintenance of cancer stem cells. The induction of EMT is associated with reprogramming of the epigenome. This review focuses on major mechanisms of epigenetic regulation mainly in lung cancer with recent data on EZH2 (enhancer of zeste 2 polycomb repressive complex 2 subunit ), the catalytic subunit of the PRC2 (Polycomb Group PcG), that behaves as an oncogene in lung cancer associated with gene repression, non-coding RNAs and the epitranscriptome.
Keywords:
chromatin modifications; EMT; Epigenetics; Epitranscriptomics; EZH2; non-coding RNAs; NSCLC; SCLC1. Introduction
Lung cancer is the leading cause of cancer deaths worldwide [1]. Non-small cell lung cancer (NSCLC) accounts for about 85% of cases and includes adenocarcinomas, characterized by RAS or EGFR mutations, squamous cell carcinoma with FGFR1 amplification, PTEN or PIK3CA mutations [2,3], and large cell carcinoma, which are highly heterogeneous. The recent WHO classification [4] now distributes large cell carcinoma into either NSCLC or small cell lung cancer (SCLC) depending on its characteristics. SCLC accounts for approximately 15% of lung tumors and is among the most aggressive tumor types because of high proliferation and early metastasis. It is observed almost exclusively in heavy smokers, and is characterized by gene inactivation of p53 (TP53) and retinoblastoma (RB1), along with expression of neuroendocrine markers [5,6,7]. Metastasis is triggered by enhanced cellular migration and invasion, and the epithelial to mesenchymal transition (EMT) described in the following paragraph, believed to play a role in this process.
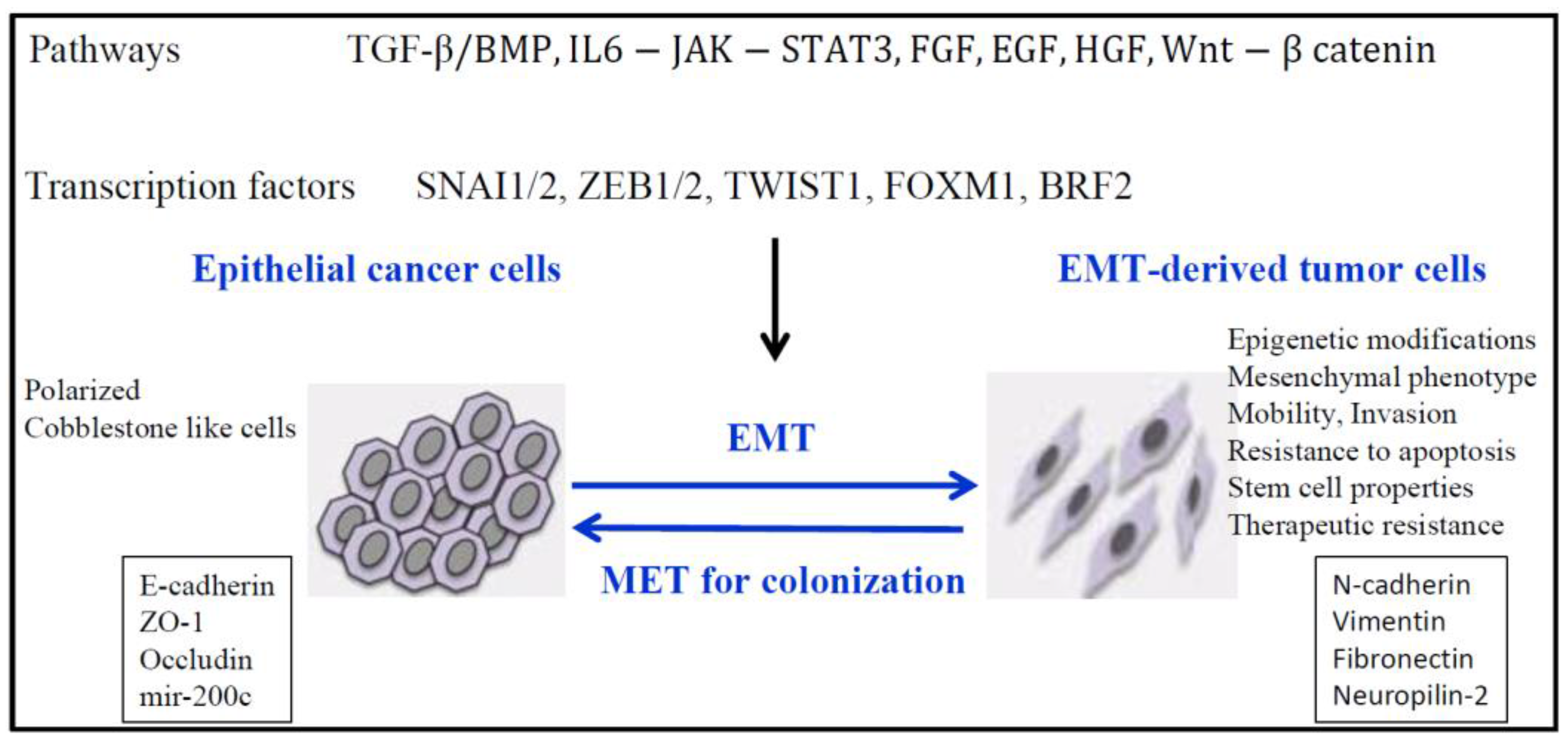
EMT is a fundamental developmental process that is reactivated in wound healing and a variety of diseases including cancer where it induces the development of metastasis, resistance to treatment, and generation and maintenance of cancer stem cells [8,9,10,11,12,13]. However, two recent studies show that EMT can be dispensable for metastasis, but nevertheless contributed substantially to chemoresistance [14,15]. These studies were recently questioned because they coincided with a time when the definition of the EMT was undergoing re-evaluation because of possible multiple partial EMT states, and the proofs that EMT did not occur during metastasis were not completely supported [13] The EMT program includes loss of intercellular adhesion with loss of tight junctions, adherens and gap junctions, increased invasion and migration, and a switch from epithelial to mesenchymal gene expression patterns. E-cadherin and ZO-1 expression is often lost, while in contrast there is a gain of N-cadherin, Vimentin and Fibronectin expression (Figure 1). In TGFβ-induced EMT in lung cancer cells, we found increased expression of Neuropilin-2 (NRP2), the receptor for class 3 semaphorins, ligands providing guidance to and control of cell movement [16]. Most recently, we found that TGFβ preferentially induced NRP2b, an understudied isoform of NRP2 that was responsible for promoting the oncogenic response in cell lines and correlated with tumor progression in patients [17]. In contrast, the level of SEMA3F (semaphorin 3F), a secreted semaphorin with potent antitumor activity that binds NRP2, was decreased in a ZEB1-induced EMT lung cancer model [18]. Of interest, neuropilins bind TGFβ suggesting a function of NRPs in TGFβ response, and additional ligands such as VEGF (Vascular Endothelial Growth Factor), HGF (Hepatocyte Growth Factor), platelet-derived growth factor (PDGF), and EGF (Epidermal Growth Factor) [19].
Many EMT studies have focused mostly on cell-based experimental models and have focused on a small number of gene promoters that are epigenetically regulated, such as E-cadherin (CDH1) [20,21,22,23]. However, the direct involvement of EMT in tumor progression and metastatic spread has been questioned. In fact, the difficulties to characterize EMT in vivo come, in part, from the fact that EMT can be transient and partial. Indeed, EMT is increasingly viewed as a program generating cells with a spectrum of multiple states between epithelial and mesenchymal extremes, and partial EMT would be frequent in tumors [12,13]. Our previous study on 22 NSCLC cell lines showed that they are clearly in different states with more epithelial traits such as NCI-H358 cells or with more mesenchymal traits such as A549, NCI-H661, and NCI-H460 cells [24]. Indeed, a recent integrative approach combining mRNA, miRNA, DNA methylation, and proteomic profiles of 38 cell lines representative of lung adenocarcinoma heterogeneity, and functional profiles consisting of cell invasiveness, adhesion, and motility, defined cell lines as epithelial (E), mesenchymal (M), or intermediate/hybrid (E/M) with mixed epithelial and mesenchymal characteristics. Aggressive hybrid cell lines were characterized with a signature shared with mesenchymal cell lines: upregulation of cytoskeletal and actin-binding proteins [25]. Such E/M hybrid cells have been observed among circulating tumors cells and are associated with metastasis [26].
In lung cancer, TGFβ is one of the most important physiologic EMT inducers, along with other factors such as HGF, FGF (Fibroblast Growth Factor), IGF (Insulin-like Growth factor-1)/PDGF, EGF/VEGF, each secreted by the tumor or/and its microenvironment and acting through downstream pathways including Wnt/β-catenin, TGFβ/SMAD, Notch, MAPK/ERK, and PI3K/Akt [27,28].
The induction of EMT is associated with reprogramming of the epigenome by the action of transcription factors (such as TWIST, SNAIL, SLUG, and ZEB1/2), non-coding RNAs such as micro-RNAs (miRNAs) and long non-coding RNAs (lncRNAs). Here, we focus on epigenetic regulation mainly in lung cancer, including recent data on non-coding RNAs and the epitranscriptome.
2. Epigenetics
Epigenetic regulation is normally dynamic and reversible, affecting chromatin structure for the regulation of gene expression without modification of DNA sequences [29,30]. It includes the incorporation of histone variants, covalent histone modifications, nucleosome re-positioning, DNA methylation, changes in the expression of non-coding RNAs, and RNA post-translational modifications. The impact of epigenetic changes in cancer is reflected by altered gene expression, reactivation of endogenous retro-elements and genomic instability.
The nucleosome is the basic chromatin unit consisting of a protein core formed by two copies of histones H2A, H2B, H3 and H4, encircled by 180–200 bp of DNA stabilized by histone H1 that binds DNA at its entry and exit points of the nucleosome. Histones are post-translationally modified by phosphorylation, acetylation, methylation and ubiquitylation (among others). DNA can be covalently modified by methylation, usually on cytosine residues immediately preceding guanosine (CpG), without altering the sequence of base pairs. The epigenome refers to this collective set of DNA and histone modifications, along with their precise distribution along chromatin and the dynamic changes in this pattern mediated by regulatory processes. DNA methylation at CpG is one of the best studied epigenetic marks. DNA methylation and histone modifications are added by enzymes called “writers” and removed by “erasers”. These modifications are further recognized by proteins called “readers” that recruit various adapters for regulation of gene expression [31] (Table 1).
DNA can be methylated at the fifth position of cytosine (5mC) in the CpG dinucleotide context by DNA methyltransferases and further demethylated by TET enzymes with sequential oxidation of 5mC to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and finally 5-carboxylcytosine (5caC). 5fC and 5caC are further cleaved by thymine-DNA glycosylases to restore unmethylated cytosine via the base-excision repair machinery [33]. In cancer, abnormal DNA methylation is observed with a global decrease of DNA methylation on repetitive sequences, and transposons, associated with chromosome anomalies and active transposition [34,35]. Of interest, gene bodies of tumor suppressor genes are undermethylated but, in contrast, their promoters are hypermethylated. Gene promoter hypermethylation is generally associated with transcriptional repression.
Histones are covalently modified primarily on their amino-terminal tails [36]. The nature and combination of histone modifications forms a “histone code” that affects transcription of nearby genes [37].
Whole genome and transcriptome sequencing established that about 70% of the genome is transcribed into non-coding RNAs. These non-coding RNAs emerged as a novel class of functional molecules with involvement in a variety of physiological processes and in tumorigenesis when misregulated. Non-coding RNAs are divided in two classes depending on their size: the small non-coding RNA group which is less than 200 nucleotides long includes microRNAs (miRNA), while the long non-coding RNA (lncRNA) group extends from 200 nucleotides to over 100 kb. More than 2000 human miRNAs have been identified and a large fraction is deregulated in cancers including lung cancer [38]. miRNAs regulate around 30% of coding genes at the post-transcriptional and translational levels and each typically targets multiple genes within a pathway. LncRNAs belong to a very heterogeneous group of transcripts (for review see [39]). Interestingly, the architecture of lncRNAs is complex: different structural domains can be combined by alternative splicing to sense or bind other RNAs, proteins and possibly DNA [40]. In addition, they undergo conformational switches that modulate their functions. Consequently, lncRNAs are considered as platforms to perform multiple functions in the cell and they act either in cis or in trans. They are involved in chromatin remodeling, transcriptional co-activation and co-repression, protein inhibition, post-transcriptional modifications (splicing), decoy binding platforms, mRNA stabilization, and more recently in nuclear organization of multichromosomal regions [41,42,43,44,45,46,47,48]. Among these functions, lncRNAs bind to chromatin-modifying proteins for their recruitment to specific sites in the genome, and lncRNAs act as competing endogenous RNAs (ceRNAs) for miRNAs. These two functions have been described in the epigenetic regulation of EMT [49,50,51].
To these classical epigenetic modifications, epitranscriptomics and the RNA code came into the spotlight recently [52]. Epigenetic modifications not only affect DNA and proteins, but also coding and non-coding RNAs. RNAs show a diverse spectrum of more than 100 modifications including N7-methylguanosine (m7G), N6-methyladenosine (m6A), m5C, pseudouridine, and queuosine [32,53]. These modifications are reversible, dynamic, and important for gene expression. Enzymes that are writers, erasers, and protein readers of these RNA modifications, analogous to the equivalent functions for histone modification, have been identified [54] (Table 1). However, additional technology development is necessary to detect, quantify, and map these modifications [52,55]. This is a challenging and exciting field still in its early stage, with much to be discovered to understand the role of RNA epigenetic modifications.
3. Epigenetics and Lung Cancer
Mutations in genes encoding epigenetic proteins and abnormalities in epigenetic regulation are clearly linked to cancer (for review see [56]). They have been described previously for lung cancer [34,57,58,59,60].
Large-scale genomic studies have identified recurrent alterations of epigenetic regulators in lung cancer (Table 2).
In SCLCs, mutations are found for histone acetyltransferases, histone methyltransferases and demethylases, and remodeling factors [7,62,66]. High frequency truncating mutations for the H3K4 histone methyltransferase KMT2D/MLL2 gene have been reported in 17% of SCLC cell lines and 8% of SCLC tumors, and KMTD2/MLL2 loss is associated with reduced H3K4me1 and impaired enhancer function [62]. Less frequent are mutations in the H3K27 histone demethylase, KDM6A/UTX gene, that occur in an exclusive fashion with KMTD2/MLL2 mutations. Of interest, bivalent promoters are characterized by the presence of both H3K4me3 active mark and H3K27me3 inactive mark that poise developmental genes, enabling them to respond rapidly to suitable stimuli [67]. Therefore, KMTD2/MLL2 or KDM6A/UTX mutations in addition to abnormal EZH2 expression (see below) lead to impaired expression of genes under control of these bivalent promoters.
For NSCLC, the most frequent mutations are found in the chromatin remodeling factors including SMARCA4/BRG1, and the H3K36 histone methyltransferase SETD2, in 6% and 9% of adenocarcinomas, respectively. BRG1 is one of the two ATPase subunits in the SWI/SNF chromatin-remodeling complex and has been reported to be frequently mutated or silenced in primary human NSCLC tumors and cell lines (for reviews [68,69]). To a lesser extent, the DOT1L methyltransferase for H3K79 is mutated in 3% of lung adenocarcinomas [64].
Abnormal expression of epigenetic players in lung cancer includes EZH2 overexpression in SCLC and NSCLC, where EZH2 acts as an oncogene in these tissues. EZH2 is the catalytic subunit of the PRC2 (Polycomb Group PcG) complex, which includes additional core components (SUZ12, EED, RBBP4). It mediates methylation of lysine 27 on histone H3 (H3K27), associated with gene repression. EZH2 overexpression in lung cancer is common and associated with aggressive tumor characteristics, advanced stage and poor prognosis [70,71,72,73]. In cell lines and patient-derived xenografts, EZH2 inhibition attenuated cell-cycle progression, growth and invasion [72,73,74,75]. Interestingly, tobacco smoke causes upregulation of Wnt signaling by recruiting EZH2 to suppress Dickkopf-1, a Wnt antagonist [76].
At the transcriptional level, EZH2 is suppressed by the pRB protein. At least in part, this explains the upregulation of EZH2 in SCLC, a disease with near-universal mutation of RB [77,78]. Of interest, the retinoblastoma protein occupies repetitive sequences that include tandem sequence repeats and interspersed repeats (endogenous retroviruses and LINE-1 elements) in somatic cells and is associated with EZH2. Consequently, H3K27 trimethylation maintains silencing of these sequences [79]. In cancer cells, these repetitive sequences can be reactivated. When pRB binding to these sequences is inhibited, the consequence is an increased susceptibility for spontaneous lymphoma in mice. Therefore, loss of pRB in SCLC or abnormal level of EZH2 would be involved in repetitive sequence expression responsible for mutagenesis and multiple possible aberrations.
Links between epitranscriptome and cancer have emerged with the discovery that FTO (Fat mass and obesity-associated protein), first identified as the m6A eraser, but recently described as the m6Am (N6, 2′-O-dimethyladenosine) eraser, is abnormally expressed in acute myeloid leukemia [80], and affects the stability and subcellular location of mRNAs [81]. FTO is also overexpressed in breast cancer compared to adjacent breast tissues [82] and overexpression is associated with higher metastatic potential and resistance to chemotherapy in an in vitro cellular breast cancer model [83]. The ncRNA epitranscriptome is also altered in cancer [32] but more data are needed for lung cancer.
4. EMT and Epigenetics in Lung Cancer
A review about epigenetic regulation of EMT in NSCLC was recently published [28]. Additional data will be described below for lung cancer.
The involvement of the Snail and ZEB1 transcription factors strongly supports epigenetic modifications during EMT. Indeed, Snail has been described as a pseudosubstrate “hook” for the histone demethylase LSD1 that demethylates H3K4 [84,85] and H3K9 (Table 1). Through its SNAG domain that mimics a histone H3 tail, Snail binds to LSD1 and the complex is stabilized by association of the co-repressor CoREST. HDAC1/2 and PRC2 are later recruited for CDH1 repression in cancer cells, including mammary epithelial tumor cells. SNAIL was also found to associate with G9a (EHMT2), a major histone methyltransferase responsible for creating the H3K9me2 repressive mark [86] and which also contributes to methylation of H3K27 [87] (Table 1). In addition, SNAIL interacts with SUV39H1 (another histone methyltransferase) during EMT induced by TGF-β and mediates silencing of CDH1 by the addition of a third methyl group to H3K9 that confers a more stable and durable repressive state than H3K9me2 [88].
ZEB1 also associates with partners involved in epigenetic regulation. For gene activation, ZEB1 associates with the histone acetyltransferases (HATs) p300, PCAF, and Tip60. In contrast, as a repressor, ZEB1 interacts with CtBP [89] and recruits class I and II histone deacetylases (HDACs) [90]. Of interest, CtBP associates with several other partners including the Polycomb complex PRC2 as described above, G9a (EHMT2), the co-repressor CoREST, and the histone demethylase LSD1 (for review see [91,92]). ZEB1 can also recruit the nicotinamide adenine dinucleotide-dependent sirtuin, SIRT1, in prostate cancer cells to repress CDH1 and to induce several EMT markers [93]. In addition, ZEB1 interacts via its N-terminal region with BRG1 to repress CDH1 in colon cancer cells [94].
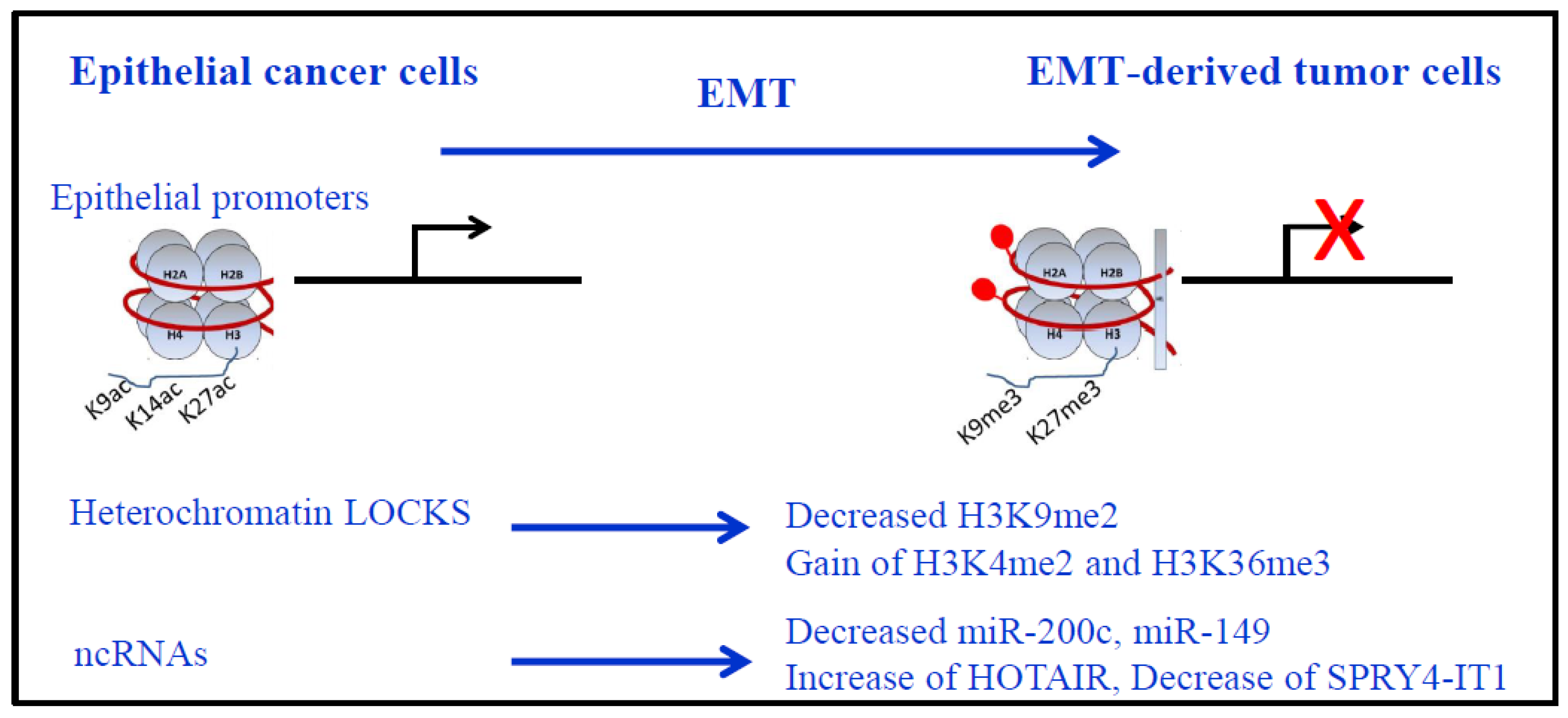
In H358 NSCLC cells where EMT was induced by ZEB1 expression, we used Western blot and immunocytochemistry to identify a global decrease in H3K27 acetylation. We found that ZEB1 binding resulted in decreased acetylation of histone H3 on residues K9 and K27 of target genes [95] (Figure 2). Our results showed that ZEB1 increased trimethylation of H3K27 on selected target genes and that H3K4me2 did not change drastically upon ZEB1 binding. These results suggested that ZEB1 recruitment of PRC2 during EMT would create bivalent domains for epithelial gene repression. Such genes would then be poised for rapid reversal of the repressed state to facilitate the mesenchymal to epithelial transition (MET) thought to be important for the subsequent growth of metastatic deposits. This hypothesis fits with the PRC2 repressive activity on promoters with the activating mark H3K4me2/3 [96].
Recent studies showed that specific “long-range” chromatin domains across the genome, called “LOCKs”, found in non-repetitive heterochromatin domains up to several megabases, are epigenetically remodeled as a major driving force during EMT [97]. A global reduction in the heterochromatin mark H3K9me2, an increase in the euchromatin mark, H3K4me3, and an increase in the transcriptional mark, H3K36me3, were described (Figure 2). These changes depended largely on LSD1. Of interest, DNA methylation was preserved across the genome during EMT.
Epigenetic modifications also affect super-enhancers that are localized to unique relatively small subsets of genes that differ between cell states during EMT. They are often found at key oncogenes, such as MYC. Loss of BRD4, a bromodomain protein that binds acetylated histones, or its pharmacological inhibition, can cause super enhancer-mediated gene expression to be lost [98]. In SCLCs, MYCL, one of the three MYC family oncogenes, is often overexpressed and treatment with JQ1, a BET bromodomain inhibitor, considerably decreased cell growth, induced cell cycle arrest and apoptosis, and reduced expression of the three MYC genes [99].
Non-coding RNAs contribute to EMT in NSCLC. For example, micro-RNAs including miR-132 and miR-149 that target ZEB2 and FOXM1 (Forkhead box M1), respectively, and downregulation of miR-149 was inversely correlated with invasive and EMT phenotypes in NSCLC [100]. The miR-200 family (that includes miR-200a, miR-200b, miR-200c, miR-141, and miR-429) received a lot of interest because of a negative transcriptional feedback loop between miR-200c and ZEB1 [101,102]. In our ZEB1-induced EMT model in the H358 NSCLC cell line, we observed a decrease of miR-200c during EMT [95]. Moreover, in the HCC4006 NSCLC cell line that became resistant to EGFR inhibitors, acquired resistance was associated with ZEB1, an EMT phenotype, and decreased miR-200c levels [103].
Several long non-coding RNAs (lncRNAs) are also associated with an EMT signature and aggressiveness [51,60]. In NSCLC, a positive correlation with lymph node metastasis was described for HOTAIR, CARLO-5, PVT1, MVIH and ZXF1, while a negative correlation was found with MEG3, SPRY4-ITI, BANCR and GAS6-AS1 [60]. Among these lncRNAs, HOTAIR and SPRY4-ITI are of interest in EMT regulation (Figure 2). HOTAIR interacts with the PRC2 complex to induce in trans H3K27 trimethylation of the HOXD locus, and the LSD1/coREST/REST complex that catalyzes H3K4me2 demethylation. However, this model of PRC2 binding to HOTAIR for targeting specific sequences was recently challenged, since PRC2 interacts with strong affinity but weak specificity to a wide set of all RNAs. Indeed, PRC2 is dispensable for HOTAIR-mediated transcriptional repression while PRC2 recruitment and H3K27 trimethylation were proposed to occur due to HOTAIR-dependent silencing [104,105]. How this happens mechanistically will require further investigation. HOTAIR is also involved in the maintenance of stemness and EMT in colon and breast cancer cell lines [106]. SPRY4-ITI is an inhibitor of the MAPK signaling pathway, and tumor suppressive functions were described in NSCLC cell lines. SPRY4-ITI inhibits NSCLC cell migration/invasion, and suppresses metastasis. SPRY4-ITI is downregulated by EZH2 and is involved in the modulation of EMT through induction of E-cadherin expression and repression of vimentin [107].
MALAT-1 (metastasis associated lung adenocarcinoma transcript-1, also called NEAT2) is a predictive marker for metastasis and shorter survival in early stage lung adenocarcinoma [51,60]. It can act as a ceRNA for several miRNAs including miR-200c, and can recruit PRC2 subunits (EZH2 and Suz12) to the CDH1 promoter. CCAT2 (colon cancer associated transcript 2) is also a predictive marker for metastasis in lung cancer. In addition, lncRNAs are associated with EMT, stem cell properties and drug resistance. For example, UCA1, BC087858 and GAS5 are associated with resistance to EGFR tyrosine kinase inhibitors (EGFR-TKIs) in lung cancer possibly through the Akt signaling pathway.
Lastly, the epitranscriptome is likely involved in EMT but to our knowledge data are missing in lung cancer. However, we suspect that modifications of lncRNAs MALAT1 and TUG1 are candidates. Indeed, for MALAT1, two major m6A sites were found at consensus sites in two hairpin stem structures in several human cell lines including, MDA-MB231 breast cancer cells [108]. The consequence of this modification would destabilize these secondary structures and would modify interactions with RNA binding proteins. One can speculate that FTO overexpression would alter m6A levels during EMT. The other lncRNA TUG1 function might be altered as well, as one site shows m6A modification [108].
5. Therapeutic Inhibition of EMT
From a therapeutic standpoint, epigenetic processes are targets for therapeutic exploitation [11,22,31], and epigenetic regulators implicated in cancer and their corresponding inhibitors are described in recent reviews [56,109]. Some of them would be appropriate to target EMT in lung cancer. EZH2 inhibitors would be good candidates because of EZH2 oncogenic status in lung cancer, and some are in clinical development [110]. We also suspect that bromodomain inhibitors such as JQ1 (known as TEN-010 in clinical trials), the BET inhibitor for BRD4, would be appropriate because JQ1 demonstrated efficacy in blocking tumor progression in several cancer models including SCLC lung cancer [98,99]. One possible mechanism would be a decrease of C-Myc recruitment to EZH2 and consequently reduced EZH2 expression, as shown in bladder cancer [111]. Combined treatments with histone deacetylase inhibitors (such as SAHA, i.e., vorinostat, an FDA-approved drug) and bromodomain inhibitors should be considered in NSCLC for their synergistic therapeutic benefit seen in animal models [112,113]. HDAC inhibitors were also shown to induce E-cadherin in lung cancer and this restoration increased sensitivity to EGFR inhibitors [114,115]. Although the mode of action of the two drugs is multifaceted, one reason for this benefit would be their immunostimulatory effects on tumor growth arrest and prolonged survival as shown in a mouse model for lung adenocarcinoma [113].
6. Conclusions
In this review, epigenetics regulation of EMT was described in lung cancer with a particular interest in EZH2, an oncogene in lung tumors. Recent data on non-coding RNAs and epitranscriptomics were introduced. Attempts to inhibit EMT should consider combined treatments with HDAC and bromodomain inhibitors. However, caution is needed with these treatments to address the mesenchymal to epithelial transition (MET) that occurs at sites of metastasis.
Acknowledgments
Joëlle Roche was supported by Région Poitou-Charentes, France; Robert M. Gemmill and Harry A. Drabkin by DOD LC150622.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| ADC | Adenocarcinoma |
| ceRNA | competing endogenous RNA |
| EGF | Epidermal Growth Factor |
| EGFR | Epidermal Growth Factor Recptor |
| EMT | Epithelial to Mesenchymal transition |
| EZH2 | enhancer of zeste 2 polycomb repressive complex 2 subunit |
| HDAC | Histone deacetylase |
| HGF | Hepatocyte Growth Factor |
| lncRNA | long non-coding RNA |
| MET | Mesenchymal to Epithelial Transition |
| NSCLC | noRsmall cell lung cancer |
| PRC2 | polycomb repressive complex 2 |
| SC | squamous cell carcinoma |
| SCLC | small cell lung cancer |
| VEGF | Vascular Endothelial Growth Factor |
References
- Torre, L.A.; Siegel, R.L.; Jemal, A. Lung cancer statistics. Adv. Exp. Med. Biol. 2016, 893, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Heist, R.S.; Engelman, J.A. SnapShot: Non-small cell lung cancer. Cancer Cell 2012, 21, 448.e2. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Travis, W.D.; Brambilla, E.; Nicholson, A.G.; Yatabe, Y.; Austin, J.H.; Beasley, M.B.; Chirieac, L.R.; Dacic, S.; Duhig, E.; Flieder, D.B.; et al. The 2015 world health organization classification of lung tumors: Impact of genetic, clinical and radiologic advances since the 2004 classification. J. Thorac. Oncol. 2015, 10, 1243–1260. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Nau, M.M.; Chiba, I.; Birrer, M.J.; Rosenberg, R.K.; Vinocour, M.; Levitt, M.; Pass, H.; Gazdar, A.F.; Minna, J.D. p53: A frequent target for genetic abnormalities in lung cancer. Science 1989, 246, 491–494. [Google Scholar] [CrossRef] [PubMed]
- Wistuba, II; Gazdar, A.F.; Minna, J.D. Molecular genetics of small cell lung carcinoma. Semin. Oncol. 2001, 28, 3–13. [Google Scholar] [CrossRef]
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretic, L.; Kong, G.; Leenders, F.; Lu, X.; Fernandez-Cuesta, L.; Bosco, G.; et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015, 524, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, F.; Stassi, G.; De Maria, R. Epithelial-mesenchymal transition: A new target in anticancer drug discovery. Nat. Rev. Drug Discov. 2016, 15, 311–325. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging biological principles of metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Nasarre, P.; Gemmill, R.M.; Potiron, V.A.; Roche, J.; Lu, X.; Baron, A.E.; Korch, C.; Garrett-Mayer, E.; Lagana, A.; Howe, P.H.; et al. Neuropilin-2 is upregulated in lung cancer cells during TGF-beta1-induced epithelial-mesenchymal transition. Cancer Res. 2013, 73, 7111–7121. [Google Scholar] [CrossRef] [PubMed]
- Gemmill, R.M.; Nasarre, P.; Nair-Menon, J.; Cappuzzo, F.; Landi, L.; D’Incecco, A.; Uramoto, H.; Yoshida, T.; Haura, E.B.; Armeson, K.; et al. The neuropilin 2 isoform NRP2b uniquely supports TGFbeta-mediated progression in lung cancer. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Clarhaut, J.; Gemmill, R.M.; Potiron, V.A.; Ait-Si-Ali, S.; Imbert, J.; Drabkin, H.A.; Roche, J. ZEB-1, a repressor of the semaphorin 3F tumor suppressor gene in lung cancer cells. Neoplasia 2009, 11, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Nasarre, P.; Gemmill, R.M.; Drabkin, H.A. The emerging role of class-3 semaphorins and their neuropilin receptors in oncology. Onco. Targets Ther. 2014, 7, 1663–1687. [Google Scholar] [CrossRef] [PubMed]
- Tam, W.L.; Weinberg, R.A. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat. Med. 2013, 19, 1438–1449. [Google Scholar] [CrossRef] [PubMed]
- Cieslik, M.; Hoang, S.A.; Baranova, N.; Chodaparambil, S.; Kumar, M.; Allison, D.F.; Xu, X.; Wamsley, J.J.; Gray, L.; Jones, D.R.; et al. Epigenetic coordination of signaling pathways during the epithelial-mesenchymal transition. Epigenet. Chromatin 2013, 6, 28. [Google Scholar] [CrossRef] [PubMed]
- Mishra, V.K.; Johnsen, S.A. Targeted therapy of epigenomic regulatory mechanisms controlling the epithelial to mesenchymal transition during tumor progression. Cell Tissue Res. 2014, 356, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Kiesslich, T.; Pichler, M.; Neureiter, D. Epigenetic control of epithelial-mesenchymal-transition in human cancer. Mol. Clin. Oncol. 2013, 1, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Gemmill, R.M.; Roche, J.; Potiron, V.A.; Nasarre, P.; Mitas, M.; Coldren, C.D.; Helfrich, B.A.; Garrett-Mayer, E.; Bunn, P.A.; Drabkin, H.A. ZEB1-responsive genes in non-small cell lung cancer. Cancer Lett. 2011, 300, 66–78. [Google Scholar] [CrossRef] [PubMed]
- Schliekelman, M.J.; Taguchi, A.; Zhu, J.; Dai, X.; Rodriguez, J.; Celiktas, M.; Zhang, Q.; Chin, A.; Wong, C.H.; Wang, H.; et al. Molecular portraits of epithelial, mesenchymal, and hybrid States in lung adenocarcinoma and their relevance to survival. Cancer Res. 2015, 75, 1789–1800. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Boareto, M.; Huang, B.; Jia, D.; Lu, M.; Ben-Jacob, E.; Onuchic, J.N.; Levine, H. Implications of the Hybrid Epithelial/Mesenchymal Phenotype in Metastasis. Front. Oncol. 2015, 5, 155. [Google Scholar] [CrossRef] [PubMed]
- Dong, N.; Shi, L.; Wang, D.C.; Chen, C.; Wang, X. Role of epigenetics in lung cancer heterogeneity and clinical implication. Semin. Cell Dev. Biol. 2017, 64, 18–25. [Google Scholar] [CrossRef] [PubMed]
- O'Leary, K.; Shia, A.; Schmid, P. Epigenetic Regulation of EMT in non-small cell lung cancer. Curr. Cancer Drug Targets 2017. [Google Scholar] [CrossRef]
- Kouzarides, T. SnapShot: Histone-modifying enzymes. Cell 2007, 128, 802. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Arrowsmith, C.H.; Bountra, C.; Fish, P.V.; Lee, K.; Schapira, M. Epigenetic protein families: A new frontier for drug discovery. Nat. Rev. Drug Discov. 2012, 11, 384–400. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M.; Pandolfi, P.P. The Epitranscriptome of noncoding RNAs in cancer. Cancer Discov. 2017, 7, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Chen, T. Tet family of 5-methylcytosine dioxygenases in mammalian development. J. Hum. Genet. 2013, 58, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Dobersch, S.; Romero-Olmedo, A.J.; Barreto, G. Epigenetics in lung cancer diagnosis and therapy. Cancer Metastasis Rev. 2015, 34, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Heyn, H.; Esteller, M. DNA methylation profiling in the clinic: Applications and challenges. Nat. Rev. Genet. 2012, 13, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Inamura, K. Diagnostic and therapeutic potential of microRNAs in lung cancer. Cancers 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.J.; Chang, H.Y. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2016, 17, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.R.; Mattick, J.S. Structure and function of long noncoding RNAs in epigenetic regulation. Nat. Struct. Mol. Biol. 2013, 20, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Cheetham, S.W.; Gruhl, F.; Mattick, J.S.; Dinger, M.E. Long noncoding RNAs and the genetics of cancer. Br. J. Cancer 2013, 108, 2419–2425. [Google Scholar] [CrossRef] [PubMed]
- Morriss, G.R.; Cooper, T.A. Protein sequestration as a normal function of long noncoding RNAs and a pathogenic mechanism of RNAs containing nucleotide repeat expansions. Hum. Genet. 2017. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Zhou, C.; Yang, J.; Li, J.; Gong, Z. Long and short noncoding RNAs in lung cancer precision medicine: Opportunities and challenges. Tumour Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, G.; Paliogiannis, P.; Sini, M.C.; Manca, A.; Palomba, G.; Doneddu, V.; Tanda, F.; Pascale, M.R.; Cossu, A. Long non-coding RNA CASC2 in human cancer. Crit. Rev. Oncol. Hematol. 2017, 111, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Yan, Y.; Qian, L.; Gong, Z. Long non-coding RNAs act as regulators of cell autophagy in diseases (Review). Oncol. Rep. 2017, 37, 1359–1366. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.W.; Huang, K.; Yang, C.; Kang, C.S. Non-coding RNAs as regulators in epigenetics (Review). Oncol. Rep. 2017, 37, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Engreitz, J.M.; Ollikainen, N.; Guttman, M. Long non-coding RNAs: Spatial amplifiers that control nuclear structure and gene expression. Nat. Rev. Mol. Cell Biol. 2016, 17, 756–770. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.M.; Zhou, G.B. Long Non-coding RNAs and their roles in non-small-cell lung cancer. Genom. Proteom. Bioinform. 2016, 14, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Khalil, A.M.; Guttman, M.; Huarte, M.; Garber, M.; Raj, A.; Rivea Morales, D.; Thomas, K.; Presser, A.; Bernstein, B.E.; van Oudenaarden, A.; et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11667–11672. [Google Scholar] [CrossRef] [PubMed]
- Hendrickson, D.G.; Kelley, D.R.; Tenen, D.; Bernstein, B.; Rinn, J.L. Widespread RNA binding by chromatin-associated proteins. Genome Biol. 2016, 17, 28. [Google Scholar] [CrossRef] [PubMed]
- Heery, R.; Finn, S.P.; Cuffe, S.; Gray, S.G. Long non-coding RNAs: Key regulators of epithelial-mesenchymal transition, tumour drug resistance and cancer stem cells. Cancers 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Chi, K.R. The RNA code comes into focus. Nature 2017, 542, 503–506. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, W.V.; Bell, T.A.; Schaening, C. Messenger RNA modifications: Form, distribution, and function. Science 2016, 352, 1408–1412. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.G.; et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887. [Google Scholar] [CrossRef] [PubMed]
- Helm, M.; Motorin, Y. Detecting RNA modifications in the epitranscriptome: Predict and validate. Nat. Rev. Genet. 2017, 18, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Pfister, S.X.; Ashworth, A. Marked for death: Targeting epigenetic changes in cancer. Nat. Rev. Drug. Discov. 2017, 16, 241–263. [Google Scholar] [CrossRef] [PubMed]
- Balgkouranidou, I.; Liloglou, T.; Lianidou, E.S. Lung cancer epigenetics: Emerging biomarkers. Biomark Med. 2013, 7, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Liloglou, T.; Bediaga, N.G.; Brown, B.R.; Field, J.K.; Davies, M.P. Epigenetic biomarkers in lung cancer. Cancer Lett. 2014, 342, 200–212. [Google Scholar] [CrossRef] [PubMed]
- Van Den Broeck, A.; Ozenne, P.; Eymin, B.; Gazzeri, S. Lung cancer: A modified epigenome. Cell Adh. Migr. 2010, 4, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Roth, A.; Diederichs, S. Long Noncoding RNAs in Lung Cancer. Curr. Top. Microbiol. Immunol. 2016, 394, 57–110. [Google Scholar] [CrossRef] [PubMed]
- Simo-Riudalbas, L.; Perez-Salvia, M.; Setien, F.; Villanueva, A.; Moutinho, C.; Martinez-Cardus, A.; Moran, S.; Berdasco, M.; Gomez, A.; Vidal, E.; et al. KAT6B is a tumor suppressor histone H3 lysine 23 acetyltransferase undergoing genomic loss in small cell lung cancer. Cancer Res. 2015, 75, 3936–3945. [Google Scholar] [CrossRef] [PubMed]
- Augert, A.; Zhang, Q.; Bates, B.; Cui, M.; Wang, X.; Wildey, G.; Dowlati, A.; MacPherson, D. Small cell lung cancer exhibits frequent inactivating mutations in the histone methyltransferase KMT2D/MLL2: CALGB 151111 (Alliance). J. Thorac. Oncol. 2017, 12, 704–713. [Google Scholar] [CrossRef] [PubMed]
- Van Haaften, G.; Dalgliesh, G.L.; Davies, H.; Chen, L.; Bignell, G.; Greenman, C.; Edkins, S.; Hardy, C.; O’Meara, S.; Teague, J.; et al. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat. Genet. 2009, 41, 521–523. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.D.; Alexandrov, A.; Kim, J.; Wala, J.; Berger, A.H.; Pedamallu, C.S.; Shukla, S.A.; Guo, G.; Brooks, A.N.; Murray, B.A.; et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 2016, 48, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Imielinski, M.; Berger, A.H.; Hammerman, P.S.; Hernandez, B.; Pugh, T.J.; Hodis, E.; Cho, J.; Suh, J.; Capelletti, M.; Sivachenko, A.; et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012, 150, 1107–1120. [Google Scholar] [CrossRef] [PubMed]
- Gardner, E.E.; Poirier, J.T.; Rudin, C.M. Histone code aberrancies in small cell lung cancer. J. Thorac. Oncol. 2017, 12, 599–601. [Google Scholar] [CrossRef] [PubMed]
- Voigt, P.; Tee, W.W.; Reinberg, D. A double take on bivalent promoters. Genes Dev. 2013, 27, 1318–1338. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.G.; Roberts, C.W. SWI/SNF nucleosome remodellers and cancer. Nat. Rev. Cancer 2011, 11, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, D.C.; Crabtree, G.R. ATP-dependent chromatin remodeling: Genetics, genomics and mechanisms. Cell Res. 2011, 21, 396–420. [Google Scholar] [CrossRef] [PubMed]
- Behrens, C.; Solis, L.M.; Lin, H.; Yuan, P.; Tang, X.; Kadara, H.; Riquelme, E.; Galindo, H.; Moran, C.A.; Kalhor, N.; et al. EZH2 protein expression associates with the early pathogenesis, tumor progression, and prognosis of non-small cell lung carcinoma. Clin. Cancer Res. 2013, 19, 6556–6565. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, J.; Kinoshita, I.; Shimizu, Y.; Kikuchi, E.; Konishi, J.; Oizumi, S.; Kaga, K.; Matsuno, Y.; Nishimura, M.; Dosaka-Akita, H. Distinctive expression of the polycomb group proteins Bmi1 polycomb ring finger oncogene and enhancer of zeste homolog 2 in nonsmall cell lung cancers and their clinical and clinicopathologic significance. Cancer 2010, 116, 3015–3024. [Google Scholar] [CrossRef] [PubMed]
- Takawa, M.; Masuda, K.; Kunizaki, M.; Daigo, Y.; Takagi, K.; Iwai, Y.; Cho, H.S.; Toyokawa, G.; Yamane, Y.; Maejima, K.; et al. Validation of the histone methyltransferase EZH2 as a therapeutic target for various types of human cancer and as a prognostic marker. Cancer Sci. 2011, 102, 1298–1305. [Google Scholar] [CrossRef] [PubMed]
- Huqun, Ishikawa, R.; Zhang, J.; Miyazawa, H.; Shimizu, Y.; Hagiwara, K.; Koyama, N. Enhancer of zeste homolog 2 is a novel prognostic biomarker in nonsmall cell lung cancer. Cancer 2012, 118, 1599–1606. [Google Scholar] [CrossRef]
- Cao, W.; Ribeiro Rde, O.; Liu, D.; Saintigny, P.; Xia, R.; Xue, Y.; Lin, R.; Mao, L.; Ren, H. EZH2 promotes malignant behaviors via cell cycle dysregulation and its mRNA level associates with prognosis of patient with non-small cell lung cancer. PLoS ONE 2012, 7, e52984. [Google Scholar] [CrossRef] [PubMed]
- Poirier, J.T.; Gardner, E.E.; Connis, N.; Moreira, A.L.; de Stanchina, E.; Hann, C.L.; Rudin, C.M. DNA methylation in small cell lung cancer defines distinct disease subtypes and correlates with high expression of EZH2. Oncogene 2015, 34, 5869–5878. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Rao, M.; Humphries, A.E.; Hong, J.A.; Liu, F.; Yang, M.; Caragacianu, D.; Schrump, D.S. Tobacco smoke induces polycomb-mediated repression of Dickkopf-1 in lung cancer cells. Cancer Res. 2009, 69, 3570–3578. [Google Scholar] [CrossRef] [PubMed]
- Coe, B.P.; Thu, K.L.; Aviel-Ronen, S.; Vucic, E.A.; Gazdar, A.F.; Lam, S.; Tsao, M.S.; Lam, W.L. Genomic deregulation of the E2F/Rb pathway leads to activation of the oncogene EZH2 in small cell lung cancer. PLoS ONE 2013, 8, e71670. [Google Scholar] [CrossRef]
- Bracken, A.P.; Pasini, D.; Capra, M.; Prosperini, E.; Colli, E.; Helin, K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003, 22, 5323–5335. [Google Scholar] [CrossRef] [PubMed]
- Ishak, C.A.; Marshall, A.E.; Passos, D.T.; White, C.R.; Kim, S.J.; Cecchini, M.J.; Ferwati, S.; MacDonald, W.A.; Howlett, C.J.; Welch, I.D.; et al. An RB-EZH2 complex mediates silencing of repetitive DNA sequences. Mol. Cell 2016, 64, 1074–1087. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Weng, H.; Su, R.; Weng, X.; Zuo, Z.; Li, C.; Huang, H.; Nachtergaele, S.; Dong, L.; Hu, C.; et al. FTO plays an oncogenic role in acute myeloid leukemia as a N6-methyladenosine RNA demethylase. Cancer Cell 2017, 31, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Mauer, J.; Luo, X.; Blanjoie, A.; Jiao, X.; Grozhik, A.V.; Patil, D.P.; Linder, B.; Pickering, B.F.; Vasseur, J.J.; Chen, Q.; et al. Reversible methylation of m6Am in the 5′ cap controls mRNA stability. Nature 2017, 541, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.; Dang, Y.; Chen, G.; Mo, Z. Overexpression of the fat mass and obesity associated gene (FTO) in breast cancer and its clinical implications. Int. J. Clin. Exp. Pathol. 2015, 8, 13405–13410. [Google Scholar] [PubMed]
- Singh, B.; Kinne, H.E.; Milligan, R.D.; Washburn, L.J.L.; Olsen, M.; Lucci, A. Important role of FTO in the survival of rare panresistant triple-negative inflammatory breast cancer cells facing a severe metabolic challenge. PLoS ONE 2016, 11, e0159072. [Google Scholar] [CrossRef]
- Christofori, G. Snail1 links transcriptional control with epigenetic regulation. EMBO J. 2010, 29, 1787–1789. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Wu, Y.; Li, J.; Dong, C.; Ye, X.; Chi, Y.I.; Evers, B.M.; Zhou, B.P. The SNAG domain of Snail1 functions as a molecular hook for recruiting lysine-specific demethylase 1. EMBO J. 2010, 29, 1803–1816. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Wu, Y.; Yao, J.; Wang, Y.; Yu, Y.; Rychahou, P.G.; Evers, B.M.; Zhou, B.P. G9a interacts with Snail and is critical for Snail-mediated E-cadherin repression in human breast cancer. J. Clin. Investig. 2012, 122, 1469–1486. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Chen, X.; Xiong, J.; Li, Y.; Li, H.; Ding, X.; Liu, S.; Chen, S.; Gao, S.; Zhu, B. Histone methyltransferase G9a contributes to H3K27 methylation in vivo. Cell Res. 2011, 21, 365–367. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Wu, Y.; Wang, Y.; Wang, C.; Kang, T.; Rychahou, P.G.; Chi, Y.I.; Evers, B.M.; Zhou, B.P. Interaction with Suv39H1 is critical for Snail-mediated E-cadherin repression in breast cancer. Oncogene 2013, 32, 1351–1362. [Google Scholar] [CrossRef] [PubMed]
- Postigo, A.A.; Dean, D.C. ZEB represses transcription through interaction with the corepressor CtBP. Proc. Natl. Acad. Sci. USA 1999, 96, 6683–6688. [Google Scholar] [CrossRef] [PubMed]
- Aghdassi, A.; Sendler, M.; Guenther, A.; Mayerle, J.; Behn, C.O.; Heidecke, C.D.; Friess, H.; Buchler, M.; Evert, M.; Lerch, M.M.; et al. Recruitment of histone deacetylases HDAC1 and HDAC2 by the transcriptional repressor ZEB1 downregulates E-cadherin expression in pancreatic cancer. Gut 2012, 61, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Chinnadurai, G. CtBP, an unconventional transcriptional corepressor in development and oncogenesis. Mol. Cell 2002, 9, 213–224. [Google Scholar] [CrossRef]
- Shi, Y.; Sawada, J.; Sui, G.; Affar el, B.; Whetstine, J.R.; Lan, F.; Ogawa, H.; Luke, M.P.; Nakatani, Y. Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature 2003, 422, 735–738. [Google Scholar] [CrossRef] [PubMed]
- Byles, V.; Zhu, L.; Lovaas, J.D.; Chmilewski, L.K.; Wang, J.; Faller, D.V.; Dai, Y. SIRT1 induces EMT by cooperating with EMT transcription factors and enhances prostate cancer cell migration and metastasis. Oncogene 2012, 31, 4619–4629. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Tillo, E.; Lazaro, A.; Torrent, R.; Cuatrecasas, M.; Vaquero, E.C.; Castells, A.; Engel, P.; Postigo, A. ZEB1 represses E-cadherin and induces an EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG1. Oncogene 2010, 29, 3490–3500. [Google Scholar] [CrossRef] [PubMed]
- Roche, J.; Nasarre, P.; Gemmill, R.; Baldys, A.; Pontis, J.; Korch, C.; Guilhot, J.; Ait-Si-Ali, S.; Drabkin, H. Global decrease of histone H3K27 acetylation in ZEB1-induced epithelial to mesenchymal transition in lung cancer cells. Cancers 2013, 5, 334–356. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, U.; Nalapareddy, K.; Saxena, M.; O'Neill, N.K.; Pinello, L.; Yuan, G.C.; Orkin, S.H.; Shivdasani, R.A. Acquired tissue-specific promoter bivalency is a basis for prc2 necessity in adult cells. Cell 2016, 165, 1389–1400. [Google Scholar] [CrossRef] [PubMed]
- McDonald, O.G.; Wu, H.; Timp, W.; Doi, A.; Feinberg, A.P. Genome-scale epigenetic reprogramming during epithelial-to-mesenchymal transition. Nat. Struct. Mol. Biol. 2011, 18, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Loven, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [PubMed]
- Kato, F.; Fiorentino, F.P.; Alibes, A.; Perucho, M.; Sanchez-Cespedes, M.; Kohno, T.; Yokota, J. MYCL is a target of a BET bromodomain inhibitor, JQ1, on growth suppression efficacy in small cell lung cancer cells. Oncotarget 2016, 7, 77378–77388. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.; Zhao, W.; Xiong, J.; Cao, R. miR-149 Inhibits non-small-cell lung cancer cells EMT by targeting FOXM1. Biochem. Res. Int. 2013, 2013, 506731. [Google Scholar] [CrossRef] [PubMed]
- De Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Burk, U.; Schubert, J.; Wellner, U.; Schmalhofer, O.; Vincan, E.; Spaderna, S.; Brabletz, T. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008, 9, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Song, L.; Bai, Y.; Kinose, F.; Li, J.; Ohaegbulam, K.C.; Munoz-Antonia, T.; Qu, X.; Eschrich, S.; Uramoto, H.; et al. ZEB1 mediates acquired resistance to the epidermal growth factor receptor-tyrosine kinase inhibitors in non-small cell lung cancer. PLoS ONE 2016, 11, e0147344. [Google Scholar] [CrossRef] [PubMed]
- Portoso, M.; Ragazzini, R.; Brencic, Z.; Moiani, A.; Michaud, A.; Vassilev, I.; Wassef, M.; Servant, N.; Sargueil, B.; Margueron, R. PRC2 is dispensable for HOTAIR-mediated transcriptional repression. EMBO J. 2017, 36, 981–994. [Google Scholar] [CrossRef] [PubMed]
- Blanco, M.R.; Guttman, M. Re-evaluating the foundations of lncRNA-Polycomb function. EMBO J. 2017, 36, 964–966. [Google Scholar] [CrossRef] [PubMed]
- Padua Alves, C.; Fonseca, A.S.; Muys, B.R.; de Barros, E.L.B.R.; Burger, M.C.; de Souza, J.E.; Valente, V.; Zago, M.A.; Silva, W.A., Jr. Brief report: The lincRNA hotair is required for epithelial-to-mesenchymal transition and stemness maintenance of cancer cell lines. Stem Cells 2013, 31, 2827–2832. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Liu, X.H.; Lu, K.H.; Nie, F.Q.; Xia, R.; Kong, R.; Yang, J.S.; Xu, T.P.; Liu, Y.W.; Zou, Y.F.; et al. EZH2-mediated epigenetic suppression of long noncoding RNA SPRY4-IT1 promotes NSCLC cell proliferation and metastasis by affecting the epithelial-mesenchymal transition. Cell Death Dis. 2014, 5, e1298. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Parisien, M.; Dai, Q.; Zheng, G.; He, C.; Pan, T. Probing N6-methyladenosine RNA modification status at single nucleotide resolution in mRNA and long noncoding RNA. RNA 2013, 19, 1848–1856. [Google Scholar] [CrossRef] [PubMed]
- Shortt, J.; Ott, C.J.; Johnstone, R.W.; Bradner, J.E. A chemical probe toolbox for dissecting the cancer epigenome. Nat. Rev. Cancer 2017, 17, 160–183. [Google Scholar] [CrossRef] [PubMed]
- Comet, I.; Riising, E.M.; Leblanc, B.; Helin, K. Maintaining cell identity: PRC2-mediated regulation of transcription and cancer. Nat. Rev. Cancer 2016, 16, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Liu, D.; Tao, D.; Xiang, W.; Xiao, X.; Wang, M.; Wang, L.; Luo, G.; Li, Y.; Zeng, F.; et al. BRD4 regulates EZH2 transcription through upregulation of C-MYC and represents a novel therapeutic target in bladder cancer. Mol. Cancer Ther. 2016, 15, 1029–1042. [Google Scholar] [CrossRef] [PubMed]
- Mazur, P.K.; Herner, A.; Mello, S.S.; Wirth, M.; Hausmann, S.; Sanchez-Rivera, F.J.; Lofgren, S.M.; Kuschma, T.; Hahn, S.A.; Vangala, D.; et al. Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nat. Med. 2015, 21, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Adeegbe, D.; Liu, Y.; Lizotte, P.H.; Kamihara, Y.; Aref, A.R.; Almonte, C.; Dries, R.; Li, Y.; Liu, S.; Wang, X.; et al. Synergistic immunostimulatory effects and therapeutic benefit of combined histone deacetylase and bromodomain inhibition in non-small cell lung cancer. Cancer Discov. 2017. [Google Scholar] [CrossRef] [PubMed]
- Witta, S.E.; Gemmill, R.M.; Hirsch, F.R.; Coldren, C.D.; Hedman, K.; Ravdel, L.; Helfrich, B.; Dziadziuszko, R.; Chan, D.C.; Sugita, M.; et al. Restoring E-cadherin expression increases sensitivity to epidermal growth factor receptor inhibitors in lung cancer cell lines. Cancer Res. 2006, 66, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Kakihana, M.; Ohira, T.; Chan, D.; Webster, R.B.; Kato, H.; Drabkin, H.A.; Gemmill, R.M. Induction of E-cadherin in lung cancer and interaction with growth suppression by histone deacetylase inhibition. J. Thorac. Oncol. 2009, 4, 1455–1465. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
EMT (Epithelial to Mesenchymal transition ) and epigenetic modifications. EMT is induced by different pathways that involve different transcription factors necessary to repress epithelial genes and to activate mesenchymal genes.
Figure 1.
EMT (Epithelial to Mesenchymal transition ) and epigenetic modifications. EMT is induced by different pathways that involve different transcription factors necessary to repress epithelial genes and to activate mesenchymal genes.
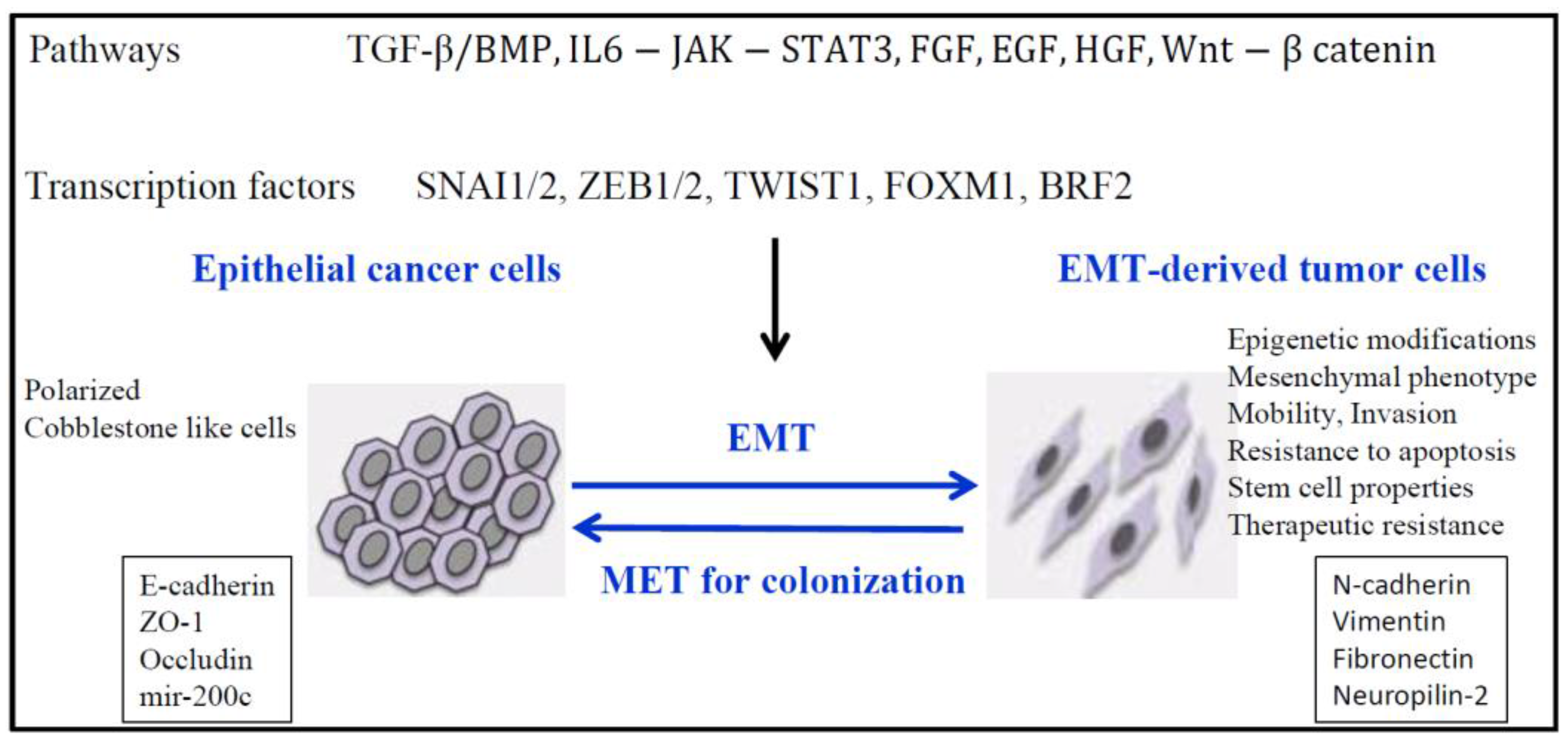
Figure 2.
Epigenetic modifications during EMT. Nucleosomes are represented with DNA in red, wrapped around the histone core (2 copies of each histone H2A, H2B, H3 and H4) with the presence of histone H1 in more compact chromatin. Repression of epithelial genes (red cross) is shown with corresponding epigenetic modifications on their promoters: more compact chromatin, decreased H3K9/14/27ac and gain of H3K9me3 and H3K27me3, associated to DNA methylation (red lollipop). Of note, only major modifications for histone H3 are presented. LOCKS (long-range chromatin domains) epigenetic marks during EMT: Global reduction in the heterochromatin mark H3K9me2, increase in the euchromatin mark H3K4me3, and increase in the transcriptional mark H3K36me3. Non-coding RNAs (ncRNAs) expressions are also modified during EMT for miRNAs (loss of miR-200c and miR-149) and lncRNAs (increased HOTAIR and decreased SPRYA-IT1 in lung cancer).
Figure 2.
Epigenetic modifications during EMT. Nucleosomes are represented with DNA in red, wrapped around the histone core (2 copies of each histone H2A, H2B, H3 and H4) with the presence of histone H1 in more compact chromatin. Repression of epithelial genes (red cross) is shown with corresponding epigenetic modifications on their promoters: more compact chromatin, decreased H3K9/14/27ac and gain of H3K9me3 and H3K27me3, associated to DNA methylation (red lollipop). Of note, only major modifications for histone H3 are presented. LOCKS (long-range chromatin domains) epigenetic marks during EMT: Global reduction in the heterochromatin mark H3K9me2, increase in the euchromatin mark H3K4me3, and increase in the transcriptional mark H3K36me3. Non-coding RNAs (ncRNAs) expressions are also modified during EMT for miRNAs (loss of miR-200c and miR-149) and lncRNAs (increased HOTAIR and decreased SPRYA-IT1 in lung cancer).

{kind=link}
{kind=link}
Table 1.
Epigenetic modifications. Only DNA and RNA methylation, histone acetylation and some epigenetic modifications for histone H3 are shown with the corresponding players (writers, erasers and readers). For H3K79 methylation, DOT1L functions are multiple: it is involved in telomeric silencing, cellular development, cell-cycle checkpoint, DNA repair, and regulation of transcription. For RNA, only m5C and m6A are described but other modifications can be found in the review by Esteller and Pandolfi (2017) [32]. ** G9a weakly methylates H3K27.
Table 1.
Epigenetic modifications. Only DNA and RNA methylation, histone acetylation and some epigenetic modifications for histone H3 are shown with the corresponding players (writers, erasers and readers). For H3K79 methylation, DOT1L functions are multiple: it is involved in telomeric silencing, cellular development, cell-cycle checkpoint, DNA repair, and regulation of transcription. For RNA, only m5C and m6A are described but other modifications can be found in the review by Esteller and Pandolfi (2017) [32]. ** G9a weakly methylates H3K27.
| Epigenetic Modification | Function | Writer | Eraser | Reader | ||
|---|---|---|---|---|---|---|
| DNA | CpG Methylation | Transcriptional repression | DNMT1/3A/3B | TET1/2/3 | MeCP, MBD1-4, UHRF1 | |
| RNA | m5C | tRNA stabilization, translation, immune response | DNMT2 (=TRDMT1) NSUN family | TET | not identified | |
| m6A | RNA splicing, export, stability, immune tolerance | METTL3/4, WTAP | FTO, ALKBH5 | YTHD family, HuR HNRNPA2B1 | ||
| Histones | Lysine Acetylation | Transcriptional activation | HAT | HDAC1-11, SIRT1-7 | BRD bromodomain | |
| Lysine Methylation | HH3K4 | Transcriptional activation | MLL1-5, SET1A/B, SET7/9, ASH1L | LSD1, JARID1a/b | Chromodomain, Tudor, MBT repeat, PHD finger | |
| HH3K9 | Transcriptional repression | G9a(EHMT2) SUV39H1/2 | LSD1, GASC1 | |||
| HH3K27 | Transcriptional repression | EZH1/2, G9a ** | UTX, JMJD3 | |||
| HH3K36 | Transcriptional activation | SETD2, ASH1L, ASF1A, NSD1-3, SMYD2 | Rph1/KDM4 Jhdm1b/Kdm2b | |||
| HH3K79 | Transcriptional regulation | DOT1L, RE-IIBP | not known | |||
Table 2.
Mutations in epigenetic regulators in lung cancers. ADC: Adenocarcinoma; SC: Squamous cell carcinoma.
Table 2.
Mutations in epigenetic regulators in lung cancers. ADC: Adenocarcinoma; SC: Squamous cell carcinoma.
| Lung Cancer | Gene | Function | Mutation | References |
|---|---|---|---|---|
| SCLC | KAT3A/CREBBP | histone acetytransferase | inactivating mutation | [7] |
| KAT3B/EP300 | histone acetytransferase | inactivating mutation | [7] | |
| KAT6B | H3K23 histone acetytransferase | genomic loss | [61] | |
| KMT2D/MLL2 | H3K4me1/2 histone methyltransferase | frequent inactivation | [7,62] | |
| KDM6A/UTX | H3K27 histone demethylase | truncating mutation in a small number of SCLC patients | [62,63] | |
| PBRM1 | chromatin remodeling factor | mutation | [62] | |
| ARID1A | mutation | [62] | ||
| ARID1B | mutation | [62] | ||
| NSCLC | KMT2D/MLL2 | H3K4me1/2 histone methyltransferase | mutation in 20% SC | [3] |
| SETD2 | H3K36 histone methyltransferase | 9% ADC | [3] | |
| DOT1L | H3K79 histone methyltransferase | 3% ADC | [64] | |
| ARID1A | chromatin remodeling factor | 7% ADC | [3] | |
| ARID1B | 6% ADC | |||
| ARID2 | 7% ADC | |||
| SMARCA4/BRG1 | 6% ADC | |||
| BRD3 | Bromodomain, binds hyperacetylated chromatin | [65] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Roche, J.; Gemmill, R.M.; Drabkin, H.A. Epigenetic Regulation of the Epithelial to Mesenchymal Transition in Lung Cancer. Cancers 2017, 9, 72. https://doi.org/10.3390/cancers9070072
AMA Style
Roche J, Gemmill RM, Drabkin HA. Epigenetic Regulation of the Epithelial to Mesenchymal Transition in Lung Cancer. Cancers. 2017; 9(7):72. https://doi.org/10.3390/cancers9070072
Chicago/Turabian StyleRoche, Joëlle, Robert M. Gemmill, and Harry A. Drabkin. 2017. "Epigenetic Regulation of the Epithelial to Mesenchymal Transition in Lung Cancer" Cancers 9, no. 7: 72. https://doi.org/10.3390/cancers9070072
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.